Introduction
Ketamine, a non-competitive N-methyl-D-aspartic acid
receptor antagonist, was discovered in 1961 and used as a clinical
anesthetic (1). Ketamine-induced
urinary tract syndrome has received increasing attention due to the
rising illicit use of ketamine as a recreational drug among young
adults. Previous studies investigated the ketamine-induced
urological sequelae (2,3), and it was demonstrated that long-term
ketamine abuse may cause lower urinary tract syndromes (LUTS) that
resemble interstitial cystitis (4). LUTS are associated with decreased
bladder capacity, incontinence, hematuria and suprapubic pain
resulting from neurological disorders (4). A number of possible factors
responsible for the association between ketamine and LUTS have been
proposed on the basis of clinical observations (5), including direct toxic damage on the
urothelium causing bladder barrier dysfunction, chronic neurogenic
inflammation and immunoglobulin E-mediated hypersensitivity.
However, the causative mechanism underlying the association between
ketamine abuse and ketamine-associated cystitis (KC) remains
unclear.
An increasing number of studies investigating the
causes of KC have used in vivo rodent models. Accumulating
evidence demonstrated that certain specific molecular factors,
including cyclooxygenase-2 and inducible nitric oxide synthase
(6,7) may interact with the submucosa
inflammatory environment caused by ketamine abuse in clinical
cases. In addition, oxidative stress mediated by mitochondria- and
endoplasmic reticulum-dependent pathways was identified to be
involved in the apoptosis of bladder cells and urothelial defects
(8). A previous study demonstrated
that long-term treatment with ketamine may lead to submucosa
fibrosis, which is associated with epithelial-mesenchymal
transition (EMT) mediated by transforming growth factor-β1 (TGF-β1)
(9). LUTS-associated voiding
dysfunctions were identified to be associated with an increased
protein expression level of P2X purinergic receptor 1 (P2RX1)
(10), and the downregulation of
phosphorylated (p-) transgelin (TAGLN) was identified to be
associated with the detrusor overactivity occurring in KC (11).
Due to the unclear etiology and the unavailability
of clinical treatments for KC, a genomic approach, using microarray
analyses to identify the molecular factors underlying KC,
represents an effective strategy for the development of adequate
therapies. Our previous studies based on microarray analysis
examined a Balb/c mouse model (12,13).
Administration of low-dosage and short-term ketamine (30 mg/kg/day
for 8 weeks) induced a significant decrease in keratin 14
expression in urothelial tissue, suggesting that ketamine was able
to damage the urothelial cell structure (12). Furthermore, using high-dosage,
long-term ketamine injections (100 mg/kg/day for 20 weeks), the
expression levels of numerous genes involved in the chronic wound
healing response were altered, suggesting the presence of an
initial stage of fibrosis from ketamine abuse (13). However, no obvious inflammatory and
histological anomalies were identified in the bladders of control
mice and mice treated with ketamine. The C57BL/6 mouse strain was
demonstrated to exhibit increased Type 1 helper T cell-dominant
responses compared with Balb/c mice, which presents the Type 2
helper T cell phenotype (14). Due
to different inflammatory responses between these two strains,
C57BL/6 mice were used in the present study to investigate the
effect of ketamine on inflammation. The present study aimed to
identify potential disease-associated genes that may help in
characterizing the pathogenesis of KC.
Materials and methods
Animals and drug treatment
A total of 40 6-week-old female C57BL/6 mice
(weighing 16.9±0.95 g) were purchased from The National Laboratory
Animal Center (Taipei, Taiwan). The animals were maintained in an
animal care facility in The Biotechnology and Health Hall of
National Chiayi University (Chiayi, Taiwan) for 1 week. A total of
20 7-week-old mice received a daily 100 mg/kg intraperitoneal
injection of ketamine (Imalgene 1000; Merial; Boehringer Ingelheim
International GmbH, Ingelheim am Rhein, Germany) for 20 consecutive
weeks to model the effects of ketamine abuse. A control group
consisting of 20 mice was injected with saline solution. The mice
were housed in polycarbonate cages, provided with food and water
ad libitum, and maintained in a 12-h light-dark cycle at
22±2°C with 60±5% humidity. The health conditions of the mice were
monitored daily for 20 weeks by observing specific behaviors,
including squeals, decreased locomotor activity and abnormal
posture. The mice were weighed weekly. Each mouse was placed in a
transparent polycarbonate cage and sacrificed by gradual-fill
CO2 exposure with a displacement rate of 20% chamber
volume/min. When impaired spontaneous breathing and unconsciousness
were observed, the CO2 flow was maintained for >1
min. Cessation of cardiac activity was confirmed via palpation. All
animal experiments were approved by The Institutional Animal Care
and Use Committee of National Chiayi University (approval no.
104047) and according to the guidelines of The Animal Research:
Reporting in vivo Experiments, recommended by The National Centre
for the Replacement, Refinement and Reduction of Animals in
Research (15).
Mouse voiding behavior analysis
At the end of the 20th week (n=16/group; 4/group
mice were excluded due a lack of voiding), urine-voiding
characteristics were investigated using a modified voided stain on
paper (VSOP) method (16). A total
of 20 µl/g distilled water was supplied to each mouse. Following 30
min, a mouse was placed in a standard polycarbonate mouse cage with
filter paper (185 mm; Advantec Toyo Kaisha, Ltd., Tokyo, Japan)
under the bottom grid. The voiding area and the number of urine
spots >0.5 cm in diameter were recorded for 2 h. The urine
stains on the filter papers were directly scanned into image files
using a scanner, and the stained areas were calculated using Image
J (version 1.47; National Institutes of Health, Bethesda, MD, USA)
software.
Total RNA extraction
The mouse urinary bladders in each group (n=5/group)
were incubated overnight in RNAlater Stabilization Solution
(Sigma-Aldrich; Merck KGaA, Darmstadt, Germany) and subsequently
homogenized in TRIzol® reagent (Invitrogen; Thermo
Fisher Scientific, Inc., Waltham, MA, USA), according to the
manufacturer's protocol. RNA concentration and purity were measured
using the NanoDrop 1,000 spectrophotometer (Thermo Fisher
Scientific, Inc., Wilmington, DE, USA), and purity was checked
using ratios of absorbance values at 260/280 and 260/230 nm. The
quality of RNA was assessed using the Bioanalyzer 2,100 (Agilent
Technologies, Inc., Santa Clara, CA, USA), and three high-quality
samples from each group were analyzed using an oligonucleotide DNA
microarray.
Scanning electron microscopy (SEM) and
histological examination
A total of seven bladders from sacrificed mice from
each group (n=7/group) were inflated in situ with Bouin's
fixative, as described in a previous study (17). Following removal, the bladders were
incubated in Bouin's fixative at room temperature for 2 h. Each
bladder was subsequently cut longitudinally into strips, rinsed
with PBS, and processed by dehydration in an ascending series of
ethanol solutions, followed by overnight immersion in 100% acetone
at 4°C. Subsequently, the samples were critical point dried using
liquid CO2 (HCP-2 critical point dryer; Hitachi, Ltd.,
Tokyo, Japan). The dried samples were subsequently mounted on an
aluminum stub, sputter-coated with gold, and imaged using a S-3500N
scanning electron microscope (magnification, ×500 or ×900)
(Hitachi, Ltd.). A portion of mouse bladders (n=8/group) was fixed
in 10% phosphate-buffered formalin for 24 h at room temperature and
subsequently embedded in paraffin and cut into 3–4 µm sections for
subsequent histopathological observation. Hematoxylin and eosin
(HE), and Masson's trichrome stainings were performed at room
temperature to examine the morphology of the tissues by light
microscope (magnification, ×200 or ×400).
Analysis of mouse oligonucleotide DNA
microarray
The Mouse Whole Genome OneArray® (version
2; Phalanx Biotech Group, Hsinchu, Taiwan) contained 27,307 DNA
oligonucleotide probes. Each probe consisted of 60 nucleotides and
was designed in the sense direction. Among these probes, 26,423
corresponded to the genes annotated in the RefSeq v51 (https://www.ncbi.nlm.nih.gov/assembly/GCF_000001635.20/)
and Ensembl v65 (https://www.ncbi.nlm.nih.gov/assembly/GCF_000001635.18/)
databases. A total of 884 control probes were additionally
included. Fluorescent antisense RNA (aRNA) targets were prepared
from 1 µg total RNA samples using the OneArray® Amino
Allyl aRNA Amplification kit (Phalanx Biotech Group) and Cy5 dye
(GE Healthcare, Chicago, IL, USA). The amplification was performed
as follows. Firstly, RNA samples were mixed with first strand cDNA
synthesized (FSS) in a PCR tube, followed by incubating for 10 min
at 70°C in a thermal cycler. FSS enzyme was added and incubated for
a further 1 h at 42°C. Next, Second Strand Master Mix was added to
each sample and incubated in the following conditions: 16°C for 15
min, 37°C for 45 min, 65°C for 15 min and 4°C for 5 min. Finally,
In Vitro Transcription Master Mix was added to each sample
and incubated for a further 16 h at 37°C. Fluorescent targets were
hybridized to the Mouse Whole Genome OneArray® with a
hybridization buffer (Phalanx Biotech Group) using a hybridization
system (Phalanx Biotech Group). Following 16 h of hybridization at
50°C, non-specific binding targets were washed away. The slides
were scanned using a DNA microarray scanner (G2505C; Agilent
Technologies, Inc.), and the Cy5 fluorescent intensity of each spot
was analyzed using GenePix software (version 4.1; Molecular
Devices, LLC, Sunnyvale, CA, USA). Each sample was analyzed using
technical and biological duplicates, with a reproducibility
>0.975 (https://www.excelfunctions.net/excel-correl-function.html).
The signal intensity data were analyzed using the Rosetta
Resolver® system (Rosetta Biosoftware, Seattle, WA, USA)
for data preprocessing and 75 percentile centering normalization.
The sample errors were additionally estimated using an
error-weighted method. The fold change and the P-value for pairwise
sample comparison were calculated for evaluation of differentially
expressed genes (DEGs). The DEGs meeting the criteria of
log2|fold change| ≥0.5 and P<0.05 were considered for
further analysis.
Reverse transcription-quantitative
polymerase chain reaction (RT-qPCR) analysis
A total of 2 µg total RNA was used for RT using the
High-Capacity cDNA RT kit (Applied Biosystems; Thermo Fisher
Scientific, Inc.). Each reaction included 20 ng cDNA, forward and
reverse primers at a concentration of 500 nM and 2× Fast SYBR Green
PCR master mix (Applied Biosystems; Thermo Fisher Scientific,
Inc.). A total of 10 µl reaction volume was used for RT-qPCR
analysis with the appropriate primers. Each sample was tested in
triplicate. A Bio-Rad CFX Connect real-time PCR machine (Bio-Rad
Laboratories, Inc., Hercules, CA, USA) and Bio-Rad CFX Manager
software (version 3.0; Bio-Rad Laboratories, Inc.) were used for
the experimental setup and data analysis. The thermocycling
conditions were as follows: 95°C for 5 min, followed by 39 cycles
of 95°C for 15 sec and 60°C for 30 sec, and finally 95°C for 10
sec. The relative quantification was performed using the
2−ΔΔCq method (18).
The RT-qPCR data of target genes were normalized to the reference
gene GAPDH. The gene analyzed with RT-qPCR and the respective
primers were the following: Fibronectin (FN1)-Forward (F),
5′-CCTCGGGAATGGAAAGGGAG-3′ and FN1-Reverse (R),
5′-ACGTGCAGGAGCAAATGGC-3′; versican (VCAN)-F,
5′-AGGAAGAGCAGACTGGAGTTGG-3′ and VCAN-R,
5′-GCTGAGACCCAGGACCATGT-3′; potassium calcium-activated channel
subfamily M α1 (KCNMA1)-F, 5′-CCATCCCGTCCACAGCAAAT-3′ and KCNMA1-R,
5′-AGGCATTATCCGGCTCATCTG-3′; potassium calcium-activated channel
subfamily M regulatory β subunit 4 (KCNMB4)-F,
5′-CTTCACCTGTGGCACCGACT-3′ and KCNMB4-R,
5′-AGGGCGGGATATAGGAGCAC-3′; ryanodine receptor 1 (RYR1)-F,
5′-TCCGCAAGCAGTATGAGGACC-3′ and RYR1-R,
5′-AGTTCAGGATCATCTGTCCCCAG-3′; protein kinase C β (PRKCB)-F,
5′-TGCCCACCGATAGAGGTACA-3′ and PRKCB-R,
5′-TGACCAGGAACATCAGCATCT-3′; GAPDH-F, 5′-AAGGTCGGTGTGAACGGATT-3′
and GAPDH-R, 5′-GTGAGTGGAGTCATACTGGAACAT-3′.
Gene pathway analysis
To investigate the biological function of the genes
differentially expressed between the control mice and the mice
treated with ketamine, the Kyoto Encyclopedia of Genes and Genomes
(KEGG) database and NABA database (19) were used to analyze the DEGs and
identify the enriched pathways. The 3,000 most significant DEGs
were analyzed. Gene set enrichment analysis (GSEA; Molecular
Signatures Database v6.2; http://software.broadinstitute.org/gsea/msigdb/collections.jsp)
was used to perform data enrichment analysis based on the Canonical
Pathway database. The pathways meeting the criterion of P<0.0005
were considered for further analysis. Pathview (v1.4.2; http://www.bioconductor.org/packages/release/bioc/html/pathview.html),
KEGGREST (v1.4.1; http://www.bioconductor.org/packages/release/bioc/html/KEGGREST.html)
and KEGGgraph (v1.22.1; http://www.bioconductor.org/packages/release/bioc/html/KEGGgraph.html)
R packages (v3.1.1; http://cran.r-project.org/bin/windows/base/old/3.1.1/NEWS.R-3.1.1.html)
were used for pathway visualization.
Statistical analysis
SigmaPlot (version 12.5; Systat Software, Inc., San
Jose, CA, USA) was used for the statistical analyses. Data are
presented as the mean ± standard deviation or as the mean ±
standard error. The RT-qPCR results were calculated from
experiments repeated three times. Statistical differences were
assessed using Student's t-test. P<0.05 was considered to
indicate a statistically significant difference.
Results
Alteration of body weight in mice
All mice (n=40) survived the 20-week treatments.
Abnormal behaviors and body weight loss were not identified.
However, the rate of weight gain in mice treated with ketamine
significantly decreased from the first week, compared with control
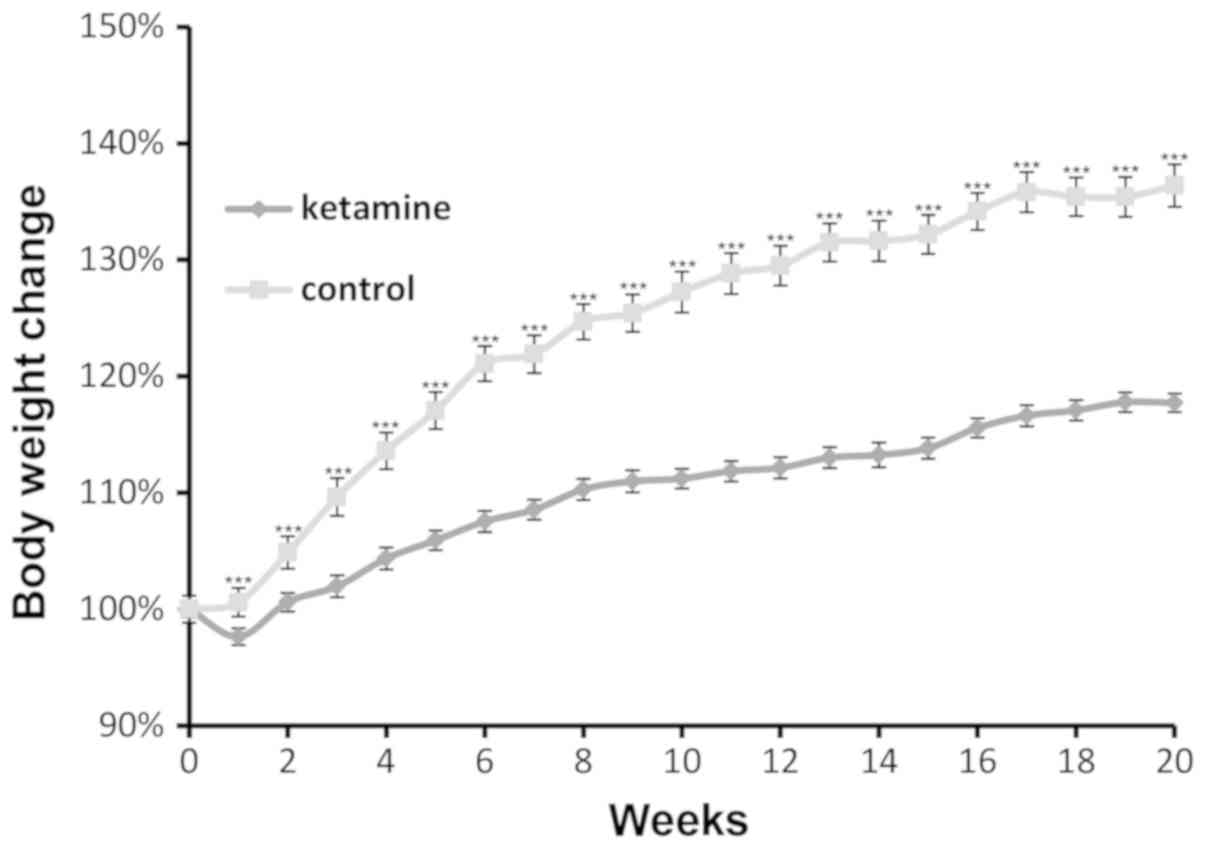
mice, and this trend continued for the remaining 19 weeks (Fig. 1). The weight of mice belonging to
the control group increased by an average of 6.41 g, whereas, the
weight of the mice treated with ketamine increased by an average of
3.05 g. The phenotype was consistent with the weight loss observed
in cases of ketamine abuse in humans (20) and in previous animal studies
(10,13), and may be caused by reduced food
consumption due to the side effects of ketamine, including
dizziness, loss of appetite or nausea (21).
Effect of treatment with ketamine on
voiding behavior of mice
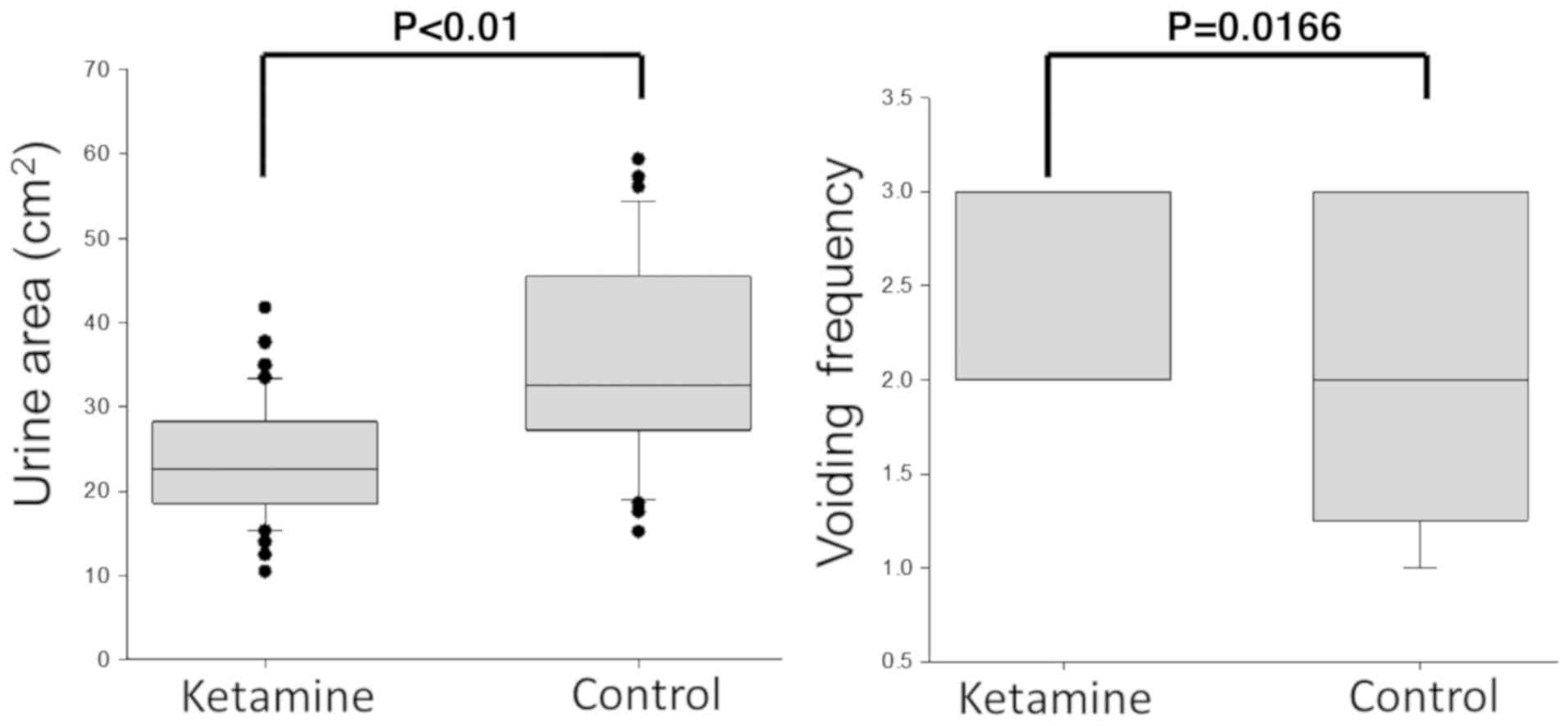
To evaluate the effect of ketamine on bladder
voiding function, the voiding area and urine spot numbers were
recorded for 2 h using a modified VSOP method at the end of the
20th week. The spot areas of mice treated with ketamine were
significantly decreased compared with control mice, whereas the
spot numbers representing voiding frequency were increased compared
with the controls (Fig. 2), thus
suggesting that altered voiding function was induced by long-term
treatment with ketamine.
Bladder histological examination
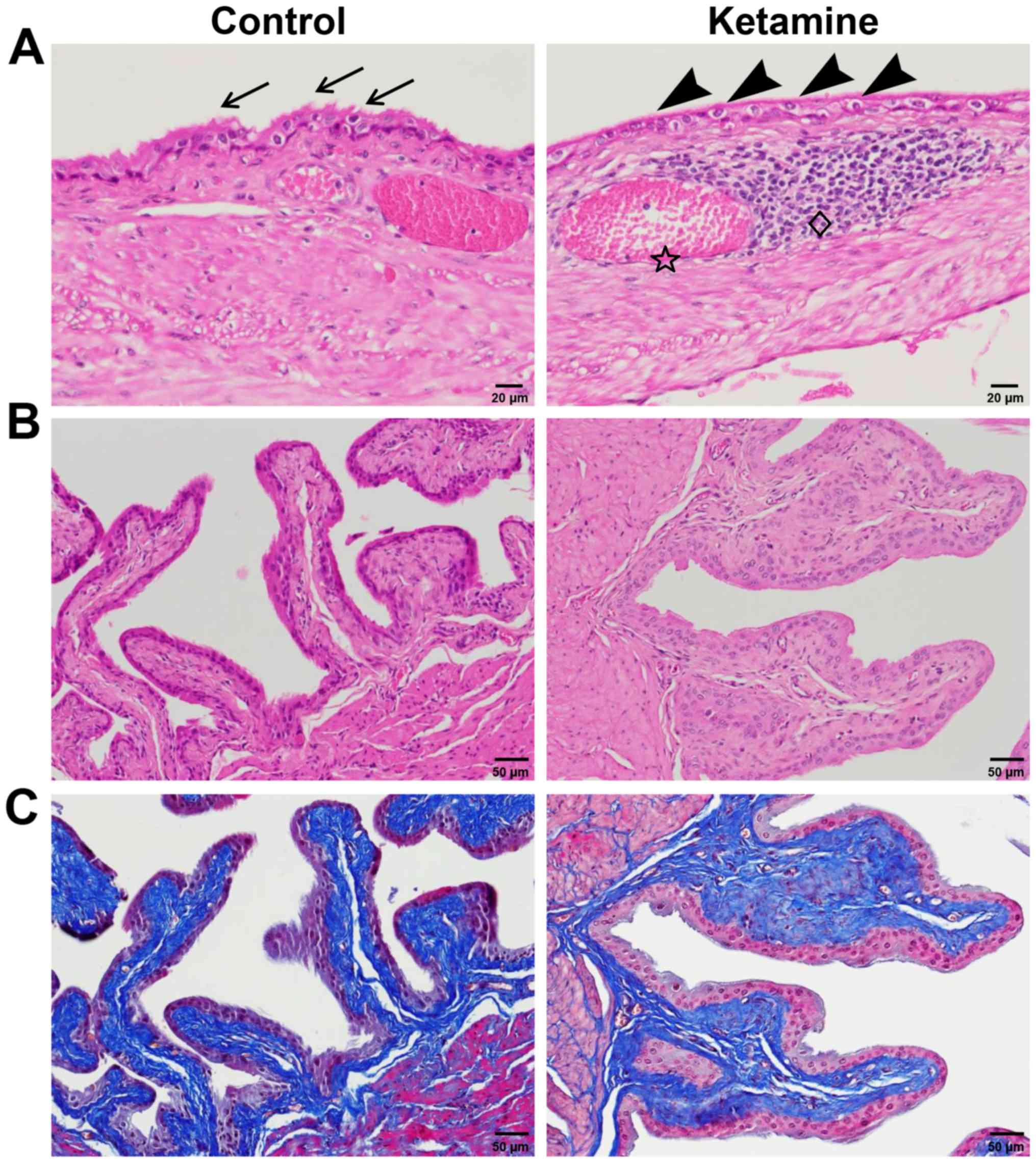
The urinary bladders of mice treated with ketamine
exhibited significant histopathological alterations compared with
the control group. At the end of the 20th week, mice treated with
ketamine exhibited a smooth apical epithelial surface and
subepithelial vascular congestion. In contrast, the apical side of
the epithelium in the control group exhibited a rough surface
(Fig. 3A). In addition, the mice
treated with ketamine exhibited protrusive and enlarged mucosal
folds resembling fibrous expansion of connective tissue stroma
(Fig. 3B and C). However, signs of
lymphoplasmacytic aggregation were detected in two bladder samples
(Fig. 3A), suggesting that
treatment with ketamine was not sufficient to induce inflammation
in the majority of C57BL/6 mice.
SEM examination of the mice bladder
epithelium
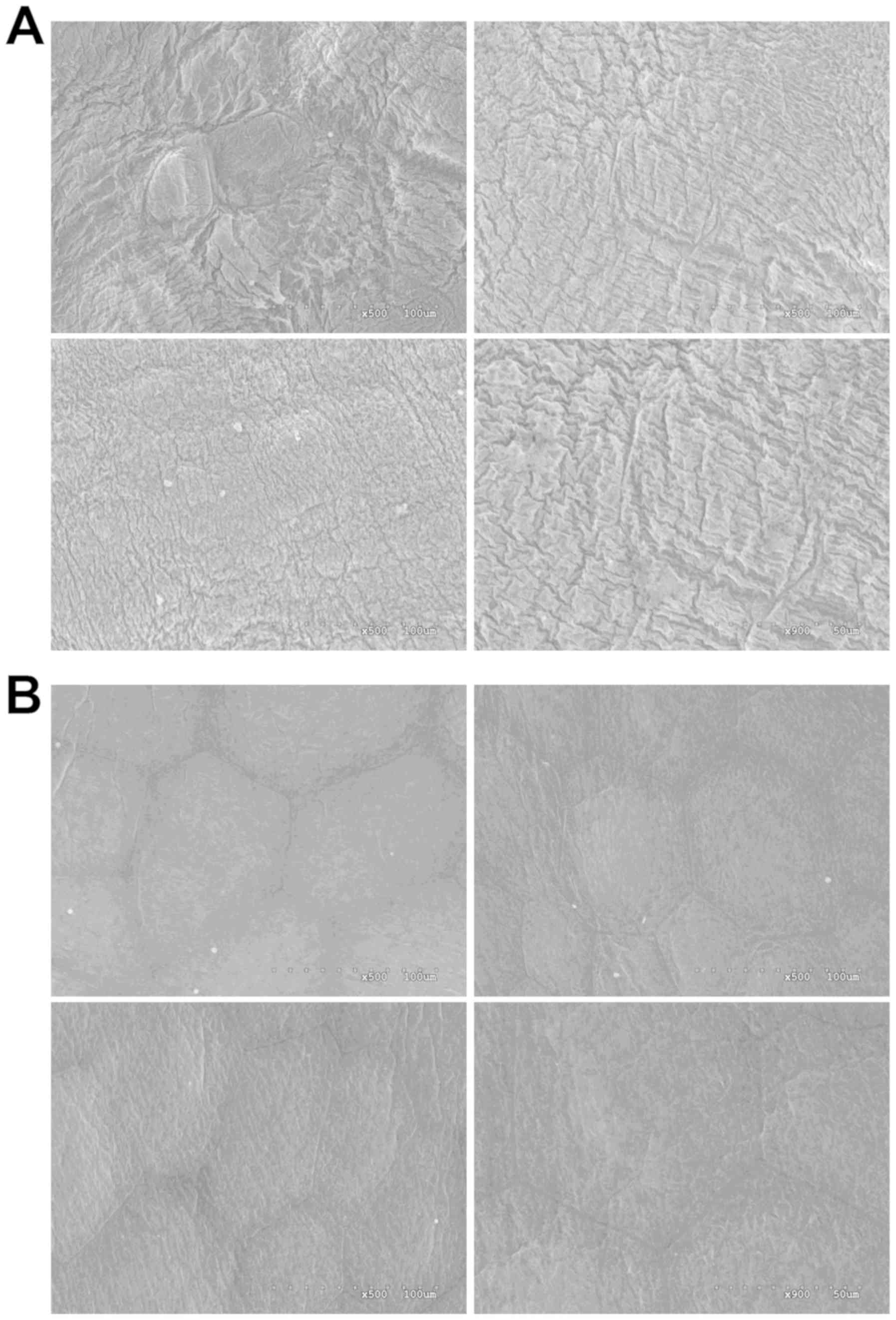
The apical surface of the mouse urinary epithelium
is covered in scallop-shaped membrane plaques consisting of
uroplakins and other integral membrane proteins (22). The SEM results suggested that the
urothelial plaques of the control mice were intact, exhibiting a
rough surface (Fig. 4A). In
contrast, the plaques on the bladders of mice treated with ketamine
were thinner and smoother, and tight junctions were identified
around hexagon-shaped umbrella cells (Fig. 4B). These morphological alterations
were consistent with the results of the HE staining, suggesting a
smooth apical epithelial surface in bladders from mice treated with
ketamine. However, no additional abnormalities, including denuded
or detached urothelium, were identified, suggesting that the
urothelial barrier function was not severely impaired following
treatment with ketamine.
DEGs and pathways identified via
microarray analysis
To analyze the gene network underlying KC
development, a Mouse Whole Genome OneArray® was
performed to determine the genes regulated by treatment with
ketamine. Mice treated with ketamine presented 284 upregulated and
527 downregulated genes compared with control mice. Subsequently,
to obtain a comprehensive overview of the identified DEGs and
investigate their biological functions, GSEA was performed to
examine the involved pathways, based on the KEGG and NABA database.
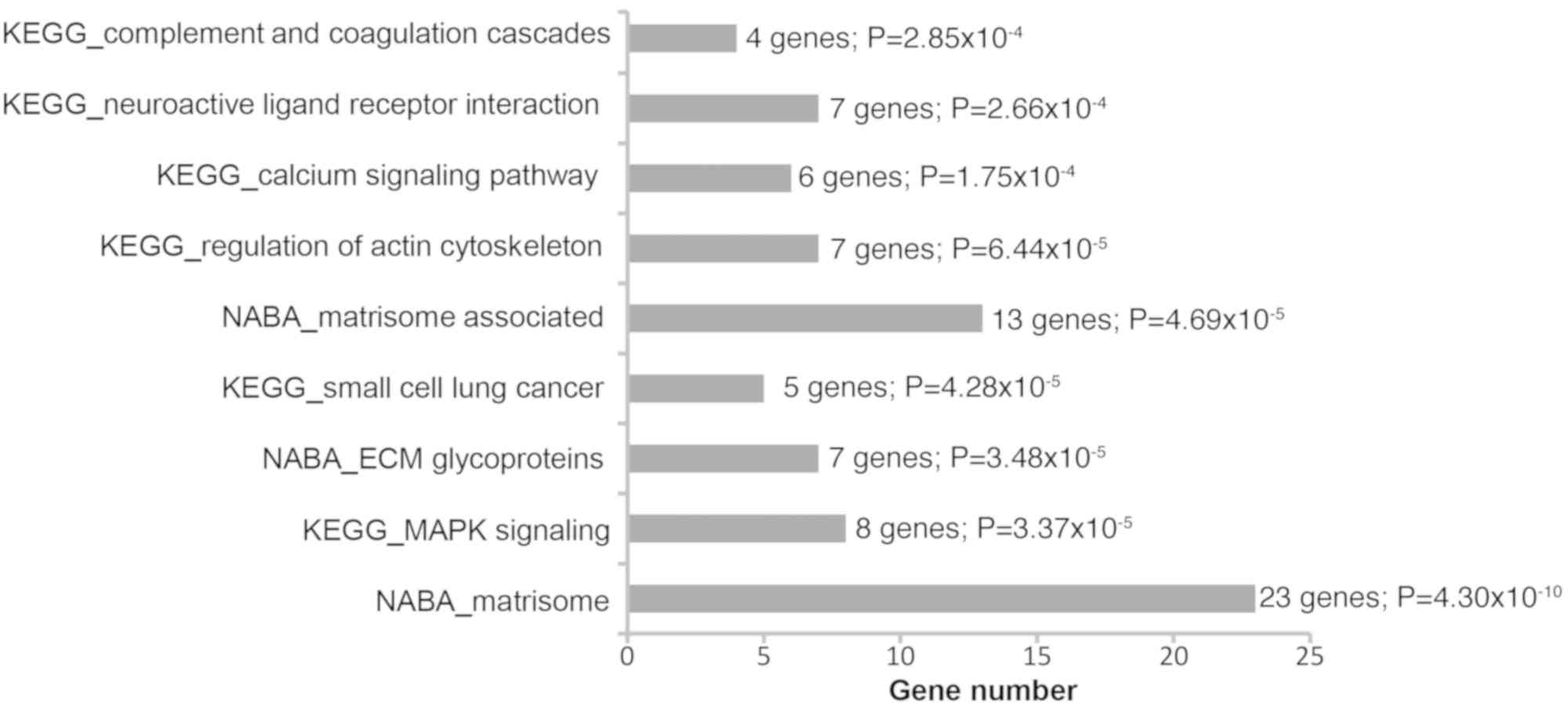
The nine most significant pathways were identified (Fig. 5), including ‘matrisome (ECM
glycoproteins, matrisome and matrisome associated)’, ‘calcium
signaling pathway’, ‘small cell lung cancer’, ‘MAPK signaling’,
‘regulation of actin cytoskeleton’, ‘neuroactive ligand receptor
interaction’ and ‘complement and coagulation cascades’. The DEGs
involved in these pathways are presented in Table I. Extracellular matrix (ECM)- and
calcium signaling-associated genes were considered for further
analysis due to the role they serve in fibrosis and dysregulated
micturition (23,24).
 | Table I.List of differentially expressed
genes and significantly enriched pathways following treatment with
ketamine. |
Table I.
List of differentially expressed
genes and significantly enriched pathways following treatment with
ketamine.
| A, Matrisome
(systematic name: M5889) |
|---|
|
|---|
| Gene symbol | NCBI gene ID | Gene name | Fold change | P-value |
|---|
| CILP | 214425 | Cartilage
intermediate layer protein | 1.12 |
3.44×10−2 |
| CLEC4D | 17474 | C-type lectin
domain family 4 member D | 1.08 |
1.44×10−2 |
| LAMC2 | 16782 | Laminin γ 2 | 0.95 |
2.24×10−2 |
| VCAN | 13003 | Versican | 0.88 |
1.30×10−3 |
| AGT | 11606 |
Angiotensinogen | 0.82 |
4.10×10−3 |
| FN1 | 14268 | Fibronectin 1 | 0.79 |
8.25×10−5 |
| FGL2 | 14190 | Fibrinogen-like
2 | 0.64 |
1.17×10−3 |
| COL1A2 | 12843 | Collagen type 1 α
2 | 0.61 |
4.46×10−2 |
| FBLN2 | 14115 | Fibulin 2 | 0.60 |
2.46×10−5 |
| S100A2 | 628324 | S100 calcium
binding protein A2 | 0.59 |
3.51×10−2 |
|
| B, Calcium
signaling pathway (systematic name: M2890) |
|
| Gene
symbol | NCBI gene
ID | Gene
name | Fold
change | P-value |
|
| CHRNA7 | 11441 | Cholinergic
receptor nicotinic α polypeptide 7 | −1.24 |
5.74×10−5 |
| CAMK2A | 12322 |
Calcium/calmodulin-dependent protein
kinase 2 α | −1.07 |
4.34×10−3 |
| RYR1 | 20190 | Ryanodine receptor
1 | −0.90 |
9.62×10−7 |
| CACNA1B | 12287 | Calcium channel
voltage-dependent N type α1 B subunit | −0.77 |
5.64×10−3 |
| BDKRB2 | 12062 | Bradykinin
receptor, β 2 | −0.62 |
3.49×10−3 |
| NOS2 | 18126 | Nitric oxide
synthase 2 | −0.61 |
1.17×10−3 |
| KCNMB4 | 58802 | Potassium
calcium-activated channel subfamily M regulatory β subunit 4 | −0.46 |
9.25×10−2 |
| KCNMA1 | 16531 | Potassium
calcium-activated channel subfamily M α 1 | −0.35 |
1.29×10−3 |
| EDNRB | 13618 | Endothelin receptor
type B | 0.53 |
9.31×10−3 |
| PLN | 18821 | Phospholamban | 0.53 |
2.91×10−2 |
| PRKCD | 18753 | Protein kinase C
δ | 0.53 |
2.84×10−2 |
| PRKCB | 18751 | Protein kinase C
β | 0.46 |
2.45×10−2 |
| EDN1 | 13614 | Endothelin 1 | 0.40 |
2.50×10−2 |
|
| C, Small cell
lung cancer (systematic name: M3228) |
|
| Gene
symbol | NCBI gene
ID | Gene
name | Fold
change | P-value |
|
| E2F1 | 13555 | E2F transcription
factor 1 | −0.69 |
3.64×10−4 |
| NFKB1 | 18033 | Nuclear factor of κ
light polypeptide gene enhancer in B cells 1 | −0.63 |
1.23×10−5 |
| NOS2 | 18126 | Nitric oxide
synthase 2 | −0.61 |
1.17×10−3 |
| LAMC2 | 16782 | Laminin γ 2 | 0.95 |
2.24×10−2 |
| FN1 | 14268 | Fibronectin 1 | 0.79 |
8.25×10−5 |
| DUSP8 | 18218 | Dual specificity
phosphatase 8 | −1.26 |
1.60×10−4 |
| HSPA1B | 15511 | Heat shock 70 kDa
protein 1B | −0.79 |
3.52×10−11 |
| CACNA1B | 12287 | Calcium channel
voltage-dependent N type α1 B subunit | −0.77 |
5.64×10−3 |
| NR4A1 | 15370 | Nuclear receptor
subfamily 4 group A member 1 | −0.70 |
1.04×10−5 |
| RRAS2 | 66922 | Related ras viral
oncogene homolog 2 | −0.68 |
1.20×10−9 |
| FGF14 | 14169 | Fibroblast growth
factor 14 | −0.65 |
3.66×10−2 |
| DUSP1 | 19252 | Dual specificity
phosphatase 1 | −0.63 |
3.99×10−5 |
| NFKB1 | 18033 | Nuclear factor of κ
light polypeptide gene enhancer in B-cells 1 | −0.63 |
1.23×10−5 |
|
| E, Regulation of
actin cytoskeleton (systematic name: M18306) |
|
| Gene
symbol | NCBI gene
ID | Gene
name | Fold
change | P-value |
|
| INSRR | 23920 | Insulin
receptor-related receptor | −0.90 |
7.72×10−4 |
| F2 | 14061 | Coagulation factor
2 | −0.74 |
4.86×10−4 |
| RRAS2 | 66922 | Related ras viral
oncogene homolog 2 | −0.68 |
1.20×10−9 |
| FGF14 | 14169 | Fibroblast growth
factor 14 | −0.65 |
3.66×10−2 |
| BDKRB2 | 12062 | Bradykinin receptor
B2 | −0.62 |
3.49×10−3 |
| FN1 | 14268 | Fibronectin 1 | 0.79 |
8.25×10−5 |
| CFL2 | 12632 | Cofilin 2 | 0.68 |
2.07×10−4 |
|
| F, Neuroactive
ligand receptor interaction (systematic name: M13380) |
|
| Gene
symbol | NCBI gene
ID | Gene
name | Fold
change | P-value |
|
| OPRD1 | 18386 | Opioid receptor δ
1 | −1.25 |
7.31×10−6 |
| CHRNA7 | 11441 | Cholinergic
receptor nicotinic α 7 | −1.24 |
5.74×10−5 |
| GHRHR | 14602 | Growth hormone
releasing hormone receptor | −1.19 |
2.09×10−3 |
| F2 | 14061 | Coagulation factor
2 | −0.74 |
4.86×10−4 |
| GRIA2 | 14800 | Glutamate receptor
ionotropic ampa 2 | −0.78 |
2.92×10−2 |
| BDKRB2 | 12062 | Bradykinin receptor
B2 | −0.62 |
3.49×10−3 |
| PPYR1 | 19065 | Pancreatic
polypeptide receptor 1 | 0.69 |
2.43×10−3 |
|
| G, Complement
and coagulation cascades (systematic name: M16894) |
|
| Gene
symbol | NCBI gene
ID | Gene
name | Fold
change | P-value |
|
| F2 | 14061 | Coagulation factor
2 | −0.74 |
4.86×10−4 |
| MASP1 | 17174 | Mannan-binding
lectin serine peptidase 1 | −0.68 |
3.67×10−2 |
| C6 | 12274 | Complement
component 6 | −0.67 |
1.82×10−2 |
| BDKRB2 | 12062 | Bradykinin receptor
B2 | −0.62 |
3.49×10−3 |
Molecular components associated with
ECM and calcium signaling are regulated by treatment with
ketamine
Genes with a role in ECM, including FN1, fibulin 2,
fibrinogen-like 2, laminin γ 2 (LAMC2) and collagen type 1 α 2
(COL1A2) were significantly upregulated (Table I), which was consistent with the
histopathological stain results, as the bladders presented
connective tissue expansion in the submucosa region (Fig. 3C). Additional ECM-associated genes
VCAN, angiotensinogen (AGT) and C-type lectin domain family 4
member D were upregulated, suggesting a role for these genes in
modulating the interactions between the ECM and neighboring cells,
promoting a profibrotic milieu. Furthermore, genes with a role in
the calcium signaling pathway were differentially expressed. In
particular, genes associated with calcium transport and
intracellular calcium storage were identified, including
calcium/calmodulin-dependent protein kinase 2 α (CAMK2A), RYR1,
PRKCB, endothelin receptor type B (EDNRB) and phospholamban.
Notably, calcium signaling may influence urinary bladder smooth
muscle (UBSM) contraction (23).
In addition, the large-conductance voltage- and
Ca2+-activated (BK) channel is the principal
K+ channel regulating local Ca2+ release by
the RYRs, located in the sarcoplasmic reticulum membrane (25). Therefore, BK channels are able to
regulate the frequency of Ca2+ sparks and subsequent
UBSM excitability (25). Notably,
two genes, KCNMA1 and KCNMB4, that constitute the α and β subunits
of the BK channels (23), were
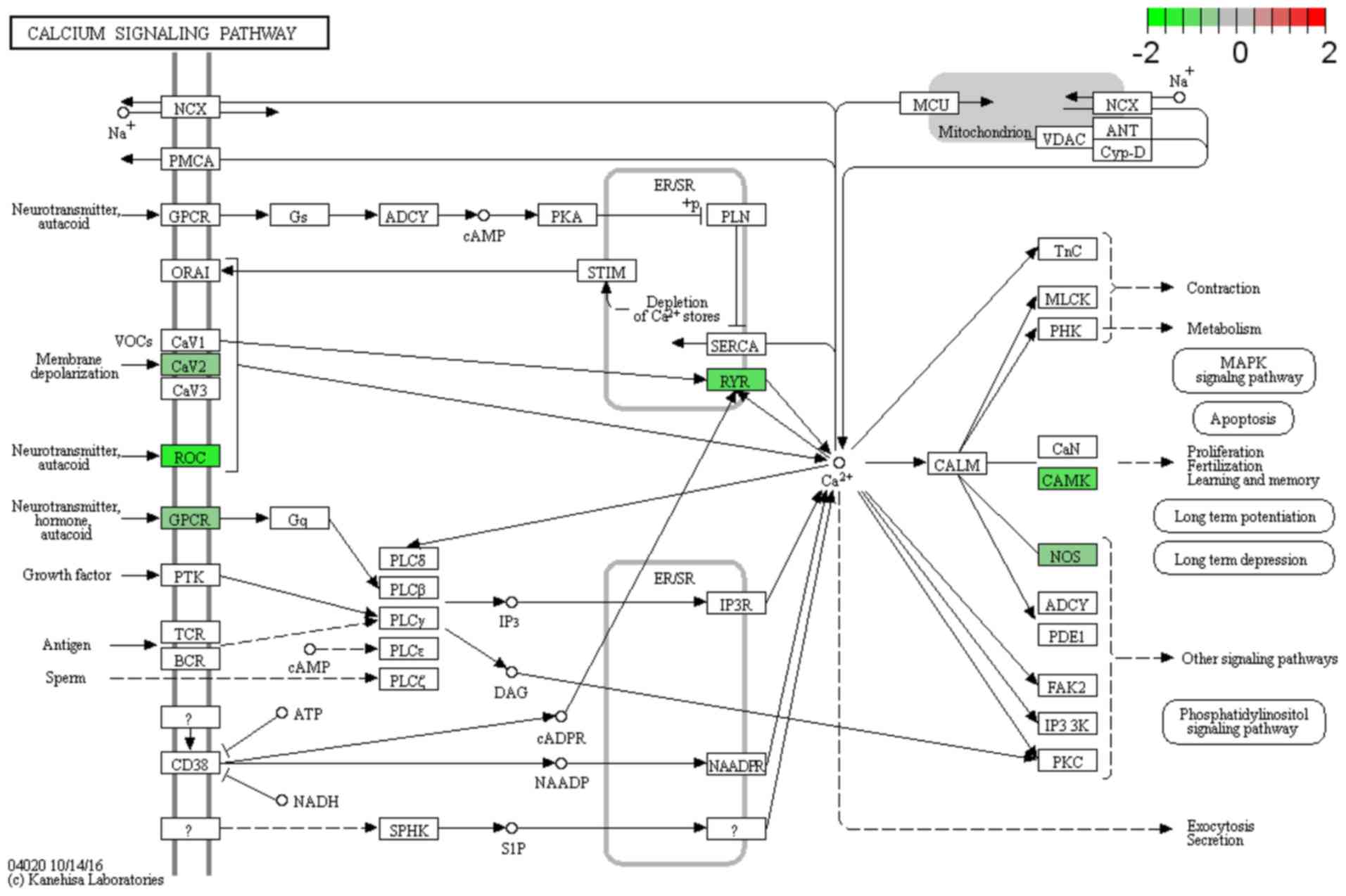
downregulated in mice treated with ketamine. To understand the role
of these genes in the calcium signaling pathway, Pathview software
was used to visualize the DEGs involved in this pathway (Fig. 6).
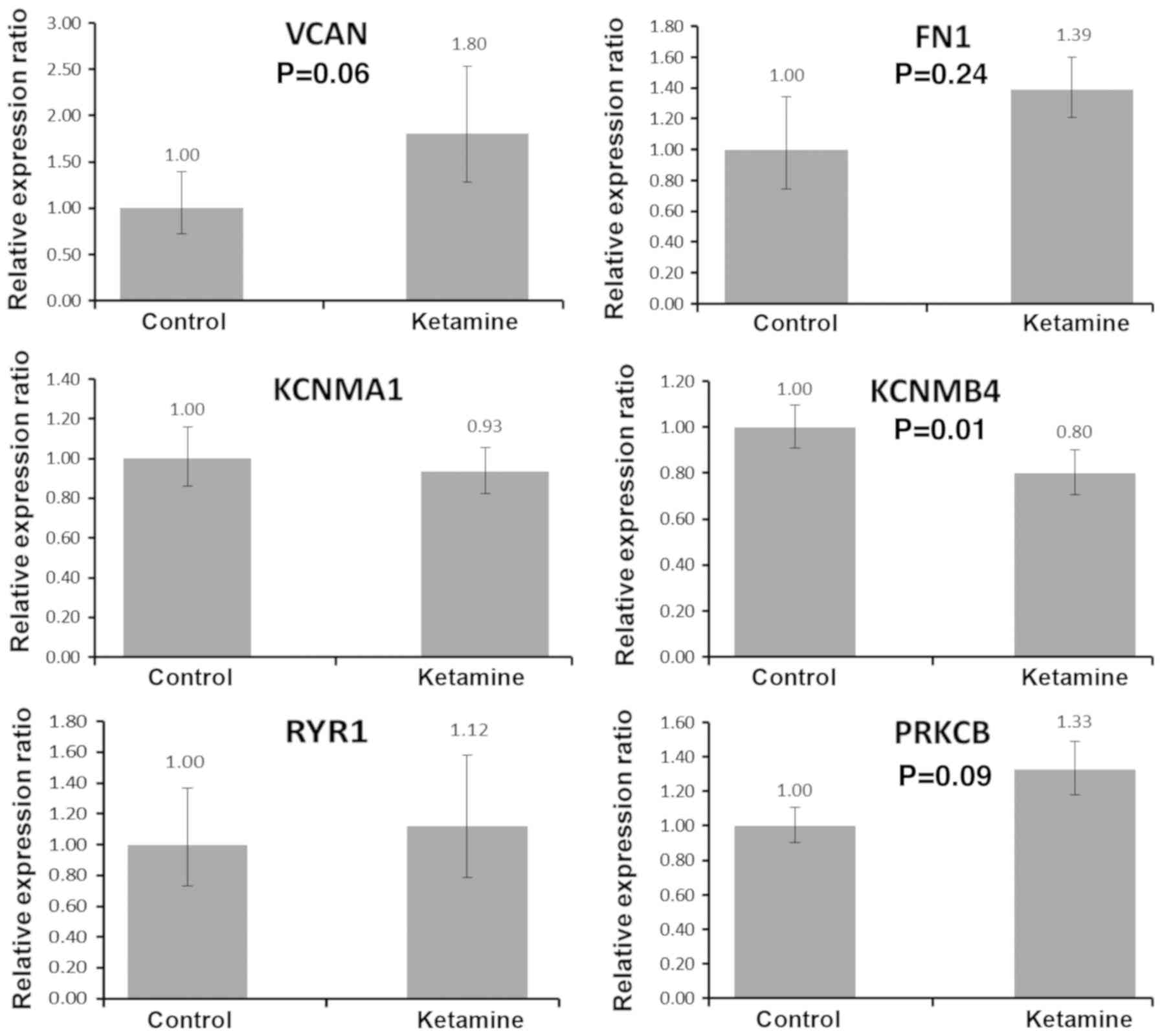
Furthermore, the expressions of six genes associated
with calcium regulation were validated by RT-qPCR (Fig. 7). The RT-qPCR results suggested
that KCNMB4 was significantly downregulated in mice treated with
ketamine, and three genes, VCAN, FN1 and PRKCB, were upregulated in
the ketamine group, although the difference was not significant.
However, the expression levels of KCNMA1 and RYR1 was not affected,
according to the RT-qPCR data. Therefore, the microarray analysis
performed represents a strategy to investigate the effects of
ketamine on the transcriptome profile.
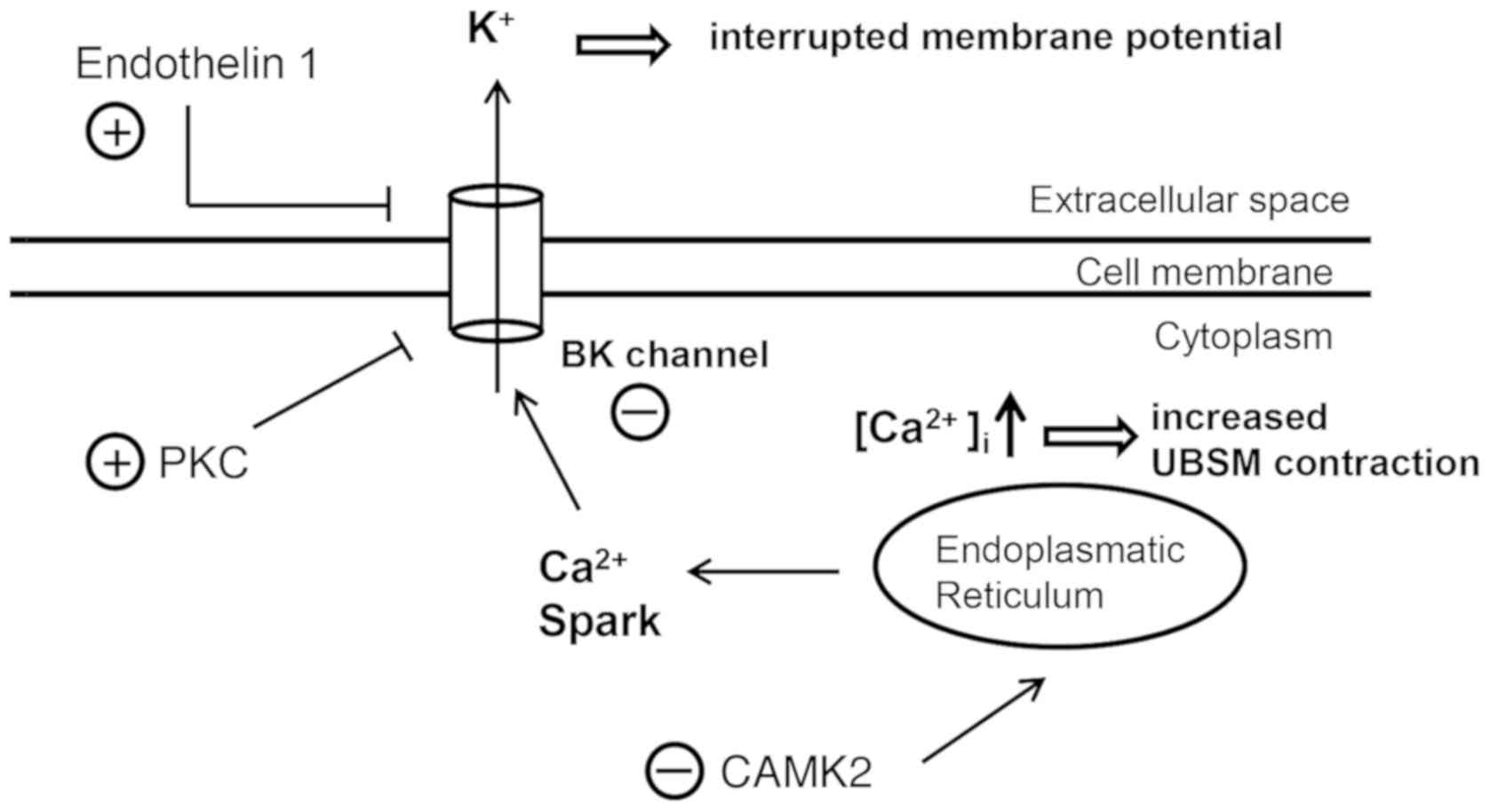
The role of calcium signaling in ketamine-induced
UBSM contraction is presented in Fig.
8. Calcium signaling represents one of the principal causes
involved in the abnormal voiding behavior identified in mice
treated with ketamine (23). The
present results provided the basis for further studies aimed to
investigate whether ketamine abuse may cause bladder dysfunction in
humans.
Discussion
Following a 20-week period, the body weights of mice
treated with ketamine were significantly lower compared with the
control mice, due to a reduced food intake and the side effects of
ketamine (21). Additionally, the
phenotype observed may be due to physiological stress caused by
chronic ketamine consumption (26). Nevertheless, mice treated with
ketamine exhibited increased micturition frequency and decreased
voiding volume compared with the control group, suggesting that
long-term treatment with ketamine may lead to urinary bladder
overactivity, a symptom previously identified to be associated with
patients with KC (27). The
present finding is consistent with a number of previous studies
using rodent models, including mice and rats (8,10).
Although the molecular mechanism of ketamine-induced bladder
hyperactivity is not fully understood, certain molecular factors,
including p-TAGLN (7) and P2RX1
(10), have been proposed to cause
detrusor dysfunction.
In the present study, the microarray data suggested
that long-term treatment with ketamine may influence detrusor
contraction, by influencing the expression of components associated
with the calcium signaling pathway. The BK channel is one of the
principal potassium channels regulating UBSM function (28). BK channels are expressed in UBSM
cells and contain four pore-forming α-subunits, KCNMA1, and four
regulatory β1-subunits, KCNMB1, or β4-subunits, KCNMB4 (29). The BK channel has been identified
to provide a negative-feedback mechanism to limit the amplitude and
duration of UBSM action potentials and associated phasic
contractions in response to intracellular Ca2+ spark
(30). Decreased gene expression
or gene knockout of KCNMB4 was identified to be associated with
increased action potential frequency and UBSM excitability
(31,32). Therefore, the downregulation of the
expression level of KCNMB4, as detected by the present microarray
analysis, may represent a cause of the ketamine-associated detrusor
overactivity. Furthermore, previous studies suggested that
endothelin-1 (EDN1) and its downstream signaling regulated by
protein kinase C (PKC) are able to induce smooth muscle contraction
by reducing the frequency of Ca2+ sparks (33,34).
PKC is additionally involved in maintaining contractility by
inhibiting the myosin light chain (MYL) phosphatase complex
activity and subsequently increasing the protein expression level
of p-MYL (35). Consequently, the
increased expression levels of components of the endothelin
pathway, including EDNRB and EDN1, in addition to the increased
expression levels of proteins belonging to the PKC family,
including protein kinase C δ and PRKCB (36), may promote detrusor contraction,
and these genes were identified to be differentially expressed in
the present study. Furthermore, the downregulation of the
expression level of CAMK2 may represent an additional cause of
detrusor contraction, since decreased CAMK2 activity was previously
identified to be associated with insufficient local Ca2+
spark and Ca2+ reuptake into the sarcoplasmic reticulum
(37). Therefore, the increased
cytosolic calcium concentration may promote
Ca2+-dependent smooth muscle contraction (38). Collectively, the present study
suggested that calcium signaling may serve a role in
ketamine-induced bladder hyperactivity.
A VSOP test suggested that ketamine was able to
significantly affect the voiding behavior of mice following a
20-week treatment. Therefore, to avoid additional secondary
effects, the treatment was not prolonged beyond the 20th week. The
present results suggested that high-dosage long-term ketamine
administration promoted inflammatory cell infiltration and vascular
congestion in the submucosal layer of the bladder, which were
observed in patients with long-term ketamine abuse (39,40).
Although the induced inflammatory response was not present in all
mice, the pathological outcome was consistent with previous studies
using an identical mouse strain, C57BL/6, and a similar ketamine
regimen, 100 mg/kg/day (10,41).
However, previous studies performed on rat models identified a
pronounced and consistent inflammatory response (7,42).
This inconsistency in the inflammatory response between rats and
mice may be due to the increased metabolic rate of mice (43). Mice may present a decreased
retention time of ketamine and its inflammation-associated
metabolites, resulting in a decreased induction of inflammation
(44). Furthermore, although
urothelial thickness was identified to be unaffected in the present
study, SEM analysis suggested that the surface of the bladders of
mice treated with ketamine was flat and smooth compared with
control mice, which exhibited a rough surface with ridge-like
structures, suggesting a decreased accumulation of uroplakins in
the urothelium. The present microarray data identified a normal
expression level of genes encoding for tight junction proteins,
including claudin and occludin, suggesting that the function of the
urothelium barrier was not affected. The present results are
similar to a previous study published by Rajandram et al
(44), demonstrating that a
low-dosage ketamine regimen (30 mg/kg), performed for 12 weeks, did
not impair the bladder mucosal surface, which exhibited defined
tight junctions. However, the present study used a high-dosage,
long-term treatment with ketamine (100 mg/kg/day for 20 weeks), and
it was hypothesized that increased ketamine consumption may affect
the morphology of the bladder apical membrane.
Ketamine is able to cause bladder interstitial
fibrosis (4), which induces an
irreversible decline in bladder function. Our previous study using
Balb/c mice suggested that certain molecular events associated with
abnormal wound healing processes may contribute to early-stage
fibrosis (13). In the present
study, using a C57BL/6 mouse model, submucosal extruding folds were
observed in the bladders of mice treated with ketamine. Notably,
the expression levels of genes involved in ECM deposition,
including FN1, LAMC2 and COL1A2, were upregulated, suggesting that
long-term treatment with ketamine may promote bladder fibrosis by
regulating the ECM. Furthermore, the expression levels of two genes
encoding for ECM components associated with fibrosis, VCAN
(45) and AGT (46,47),
were upregulated. The present results suggested that active
fibroblasts, following treatment with ketamine, may secrete these
ECM modulators, subsequently inducing fibrogenic response. Previous
studies using Sprague Dawley (SD) rats have demonstrated that
TGF-β1 signaling (48) and EMT
(9) are associated with
ketamine-induced fibrosis. Furthermore, injection of human
umbilical cord blood-derived mesenchymal stem cells or antifibrotic
compounds, including N-acetylcysteine (49) and hyaluronic acid (50), in SD rat bladder tissue have been
demonstrated to decrease fibrosis by inactivating TGF-β signaling.
However, TGF-β-associated genes were unaltered in the present
microarray analysis. The inconsistent role of TGF-β may be due to
development- and species-specific responses. In addition,
ketamine-induced fibrosis may be mediated by TGF-β-independent
pathways (42). Therefore, further
investigation is required to elucidate the discrepancies between
rats and mice in response to treatment with ketamine.
In the present study, C57BL/6 mice were treated with
ketamine for 20 weeks. To examine the effects of long-term ketamine
abuse on urinary bladders, the transcriptional profiles were
investigated. The calcium signaling pathway was identified as a
possible mechanism underlying detrusor contractility and urinary
bladder overactivity. The present findings may help in
understanding the molecular mechanism underlying KC and in
identifying novel molecular targets for the development of
effective therapies to treat ketamine-induced urinary tract
syndrome.
Acknowledgements
Not applicable.
Funding
The present study was supported by grants from The
Ministry of Science and Technology of the Republic of China, Taiwan
(grant no. MOST 104-2320-B-415-001-MY3; Taiwan) and from The
Ditmanson Medical Foundation Chiayi Christian Hospital Research
Program (grant no. R105-008; Taiwan).
Availability of data and materials
The datasets used and/or analyzed during the present
study are available from the corresponding author upon reasonable
request.
Authors' contributions
CHS, STW, and YWL were involved in the design of the
study, acquisition and interpretation of data, and drafting the
article. STW and SCW performed the experiments. SML and LCL
assisted with the SEM experiment. YCD assisted with the
histopathological analysis and interpretation. CHS, STW and YWL
analyzed and discussed the data.
Ethics approval and consent to
participate
All animal experiments were approved by The
Institutional Animal Care and Use Committee of National Chiayi
University (approval no. 104047) and according to the guidelines of
The Animal Research: Reporting in vivo Experiments,
recommended by The National Centre for the Replacement, Refinement
and Reduction of Animals in Research.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Domino EF, Chodoff P and Corssen G:
Pharmacologic effects of CI-581, a new dissociative anesthetic, in
man. Clin Pharmacol Ther. 6:279–291. 1965. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Shahani R, Streutker C, Dickson B and
Stewart RJ: Ketamine-associated ulcerative cystitis: A new clinical
entity. Urology. 69:810–812. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Chu PS, Kwok SC, Lam KM, Chu TY, Chan SW,
Man CW, Ma WK, Chui KL, Yiu MK, Chan YC, et al: 'Street
ketamine'-associated bladder dysfunction: A report of ten cases.
Hong Kong Med J. 13:311–313. 2007.PubMed/NCBI
|
|
4
|
Chu PS, Ma WK, Wong SC, Chu RW, Cheng CH,
Wong S, Tse JM, Lau FL, Yiu MK and Man CW: The destruction of the
lower urinary tract by ketamine abuse: A new syndrome? BJU Int.
102:1616–1622. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Jhang JF, Hsu YH and Kuo HC: Possible
pathophysiology of ketamine-related cystitis and associated
treatment strategies. Int J Urol. 22:816–825. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Chuang SM, Liu KM, Li YL, Jang MY, Lee HH,
Wu WJ, Chang WC, Levin RM and Juan YS: Dual involvements of
cyclooxygenase and nitric oxide synthase expressions in
ketamine-induced ulcerative cystitis in rat bladder. Neurourol
Urodyn. 32:1137–1143. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Gu D, Huang J, Yin Y, Shan Z, Zheng S and
Wu P: Long-term ketamine abuse induces cystitis in rats by
impairing the bladder epithelial barrier. Mol Biol Rep.
41:7313–7322. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Liu KM, Chuang SM, Long CY, Lee YL, Wang
CC, Lu MC, Lin RJ, Lu JH, Jang MY, Wu WJ, et al: Ketamine-induced
ulcerative cystitis and bladder apoptosis involve oxidative stress
mediated by mitochondria and the endoplasmic reticulum. Am J
Physiol Renal Physiol. 309:F318–F331. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Wang J, Chen Y, Gu D, Zhang G, Chen J,
Zhao J and Wu P: Ketamine-induced bladder fibrosis involves
epithelial-to-mesenchymal transition mediated by transforming
growth factor-β1. Am J Physiol Renal Physiol. 313:F961–F972. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Meng E, Chang HY, Chang SY, Sun GH, Yu DS
and Cha TL: Involvement of purinergic neurotransmission in ketamine
induced bladder dysfunction. J Urol. 186:1134–1141. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Gu D, Huang J, Shan Z, Yin Y, Zheng S and
Wu P: Effects of long-term ketamine administration on rat bladder
protein levels: A proteomic investigation using two-dimensional
difference gel electrophoresis system. Int J Urol. 20:1024–1031.
2013.PubMed/NCBI
|
|
12
|
Shen CH, Wang ST, Lee YR, Liu SY, Li YZ,
Wu JD, Chen YJ and Liu YW: Biological effect of ketamine in
urothelial cell lines and global gene expression analysis in the
bladders of ketamine-injected mice. Mol Med Rep. 11:887–895. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Shen CH, Wang SC, Wang ST, Lin SM, Wu JD,
Lin CT and Liu YW: Evaluation of urinary bladder fibrogenesis in a
mouse model of long-term ketamine injection. Mol Med Rep.
14:1880–1890. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Watanabe H, Numata K, Ito T, Takagi K and
Matsukawa A: Innate immune response in Th1- and Th2-dominant mouse
strains. Shock. 22:460–466. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Kilkenny C, Browne W, Cuthill IC, Emerson
M and Altman DG; National Centre for the Replacement and Refinement
Reduction of Amimals in Research, : Animal research: Reporting in
vivo experiments-the ARRIVE guidelines. J Cereb Blood Flow Metab.
31:991–993. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Sugino Y, Kanematsu A, Hayashi Y, Haga H,
Yoshimura N, Yoshimura K and Ogawa O: Voided stain on paper method
for analysis of mouse urination. Neurourol Urodyn. 27:548–552.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Cohen SM, Ohnishi T, Clark NM, He J and
Arnold LL: Investigations of rodent urinary bladder carcinogens:
Collection, processing, and evaluation of urine and bladders.
Toxicol Pathol. 35:337–347. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Naba A, Clauser KR, Ding H, Whittaker CA,
Carr SA and Hynes RO: The extracellular matrix: Tools and insights
for the ‘omics’ era. Matrix Biol. 49:10–24. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Corazza O, Assi S and Schifano F: From
‘Special K’ to ‘Special M’: The evolution of the recreational use
of ketamine and methoxetamine. CNS Neurosci Ther. 19:454–460. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Cvrcek P: Side effects of ketamine in the
long-term treatment of neuropathic pain. Pain Med. 9:253–257. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Lee G: Uroplakins in the lower urinary
tract. Int Neurourol J. 15:4–12. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Petkov GV: Central role of the BK channel
in urinary bladder smooth muscle physiology and pathophysiology. Am
J Physiol Regul Integr Comp Physiol. 307:R571–R584. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Wynn TA: Cellular and molecular mechanisms
of fibrosis. J Pathol. 214:199–210. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Petkov GV: Role of potassium ion channels
in detrusor smooth muscle function and dysfunction. Nat Rev Urol.
9:30–40. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Everds NE, Snyder PW, Bailey KL, Bolon B,
Creasy DM, Foley GL, Rosol TJ and Sellers T: Interpreting stress
responses during routine toxicity studies: A review of the biology,
impact, and assessment. Toxicol Pathol. 41:560–614. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Tsai TH, Cha TL, Lin CM, Tsao CW, Tang SH,
Chuang FP, Wu ST, Sun GH, Yu DS and Chang SY: Ketamine-associated
bladder dysfunction. Int J Urol. 16:826–829. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Heppner TJ, Bonev AD and Nelson MT:
Ca(2+)-activated K+ channels regulate action potential
repolarization in urinary bladder smooth muscle. Am J Physiol.
273:C110–C117. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Hristov KL, Chen M, Kellett WF, Rovner ES
and Petkov GV: Large-conductance voltage- and Ca2+-activated K+
channels regulate human detrusor smooth muscle function. Am J
Physiol Cell Physiol. 301:C903–C912. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Hayase M, Hashitani H, Kohri K and Suzuki
H: Role of K+ channels in regulating spontaneous activity in
detrusor smooth muscle in situ in the mouse bladder. J Urol.
181:2355–2365. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Kita M, Yunoki T, Takimoto K, Miyazato M,
Kita K, de Groat WC, Kakizaki H and Yoshimura N: Effects of bladder
outlet obstruction on properties of Ca2+-activated K+ channels in
rat bladder. Am J Physiol Regul Integr Comp Physiol.
298:R1310–R1319. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Brenner R, Chen QH, Vilaythong A, Toney
GM, Noebels JL and Aldrich RW: BK channel beta4 subunit reduces
dentate gyrus excitability and protects against temporal lobe
seizures. Nat Neurosci. 8:1752–1759. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Khimji AK and Rockey DC:
Endothelin-biology and disease. Cell Signal. 22:1615–1625. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Jaggar JH, Porter VA, Lederer WJ and
Nelson MT: Calcium sparks in smooth muscle. Am J Physiol Cell
Physiol. 278:C235–C256. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Wang T, Kendig DM, Smolock EM and Moreland
RS: Carbachol-induced rabbit bladder smooth muscle contraction:
Roles of protein kinase C and Rho kinase. Am J Physiol Renal
Physiol. 297:F1534–F1542. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Wang T, Kendig DM, Trappanese DM, Smolock
EM and Moreland RS: Phorbol 12,13-dibutyrate-induced, protein
kinase C-mediated contraction of rabbit bladder smooth muscle.
Front Pharmacol. 2:832012. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Picht E, DeSantiago J, Huke S, Kaetzel MA,
Dedman JR and Bers DM: CaMKII inhibition targeted to the
sarcoplasmic reticulum inhibits frequency-dependent acceleration of
relaxation and Ca2+ current facilitation. J Mol Cell Cardiol.
42:196–205. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Kuo IY and Ehrlich BE: Signaling in muscle
contraction. Cold Spring Harb Perspect Biol. 7:a0060232015.
View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Lin HC, Lee HS, Chiueh TS, Lin YC, Lin HA,
Lin YC, Cha TL and Meng E: Histopathological assessment of
inflammation and expression of inflammatory markers in patients
with ketamine-induced cystitis. Mol Med Rep. 11:2421–2428. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Jhang JF, Hsu YH, Jiang YH, Lee CL and Kuo
HC: Histopathological characteristics of ketamine-associated
uropathy and their clinical association. Neurourol Urodyn.
37:1764–1772. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Duan Q, Wu T, Yi X, Liu L, Yan J and Lu Z:
Changes to the bladder epithelial barrier are associated with
ketamine-induced cystitis. Exp Ther Med. 14:2757–2762. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Hills CE, Jin T, Siamantouras E, Liu IK,
Jefferson KP and Squires PE: ‘Special k’ and a loss of cell-to-cell
adhesion in proximal tubule-derived epithelial cells: modulation of
the adherens junction complex by ketamine. PLoS One. 8:e718192013.
View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Martignoni M, Groothuis G and de Kanter R:
Comparison of mouse and rat cytochrome P450-mediated metabolism in
liver and intestine. Drug Metab Dispos. 34:1047–1054.
2006.PubMed/NCBI
|
|
44
|
Rajandram R, Ong TA, Razack AH, MacIver B,
Zeidel M and Yu W: Intact urothelial barrier function in a mouse
model of ketamine-induced voiding dysfunction. Am J Physiol Renal
Physiol. 310:F885–F894. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Wight TN: Provisional matrix: A role for
versican and hyaluronan. Matrix Biol. 60-61:38–56. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Thanigaimani S, Lau DH, Agbaedeng T,
Elliott AD, Mahajan R and Sanders P: Molecular mechanisms of atrial
fibrosis: Implications for the clinic. Expert Rev Cardiovasc Ther.
15:247–256. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Nogueira A, Pires MJ and Oliveira PA:
Pathophysiological mechanisms of renal fibrosis: A review of animal
models and therapeutic strategies. In Vivo. 31:1–22. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Juan YS, Lee YL, Long CY, Wong JH, Jang
MY, Lu JH, Wu WJ, Huang YS, Chang WC and Chuang SM: Translocation
of NF-κB and expression of cyclooxygenase-2 are enhanced by
ketamine-induced ulcerative cystitis in rat bladder. Am J Pathol.
185:2269–2285. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Kim A, Yu HY, Heo J, Song M, Shin JH, Lim
J, Yoon SJ, Kim Y, Lee S, Kim SW, et al: Mesenchymal stem cells
protect against the tissue fibrosis of ketamine-induced cystitis in
rat bladder. Sci Rep. 6:308812016. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Lee YL, Lin KL, Chuang SM, Lee YC, Lu MC,
Wu BN, Wu WJ, Yuan SF, Ho WT and Juan YS: Elucidating mechanisms of
bladder repair after hyaluronan instillation in ketamine-induced
ulcerative cystitis in animal model. Am J Pathol. 187:1945–1959.
2017. View Article : Google Scholar : PubMed/NCBI
|