Introduction
Earthworms are terrestrial annelids in the
oligochaeta subclass, with a generally preferred habitat of damp
and loose soil. They include ~3,000 species worldwide, with 229
species in China (1,2). In a wide variety of soil types,
earthworms serve vital roles in converting large pieces of organic
matter into rich humus to enrich soil fertility. The earthworms are
the highest evolutionary species capable of regenerating an
anterior portion containing the central nerve system, heart and
clitellum (3). The anterior
regeneration is a unique developmental process that requires cell
proliferation, re-differentiation and sophisticated cell-cell
communication. This process can serve as a useful model for
investigating normal development and differentiation (4).
Over the past several years, cDNA library
construction and analysis have become established as indispensable
methods for functional genome analysis since they provide detailed
information about the genomic mechanisms underlying the diverse
processes of an organism (5).
However, conventionally generated cDNA libraries contain a high
percentage of 5′-truncated clones, limiting the utility of such
libraries. The Switching Mechanism at 5′End of RNA Template (SMART)
technique (6) amplifies and
enriches the full-length mRNA, and thus generates cDNA libraries
with a significantly improved ratio of full-length to partial cDNA
sequences. In the present study, the SMART technique was adopted to
construct a high quality library of full-length cDNAs
representative of adult earthworms, namely of the earthworm
Eisenia fetida (Savigny, 1826).
Unlike other model organisms, none of the
oligochaete genomes have been sequenced to the best of our
knowledge, and genomic research on earthworms lags behind that of
other model species such as Mus musculus. In the absence of
the full genome sequences, expressed sequence tags (ESTs) aid the
rapid detection of expressed genes via sequence analysis, and are a
significant resource for comparative and functional genomic studies
(2). In addition, among the
biological techniques for transcriptome analysis, the determination
of ESTs is considered the simplest method for profiling the
transcriptome, which is also particularly useful in the development
of cDNA microarrays for systematic identification of differentially
expressed genes (7). Analysis of
ESTs is an effective method for rapidly analyzing gene expression,
characterizing gene functions and discovering new genes that are
important for specific developmental and physiological processes
(8). The present study established
593 ESTs, representing 168 genes and 425 unknown tags, providing a
gene expression profile of earthworm development. This collection
of ESTs may provide a valuable basis for future research on the
physiology of earthworms.
Materials and methods
Isolation of total RNA and mRNA
Eisenia fetida earthworms were purchased from
Beijing Shuangqiao Farm (Beijing, China). Fully developed adult
Eisenia fetida earthworms weighing 0.3–0.6 g (live weight)
were selected for all experiments. All earthworm tissues were
harvested and total RNA was isolated using TRIzol reagent
(Invitrogen; Thermo Fisher Scientific, Inc., Waltham, MA, USA). The
integrity of total RNA was analyzed by electrophoresis using 1%
agarose gels. Isolation of poly(A) mRNA from total RNA was carried
out using an MN-NucleoTrap®mRNA kit (Machery-Nagel GmbH
& Co. KG, Düren, Germany) according to the manufacturer's
protocol. Oligo(dT) beads suspension was applied to total RNA and
incubated at 68°C for 5 min before eluting mRNA. Then ethidium
bromide (EB) staining was applied and 1% agarose gel was used to
visualize the result. The isolated mRNA was further vacuum
concentrated using Concentrator plus™ (Eppendorf, Hamburg,
Germany). The quantity and integrity of isolated mRNA were
determined using a nanodrop spectrophotometer and agarose gel
electrophoresis, respectively.
cDNA library construction
A total of ~8,048.4 ng mRNA was used for
single-stranded cDNA synthesis. The purified mRNA was used as a
template, Oligod(T)18 with XhoI cleavage site was used as the
primer, and first strand cDNA was transcribed at 42°C using
SuperScript™ II RnaseH-Reverse (Thermo Fisher Scientific, Inc.).
Then the mRNA was digested using RNaseH, and the resultant mRNA
fragments were used as further primers. The first cDNA chain was
used as a template for double-stranded cDNA synthesis, using DNA
Polymerase I (Takara Biotechnology Co., Ltd., Beijing, China). The
ends of the double-stranded cDNA were ligated by T4 DNA polymerase
and the ligation products were purified by
phenol/chloroform/isoamyl alcohol to remove excess impurities such
as protein. Subsequently, the double-stranded DNA fragments were
ligated into EcoRI Adaptor using T4 DNA ligase at 4°C overnight.
Then the double-stranded DNA fragments were phosphorylated with T4
Polynucleotide Kinase and digested with XhoI. Following XhoI
digestion of the double-stranded cDNA, producing XhoI sticky ends,
a QIAEXII Gel Extraction kit (Beijing BioDev-Tech, Beijing, China)
was used to recycle 0.5–4 Kb fragments. The recycled cDNA was
preserved at −20°C. Then the cDNA was ligated into the pBluescript
II SK(+) XR vector (Promega Corporation, Madison, WI, USA) in a 3:1
molar ratio with T4 DNA ligase at 4°C overnight. To reduce the
redundancy and avoid the underrepresentation of different
transcript species, cDNA fragments with different fractionated
sizes were balanced and subjected to library construction (9,10).
Prior to transformation, mixing of all ligated products with
microporous membranes was performed to remove salt ions.
Subsequently the products were transformed into 5×107/ml
DH10B competent cells (Thermo Fisher Scientific, Inc.), plated on
agar plates (10 cm diameter) by pipetting the cells onto the middle
of the plate and spreading, and monoclonal colonies were selected
for PCR amplification. The inserted sequences in the plasmids were
amplified by PCR using T3 primers (5′-ATTAACCCTCACTAAAGGGA-3′) and
T7 primers (5′-TAATACGACTCACTATAGGG-3′). The total volume of PCR
reaction mixture was 20 µl, containing 1 µl template, 10 µl 2XTaq
MasterMix (CWBIO, Beijing, China), 1 µl T3 primers (10 pmol), 1 µl
T7 primers (10 pmol) and 7 µl ddH2O (CWBIO). Cycling
conditions were: 94°C for 5 min, followed by 30 cycles of 94°C for
30 sec, 55°C for 40 sec and 72°C for 60 sec, followed by 72°C for 5
min.
Bioinformatic analysis
cDNA clones were selected randomly from the cDNA
library and the vector sequences were trimmed from the raw sequence
data using Vecscreen tool (www.ncbi.nlm.nih.gov/tools/vecscreen/) from the
National Center for Biotechnology Information. The sequence of each
EST was also edited, mainly to remove ambiguous bases and
poor-quality sequences (nucleotide sequences <100 bp). All
edited sequences were assembled into groups using SeqMan software
version 8.0 (DNASTAR, Madison, WI, USA). The processed cDNA
sequences were used to perform a BLAST search in the GenBank
database to compare all available ESTs and genes to date (11). The Basic Local Alignment Search
Tool (BLAST; blast.ncbi.nlm.nih.gov/Blast.cgi) results with
P-values <1×10−8 were generally regarded as a
significant match (12,13). A large-scale Unigene assembly of
the ESTs was initiated to identify and functionally annotate as
many unique transcripts as possible. BLAST analysis against the
Kyoto Encyclopedia of Genes and Genomes (KEGG) database, and
protein and nucleic acid databases was conducted for examination of
biological functions. The ESTs homologous to known proteins were
further annotated for Gene Ontology (GO; www.geneontology.org) terms and the GO analysis was
carried out using WEB-based GEne SeT AnaLysis Toolkit (WebGestalt;
www.webgestalt.org/option.php)
(14,15).
Results
Construction of cDNA library
Obtaining an adequate quantity of high quality mRNA
initially is the key to yielding a sufficient quantity of
first-strand full-length cDNA by reverse transcription. In the
present study, total RNA was extracted from the tissues of
earthworms. As shown in Fig. 1,
28s and 18s bands were clearly visible in the electrophoresis gel
of total RNA, indicating that the total RNA was obtained. The
optical density (OD)260/OD280 ratio for the
total RNA was 2.04, well within the range of 1.8–2.1, indicating
that the isolated total RNA was suitable for cDNA library
construction.
Once the double-stranded cDNA was synthesized as
described in the Materials and methods, the present study
determined the size distribution of the products. Diffuse strips
between 0.5–4.0 kb could be detected by 1% agarose gel
electrophoresis, which indicated that double-stranded cDNAs were
successfully synthesized. A cDNA library of 4.12×105
clones was obtained and half of the bacteria were cultured for
amplification, which produced a total of 1.4×1011
clones. Several colonies were selected, and the inserted sequences
in the plasmids were amplified by PCR using T3 and T7 primers. The
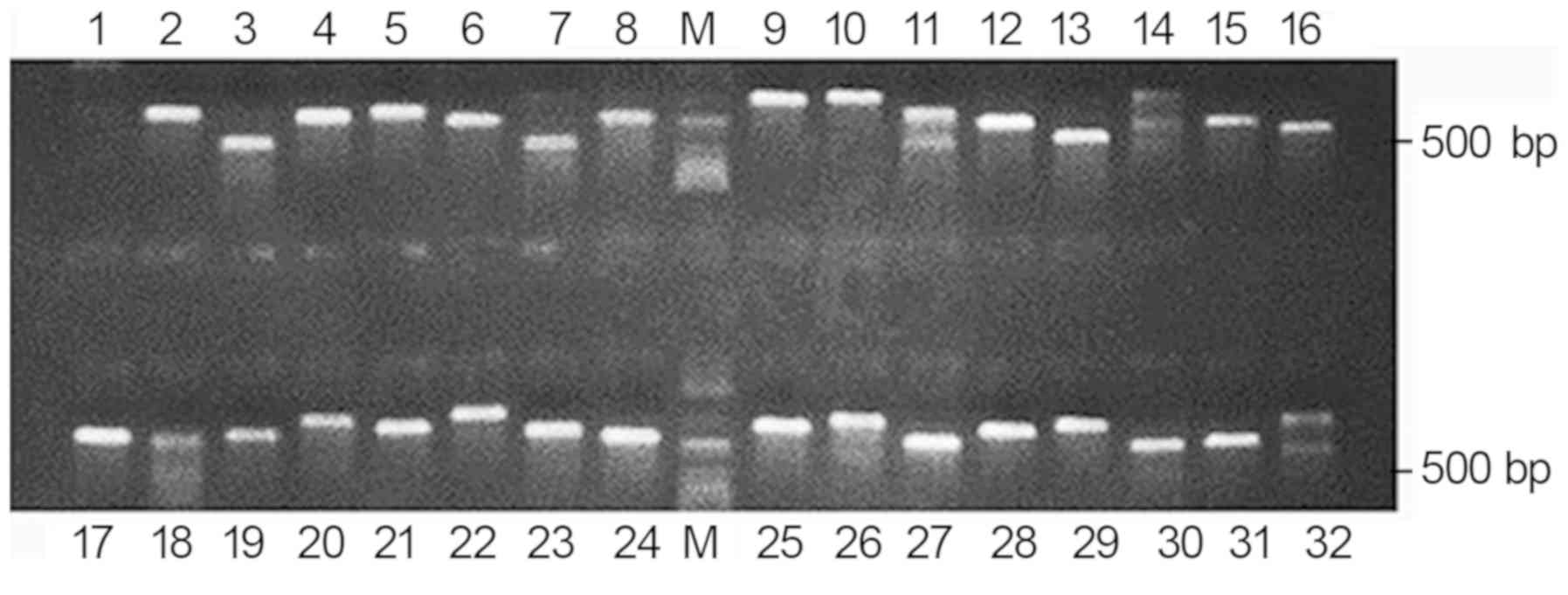
PCR products were detected using 1% agarose gel electrophoresis as
clear bands. No nonspecific bands were identified and the
recombinant rate was 97% (Fig.
2).
To investigate the quality of the full-length cDNA
library, the lengths of the cDNA inserts were assessed. Sequence
outputs were manually edited to remove vector and ambiguous
sequences. Then, the sequence data of the cDNA clones obtained by
random partial sequencing were searched in the NCBI GenBank using
BLAST to identify similarities with sequences in the nucleic acid
databases. An evaluation of cDNA insert size and its distribution
revealed a low level of insert size bias in the final cDNA library.
The majority of the cDNA inserts were larger than 500 bp.
EST analysis
Instead of the amplified library, the primary cDNA
library was used to generate ESTs to reduce the redundancy of cDNA
clones. Following the removal of the redundant sequences and
low-quality sequences (<100 bp), 1,148 effective sequences
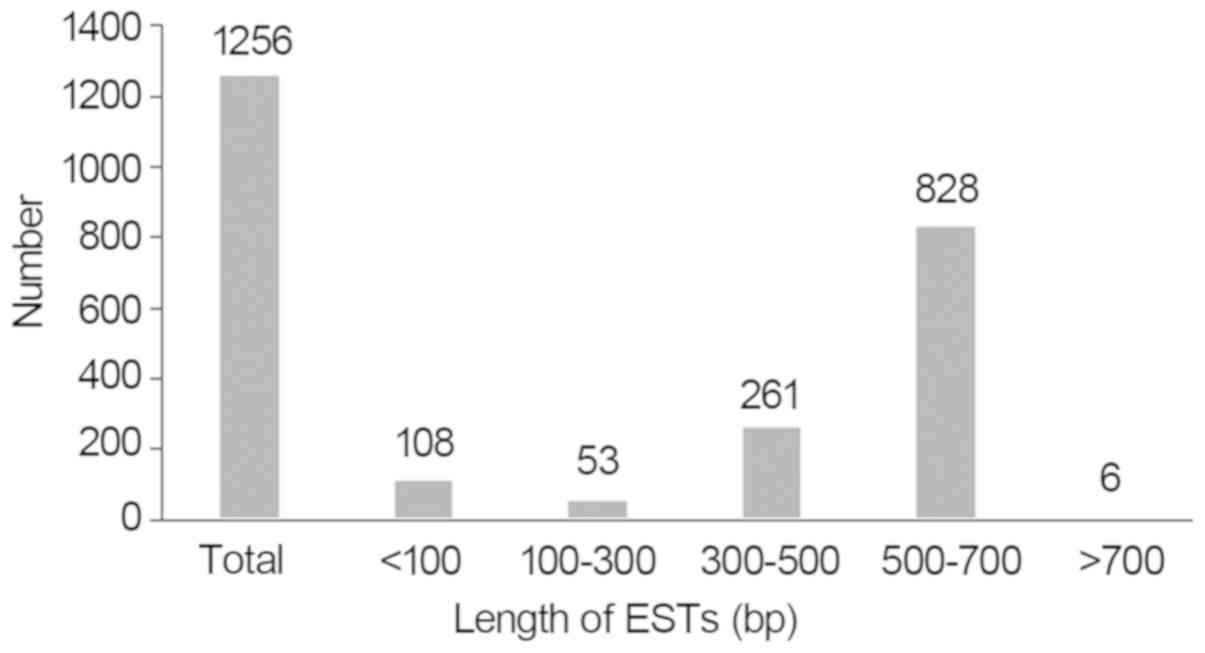
(>100 bp) from the total cDNA sequences were obtained. As shown
in Fig. 3, 53 ESTs were 100–300
bp, 261 ESTs were 300–500 bp, 828 ESTs were 500–700 bp and 6 ESTs
were larger than 700 bp. Taken together, 1,148 ESTs were larger
than 100 bp. Among them, the shortest sequence was 100 bp, the
longest was 718 bp and the average length was ~452 bp. Following
sequencing, a homology BLAST search and assembling of the data, 368
singletons and 225 contigs were obtained out of the 1,140
high-quality ESTs, as shown in Table
I. Additionally, a total of 593 individual ESTs were analyzed
and 168 ESTs annotated in GenBank with nematode homology (Table I).
 | Table I.Summary of ESTs obtained from the
cDNA library of earthworms. |
Table I.
Summary of ESTs obtained from the
cDNA library of earthworms.
| ESTs | Number |
|---|
| Total number of
ESTs | 1,256 |
| Total length of
ESTs (bp) | 568,140 |
| Average length of
ESTs (bp) | 452.34 |
| Unique genes | 593 |
| Contigs | 225 |
| Singletons | 368 |
| Annotation | 168 |
GO annotation and bioinformatic
analysis
The cDNA functions were classified using the GO
database into the three main categories of molecular functions,
cell components and biological processes.
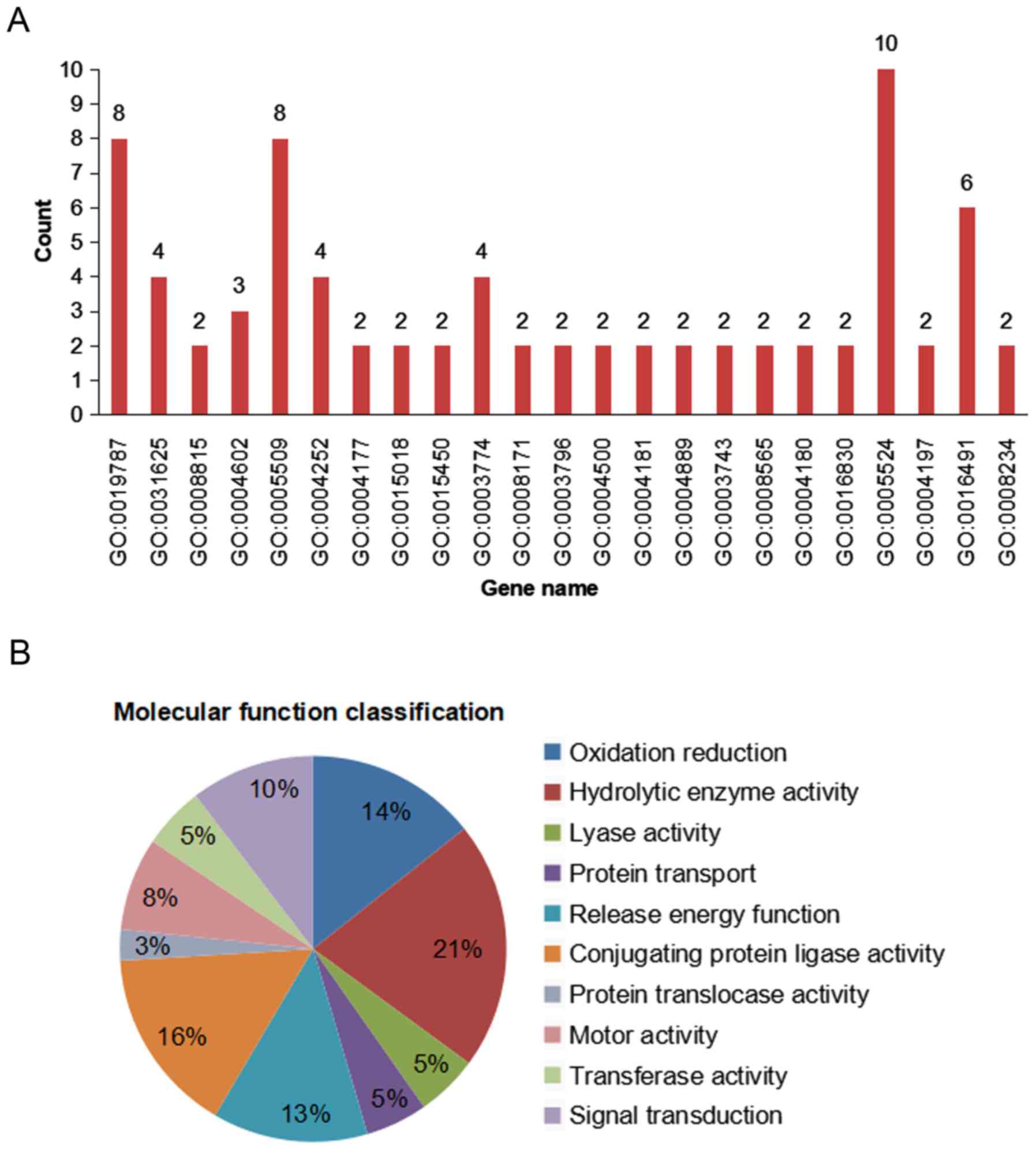
GO annotation of genes associated with molecular
functions indicated that among the 168 ESTs, 46% (77/168 ESTs) were
associated with growth and metabolic pathways, with the
distribution of the 77 ESTs shown in Fig. 4A. Out of the 77 ESTs, 21% (16 ESTs)
were associated with ‘proteolytic enzymes’, 16% (12 ESTs) with
‘protein ligases’, 14% (11 ESTs) with ‘oxido-reductases’, 13% (10
ESTs) with ‘energy release’, 10% (8 ESTs) with ‘signal transduction
and cell communication’, 5% (4 ESTs) with ‘transport’ and only 3%
(2 ESTs) with ‘post-translational modification’, ‘protein turnover’
and ‘chaperones’ (Fig. 4B).
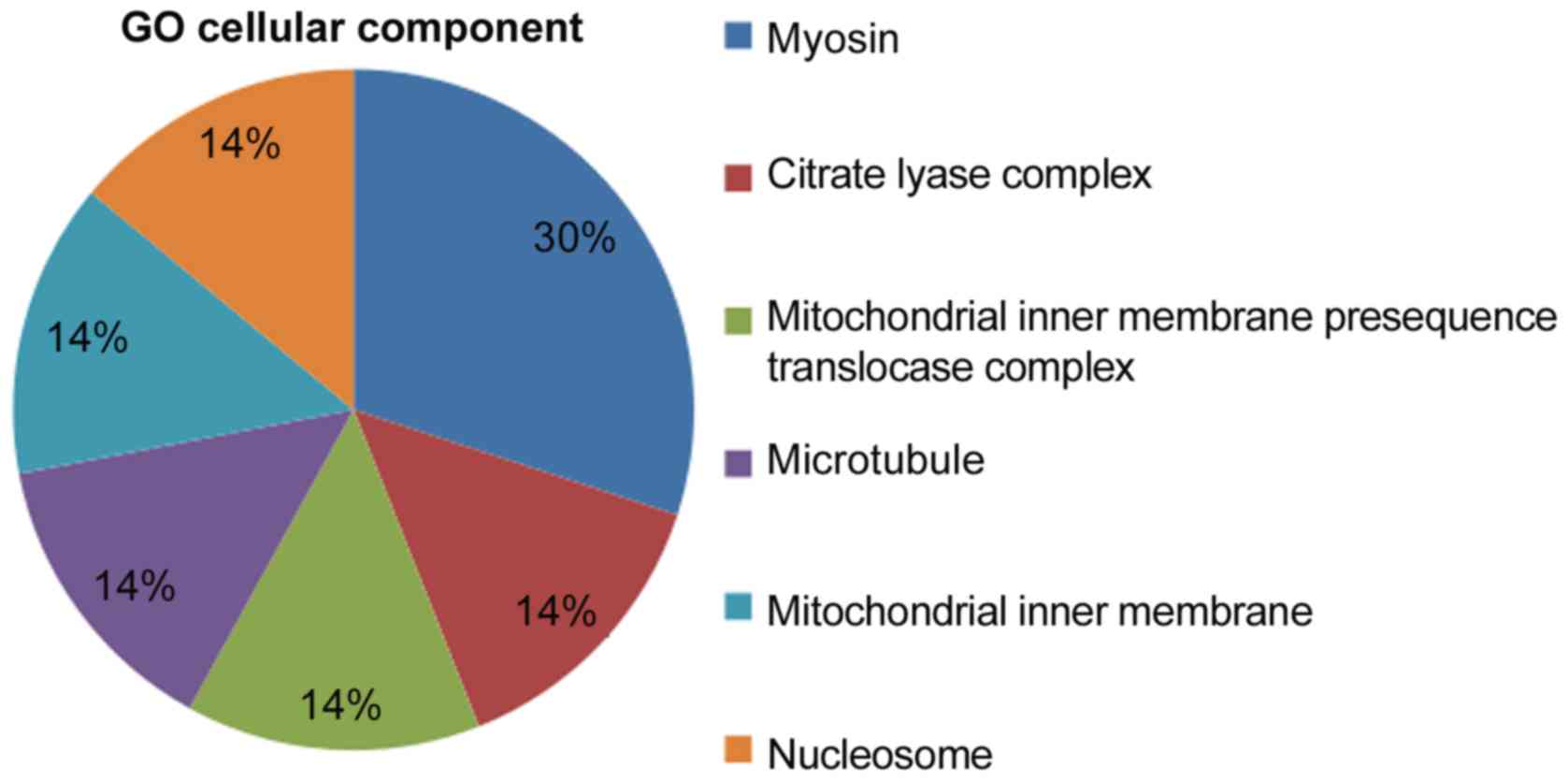
Cellular components associated with the cDNAs
included ‘myosin’, the ‘citrate lyase compound’, the ‘mitochondrial
inner membrane translocase compound’, ‘microtubules’, the
‘mitochondrial inner membrane’ and ‘nucleosomes’. The proportions
of cellular components are presented in Fig. 5. It can be observed that the
proportion of ‘myosin’ among cellular components was the largest.
Myosin, an actin-dependent molecular motor, is involved in a number
of important functions in earthworms. In particular, the myosin
network can drive movement and support different moving speeds of
earthworms. This specific feature is closely related to the free
moving ability of earthworms (16).
Regarding biological processes, known genes were
determined as those presenting significant matches to protein
sequences with known functions in non-redundant nucleotide
databases. According to these biological functions, the biological
processes component was divided into different functions including
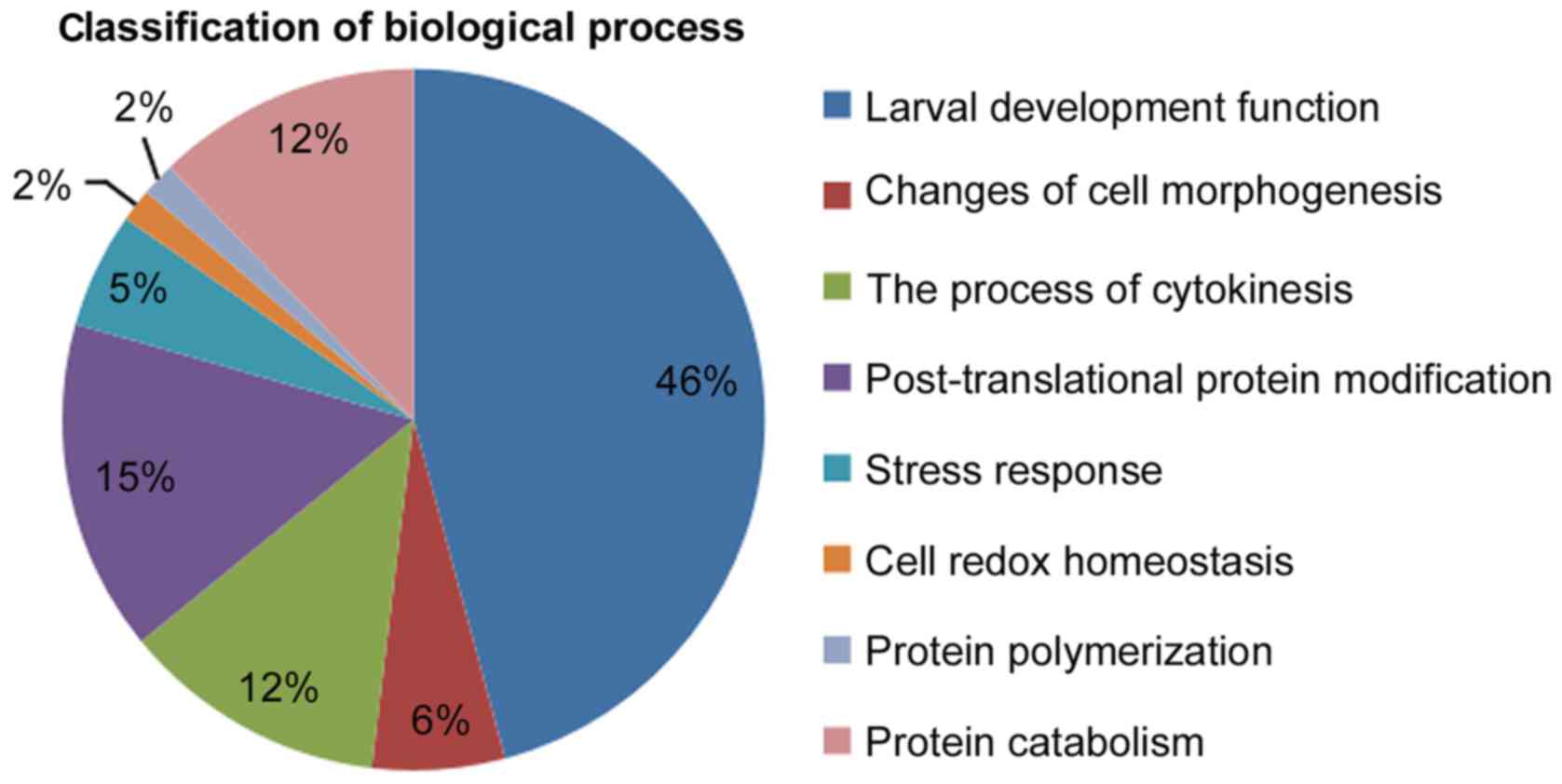
‘larval development’ (46%), ‘changes of cell morphogenesis’ (6%),
‘the process of cytokinesis’ (12%), ‘post-translational protein
modification’ (15%), ‘stress response’ (5%), ‘cell redox
homeostasis’ (2%), ‘protein polymerization’ (2%) and ‘protein
catabolism’ (12%), as shown in Fig.
6. It was concluded from the above data that promoting growth
was considered to be an important biological function of genes
associated with biological processes.
KEGG pathway annotation
KEGG is a collection of online databases describing
pathways associated with biochemical, genomic and enzymatic
processes. Furthermore, it provides annotations of biochemical
pathways for the species in which the genome has been sequenced
(17). In this analysis, proteins
are not viewed as individual gene products but are organized into
pathways and networks according to their biological function(s). In
the present study, from the data in Table II, 15 of the 168 ESTs were
revealed to be involved in metabolism. Notably, 4 of the 15 ESTs
(27%) were involved in the ubiquitin-mediated proteolysis pathway,
which was the most represented. The glutathione metabolism (3 ESTs)
and arachidonic acid metabolism (2 ESTs) pathways were the second
and third most represented pathways, respectively. Additionally,
chondroitin sulfate biosynthesis, heparan sulfate biosynthesis,
riboflavin metabolism, selenoamino acid metabolism,
γ-hexachlorocyclohexane degradation, and the fructose and mannose
metabolism pathways were also represented.
| Table II.Pathway analysis based on the Kyoto
Encyclopedia of Genes and Genomes database. |
Table II.
Pathway analysis based on the Kyoto
Encyclopedia of Genes and Genomes database.
| Pathway | Count | P-value | Q-value | Protein | Input symbol |
|---|
| Ubiquitin-mediated
proteolysis | 4 |
1.12×10−5 |
1.73×10−4 | ubc-14;ubc-13;
ubc-18;let-70 | Y87G2A.9;Y54G2A.31;
R01H2.6;M7.1 |
| Glutathione
metabolism | 3 |
2.55×10−5 |
2.63×10−4 | W07G4.4;F26E4.12;
C11E4.1 | W07G4.4;F26E4.12;
C11E4.1 |
| Arachidonic acid
metabolism | 2 |
2.01×10−4 |
1.25×10−3 | F26E4.12;
C11E4.1 | F26E4.12;
C11E4.1 |
| Chondroitin sulfate
biosynthesis | 1 |
6.54×10−3 |
2.03×10−2 | sqv-8 | ZK1307.5 |
| Heparan sulfate
biosynthesis | 1 |
1.14×10−2 |
2.75×10−2 | sqv-8 | ZK1307.5 |
| Riboflavin
metabolism | 1 |
1.14×10−2 |
2.75×10−2 | F02E9.7 | F02E9.7 |
| Selenoamino acid
metabolism | 1 |
3.39×10−2 |
4.62×10−2 | seld-1 | Y45F10A.4 |
|
γ-hexachlorocyclohexane degradation | 1 |
4.02×10−2 |
4.62×10−2 | F02E9.7 | F02E9.7 |
| Fructose and
mannose metabolism | 1 |
4.18×10−2 |
4.62×10−2 | R04B5.5 | R04B5.5 |
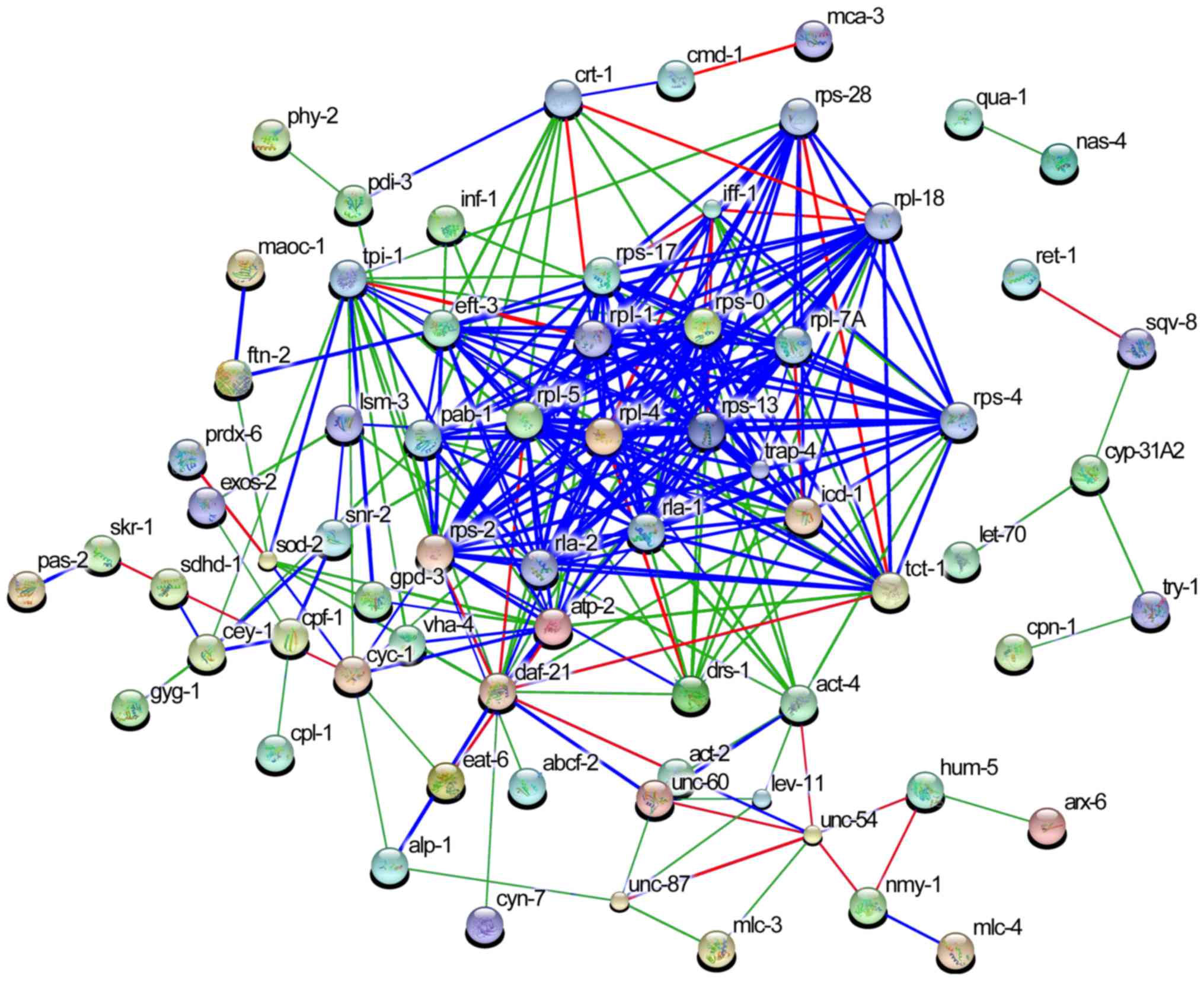
Mutual relationship between 287
genes
Relationships among the 168 ESTs were analyzed using
the STRING database, and a functional association network was
determined with 287 nodes, as shown in Table III. Each node corresponds to a
gene and each (weighted) edge represents the evidence of a
functional association between the gene pair. Predicted potential
regulators are presented in Fig.
7. It can be seen from the STRING results that the most
associated nodes (blue) included the 19 genes: Ribosomal protein L
(rpl)-1, ribosomal protein S (rps)-0, rpl-4, rpl-5, rps-13, rps-2,
acidic ribosomal protein (rla)-1, translocon-associated protein-4,
transcription factor BTF3 homolog, rpl-7A, iff-1, rps-17,
elongation factor 1α3, polyadenylate-binding protein-1, rps-28,
translationally-controlled tumor protein homolog-1, rpl-18, rps-4
and adenosine triphosphate synthase subunit β mitochondrial
precursor-2. Of note, the genes rpl-4, rps-13, rpl-5, rpl-1, rps-0,
rla-1 and rpl-7A served critical roles in overall gene expression
networks.
| Table III.Relationships between the 287
associated genes, including several important parameters. |
Table III.
Relationships between the 287
associated genes, including several important parameters.
| node1 | node2 |
node1_string_id |
node2_string_id |
node1_external_id |
node2_external_id | coexpression | combined_score |
|---|
| rpl-5 | atp-2 | 502,561 | 496,479 | F54C9.5.1 | C34E10.6.3 | 0.866 | 0.871 |
| rpl-18 | snr-2 | 510,406 | 508,903 | Y45F10D.12.2 | W08E3.1 | 0.426 | 0.468 |
| trap-4 | rps-2 | 511,330 | 497,379 | Y56A3A.21.2 | C49H3.11.1 | 0.243 | 0.919 |
| sod-2 | daf-21 | 498,735 | 497,248 | F10D11.1.1 | C47E8.5.1 | 0 | 0.572 |
| rla-2 | rps-4 | 511,588 | 510,280 | Y62E10A.1.1 | Y43B11AR.4.2 | 0.981 | 0.998 |
| rpl-1 | drs-1 | 511,889 | 493,869 | Y71F9AL.13a.1 | B0464.1.1 | 0.492 | 0.523 |
| skr-1 | sdhd-1 | 501,798 | 500,588 | F46A9.5.3 | F33A8.5.2 | 0 | 0.587 |
| prdx-6 | sod-2 | 509,858 | 498,735 | Y38C1AA.11 | F10D11.1.1 | 0.233 | 0.726 |
| act-4 | rps-0 | 505,013 | 493,798 | M03F4.2a | B0393.1.1 | 0.426 | 0.425 |
| daf-21 | rps-13 | 497,248 | 495,308 | C47E8.5.1 | C16A3.9.1 | 0.402 | 0.474 |
| rpl-1 | rpl-4 | 511,889 | 493,528 | Y71F9AL.13a.1 | B0041.4.1 | 0.8 | 0.999 |
| cyc-1 | atp-2 | 497,719 | 496,479 | C54G4.8.1 | C34E10.6.3 | 0.849 | 0.873 |
| rla-1 | act-4 | 509,817 | 505,013 | Y37E3.7.2 | M03F4.2a | 0.538 | 0.538 |
| rpl-7A | rps-17 | 509,604 | 506,860 | Y24D9A.4a | T08B2.10.1 | 0.872 | 0.923 |
| rps-28 | rpl-5 | 510,228 | 502,561 | Y41D4B.5.2 | F54C9.5.1 | 0.77 | 0.887 |
| rpl-18 | cyc-1 | 510,406 | 497,719 | Y45F10D.12.2 | C54G4.8.1 | 0.387 | 0.411 |
| tpi-1 | rps-0 | 509,457 | 493,798 | Y17G7B.7.2 | B0393.1.1 | 0.477 | 0.49 |
| daf-21 | drs-1 | 497,248 | 493,869 | C47E8.5.1 | B0464.1.1 | 0.281 | 0.543 |
| lsm-3 | rla-2 | 511,591 | 511,588 | Y62E10A.12.2 | Y62E10A.1.1 | 0 | 0.408 |
| rpl-1 | rps-4 | 511,889 | 510,280 | Y71F9AL.13a.1 | Y43B11AR.4.2 | 0.819 | 0.989 |
| tpi-1 | atp-2 | 509,457 | 496,479 | Y17G7B.7.2 | C34E10.6.3 | 0.448 | 0.877 |
| lev-11 | unc-60 | 509,147 | 496,717 | Y105E8B.1d | C38C3.5b.1 | 0 | 0.431 |
| rpl-5 | icd-1 | 502,561 | 497,806 | F54C9.5.1 | C56C10.8.1 | 0.864 | 0.87 |
| rla-2 | pab-1 | 511,588 | 509,182 | Y62E10A.1.1 | Y106G6H.2a.4 | 0.102 | 0.904 |
| unc-54 | mlc-3 | 498,853 | 498,666 | F11C3.3.1 | F09F7.2a.2 | 0.452 | 0.452 |
| alp-1 | cyc-1 | 507,123 | 497,719 | T11B7.4d | C54G4.8.1 | 0 | 0.409 |
| mca-3 | cmd-1 | 511,739 | 507,723 | Y67D8C.10b | T21H3.3.1 | 0 | 0.581 |
| cyc-1 | rps-2 | 497,719 | 497,379 | C54G4.8.1 | C49H3.11.1 | 0.579 | 0.579 |
| rpl-1 | daf-21 | 511,889 | 497,248 | Y71F9AL.13a.1 | C47E8.5.1 | 0.251 | 0.415 |
| rla-2 | iff-1 | 511,588 | 506,602 | Y62E10A.1.1 | T05G5.10 | 0.436 | 0.521 |
| rpl-1 | rla-1 | 511,889 | 509,817 | Y71F9AL.13a.1 | Y37E3.7.2 | 0.794 | 0.997 |
| tpi-1 | vha-4 | 509,457 | 506,234 | Y17G7B.7.2 | T01H3.1.1 | 0.483 | 0.483 |
| vha-4 | atp-2 | 506,234 | 496,479 | T01H3.1.1 | C34E10.6.3 | 0.304 | 0.724 |
| tpi-1 | sod-2 | 509,457 | 498,735 | Y17G7B.7.2 | F10D11.1.1 | 0.213 | 0.84 |
| rps-4 | act-4 | 510,280 | 505,013 | Y43B11AR.4.2 | M03F4.2a | 0.429 | 0.469 |
| rpl-18 | rps-2 | 510,406 | 497,379 | Y45F10D.12.2 | C49H3.11.1 | 0.999 | 0.999 |
| rpl-18 | rps-13 | 510,406 | 495,308 | Y45F10D.12.2 | C16A3.9.1 | 0.999 | 0.999 |
| rps-17 | tct-1 | 506,860 | 499,979 | T08B2.10.1 | F25H2.11.2 | 0.874 | 0.884 |
| nmy-1 | mlc-4 | 502,225 | 497,828 | F52B10.1 | C56G7.1.2 | 0 | 0.698 |
| drs-1 | rpl-4 | 493,869 | 493,528 | B0464.1.1 | B0041.4.1 | 0.581 | 0.74 |
| rpl-5 | rps-0 | 502,561 | 493,798 | F54C9.5.1 | B0393.1.1 | 0.999 | 0.999 |
| sdhd-1 | cey-1 | 500,588 | 500,586 | F33A8.5.2 | F33A8.3.2 | 0.224 | 0.624 |
| rpl-18 | rps-28 | 510,406 | 510,228 | Y45F10D.12.2 | Y41D4B.5.2 | 0.78 | 0.928 |
| rps-28 | rps-13 | 510,228 | 495,308 | Y41D4B.5.2 | C16A3.9.1 | 0.794 | 0.979 |
| pab-1 | rpl-5 | 509,182 | 502,561 | Y106G6H.2a.4 | F54C9.5.1 | 0.762 | 0.978 |
| rps-4 | icd-1 | 510,280 | 497,806 | Y43B11AR.4.2 | C56C10.8.1 | 0.826 | 0.833 |
| eft-3 | atp-2 | 505,324 | 496,479 | R03G5.1a.2 | C34E10.6.3 | 0.478 | 0.602 |
| iff-1 | rps-0 | 506,602 | 493,798 | T05G5.10 | B0393.1.1 | 0.604 | 0.623 |
| rpl-18 | pab-1 | 510,406 | 509,182 | Y45F10D.12.2 | Y106G6H.2a.4 | 0.522 | 0.95 |
| iff-1 | rpl-4 | 506,602 | 493,528 | T05G5.10 | B0041.4.1 | 0.603 | 0.656 |
| rla-2 | rpl-18 | 511,588 | 510,406 | Y62E10A.1.1 | Y45F10D.12.2 | 0.967 | 0.997 |
| crt-1 | rpl-5 | 509,852 | 502,561 | Y38A10A.5.1 | F54C9.5.1 | 0.452 | 0.465 |
| rps-28 | tpi-1 | 510,228 | 509,457 | Y41D4B.5.2 | Y17G7B.7.2 | 0.278 | 0.451 |
| rps-4 | iff-1 | 510,280 | 506,602 | Y43B11AR.4.2 | T05G5.10 | 0.64 | 0.888 |
| icd-1 | rps-13 | 497,806 | 495,308 | C56C10.8.1 | C16A3.9.1 | 0.856 | 0.856 |
| trap-4 | rla-1 | 511,330 | 509,817 | Y56A3A.21.2 | Y37E3.7.2 | 0.176 | 0.923 |
| nmy-1 | unc-54 | 502,225 | 498,853 | F52B10.1 | F11C3.3.1 | 0.102 | 0.565 |
| trap-4 | rps-13 | 511,330 | 495,308 | Y56A3A.21.2 | C16A3.9.1 | 0.148 | 0.942 |
| iff-1 | rps-13 | 506,602 | 495,308 | T05G5.10 | C16A3.9.1 | 0.614 | 0.614 |
| maoc-1 | ftn-2 | 498,218 | 497,902 | E04F6.3 | D1037.3.4 | 0.997 | 0.996 |
| rpl-18 | eft-3 | 510,406 | 505,324 | Y45F10D.12.2 | R03G5.1a.2 | 0.428 | 0.938 |
| exos-2 | cpf-1 | 512,052 | 500,247 | Y73B6BL.3 | F28C6.3 | 0.403 | 0.403 |
| rla-2 | rla-1 | 511,588 | 509,817 | Y62E10A.1.1 | Y37E3.7.2 | 0.999 | 0.999 |
| rla-2 | eft-3 | 511,588 | 505,324 | Y62E10A.1.1 | R03G5.1a.2 | 0.104 | 0.91 |
| iff-1 | rps-2 | 506,602 | 497,379 | T05G5.10 | C49H3.11.1 | 0.696 | 0.903 |
| rpl-7A | rpl-4 | 509,604 | 493,528 | Y24D9A.4a | B0041.4.1 | 0.955 | 0.996 |
| tpi-1 | daf-21 | 509,457 | 497,248 | Y17G7B.7.2 | C47E8.5.1 | 0.313 | 0.404 |
| crt-1 | rps-0 | 509,852 | 493,798 | Y38A10A.5.1 | B0393.1.1 | 0.402 | 0.462 |
| rpl-7A | iff-1 | 509,604 | 506,602 | Y24D9A.4a | T05G5.10 | 0.477 | 0.536 |
| pab-1 | rps-17 | 509,182 | 506,860 | Y106G6H.2a.4 | T08B2.10.1 | 0.217 | 0.936 |
| tpi-1 | rps-17 | 509,457 | 506,860 | Y17G7B.7.2 | T08B2.10.1 | 0.341 | 0.412 |
| act-4 | rpl-4 | 505,013 | 493,528 | M03F4.2a | B0041.4.1 | 0.471 | 0.572 |
| crt-1 | rps-2 | 509,852 | 497,379 | Y38A10A.5.1 | C49H3.11.1 | 0.507 | 0.523 |
| rpl-7A | tct-1 | 509,604 | 499,979 | Y24D9A.4a | F25H2.11.2 | 0.793 | 0.794 |
| exos-2 | lsm-3 | 512,052 | 511,591 | Y73B6BL.3 | Y62E10A.12.2 | 0.164 | 0.449 |
| rla-1 | icd-1 | 509,817 | 497,806 | Y37E3.7.2 | C56C10.8.1 | 0.835 | 0.835 |
| crt-1 | cmd-1 | 509,852 | 507,723 | Y38A10A.5.1 | T21H3.3.1 | 0.116 | 0.609 |
| pab-1 | atp-2 | 509,182 | 496,479 | Y106G6H.2a.4 | C34E10.6.3 | 0.432 | 0.432 |
| inf-1 | rpl-5 | 503,072 | 502,561 | F57B9.6a.3 | F54C9.5.1 | 0.409 | 0.409 |
| trap-4 | rps-0 | 511,330 | 493,798 | Y56A3A.21.2 | B0393.1.1 | 0.174 | 0.911 |
| rpl-18 | atp-2 | 510,406 | 496,479 | Y45F10D.12.2 | C34E10.6.3 | 0.476 | 0.659 |
| rps-2 | drs-1 | 497,379 | 493,869 | C49H3.11.1 | B0464.1.1 | 0.664 | 0.663 |
| rpl-5 | drs-1 | 502,561 | 493,869 | F54C9.5.1 | B0464.1.1 | 0.332 | 0.403 |
| rpl-5 | daf-21 | 502,561 | 497,248 | F54C9.5.1 | C47E8.5.1 | 0.559 | 0.676 |
| rpl-18 | rpl-5 | 510,406 | 502,561 | Y45F10D.12.2 | F54C9.5.1 | 0.999 | 0.999 |
| trap-4 | rpl-5 | 511,330 | 502,561 | Y56A3A.21.2 | F54C9.5.1 | 0.273 | 0.949 |
| snr-2 | rps-0 | 508,903 | 493,798 | W08E3.1 | B0393.1.1 | 0.35 | 0.414 |
| unc-54 | daf-21 | 498,853 | 497,248 | F11C3.3.1 | C47E8.5.1 | 0 | 0.674 |
| rpl-18 | rps-0 | 510,406 | 493,798 | Y45F10D.12.2 | B0393.1.1 | 0.999 | 0.999 |
| rpl-7A | icd-1 | 509,604 | 497,806 | Y24D9A.4a | C56C10.8.1 | 0.737 | 0.756 |
| tpi-1 | rps-2 | 509,457 | 497,379 | Y17G7B.7.2 | C49H3.11.1 | 0.484 | 0.521 |
| qua-1 | nas-4 | 506,538 | 494,442 | T05C12.10 | C05D11.6 | 0.42 | 0.419 |
| try-1 | cpn-1 | 513,205 | 501,580 | ZK546.15 | F43G9.9.1 | 0.408 | 0.408 |
| rla-1 | iff-1 | 509,817 | 506,602 | Y37E3.7.2 | T05G5.10 | 0.551 | 0.691 |
| rpl-18 | iff-1 | 510,406 | 506,602 | Y45F10D.12.2 | T05G5.10 | 0.564 | 0.577 |
| pab-1 | eft-3 | 509,182 | 505,324 | Y106G6H.2a.4 | R03G5.1a.2 | 0.398 | 0.708 |
| abcf-2 | rps-2 | 508,270 | 497,379 | T27E9.7.1 | C49H3.11.1 | 0.268 | 0.452 |
| tct-1 | rps-0 | 499,979 | 493,798 | F25H2.11.2 | B0393.1.1 | 0.987 | 0.987 |
| alp-1 | daf-21 | 507,123 | 497,248 | T11B7.4d | C47E8.5.1 | 0 | 0.998 |
| daf-21 | rps-0 | 497,248 | 493,798 | C47E8.5.1 | B0393.1.1 | 0.44 | 0.445 |
| lev-11 | act-4 | 509,147 | 505,013 | Y105E8B.1d | M03F4.2a | 0.244 | 0.441 |
| daf-21 | atp-2 | 497,248 | 496,479 | C47E8.5.1 | C34E10.6.3 | 0.415 | 0.745 |
| rla-1 | tct-1 | 509,817 | 499,979 | Y37E3.7.2 | F25H2.11.2 | 0.998 | 0.999 |
| rps-17 | iff-1 | 506,860 | 506,602 | T08B2.10.1 | T05G5.10 | 0.569 | 0.678 |
| rps-28 | rla-1 | 510,228 | 509,817 | Y41D4B.5.2 | Y37E3.7.2 | 0.785 | 0.963 |
| try-1 | cyp-31A2 | 513,205 | 503,484 | ZK546.15 | H02I12.8 | 0.543 | 0.543 |
| rpl-1 | rpl-5 | 511,889 | 502,561 | Y71F9AL.13a.1 | F54C9.5.1 | 0.799 | 0.995 |
| rps-28 | rps-0 | 510,228 | 493,798 | Y41D4B.5.2 | B0393.1.1 | 0.76 | 0.966 |
| act-4 | unc-54 | 505,013 | 498,853 | M03F4.2a | F11C3.3.1 | 0.168 | 0.474 |
| rpl-5 | tct-1 | 502,561 | 499,979 | F54C9.5.1 | F25H2.11.2 | 0.996 | 0.996 |
| rla-1 | rps-13 | 509817 | 495,308 | Y37E3.7.2 | C16A3.9.1 | 0.999 | 0.999 |
| rpl-1 | rps-17 | 511,889 | 5068,60 | Y71F9AL.13a.1 | T08B2.10.1 | 0.799 | 0.993 |
| rpl-1 | rla-2 | 511,889 | 511,588 | Y71F9AL.13a.1 | Y62E10A.1.1 | 0.772 | 0.996 |
| cey-1 | cpf-1 | 500,586 | 500,247 | F33A8.3.2 | F28C6.3 | 0 | 0.904 |
| pab-1 | rps-13 | 509,182 | 495,308 | Y106G6H.2a.4 | C16A3.9.1 | 0.327 | 0.928 |
| sqv-8 | ret-1 | 512,932 | 508,785 | ZK1307.5 | W06A7.3f | 0 | 0.579 |
| rps-28 | rpl-7A | 510,228 | 509,604 | Y41D4B.5.2 | Y24D9A.4a | 0.752 | 0.838 |
| icd-1 | rps-0 | 497,806 | 493,798 | C56C10.8.1 | B0393.1.1 | 0.875 | 0.88 |
| rla-1 | tpi-1 | 509,817 | 509,457 | Y37E3.7.2 | Y17G7B.7.2 | 0.501 | 0.501 |
| icd-1 | rps-2 | 497,806 | 497,379 | C56C10.8.1 | C49H3.11.1 | 0.913 | 0.913 |
| sod-2 | atp-2 | 498,735 | 496,479 | F10D11.1.1 | C34E10.6.3 | 0.271 | 0.595 |
| hum-5 | arx-6 | 506,256 | 496,567 | T02C12.1 | C35D10.16 | 0 | 0.421 |
| rla-1 | eft-3 | 509,817 | 505,324 | Y37E3.7.2 | R03G5.1a.2 | 0.189 | 0.92 |
| iff-1 | rpl-5 | 506,602 | 502,561 | T05G5.10 | F54C9.5.1 | 0.58 | 0.61 |
| atp-2 | rpl-4 | 496,479 | 493,528 | C34E10.6.3 | B0041.4.1 | 0.828 | 0.835 |
| rpl-5 | rps-13 | 502,561 | 495,308 | F54C9.5.1 | C16A3.9.1 | 0.999 | 0.999 |
| unc-87 | unc-60 | 498,495 | 496,717 | F08B6.4a | C38C3.5b.1 | 0 | 0.407 |
| mlc-3 | unc-87 | 498,666 | 498,495 | F09F7.2a.2 | F08B6.4a | 0.516 | 0.516 |
| gpd-3 | atp-2 | 504,646 | 496,479 | K10B3.7.2 | C34E10.6.3 | 0.214 | 0.523 |
| rpl-5 | rps-2 | 502,561 | 497,379 | F54C9.5.1 | C49H3.11.1 | 0.999 | 0.999 |
| tct-1 | icd-1 | 499,979 | 497,806 | F25H2.11.2 | C56C10.8.1 | 0.746 | 0.751 |
| rpl-1 | rps-2 | 511,889 | 497,379 | Y71F9AL.13a.1 | C49H3.11.1 | 0.798 | 0.995 |
| pab-1 | icd-1 | 509,182 | 497,806 | Y106G6H.2a.4 | C56C10.8.1 | 0.458 | 0.458 |
| rps-4 | rps-13 | 510,280 | 495,308 | Y43B11AR.4.2 | C16A3.9.1 | 0.999 | 0.999 |
| rps-4 | rpl-7A | 510,280 | 509,604 | Y43B11AR.4.2 | Y24D9A.4a | 0.949 | 0.989 |
| pab-1 | rpl-4 | 509,182 | 493,528 | Y106G6H.2a.4 | B0041.4.1 | 0.69 | 0.966 |
| rps-17 | rps-0 | 506,860 | 493,798 | T08B2.10.1 | B0393.1.1 | 0.999 | 0.999 |
| rps-17 | icd-1 | 506,860 | 497,806 | T08B2.10.1 | C56C10.8.1 | 0.856 | 0.861 |
| rpl-7A | drs-1 | 509,604 | 493,869 | Y24D9A.4a | B0464.1.1 | 0.451 | 0.519 |
| rpl-18 | rla-1 | 510,406 | 509,817 | Y45F10D.12.2 | Y37E3.7.2 | 0.996 | 0.999 |
| rpl-1 | rpl-18 | 511,889 | 510,406 | Y71F9AL.13a.1 | Y45F10D.12.2 | 0.832 | 0.993 |
| daf-21 | unc-60 | 497,248 | 496,717 | C47E8.5.1 | C38C3.5b.1 | 0 | 0.911 |
| sqv-8 | cyp-31A2 | 512,932 | 503,484 | ZK1307.5 | H02I12.8 | 0.411 | 0.411 |
| act-4 | rps-13 | 505,013 | 495,308 | M03F4.2a | C16A3.9.1 | 0.53 | 0.53 |
| tct-1 | rps-13 | 499,979 | 495,308 | F25H2.11.2 | C16A3.9.1 | 0.996 | 0.996 |
| crt-1 | eft-3 | 509,852 | 505,324 | Y38A10A.5.1 | R03G5.1a.2 | 0.573 | 0.573 |
| rps-2 | daf-21 | 497,379 | 497,248 | C49H3.11.1 | C47E8.5.1 | 0.658 | 0.721 |
| let-70 | cyp-31A2 | 505,189 | 503,484 | M7.1 | H02I12.8 | 0 | 0.522 |
| rps-13 | drs-1 | 495,308 | 493,869 | C16A3.9.1 | B0464.1.1 | 0.528 | 0.528 |
| crt-1 | pdi-3 | 509,852 | 503,539 | Y38A10A.5.1 | H06O01.1.3 | 0.726 | 0.739 |
| rpl-18 | rpl-7A | 510,406 | 509,604 | Y45F10D.12.2 | Y24D9A.4a | 0.925 | 0.984 |
| rps-13 | rps-0 | 495,308 | 493,798 | C16A3.9.1 | B0393.1.1 | 0.999 | 0.999 |
| rps-4 | rpl-4 | 510,280 | 493,528 | Y43B11AR.4.2 | B0041.4.1 | 0.999 | 0.999 |
| rps-4 | rps-2 | 510,280 | 497,379 | Y43B11AR.4.2 | C49H3.11.1 | 0.999 | 0.999 |
| rla-1 | rpl-7A | 509,817 | 509,604 | Y37E3.7.2 | Y24D9A.4a | 0.872 | 0.912 |
| rps-28 | rps-17 | 510,228 | 506,860 | Y41D4B.5.2 | T08B2.10.1 | 0.805 | 0.989 |
| rpl-18 | tct-1 | 510,406 | 499,979 | Y45F10D.12.2 | F25H2.11.2 | 0.963 | 0.966 |
| rla-2 | rps-28 | 511,588 | 510,228 | Y62E10A.1.1 | Y41D4B.5.2 | 0.799 | 0.879 |
| eft-3 | rpl-4 | 505,324 | 493,528 | R03G5.1a.2 | B0041.4.1 | 0.713 | 0.97 |
| tpi-1 | rpl-4 | 509,457 | 493,528 | Y17G7B.7.2 | B0041.4.1 | 0.489 | 0.498 |
| tpi-1 | gpd-3 | 509,457 | 504,646 | Y17G7B.7.2 | K10B3.7.2 | 0.608 | 0.998 |
| unc-54 | unc-60 | 498,853 | 496,717 | F11C3.3.1 | C38C3.5b.1 | 0 | 0.587 |
| tpi-1 | cey-1 | 509,457 | 500,586 | Y17G7B.7.2 | F33A8.3.2 | 0 | 0.403 |
| hum-5 | unc-54 | 506,256 | 498,853 | T02C12.1 | F11C3.3.1 | 0 | 0.54 |
| rps-4 | pab-1 | 510,280 | 509,182 | Y43B11AR.4.2 | Y106G6H.2a.4 | 0.344 | 0.955 |
| rla-2 | icd-1 | 511,588 | 497,806 | Y62E10A.1.1 | C56C10.8.1 | 0.809 | 0.81 |
| rpl-7A | rpl-5 | 509,604 | 502,561 | Y24D9A.4a | F54C9.5.1 | 0.934 | 0.994 |
| rps-17 | eft-3 | 506,860 | 505,324 | T08B2.10.1 | R03G5.1a.2 | 0.187 | 0.916 |
| rpl-18 | rpl-4 | 510,406 | 493,528 | Y45F10D.12.2 | B0041.4.1 | 0.999 | 0.999 |
| rla-2 | daf-21 | 511,588 | 497,248 | Y62E10A.1.1 | C47E8.5.1 | 0.38 | 0.548 |
| rps-4 | tct-1 | 510,280 | 499,979 | Y43B11AR.4.2 | F25H2.11.2 | 0.941 | 0.946 |
| rps-4 | rps-28 | 510,280 | 510,228 | Y43B11AR.4.2 | Y41D4B.5.2 | 0.76 | 0.971 |
| rla-2 | rpl-5 | 511,588 | 502,561 | Y62E10A.1.1 | F54C9.5.1 | 0.967 | 0.999 |
| rps-4 | rpl-5 | 510,280 | 502,561 | Y43B11AR.4.2 | F54C9.5.1 | 0.999 | 0.999 |
| rla-1 | pab-1 | 509,817 | 509,182 | Y37E3.7.2 | Y106G6H.2a.4 | 0.29 | 0.929 |
| rla-2 | rps-2 | 511,588 | 497,379 | Y62E10A.1.1 | C49H3.11.1 | 0.991 | 0.999 |
| iff-1 | tct-1 | 506,602 | 499,979 | T05G5.10 | F25H2.11.2 | 0.483 | 0.483 |
| rla-1 | rps-2 | 509,817 | 497,379 | Y37E3.7.2 | C49H3.11.1 | 0.997 | 0.999 |
| rpl-1 | pab-1 | 511,889 | 509,182 | Y71F9AL.13a.1 | Y106G6H.2a.4 | 0.237 | 0.918 |
| rps-28 | rps-2 | 510,228 | 497,379 | Y41D4B.5.2 | C49H3.11.1 | 0.732 | 0.981 |
| rpl-5 | rpl-4 | 502,561 | 493,528 | F54C9.5.1 | B0041.4.1 | 0.999 | 0.999 |
| rps-4 | rps-17 | 510,280 | 506,860 | Y43B11AR.4.2 | T08B2.10.1 | 0.995 | 0.999 |
| rpl-18 | rps-17 | 510,406 | 506,860 | Y45F10D.12.2 | T08B2.10.1 | 0.956 | 0.996 |
| rpl-1 | rpl-7A | 511,889 | 509,604 | Y71F9AL.13a.1 | Y24D9A.4a | 0.796 | 0.96 |
| inf-1 | rps-0 | 503,072 | 493,798 | F57B9.6a.3 | B0393.1.1 | 0.414 | 0.433 |
| alp-1 | unc-87 | 507,123 | 498,495 | T11B7.4d | F08B6.4a | 0.413 | 0.421 |
| rla-1 | rpl-4 | 509,817 | 493,528 | Y37E3.7.2 | B0041.4.1 | 0.997 | 0.999 |
| rps-2 | rps-13 | 497,379 | 495,308 | C49H3.11.1 | C16A3.9.1 | 0.998 | 0.999 |
| pdi-3 | rps-2 | 503,539 | 497,379 | H06O01.1.3 | C49H3.11.1 | 0.457 | 0.457 |
| cpl-1 | cpf-1 | 506,335 | 500,247 | T03E6.7.2 | F28C6.3 | 0 | 0.408 |
| gyg-1 | cey-1 | 502,889 | 500,586 | F56B6.4a | F33A8.3.2 | 0 | 0.55 |
| hum-5 | nmy-1 | 506,256 | 502,225 | T02C12.1 | F52B10.1 | 0 | 0.544 |
| gpd-3 | sod-2 | 504,646 | 498,735 | K10B3.7.2 | F10D11.1.1 | 0 | 0.588 |
| rps-4 | tpi-1 | 510,280 | 509,457 | Y43B11AR.4.2 | Y17G7B.7.2 | 0.444 | 0.444 |
| rla-2 | trap-4 | 511,588 | 511,330 | Y62E10A.1.1 | Y56A3A.21.2 | 0.076 | 0.916 |
| rps-28 | rpl-4 | 510,228 | 493,528 | Y41D4B.5.2 | B0041.4.1 | 0.756 | 0.847 |
| drs-1 | rps-0 | 493,869 | 493,798 | B0464.1.1 | B0393.1.1 | 0.464 | 0.494 |
| sdhd-1 | cyc-1 | 500,588 | 497,719 | F33A8.5.2 | C54G4.8.1 | 0.382 | 0.54 |
| rpl-1 | icd-1 | 511,889 | 497,806 | Y71F9AL.13a.1 | C56C10.8.1 | 0.777 | 0.787 |
| rps-17 | rps-13 | 506,860 | 495,308 | T08B2.10.1 | C16A3.9.1 | 0.997 | 0.999 |
| rla-1 | rps-0 | 509,817 | 493,798 | Y37E3.7.2 | B0393.1.1 | 0.999 | 0.999 |
| daf-21 | rpl-4 | 497,248 | 493,528 | C47E8.5.1 | B0041.4.1 | 0.59 | 0.634 |
| tpi-1 | cyc-1 | 509,457 | 497,719 | Y17G7B.7.2 | C54G4.8.1 | 0.442 | 0.444 |
| trap-4 | rpl-4 | 511,330 | 493,528 | Y56A3A.21.2 | B0041.4.1 | 0.308 | 0.951 |
| rpl-1 | rps-28 | 511,889 | 510,228 | Y71F9AL.13a.1 | Y41D4B.5.2 | 0.793 | 0.939 |
| rps-28 | tct-1 | 510,228 | 499,979 | Y41D4B.5.2 | F25H2.11.2 | 0.683 | 0.683 |
| eft-3 | rpl-5 | 505,324 | 502,561 | R03G5.1a.2 | F54C9.5.1 | 0.701 | 0.974 |
| sod-2 | cyc-1 | 498,735 | 497,719 | F10D11.1.1 | C54G4.8.1 | 0.456 | 0.604 |
| pdi-3 | phy-2 | 503,539 | 500,798 | H06O01.1.3 | F35G2.4.1 | 0 | 0.4 |
| rpl-18 | daf-21 | 510,406 | 497,248 | Y45F10D.12.2 | C47E8.5.1 | 0.387 | 0.401 |
| rpl-1 | tct-1 | 511,889 | 499,979 | Y71F9AL.13a.1 | F25H2.11.2 | 0.751 | 0.757 |
| rpl-1 | iff-1 | 511,889 | 506,602 | Y71F9AL.13a.1 | T05G5.10 | 0.612 | 0.624 |
| rpl-1 | tpi-1 | 511,889 | 509,457 | Y71F9AL.13a.1 | Y17G7B.7.2 | 0.396 | 0.614 |
| rps-0 | rpl-4 | 493,798 | 493,528 | B0393.1.1 | B0041.4.1 | 0.999 | 0.999 |
| eft-3 | rps-13 | 505,324 | 495,308 | R03G5.1a.2 | C16A3.9.1 | 0.362 | 0.956 |
| act-4 | unc-60 | 505,013 | 496,717 | M03F4.2a | C38C3.5b.1 | 0 | 0.863 |
| crt-1 | pab-1 | 509,852 | 509,182 | Y38A10A.5.1 | Y106G6H.2a.4 | 0.425 | 0.453 |
| tct-1 | atp-2 | 499,979 | 496,479 | F25H2.11.2 | C34E10.6.3 | 0.575 | 0.583 |
| rps-13 | rpl-4 | 495,308 | 493,528 | C16A3.9.1 | B0041.4.1 | 0.998 | 0.999 |
| rps-2 | rpl-4 | 497,379 | 493,528 | C49H3.11.1 | B0041.4.1 | 0.999 | 0.999 |
| rla-2 | rps-17 | 511,588 | 506,860 | Y62E10A.1.1 | T08B2.10.1 | 0.997 | 0.999 |
| rla-1 | rps-17 | 509,817 | 506,860 | Y37E3.7.2 | T08B2.10.1 | 0.966 | 0.997 |
| rps-17 | rps-2 | 506,860 | 497,379 | T08B2.10.1 | C49H3.11.1 | 0.969 | 0.999 |
| cyc-1 | eat-6 | 497,719 | 493,780 | C54G4.8.1 | B0365.3.2 | 0.295 | 0.425 |
| trap-4 | rpl-18 | 511,330 | 510,406 | Y56A3A.21.2 | Y45F10D.12.2 | 0.37 | 0.956 |
| tpi-1 | rps-13 | 509,457 | 495,308 | Y17G7B.7.2 | C16A3.9.1 | 0.51 | 0.556 |
| rla-2 | rps-0 | 511,588 | 493,798 | Y62E10A.1.1 | B0393.1.1 | 0.994 | 0.999 |
| icd-1 | drs-1 | 497,806 | 493,869 | C56C10.8.1 | B0464.1.1 | 0.403 | 0.509 |
| sod-2 | ftn-2 | 498,735 | 497,902 | F10D11.1.1 | D1037.3.4 | 0 | 0.494 |
| pab-1 | cey-1 | 509,182 | 500,586 | Y106G6H.2a.4 | F33A8.3.2 | 0.317 | 0.447 |
| pab-1 | rps-0 | 509,182 | 493,798 | Y106G6H.2a.4 | B0393.1.1 | 0.546 | 0.958 |
| rla-1 | rpl-5 | 509,817 | 502,561 | Y37E3.7.2 | F54C9.5.1 | 0.996 | 0.999 |
| rps-17 | rpl-4 | 506,860 | 493,528 | T08B2.10.1 | B0041.4.1 | 0.995 | 0.999 |
| lsm-3 | rpl-4 | 511,591 | 493,528 | Y62E10A.12.2 | B0041.4.1 | 0.126 | 0.622 |
| tct-1 | rpl-4 | 499,979 | 493,528 | F25H2.11.2 | B0041.4.1 | 0.919 | 0.926 |
| icd-1 | daf-21 | 497,806 | 497,248 | C56C10.8.1 | C47E8.5.1 | 0.439 | 0.478 |
| rpl-1 | rps-0 | 511,889 | 493,798 | Y71F9AL.13a.1 | B0393.1.1 | 0.808 | 0.992 |
| rpl-1 | trap-4 | 511,889 | 511,330 | Y71F9AL.13a.1 | Y56A3A.21.2 | 0.258 | 0.92 |
| rps-4 | eft-3 | 510,280 | 505,324 | Y43B11AR.4.2 | R03G5.1a.2 | 0.402 | 0.939 |
| rps-4 | rla-1 | 510,280 | 509,817 | Y43B11AR.4.2 | Y37E3.7.2 | 0.997 | 0.999 |
| atp-2 | rps-0 | 496,479 | 493,798 | C34E10.6.3 | B0393.1.1 | 0.432 | 0.763 |
| rpl-7A | rps-2 | 509,604 | 497,379 | Y24D9A.4a | C49H3.11.1 | 0.914 | 0.978 |
| rla-2 | rpl-7A | 511,588 | 509,604 | Y62E10A.1.1 | Y24D9A.4a | 0.936 | 0.958 |
| rps-28 | icd-1 | 510,228 | 497,806 | Y41D4B.5.2 | C56C10.8.1 | 0.772 | 0.772 |
| rpl-18 | icd-1 | 510,406 | 497,806 | Y45F10D.12.2 | C56C10.8.1 | 0.857 | 0.863 |
| inf-1 | rps-2 | 503,072 | 497,379 | F57B9.6a.3 | C49H3.11.1 | 0.395 | 0.417 |
| tpi-1 | tct-1 | 509,457 | 499,979 | Y17G7B.7.2 | F25H2.11.2 | 0.428 | 0.476 |
| rps-17 | rpl-5 | 506,860 | 502,561 | T08B2.10.1 | F54C9.5.1 | 0.972 | 0.998 |
| snr-2 | cpf-1 | 508,903 | 500,247 | W08E3.1 | F28C6.3 | 0.245 | 0.919 |
| pab-1 | rps-2 | 509,182 | 497,379 | Y106G6H.2a.4 | C49H3.11.1 | 0.704 | 0.968 |
| skr-1 | pas-2 | 501,798 | 497,931 | F46A9.5.3 | D1054.2.1 | 0 | 0.899 |
| rla-2 | act-4 | 511,588 | 505,013 | Y62E10A.1.1 | M03F4.2a | 0.508 | 0.508 |
| icd-1 | rpl-4 | 497,806 | 493,528 | C56C10.8.1 | B0041.4.1 | 0.826 | 0.858 |
| eft-3 | rps-2 | 505,324 | 497,379 | R03G5.1a.2 | C49H3.11.1 | 0.66 | 0.971 |
| rpl-1 | rps-13 | 511,889 | 495,308 | Y71F9AL.13a.1 | C16A3.9.1 | 0.8 | 0.997 |
| rps-2 | atp-2 | 497,379 | 496,479 | C49H3.11.1 | C34E10.6.3 | 0.943 | 0.947 |
| unc-54 | unc-87 | 498,853 | 498,495 | F11C3.3.1 | F08B6.4a | 0.566 | 0.776 |
| tct-1 | rps-2 | 499,979 | 497,379 | F25H2.11.2 | C49H3.11.1 | 0.999 | 0.999 |
| trap-4 | rps-4 | 511,330 | 510,280 | Y56A3A.21.2 | Y43B11AR.4.2 | 0.245 | 0.921 |
| rps-28 | iff-1 | 510,228 | 506,602 | Y41D4B.5.2 | T05G5.10 | 0.432 | 0.635 |
| rla-1 | daf-21 | 509,817 | 497,248 | Y37E3.7.2 | C47E8.5.1 | 0.394 | 0.543 |
| rps-4 | crt-1 | 510,280 | 509,852 | Y43B11AR.4.2 | Y38A10A.5.1 | 0.491 | 0.5 |
| rpl-18 | crt-1 | 510,406 | 509,852 | Y45F10D.12.2 | Y38A10A.5.1 | 0.595 | 0.597 |
| rps-2 | rps-0 | 497,379 | 493,798 | C49H3.11.1 | B0393.1.1 | 0.999 | 0.999 |
| lev-11 | unc-87 | 509,147 | 498,495 | Y105E8B.1d | F08B6.4a | 0.272 | 0.454 |
| trap-4 | rps-17 | 511,330 | 506,860 | Y56A3A.21.2 | T08B2.10.1 | 0.308 | 0.929 |
| tct-1 | daf-21 | 499,979 | 497,248 | F25H2.11.2 | C47E8.5.1 | 0.648 | 0.713 |
| crt-1 | rpl-4 | 509,852 | 493,528 | Y38A10A.5.1 | B0041.4.1 | 0.472 | 0.604 |
| rpl-18 | rps-4 | 510,406 | 510,280 | Y45F10D.12.2 | Y43B11AR.4.2 | 0.999 | 0.999 |
| eft-3 | rps-0 | 505,324 | 493,798 | R03G5.1a.2 | B0393.1.1 | 0.523 | 0.959 |
| rpl-1 | eft-3 | 511,889 | 505,324 | Y71F9AL.13a.1 | R03G5.1a.2 | 0.348 | 0.948 |
| iff-1 | icd-1 | 506,602 | 497,806 | T05G5.10 | C56C10.8.1 | 0.47 | 0.47 |
| rpl-7A | rps-13 | 509,604 | 495,308 | Y24D9A.4a | C16A3.9.1 | 0.944 | 0.979 |
| eft-3 | ftn-2 | 505,324 | 497,902 | R03G5.1a.2 | D1037.3.4 | 0 | 0.999 |
| rps-4 | drs-1 | 510,280 | 493,869 | Y43B11AR.4.2 | B0464.1.1 | 0.422 | 0.434 |
| snr-2 | cey-1 | 508,903 | 500,586 | W08E3.1 | F33A8.3.2 | 0 | 0.899 |
| rpl-1 | atp-2 | 511,889 | 496,479 | Y71F9AL.13a.1 | C34E10.6.3 | 0.144 | 0.403 |
| rla-2 | rpl-4 | 511,588 | 493,528 | Y62E10A.1.1 | B0041.4.1 | 0.995 | 0.999 |
| act-2 | act-4 | 506,426 | 505,013 | T04C12.5 | M03F4.2a | 0 | 0.481 |
| cyn-7 | daf-21 | 512,151 | 497,248 | Y75B12B.2.2 | C47E8.5.1 | 0.359 | 0.408 |
| rla-2 | tct-1 | 511,588 | 499,979 | Y62E10A.1.1 | F25H2.11.2 | 0.96 | 0.963 |
| rps-4 | rps-0 | 510,280 | 493,798 | Y43B11AR.4.2 | B0393.1.1 | 0.999 | 0.999 |
| rpl-18 | drs-1 | 510,406 | 493,869 | Y45F10D.12.2 | B0464.1.1 | 0.408 | 0.474 |
| atp-2 | eat-6 | 496,479 | 493,780 | C34E10.6.3 | B0365.3.2 | 0.666 | 0.707 |
| rpl-7A | rps-0 | 509,604 | 493,798 | Y24D9A.4a | B0393.1.1 | 0.942 | 0.947 |
| lsm-3 | snr-2 | 511,591 | 508,903 | Y62E10A.12.2 | W08E3.1 | 0.323 | 0.625 |
| rla-2 | rps-13 | 511,588 | 495,308 | Y62E10A.1.1 | C16A3.9.1 | 0.996 | 0.999 |
Discussion
The present study reported on the construction of a
high-quality cDNA library from tissues of the earthworm Eisenia
fetida (Savigny, 1826), following preliminary analysis of ESTs,
putative functional analysis of the ESTs and the gene expression
pattern associated with the physiological functions of this
organism. cDNA libraries are widely used to identify genes and
splice variants and are considered to be a physical resource for
the construction of full-length clones (18,19).
In the present study, a cDNA library was utilized to provide a
molecular resource for the analysis of genes involved in the
specific biology of earthworms in terms of their development,
survival, pathogenicity and virulence. There are two main factors
to consider when assessing the quality of a cDNA library:
Representation and cDNA lengths. According to Clarke-Carbon's
formula (20), a cDNA library
should contain at least 1.7×105 independent clones to
ensure that 99% of low-abundance mRNAs will be represented in the
library (21). Furthermore, the
average length of the inserted cDNAs should be no less than 1.0 kb
to ensure the integrity of cDNAs, indicating that in the present
study the fragment sizes were effective for ensuring full-length
cDNAs in the cDNA library. Since selection bias could favor the
smaller cDNAs, the present study used fewer PCR cycles to minimize
such bias as previously suggested (10). In addition, up to 25 PCR
amplification cycles were used to generate an adequate amount of
cDNA for cloning.
The generation of ESTs is an effective and unique
approach in molecular studies as it allows for the analysis and
measurement of gene expression, as well as simultaneous discovery
of new genes. As each EST represents a copy of the functional part
of a genome, the study of ESTs is believed to be a more effective
way to discover functional genes (22). Furthermore, analysis of the
expression of a large number of genes combined with the knowledge
of their functions enables insight into the overall situation in
terms of biological processes (23), for the current purposes in
earthworms. In the present study, ~91% of the ESTs generated were
sequences with known or putative functions, while the remainder
represented unknown proteins or sequences with no similarities to
those in the databases. Although close to 600 ESTs were reported,
this is actually far from what could be considered as a ‘complete’
transcriptome (which usually includes between 15,000–20,000 to
>100,000 ESTs). Therefore, the present characterization of this
seemingly partial transcriptome may far from reflect the full
transcriptomic profile of tissues in earthworms.
A comparison of the classification of ESTs with a
C. elegans cDNA library based on their putative functions
was conducted. Based on identification of clusters via GO analysis,
168 ESTs were matched to C. elegans genes by BLASTx. It is
well known that earthworms serve significant roles in organic
matter decomposition and mineral cycling, and thus are considered
to be important contributors to soil fertility and humification
processes (24). In the present
study, hydrolytic enzyme activity, conjugating protein ligase
activity, oxidation reduction and energy release activity of
metabolic enzymes accounted for a large proportion of the molecular
functions component. These molecular functions are considered to be
a key part of the physiological functions of earthworms, which
allow earthworms to survive in different soil environments.
Cell components, as part of GO annotation, are
mainly categorized based on subcellular location (including cell
cytoplasm, mitochondria, lysosomes, nucleus, microtubules, plasma
membrane and myosin), which is highly important for the study of
protein functions (25). The
results of subcellular localization analysis can provide
significant clues to aid the understanding of protein functions. In
the present study, several cell components were determined through
GO analysis of the 168 ESTs (annotated genes). It was evident that
these cell components had strong associations with the regulation
of gene expression during the biological development of earthworms,
enabling the regeneration of the anterior portion, alterations in
movement ability and tissue differentiation.
Due to the temporal specificity of gene expression
and interactions with other gene products, the specific pathway
undertaken, sequence of gene expression and expression pattern may
ultimately change the effects of multiple pathways in earthworms
(2). Therefore, the cDNA library
and transcription profile of genes representative of fully
developed adult earthworms may differ markedly to those of juvenile
earthworms. In the present study, gene expression profiles
representative of adult earthworm development were generated;
however, gene expression profiling of juvenile earthworms was not
performed, nor were analyses of the expected differential gene
expression between juveniles and adults. In the present study, 168
individual ESTs of earthworms were analyzed in terms of KEGG
pathway annotation, which identified 9 corresponding categories.
Among them, glutathione metabolism is involved in antioxidant
defense systems in Eisenia Andrei, and the associated
enzymes are mainly identified in cytosolic fractions (26). Chondroitin sulfate and heparan
sulfate biosynthesis are involved in biosynthesis pathways, which
have important effects on growth and regeneration. In addition,
heparan sulfate is also considered to be a type of anticoagulant,
which may also be a function of arachidonic acid (27). Therefore, understanding the
molecular function of earthworms may provide some basis for the
treatment of thrombotic disorders.
Information on functional annotation and relevant
biological interactions associated with a particular gene is
available from many online resources. The gene network comprises a
collection of genes that cooperate with each other to control the
main biological processes. The STRING database suggested a
functional context for earthworm lumbrokinase with unknown
specificity. A previous study revealed that lumbrokinase and dilong
administration can efficiently reduce the incidence of cardiac
disease among nonsmokers exposed to second-hand smoke (28). In this regard, the discovery of
genes and protein interactions in earthworms has provided a basis
for further investigation into human diseases.
Acknowledgements
Not applicable.
Funding
The present study was supported by the National
Natural Science Foundation of China (grant nos. 30472251, 30901821
and 81370312), the Natural Science Foundation for Young Scientists
of Shanxi Province, China (grant no. 2010021035-2), Natural Science
Foundation of Shanxi Province, China (grant no. 2015011113),
International Scientific and Technological Cooperative Foundation
of Shanxi Province, China (grant no. 201703D421023), the Fund for
Shanxi ‘1331 Project’ Key Subjects Construction and the Fund for
Shanxi Key Subjects Construction.
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
BFY, BN and JX conceived and designed the
experiments and bioinformatics analysis. CL and FXM performed
bioinformatics analysis, and wrote the manuscript. XW, PYM, QZ and
JBT performed the experiments. RG, ZZL, HLW and NLC contributed to
designing the present study and revising the manuscript. JHW and
GQS analyzed and interpreted the data.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Glossary
Abbreviations
Abbreviations:
|
EST
|
expressed sequence tags
|
|
GO
|
Gene Ontology
|
|
KEGG
|
Kyoto Encyclopedia of Genes and
Genomes
|
References
|
1
|
Torres-Leguizamon M, Mathieu J, Decaens T
and Dupont L: Genetic structure of earthworm populations at a
regional scale: Inferences from mitochondrial and microsatellite
molecular markers in Aporrectodea icterica (Savigny 1826). PLoS
One. 9:e1015972014. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Pirooznia M, Gong P, Guan X, Inouye LS,
Yang K, Perkins EJ and Deng Y: Cloning, analysis and functional
annotation of expressed sequence tags from the Earthworm Eisenia
fetida. BMC Bioinformatics. 8 (Suppl 7):S72007. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Cho SJ, Koh KS, Lee E and Park SC:
Differential expression of threelabial genes during earthworm head
regeneration. Biosci Biotechnol Biochem. 73:2609–2614. 2014.
View Article : Google Scholar
|
|
4
|
Zheng P, Shao Q, Diao X, Li Z and Han Q:
Expression of stem cell pluripotency factors during regeneration in
the earthworm Eisenia foetida. Gene. 575:58–65. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Kim TH, Kim NS, Lim D, Lee KT, Oh JH, Park
HS, Jang GW, Kim HY, Jeon M, Choi BH, et al: Generation and
analysis of large-scale expressed sequence tags (ESTs) from a
full-length enriched cDNA library of porcine backfat tissue. BMC
Genomics. 7:362006. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Wellenreuther R, Schupp I, Poustka A and
Wiemann S; German cDNA Consortium, : SMART amplication combined
with cDNA size fractionation in order to obtain large full-length
clones. BMC Genomics. 5:362004. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Dai Y, Su W, Yang C, Song B, Li Y and Fu
Y: Development of novel polymorphic EST-SSR markers in bailinggu
(Pleurotus tuoliensis) for crossbreeding. Genes (Basel). 8(pii):
E3252017. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Liu C, Liu D, Guo Y, Lu T, Li X, Zhang M,
Ma J, Ma Y and Guan W: Construction of a full-length enriched cDNA
library and preliminary analysis of expressed sequence tags from
Bengal Tiger Panthera tigris tigris. Int J Mol Sci. 14:11072–11083.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Carninci P, Shibata Y, Hayatsu N, Itoh M,
Shiraki T, Hirozane T, Watahiki A, Shibata K, Konno H, Muramatsu M
and Hayashizaki Y: Balanced-size and long-size cloning of
full-length, cap-trapped cDNAs into vectors of the novel lambda-FLC
family allows enhanced gene discovery rate and functional analysis.
Genomics. 77:79–90. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Ling P, Wang M, Chen X and Campbell KG:
Construction and characterization of a full-length cDNA library for
the wheat stripe rust pathogen (Puccinia striiformis f. sp.
tritici). BMC Genomics. 8:1452007. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Ye J, McGinnis S and Madden TL: BLAST:
Improvements for better sequence analysis. Nucleic Acids Res 34
(Web Server Issue). W6–W9. 2006. View Article : Google Scholar
|
|
12
|
NCBI Resource Coordinators: Database
resources of the national center for biotechnology information.
Nucleic Acids Res. 41:(Database Issue). D8–D20. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Thompson JD, Higgins DG and Gibson TJ:
CLUSTAL W: Improving the sensitivity of progressive multiple
sequence alignment through sequence weighting, position-specific
gap penalties and weight matrix choice. Nucleic Acids Res.
22:4673–4680. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Wang J, Duncan D, Shi Z and Zhang B:
WEB-based GEne SeT AnaLysis Toolkit (WebGestalt): Update 2013.
Nucleic Acids Res 41 (Web Server Issue). W77–W83. 2013. View Article : Google Scholar
|
|
15
|
Zhang B, Kirov S and Snoddy J: WebGestalt:
An integrated system for exploring gene sets in various biological
contexts. Nucleic Acids Res 33 (Web Server Issue). W741–W748. 2005.
View Article : Google Scholar
|
|
16
|
Ravaux J, Hassanin A, Deutsch J, Gaill F
and Markmann- Mulisch U: Sequence analysis of the myosin regulatory
light chain gene of the vestimentiferan Riftia pachyptila. Gene.
263:141–149. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Kanehisa M, Goto S, Kawashima S and Nakaya
A: The KEGG databases at GenomeNet. Nucleic Acids Res. 30:42–46.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Wiemann S, Mehrle A, Bechtel S,
Wellenreuther R, Pepperkok R and Poustka A; German cDNA Consortium,
: cDNAs for functional genomics and proteomics: The German
Consortium. Comptes Rendus Biologies. 326:1003–1009. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Blair MW, Fernandez AC, Ishitani M, Moreta
D, Seki M, Ayling S and Shinozaki K: Construction and EST
sequencing of full-length, drought stress cDNA libraries for common
beans (Phaseolus vulgaris L.). BMC Plant Biol. 11:1712011.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Clarke L and Carbon J: A colony bank
containing synthetic CoI EI hybrid plasmids representative of the
entire E. coli genome. Cell. 9:91–99. 1976. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Li YP, Xia RX, Wang H, Li XS, Liu YQ, Wei
ZJ, Lu C and Xiang ZH: Construction of a full-length cDNA Library
from Chinese oak silkworm pupa and identification of a
KK-42-binding protein gene in relation to pupa-diapause
termination. Int J Biol Sci. 5:451–457. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Hatey F, Tosser-Klopp G,
Clouscard-Martinato K, Mulsant P and Gasser F: Expressed sequence
tags for genes: A review. Genet Selec Evol. 30:521–524. 1998.
View Article : Google Scholar
|
|
23
|
Thanh T, Chi VT, Abdullah MP, Omar H,
Noroozi M, Ky H and Napis S: Construction of cDNA library and
preliminary analysis of expressed sequence tags from green
microalga Ankistrodesmus convolutus Corda. Mol Biol Rep.
38:177–182. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Duo L, Yin L, Zhang C and Zhao S:
Ecotoxicological responses of the earthworm Eisenia fetida
to EDTA addition under turfgrass growing conditions. Chemosphere.
220:56–60. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Ashburner M, Ball CA, Blake JA, Botstein
D, Butler H, Cherry JM, Davis AP, Dolinski K, Dwight SS, Eppig JT,
et al: Gene ontology: Tool for the unification of biology. The Gene
Ontology Consortium. Nat Genet. 25:25–29. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Homa J, Stalmach M, Wilczek G and
Kolaczkowska E: Effective activation of antioxidant system by
immune-relevant factors reversely correlates with apoptosis of
Eisenia andrei coelomocytes. J Comp Physiol B. 186:417–430. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Li P, Sheng J, Liu Y, Li J, Liu J and Wang
F: Heparosan-derived heparan sulfate/heparin-like compounds: One
kind of potential therapeutic agents. Med Res Rev. 33:665–692.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Lai CH, Han CK, Shibu MA, Pai PY, Ho TJ,
Day CH, Tsai FJ, Tsai CH, Yao CH and Huang CY: Lumbrokinase from
earthworm extract ameliorates second-hand smoke-induced cardiac
fibrosis. Environ Toxicol. 30:1216–1225. 2015. View Article : Google Scholar : PubMed/NCBI
|