Introduction
The latest data released by the World Health
Organization revealed that lung cancer has the highest global
morbidity and mortality rates of all malignant tumors, and this
trend is increasing yearly (1).
Based on biological characteristics, treatment and prognosis, lung
cancer is classified as non-small-cell lung cancer (NSCLC) or
small-cell lung cancer (SCLC). NSCLC accounts for ~85% of all lung
cancer cases, of which lung squamous cell cancer (LUSC) accounts
for 20–30%, and has a five-year survival rate of <15% (2,3).
With the recent rapid development of gene detection methods and
targeted drugs, the overall survival time (OS) of patients with
NSCLC has significantly improved (4). However, not all patients benefit from
targeted therapy; in LUSC, the frequency of gene mutations
sensitive to targeted drugs is relatively low, and this is
accompanied by poor efficacy and the occurrence of drug resistance.
In addition, the prognosis and OS of patients with early-stage lung
cancer are markedly more favorable compared with those of patients
at an advanced disease stage. Therefore, the identification of
novel biomarkers and therapeutic targets is important for improving
early diagnosis, treatment strategies and prognostic detection in
patients with LUSC.
At present, the pathogenesis and progressive
mechanisms of LUSC remain unclear, though the two most important
mechanisms of tumorigenesis are gene mutations and epigenetic
alterations (5,6). Relatively few gene mutation sites
exist, particularly for early-stage patients; owing to severe
fragmentization of tumor gene fragments in the blood, gene
mutations are not suitable for the monitoring and diagnosis of
early-stage cancer, and epigenetic changes provide a more suitable
target (7,8). DNA methylation is easily detectable,
and therefore the most studied, epigenetic modification, mediating
the occurrence and development of cancer by regulating gene
expression (8–10). It has also been indicated that DNA
methylation may occur prior to gene mutation, deeming it more
suitable for the early detection of cancer. Studies examining
methylation and tumors have recently attracted increased attention,
including a series of studies concerning targeted epigenetic
therapy approaches for acute myeloid leukemia (11). Even in solid tumors, methylated or
epigenetic signatures have become an area of increasing interest,
in such malignancies as breast cancer (12), esophageal carcinoma (13,14),
epithelial ovarian (15) and liver
cancer (16). These studies
indicated that the methylation of some specific genes may affect
gene expression, and is closely associated with the diagnosis and
prognosis of some types of cancer. Therefore, the identification of
abnormal gene methylation signatures may provide a basis for the
early diagnosis, prognosis and targeted therapy for patients with
tumors.
In the context of the era of big data,
bioinformatics analysis serves an important role in the
comprehensive research of carcinomas, utilizing high-throughput
databases such as The Cancer Genome Atlas (TCGA). The present study
utilized data from TCGA, in which the genetic information profiles
and corresponding clinical data of multiple cancer types can be
effectively extracted for analysis, bridging the gap between
molecular biology research and clinical application (17,18).
Methylation-driven genes (MDGs) were the primary focus of the
present study, defined as genes of differentially methylated states
and significant predictive transcriptional function; as such, MDGs
were identified by dissecting and integrating the correlation
between methylation state and the level of gene expression.
Previous studies have confirmed that MDGs are more comprehensive
and representative tumor biomarkers compared with differentially
methylated genes (DMGs) (14). In
the present study, methylation, gene expression and patient
survival information were extracted from TCGA, and the MethylMix
algorithm and survival analysis were used to identify hub MDGs and
their prognostic signatures, with a view to providing a basis for
individualized precision treatment in patients with LUSC.
Materials and methods
Data acquisition and
preprocessing
Firstly, the DNA methylation and gene expression
quantification data of patients with LUSC were downloaded from TCGA
(https://www.cancer.gov/tcga/), along
with the corresponding clinical information, which included details
of prognostic or survival analysis. According to the TCGA data
number, the data were divided into two groups, LUSC samples and
normal samples. In these data, the normal samples were tissues
adjacent to the tumor. The Illumina Human Methylation 450 k
platform was used to transform and normalize the initial DNA
methylation data, which were expressed as β-values (range, 0–1)
corresponding with low to high methylation states (19). Gene expression quantification data
were obtained in RNA-Seq format.
Screening of differentially expressed
genes (DEGs), DMGs and MDGs in LUSC
The R edge package (http://bioconductor.org/packages/edgeR/) was used to
identify and analyze DEGs by comparing gene expression
quantification data between normal and cancerous specimens, with a
fold change (FC)=5 and adjusted P-value (padj)=0.01 as
the threshold. The limma package (http://bioinf.wehi.edu.au/limma/) was used to compare
the methylation states of normal and cancerous specimens, and DMGs
were screened out using a false discovery rate of 0.01 and
log2FC=1.
Next, MDGs were identified using the R MethylMix
package (http://bioconductor.org/packages/3.9/bioc/html/MethylMix.html).
MethylMix is a new algorithm developed by Gevaert et al
(20,21), which uses univariate b mixture
modeling to determine the methylation state of genes in cancer
samples, and the Wilcoxon rank test to categorize these into hyper-
and hypomethyled groups compared with the methylation state in
normal tissues. It may also be used to determine the correlation
between DNA methylation state and gene expression level, the
absolute value of correlation coefficient (|Cor|) representing the
degree of correlation. In the present study, the screening
conditions of MDGs with significant inverse correlation were set as
padj<0.05, log2FC=0 and Cor<-0.5.
Pathway analysis of MDGs in LUSC
To further investigate the dominating functional
pathways of MDGs in LUSC, ConsensusPathDB (http://cpdb.molgen.mpg.de/) and Cytoscape.js was
utilized to analyze and visualize the genetic interaction of
high-throughput expression data (22,23).
ConsensusPathDB is currently the most comprehensive database of
functional interaction networks, integrating the functional aspects
of genes, proteins, complexes and metabolites. Cytoscape.js is a
graph library written in JavaScript that is used as visualization
software for graph analysis (24).
P<0.05 was set as the cut-off for minimum overlap criterion.
Survival and joint survival analysis
of MDGs in LUSC
It is important to note that not all MDGs are
significantly associated with cancer prognosis. In order to improve
the understanding of the association between MDGs and patient
survival, the MDGs independently associated with prognosis,
classified as hub MDGs, were identified. Firstly, using the R
package survival, Kaplan-Meier survival analysis and the log-rank
test were conducted to determine the association between the
methylation state of MDGs and the survival of patients with LUSC.
P<0.05 was considered to indicate a statistically significant
correlation.
Owing to the complexity of tumor tissue regulation
by the combination of multiple factors, it was necessary and
important to conduct joint survival analysis between the degree of
methylation, the corresponding levels of MDG expression and patient
survival. A joint survival curve was generated using the survival
package, with P<0.05 as the cutoff value. It should be noted
that the MDGs screened out using MethylMix were all characterized
by a significant inverse correlation (Cor <-0.5). Therefore, of
the joint survival analysis, there were only two cases to determine
survival: Hypermethylation and low expression; and hypomethylation
and high expression.
Finally, using the first two steps the common genes
were identified. PMPCAP1, SOWAHC, ZNF454 AND LINC00668 were
statistically significant in the survival analysis, while PMPCAP1,
SOWAHC, ZNF454 and ADH7 were statistically significant in the joint
survival analysis. So, PMPCAP1, SOWAHC and ZNF454 were taken as the
hub MDGs.
Correlation analysis between
methylation sites and the expression of hub MDGs
Finally, to further examine the internal mechanisms
and more precise targets of the 3 identified hub MDGs, data
corresponding to the initial methylation sites of these genes were
downloaded. The present study focused on the correlation between
the methylation of abnormal methylation sites and the corresponding
gene expression of hub MDGs. Both P<0.05 and |Cor|>0.5 used
as the cut-offs for the identification of key methylation
sites.
Results
Identification of MDGs in LUSC
Firstly, a total of 370 LUSC samples and 42 normal
samples (from 372 cases) with DNA methylation data were downloaded
from TCGA database; 502 LUSC samples and 49 normal samples (from
501 cases) with gene expression quantification data were also
downloaded. Additionally, 366 of the patients with LUSC also
possessed clinical data for prognostic and survival analysis.
Secondly, the R edge and limma packages were used to compare data
between cancerous and normal samples, respectively, and to screen
out 994 DEGs and 356 DMGs. Finally, the MethylMix algorithm
(padj<0.05, log2FC=0 and Cor <-0.5) was
used to identify 30 MDGs with strong inverse correlation between
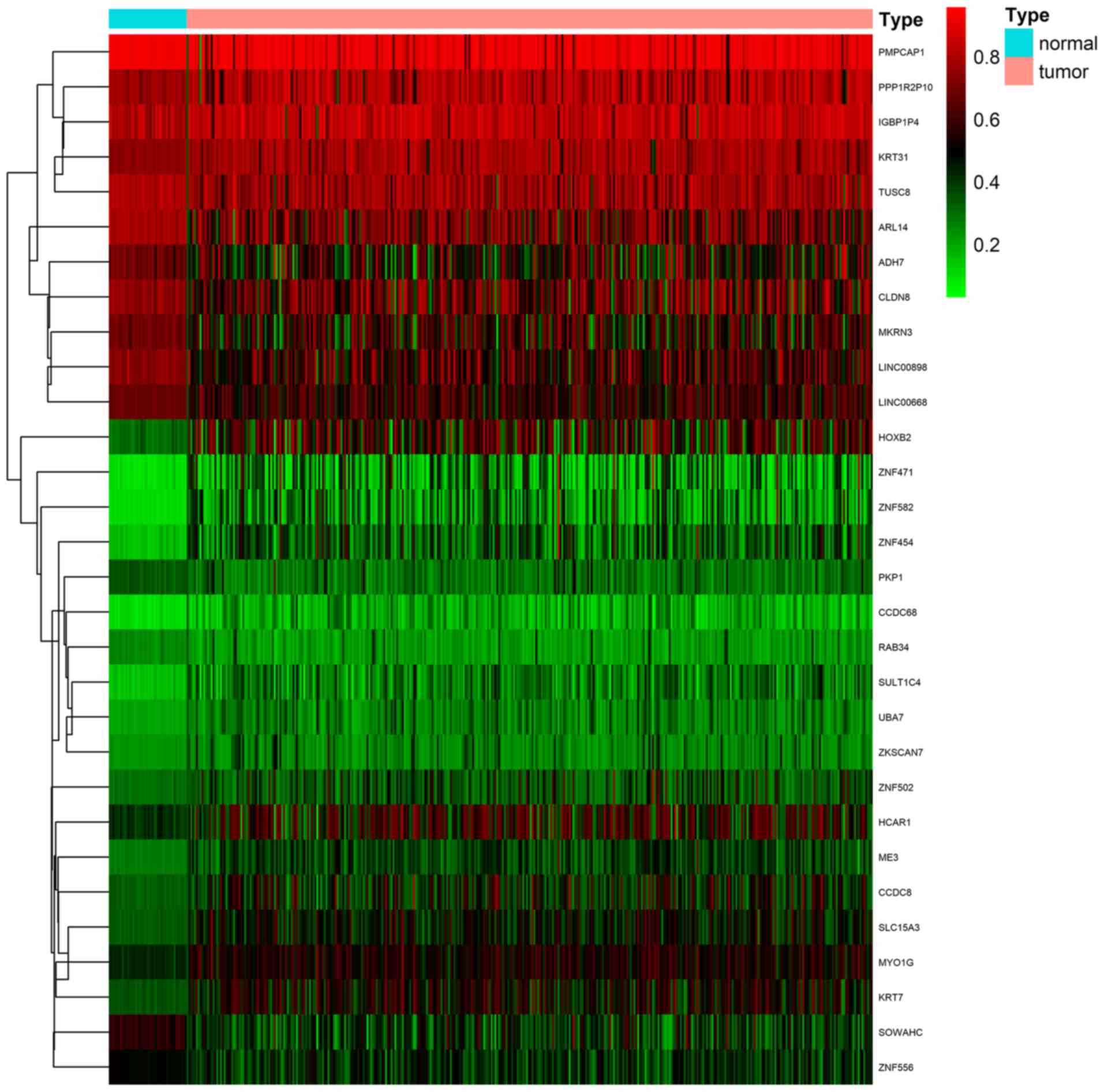
DNA methylation state and gene expression (Fig. 1; Table
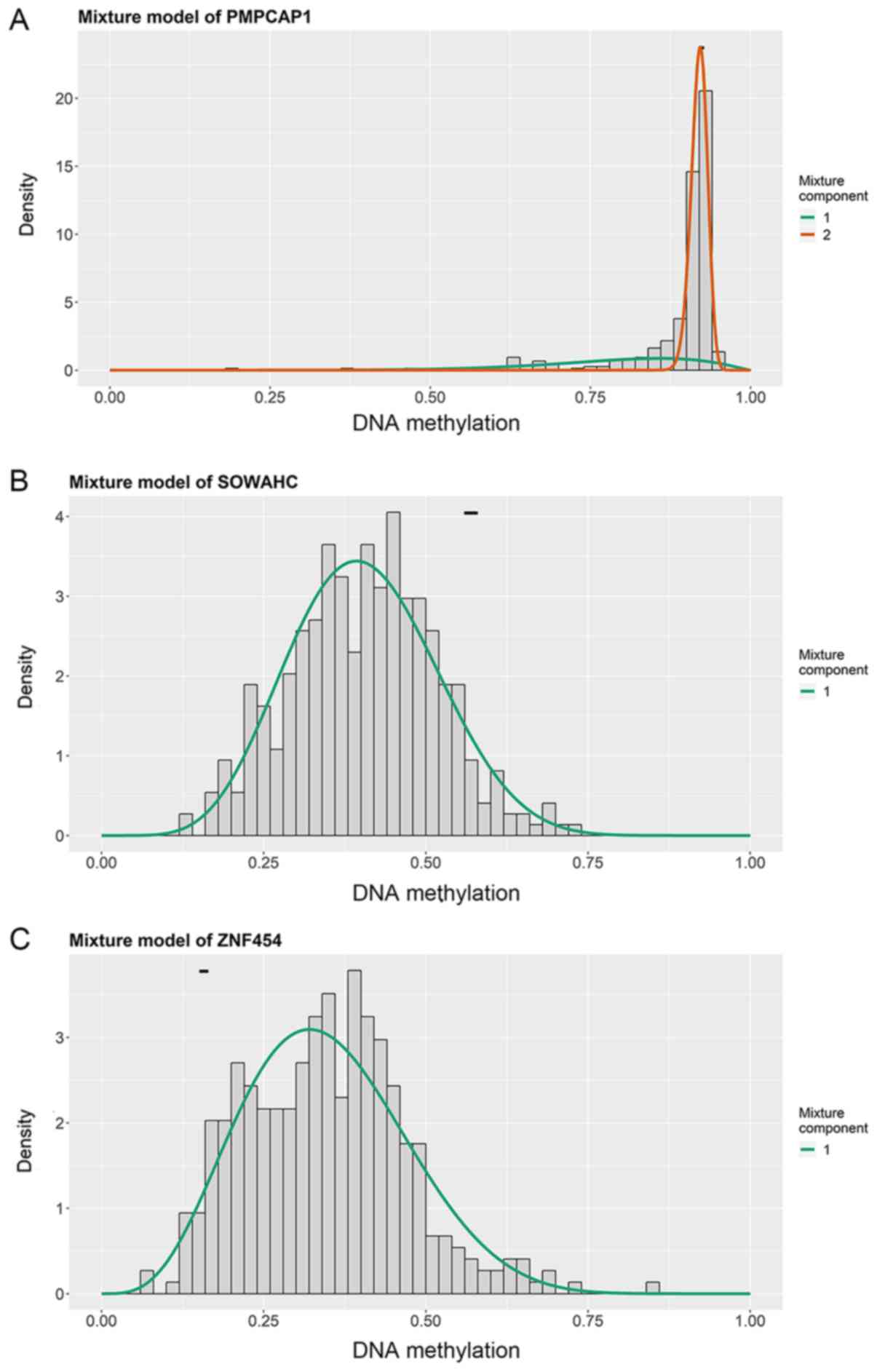
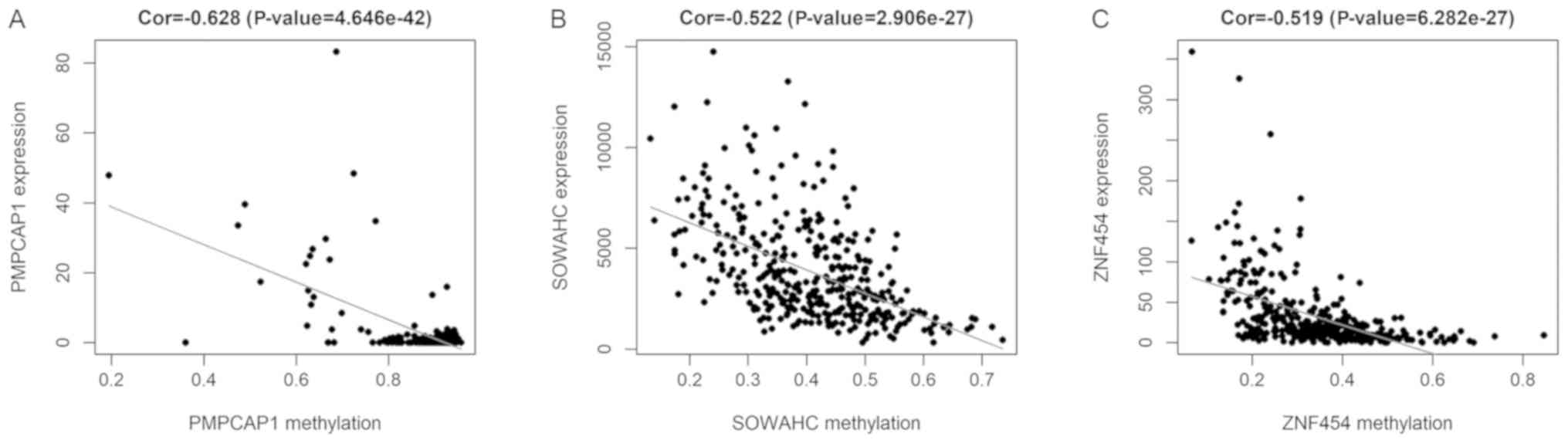
I). The methylation models are presented in Figs. 2 and S1, and the correlation plots are
demonstrated in Figs. 3 and
S2.
 | Table I.Output results of 30 MDGs using the
MethylMix algorithm. |
Table I.
Output results of 30 MDGs using the
MethylMix algorithm.
| Gene | Normal
meana | Tumor
meana | logFC | P-value |
padj | Cor | P-value of Cor |
|---|
| ZNF582 | 0.102781 | 0.275603 | 1.423022 |
5.87×10−22 |
1.48×10−19 | −0.564313 |
1.69×10−32 |
| ME3 | 0.277294 | 0.382910 | 0.465587 |
7.11×10−22 |
1.79×10−19 | −0.553976 |
3.80×10−31 |
| ZNF454 | 0.157826 | 0.346334 | 1.133824 |
1.64×10−21 |
4.12×10−19 | −0.519207 |
6.28×10−27 |
| SLC15A3 | 0.333165 | 0.467888 | 0.489927 |
1.74×10−20 |
4.39×10−18 | −0.556532 |
1.78×10−31 |
| MYO1G | 0.426675 | 0.533590 | 0.322595 |
3.87×10−20 |
9.76×10−18 | −0.551080 |
8.92×10−31 |
| SOWAHC | 0.569583 | 0.405071 | −0.491730 |
1.12×10−19 |
2.82×10−17 | −0.522119 |
2.91×10−27 |
| CCDC68 | 0.103194 | 0.199258 | 0.949279 |
3.02×10−19 |
7.61×10−17 | −0.503257 |
3.77×10−25 |
| SULT1C4 | 0.152713 | 0.285577 | 0.903061 |
9.50×10−19 |
2.39×10−16 | −0.500977 |
6.65×10−25 |
| KRT7 | 0.350295 | 0.504885 | 0.527384 |
1.37×10−17 |
3.44×10−15 | −0.574797 |
6.43×10−34 |
| UBA7 | 0.202113 | 0.276516 | 0.452201 |
5.21×10−16 |
1.31×10−13 | −0.540067 |
2.12×10−29 |
| HOXB2 | 0.305355 | 0.536591 | 0.813338 |
3.38×10−14 |
8.52×10−12 | −0.611253 |
2.80×10−29 |
| LINC00898 | 0.734546 | 0.587677 | −0.321829 |
1.89×10−13 |
4.76×10−11 | −0.557094 |
1.50×10−31 |
| LINC00668 | 0.676192 | 0.576143 | −0.231005 |
2.25×10−13 |
5.66×10−11 | −0.584201 |
3.09×10−35 |
| ARL14 | 0.790435 | 0.661123 | −0.257730 |
3.39×10−12 |
8.53×10−10 | −0.505203 |
2.31×10−25 |
| MKRN3 | 0.691598 | 0.542513 | −0.350275 |
4.42×10−12 |
1.11×10−9 | −0.704902 |
7.51×10−57 |
| CCDC8 | 0.327849 | 0.448283 | 0.451379 |
5.46×10−12 |
1.38×10−9 | −0.579013 |
1.67×10−34 |
| ZNF471 | 0.101118 | 0.267232 | 1.402048 |
5.90×10−12 |
1.49×10−9 | −0.551843 |
7.13×10−31 |
| PKP1 | 0.341899 | 0.298902 | −0.193899 |
6.88×10−12 |
1.73×10−9 | −0.536918 |
5.14×10−29 |
| ADH7 | 0.680263 | 0.529756 | −0.360765 |
6.46×10−11 |
1.63×10−8 | −0.572790 |
1.21×10−33 |
| ZNF556 | 0.487939 | 0.414088 | −0.236764 |
2.67×10−10 |
6.74×10−8 | −0.638623 |
8.72×10−44 |
| KRT31 | 0.742604 | 0.772890 | 0.057671 |
6.79×10−10 |
1.71×10−7 | −0.542682 |
1.01×10−29 |
| RAB34 | 0.253476 | 0.227666 | −0.154930 |
5.53×10−9 |
1.39×10−6 | −0.523058 |
2.26×10−27 |
| CLDN8 | 0.746796 | 0.626198 | −0.254095 |
8.60×10−8 |
2.17×10−5 | −0.513212 |
3.00×10−26 |
| ZNF502 | 0.294033 | 0.369115 | 0.328093 |
3.42×10−7 |
8.61×10−5 | −0.668828 |
2.54×10−49 |
| PPP1R2P10 | 0.764206 | 0.790535 | 0.048868 |
7.83×10−7 |
1.97×10−4 | −0.568402 |
4.79×10−33 |
| HCAR1 | 0.408911 | 0.525195 | 0.361067 |
1.54×10−6 |
3.89×10−4 | −0.632664 |
9.13×10−43 |
| TUSC8 | 0.805850 | 0.753565 | −0.096780 |
2.95×10−6 |
7.43×10−4 | −0.515211 |
1.79×10−26 |
| PMPCAP1 | 0.925864 | 0.889418 | −0.057939 |
5.71×10−6 |
1.44×10−3 | −0.628458 |
4.65×10−42 |
| ZKSCAN7 | 0.229582 | 0.273455 | 0.252291 |
8.69×10−6 |
2.19×10−3 | −0.515312 |
1.74×10−26 |
| IGBP1P4 | 0.800055 | 0.826529 | 0.046965 |
1.59×10−4 |
4.00×10−2 | −0.556656 |
1.72×10−31 |
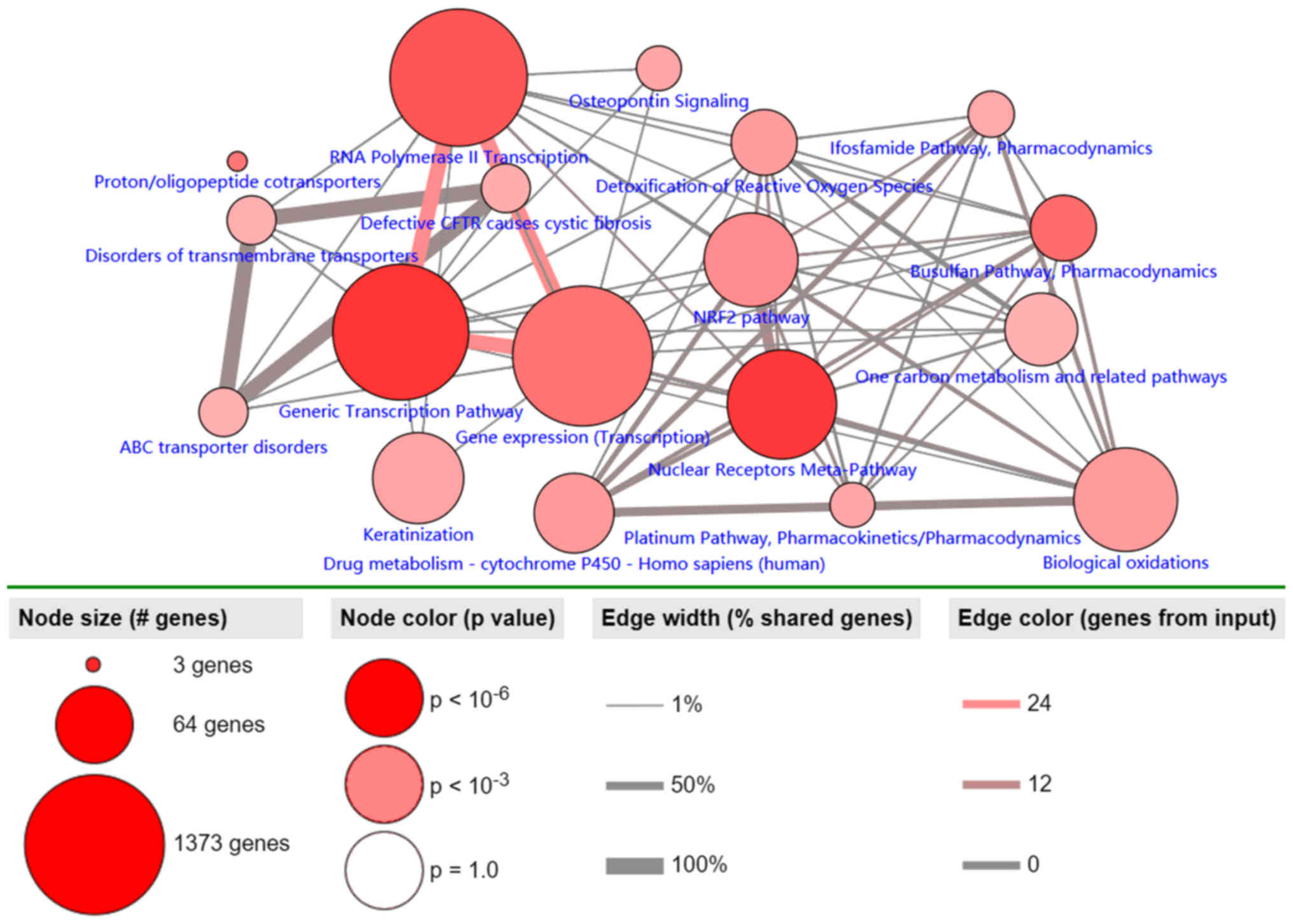
Pathway analysis of MDGs in LUSC
Pathway analysis of the 30 identified MDGs was
conducted using the ConsensusPathDB database (Fig. 4). The results revealed 3 primary
pathways: ‘generic transcription’, ‘RNA polymerase II
transcription’ and ‘gene expression (transcription)’. The largest
numbers of genes were associated with these 3 pathways, and ~100%
of all shared genes, and to the most genes from input
(P<0.001).
Recognition of hub MDGs in LUSC
Initially, survival analysis between hyper- and
hypomethylated MDGs revealed 4 genes with statistical importance:
Peptidase, mitochondrial processing a subunit pseudogene 1 (PMPCAP;
P=0.00173), sosondowah ankyrin repeat domain family member C
(SOWAHC; P=0.04), zinc finger protein (ZNF) 454 (P=0.023) and
LINC00668 (P=0.046; Figs. 5A-D and
S3). Joint survival analysis was
then conducted between the degree of methylation and the
corresponding gene expression level of MDGs and survival, and
PMPCAP1 (P=0.041), SOWAHC (P=0.028), ZNF454 (P=0.00935) and ADH7
(P=0.033) were identified as statistically significant (Figs. 5E-H and S4). By taking the common genes of the
first two steps, 3 hub MDGs (PMPCAP1, SOWAHC and ZNF454) were
deemed to be independently associated with prognosis in LUSC. Of
these 3 hub MDGs, PMPCAP1 and SOWAHC, characterized by
hypomethylation and high expression levels, were associated with
poor prognosis in patients with LUSC, whilst ZNF454, characterized
by hypermethylation and low expression level, was associated with
an improved prognosis.
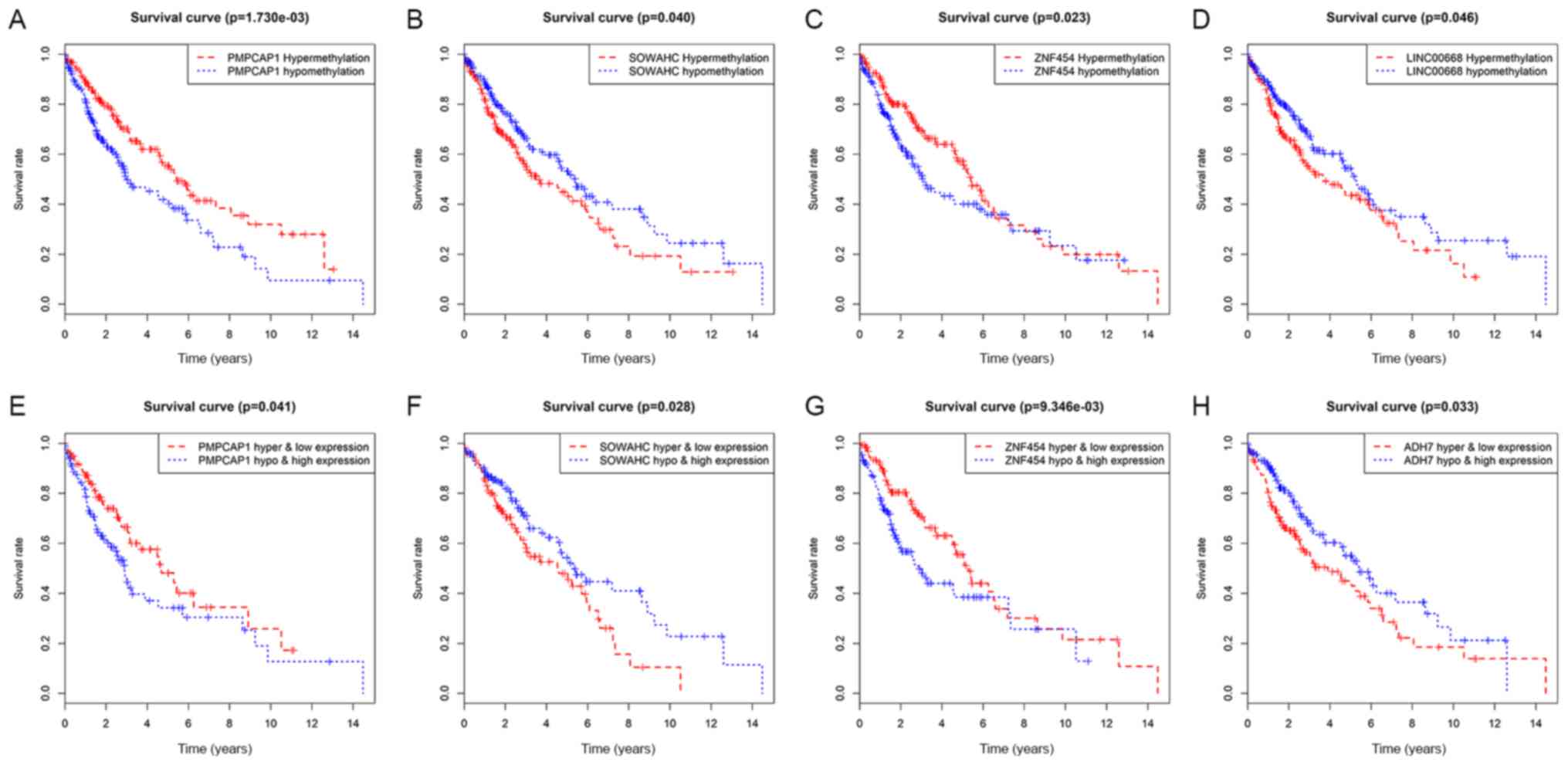 | Figure 5.Survival analysis curves of MDGs with
statistical significance in LUSC. Kaplan-Meier survival curves,
where the x-axis represents the overall survival time and the
y-axis represents the survival rate. (A-D) Survival analysis
comparing overall survival and the methylation state of (A)
PMPCAP1, (B) SOWAHC, (C) ZNF454 and (D) LINC00668, respectively.
(E-H) Joint survival analysis comparing overall survival between
hypermethylation/low expression and hypomethylation/high expression
of (E) PMPCAP1, (F) SOWAHC, (G) ZNF454 and (H) ADH7, respectively.
P<0.05. MDGs, methylation-driven genes; LUSC, lung squamous cell
cancer; PMPCAP1, peptidase, mitochondrial processing a subunit
pseudogene 1; SOWAHC, sosondowah ankyrin repeat domain family
member C; ZNF454, zinc finger protein 454. |
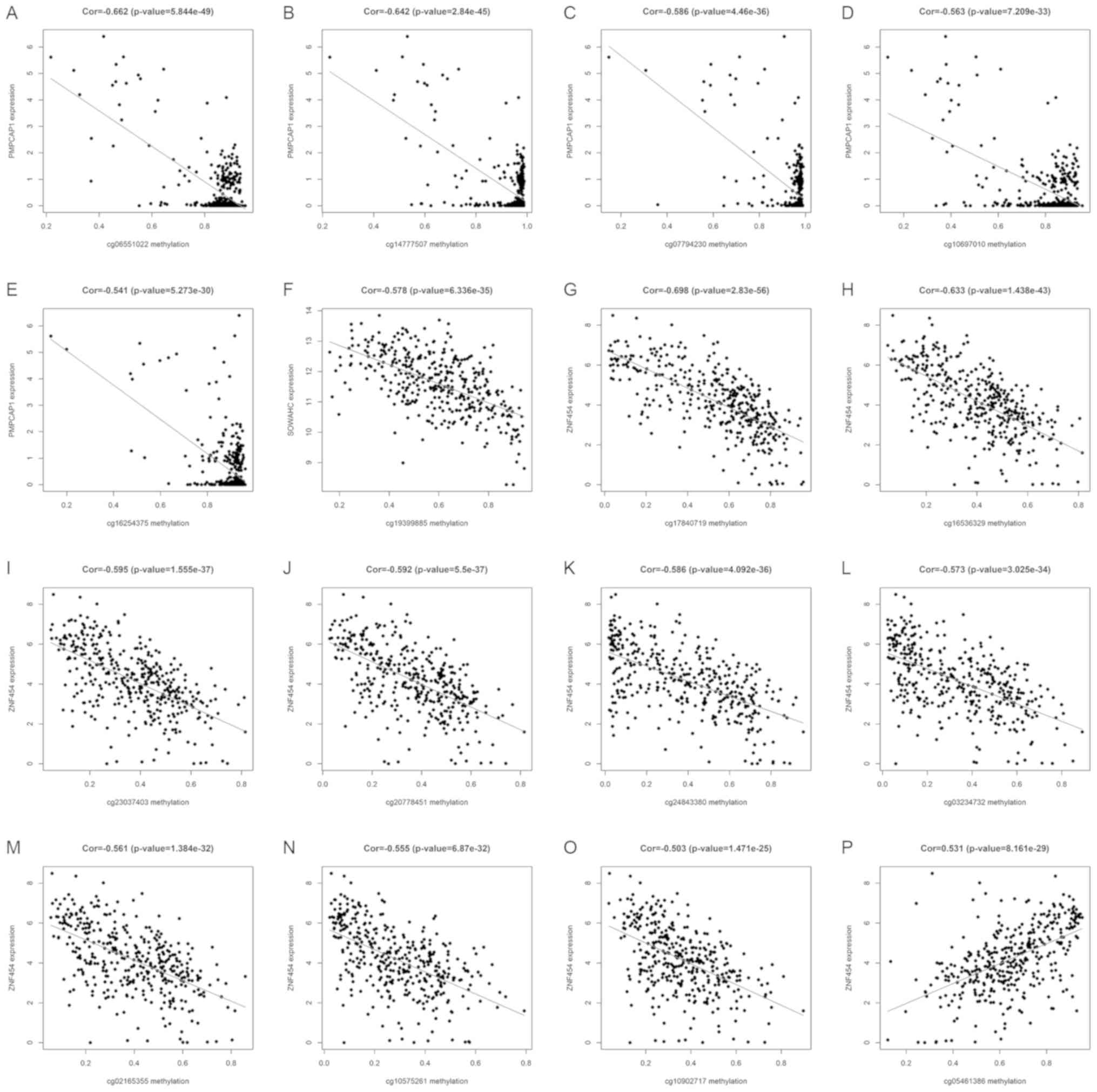
Key methylation sites of hub MDGs in
LUSC
Using the associated R packages, key methylation
sites statistically relevant to the expression of hub MDGs in LUSC
were identified. The results of correlation analysis revealed 5 key
methylation sites of the PMPCAP1 gene (cg06551022, cg14777507,
cg07794230, cg10697010 and cg16254375), 1 key methylation site of
the SOWAHC gene (cg19399885) and 10 key methylation sites of the
ZNF454 gene (cg17840719, cg16536329, cg23037403, cg20778451,
cg24843380, cg03234732, cg02165355, cg10575261, cg10902717 and
cg05461386; Fig. 6 and Table II; P<0.001).
| Table II.Correlation between key methylation
sites and expression of PMPCAP1, SOWAHC and ZNF454. |
Table II.
Correlation between key methylation
sites and expression of PMPCAP1, SOWAHC and ZNF454.
| Gene | Methylation
site | Cor | P-value |
|---|
| PMPCAP1 | cg06551022 | −0.662 |
5.84×10−49 |
|
| cg14777507 | −0.642 |
2.84×10−45 |
|
| cg07794230 | −0.586 |
4.46×10−36 |
|
| cg10697010 | −0.563 |
7.21×10−33 |
|
| cg16254375 | −0.541 |
5.27×10−30 |
| SOWAHC | cg19399885 | −0.578 |
6.34×10−35 |
| ZNF454 | cg17840719 | −0.698 |
2.83×10−56 |
|
| cg16536329 | −0.633 |
1.44×10−43 |
|
| cg23037403 | −0.595 |
1.56×10−37 |
|
| cg20778451 | −0.592 |
5.50×10−37 |
|
| cg24843380 | −0.586 |
4.09×10−36 |
|
| cg03234732 | −0.573 |
3.03×10−34 |
|
| cg02165355 | −0.561 |
1.38×10−32 |
|
| cg10575261 | −0.555 |
6.87×10−32 |
|
| cg10902717 | −0.503 |
1.47×10−25 |
|
| cg05461386 |
0.531 |
8.16×10−29 |
Discussion
The morbidity and mortality rates of LUSC are the
second highest of all the pathological types of lung carcinoma,
with poor prognosis depending on the biological characteristics of
the specific subtype. Furthermore, >70% of patients with LUSC
present with late-stage disease at the diagnosis, for which
treatment options are limited, and clinical outcomes are far poorer
compared with those in patients with early-stage disease (25). In order to decrease the mortality
rate of LUSC, novel approaches for early diagnosis and treatment
are required, as well as the identification of novel predictive
biomarkers and therapeutic targets.
Compared with the rapid development of precise
gene-targeted therapy in lung adenocarcinoma, there are a limited
number of effective and distinctive targets to improve the
prognosis of patients with LUSC. In addition to studies into gene
mutations, the association between epigenetic changes (particularly
DNA methylation) and LUSC has also attracted great attention.
Epigenetic studies have revealed that genome-scale epigenetic
modifications, including DNA methylation, histone modification and
microRNA interference, are involved in the pathogenic mechanisms of
malignancy (26). Due to its
stability and ease of detection, accumulating evidence has
demonstrated that aberrant gene methylation may serve as an
effective, non-invasive diagnostic biomarker and therapeutic in
carcinoma (27,28). Reports have indicated that the
abnormal methylation of certain genes, such as ZNF671 (29), ADAMTS1 (30) and CD36 (31), may alter their functions, including
the regulation of the cell cycle and signal transduction pathways,
as well as transcriptional inhibition. Kiyozumi et al
(32) demonstrated that
indoleamine 2,3-dioygenase 1 promoter hypomethylation is associated
with poor prognosis in esophageal cancer. Therefore, the accurate
detection of methylated genes is likely to improve the clinical
management of LUSC.
Studies have previously identified DMGs in LUSC
(33–35); however, not all genes can be
transcriptionally expressed. Therefore, as DMGs are not able to
precisely demonstrate the relevance between genetic methylation and
oncogenesis, MDGs are considered to be more representative
(14,36). In the present study,
high-throughput bioinformatics tools were used to identify and
analyze MDGs associated with the prognosis of LUSC. Data extracted
from TCGA were analyzed using packages from R, including edge,
limma and MethylMix, and 30 LUSC-associated MDGs were derived. To
improve the understanding of the functional pathways involving
these MDGs, significant pathways were visualized using the
ConsensusPathDB and Cytoscape.js library in the present study. The
results identified 3 primary pathways: ‘generic transcription’;
‘RNA polymerase II transcription’; and ‘gene expression
(transcription)’, which were affected by MDG interactions at a
functional level. In other words, differential methylation of
specific MDGs is able affect their expression and transcription.
Furthermore, considering that not all MDGs are significantly
associated with cancer prognosis, Kaplan-Meier survival and joint
survival analyses were conducted using the R survival package,
yielding 5 candidate prognosis-associated MDGs: PMPCAP1; SOWAHC;
ZNF454; LINC00668; and ADH7 (P<0.05). The common genes showing
significance in survival and joint survival analyses were chosen as
the 3 hub MDGs (PMPCAP1, SOWAHC and ZNF454), which were identified
to function as potential independent prognosis-associated markers
for LUSC. The hub MDGs were determined by analyzing the association
between hyper- or hypomethylation and survival, but also by
integrating the degree of methylation and the expression of MDGs
with survival.
PMPCAP1, a pseudogene of PMPCA1, is located on
chromosome 4q22.1. To the best of our knowledge, the function of
PMPCAP1 has not been investigated thus far. However, a number of
studies have suggested that the functions of certain pseudogenes
differ from those of normal homologous genes, but that the
expression of associated non-coding (nc)RNAs plays an important
regulatory role in the development of certain diseases (37–39).
For example, PTENP expression may generate ncRNAs that
competitively inhibit the function of PTEN, a known
tumor-suppressor gene, and therefore inhibit cancer cell
proliferation (40). Pseudogenes
may also affect oncogenesis through epigenetic changes. The present
study indicated that PMPCAP1 was hypomethylated in LUSC compared
with normal tissues, which was associated with high expression
levels, and ultimately, poor prognosis (P=0.041). Therefore, it may
be speculated that the hypomethylation of PMPCAP1 in cancer tissue
(and the subsequent increase in RNA expression) is an indicator of
poor clinical outcome in patients with LUSC, though further
investigation is required to confirm this hypothesis.
SOWAHC, also known as ankyrin repeat domain (ANKRD)
57, is a protein-coding gene. The principle biological function of
the ANKRD family is to mediate interactions between proteins
(41). Takahashi et al
(42) identified that ANKRD1 was
overexpressed in EGFR-TKIs-resistant NSCLC with EGFR mutation, and
that by inhibiting ANKRD1 expression, resistant cells were
re-sensitized to afatinib and osimertinib. Lei et al
(43) also demonstrated that
ANKRD1 regulated apoptosis in ovarian cancer cells and functioned
as a potential target to increase sensitivity to chemotherapy in
ovarian cancer. In addition, the prognostic value of SOWAHC has
been confirmed in bladder cancer (41); however, its value in LUSC has not
been elucidated thus far, to the best of our knowledge. In the
present study, the results of the correlation analysis indicated
that the methylation of SOWAHC was negatively associated with its
expression, commonly presenting as hypomethylation and high
expression. Joint survival analysis revealed a significant
association between the combined methylation and expression data
and survival (P=0.028), suggesting that hypomethylation and high
expression levels denoted improved prognosis in LUSC. Therefore,
SOWAHC may be a potential biomarker of LUSC.
ZNF454 is a protein-coding gene, which expresses a
protein measuring 522 amino acids. The ZNF454 protein is primarily
involved in functional pathways associated with gene expression and
transcription, namely ‘DNA binding’, ‘DNA-binding transcription
factor activity by RNA polymerase II-specific’, ‘nucleic acid
binding’ and ‘metal ion binding’. To the best of our knowledge, no
published studies of ZNF454 were available until now, while a
number of other members of the ZNF family have been investigated.
Previous studies have suggested that, as the largest family of
transcription factors in humans, ZNFs serve numerous important
roles, and were recently confirmed as potential tumor suppressors
(44). For example, through ZNF545
promoter methylation-associated deactivation, ZNF545 inhibited
tumor proliferation in colorectal cancer via the PI3K/AKT and
MAPK/ERK signaling pathways (45).
In the present study, ZNF454 was generally hypermethylated and
expressed to a low degree in LUSC, and was associated with
favorable prognoses. Therefore, ZNF454 may be a potential
tumor-suppressor gene that functions as a transcriptional
regulator, with potential use as a prognostic indicator.
Previous studies have demonstrated that
site-specific methylation, such as that at the promoter or
enhancer, particularly of CpG sites, may notably affect gene
expression (46,47). In the present study, the specific
methylation sites of 3 hub MDGs that were associated with gene
expression were identified. The results indicated that the
expression of PMPCAP1 is negatively associated with the methylation
of 5 sites (cg06551022, cg14777507, cg07794230, cg10697010 and
cg16254375). A single methylation site was associated with SOWAHC
expression (cg19399885) and the expression of ZNF454 was associated
with 10 methylation sites, including 9 negatively related sites
(cg17840719, cg16536329, cg23037403, cg20778451, cg24843380,
cg03234732, cg02165355, cg10575261 and cg10902717) and 1 positive
site (cg05461386). Further studies on the effects of these
methylation sites on gene expression are required, which may assist
in identifying more precise diagnostic and therapeutic targets to
improve the prognosis of patients with LUSC.
There were certain limitations to the present study:
Firstly, due to the lack data from other databases, the results
were not externally validated, which may have partially decreased
reliability. Secondly, limited financial support prevented further
mechanistic studies with lung cancer cell lines or human tissue
samples, which is a potential future research prospect.
In conclusion, using the MethylMix algorithm, the
present study identified 3 hub MDGs (PMPCAP1, SOWAHC and ZNF454)
with independent prognostic values in LUSC. In patients with LUSC,
PMPCAP1 and SOWAHC were hypomethylated and highly expressed, which
was determined to be an indication of poor prognosis. By contrast,
ZNF454 was hypermethylated and expressed to a low degree, which was
associated with improved prognosis. In addition, specific sites of
aberrant methylation were investigated to identify more precise
targets for clinical application. Although the results require
further experimental validation, the present study provides
diagnostic, therapeutic and prognostic value for patients with
LUSC, and may guide future clinical applications to some
extent.
Supplementary Material
Supporting Data
Acknowledgements
The results published here are in whole based upon
data generated by the TCGA Research Network: https://www.cancer.gov/tcga.
Funding
The present study was supported by the Key
Technology Research and Development Program of Shandong Province
(grant no. GG201710060039).
Availability of data and materials
The authors declare that the initial data are
available from the TCGA database, and that all data generated and
analyzed during the present study are included in this published
article.
Authors' contributions
QZhu, JW and JL conceived and designed the study. JW
and QZha performed the data analysis and generated the figures. FW
and LF contributed analysis tools. QZhu drafted the manuscript. BS
and CX also performed data analysis and critically revised the
manuscript. All authors read and approved the final manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Glossary
Abbreviations
Abbreviations:
|
NSCLC
|
non-small-cell lung cancer
|
|
LUSC
|
lung squamous cell cancer
|
|
TCGA
|
The Cancer Genome Atlas
|
|
MDG
|
methylation-driven gene
|
|
DEG
|
differentially expressed gene
|
|
DMG
|
differentially methylated gene
|
|
FC
|
fold change
|
|
padj
|
adjusted P-value
|
|
Cor
|
correlation coefficient
|
|
|Cor|
|
absolute correlation coefficient
value
|
|
PMPCAP1
|
peptidase, mitochondrial processing a
subunit pseudogene 1
|
|
ZNF
|
zinc finger protein
|
|
SOWAHC
|
sosondowah ankyrin repeat domain
family member C
|
|
ANKRD
|
ankyrin repeat domain
|
References
|
1
|
Bray F, Ferlay J, Soerjomataram I, Siegel
RL, Torre LA and Jemal A: Global cancer statistics 2018: GLOBOCAN
estimates of incidence and mortality worldwide for 36 cancers in
185 countries. CA Cancer J Clin. 68:394–424. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Lim SL, Jia Z, Lu Y, Zhang H, Ng CT, Bay
BH, Shen HM and Ong CN: Metabolic signatures of four major
histological types of lung cancer cells. Metabolomics. 14:1182018.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Choi M, Kadara H, Zhang J, Parra ER,
Rodriguez-Canales J, Gaffney SG, Zhao Z, Behrens C, Fujimoto J,
Chow C, et al: Mutation profiles in early-stage lung squamous cell
carcinoma with clinical follow-up and correlation with markers of
immune function. Ann Oncol. 28:83–89. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Dong J, Li B, Lin D, Zhou Q and Huang D:
Advances in targeted therapy and immunotherapy for non-small cell
lung cancer based on accurate molecular typing. Front Pharmacol.
10:2302019. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Chakravarthi BV, Nepal S and Varambally S:
Genomic and epigenomic alterations in cancer. Am J Pathol.
186:1724–1735. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Mehta A, Dobersch S, Romero-Olmedo AJ and
Barreto G: Epigenetics in lung cancer diagnosis and therapy. Cancer
Metastasis Rev. 34:229–241. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Pfister SX and Ashworth A: Marked for
death: Targeting epigenetic changes in cancer. Nat Rev Drug Discov.
16:241–263. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Baylin SB and Jones PA: Epigenetic
determinants of cancer. Cold Spring Harb Perspect Biol.
8:a0195052016. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Dawson MA and Kouzarides T: Cancer
epigenetics: From mechanism to therapy. Cell. 150:12–27. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Bernstein BE, Meissner A and Lander ES:
The mammalian epigenome. Cell. 128:669–681. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Wouters BJ and Delwel R: Epigenetics and
approaches to targeted epigenetic therapy in acute myeloid
leukemia. Blood. 127:42–52. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Győrffy B, Bottai G, Fleischer T, Munkácsy
G, Budczies J, Paladini L, Børresen-Dale AL, Kristensen VN and
Santarpia L: Aberrant DNA methylation impacts gene expression and
prognosis in breast cancer subtypes. Int J Cancer. 138:87–97. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Lin DC, Wang MR and Koeffler HP: Genomic
and epigenomic aberrations in esophageal squamous cell carcinoma
and implications for patients. Gastroenterology. 154:374–389. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Lu T, Chen D, Wang Y, Sun X, Li S, Miao S,
Wo Y, Dong Y, Leng X, Du W and Jiao W: Identification of DNA
methylation-driven genes in esophageal squamous cell carcinoma: A
study based on the cancer genome atlas. Cancer Cell Int. 19:522019.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Gloss BS and Samimi G: Epigenetic
biomarkers in epithelial ovarian cancer. Cancer Lett. 342:257–263.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Villanueva A, Portela A, Sayols S,
Battiston C, Hoshida Y, Méndez-González J, Imbeaud S, Letouzé E,
Hernandez-Gea V, Cornella H, et al: DNA methylation-based prognosis
and epidrivers in hepatocellular carcinoma. Hepatology.
61:1945–1956. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Tomczak K, Czerwińska P and Wiznerowicz M:
The cancer genome atlas (TCGA): An immeasurable source of
knowledge. Contemp Oncol (Pozn). 19:A68–A77. 2015.PubMed/NCBI
|
|
18
|
No authors listed, . The TCGA Legacy.
Cell. 173:281–282. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Bibikova M, Barnes B, Tsan C, Ho V,
Klotzle B, Le JM, Delano D, Zhang L, Schroth GP, Gunderson KL, et
al: High density DNA methylation array with single CpG site
resolution. Genomics. 98:288–295. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Gevaert O: MethylMix: An R package for
identifying DNA methylation-driven genes. Bioinformatics.
31:1839–1841. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Gevaert O, Tibshirani R and Plevritis SK:
Pancancer analysis of DNA methylation-driven genes using MethylMix.
Genome Biol. 16:172015. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Kamburov A, Stelzl U, Lehrach H and Herwig
R: The ConsensusPathDB interaction database: 2013 update. Nucleic
Acids Res. 41:D793–D800. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Kamburov A, Pentchev K, Galicka H,
Wierling C, Lehrach H and Herwig R: ConsensusPathDB: Toward a more
complete picture of cell biology. Nucleic Acids Res. 39:D712–D717.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Franz M, Lopes CT, Huck G, Dong Y, Sumer O
and Bader GD: Cytoscape.js: A graph theory library for
visualisation and analysis. Bioinformatics. 32:309–311.
2016.PubMed/NCBI
|
|
25
|
Blandin Knight S, Crosbie PA, Balata H,
Chudziak J, Hussell T and Dive C: Progress and prospects of early
detection in lung cancer. Open Biol. 7:2017. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Nebbioso A, Tambaro FP, Dell'Aversana C
and Altucci L: Cancer epigenetics: Moving forward. PLoS Genet.
14:e10073622018. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Zhu J and Yao X: Use of DNA methylation
for cancer detection: Promises and challenges. Int J Biochem Cell
Biol. 41:147–154. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Leygo C, Williams M, Jin HC, Chan MWY, Chu
WK, Grusch M and Cheng YY: DNA methylation as a noninvasive
epigenetic biomarker for the detection of cancer. Dis Markers.
2017:37265952017. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Mase S, Shinjo K, Totani H, Katsushima K,
Arakawa A, Takahashi S, Lai HC, Lin RI, Chan MWY, Sugiura-Ogasawara
M and Kondo Y: ZNF671 DNA methylation as a molecular predictor for
the early recurrence of serous ovarian cancer. Cancer Sci.
110:1105–1116. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Eissa MAL, Lerner L, Abdelfatah E, Shankar
N, Canner JK, Hasan NM, Yaghoobi V, Huang B, Kerner Z, Takaesu F,
et al: Promoter methylation of ADAMTS1 and BNC1 as potential
biomarkers for early detection of pancreatic cancer in blood. Clin
Epigenetics. 11:592019. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Sun Q, Zhang W, Wang L, Guo F, Song D,
Zhang Q, Zhang D, Fan Y and Wang J: Hypermethylated CD36 gene
affected the progression of lung cancer. Gene. 678:395–406. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Kiyozumi Y, Baba Y, Okadome K, Yagi T,
Ogata Y, Eto K, Hiyoshi Y, Ishimoto T, Iwatsuki M, Iwagami S, et
al: Indoleamine 2,3-dioxygenase 1 promoter hypomethylation is
associated with a poor prognosis in patients with esophageal
cancer. Cancer Sci. 110:1863–1871. 2019.PubMed/NCBI
|
|
33
|
Carvalho RH, Hou J, Haberle V, Aerts J,
Grosveld F, Lenhard B and Philipsen S: Genomewide DNA methylation
analysis identifies novel methylated genes in non-small-cell lung
carcinomas. J Thorac Oncol. 8:562–573. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Gao C, Zhuang J, Zhou C, Ma K, Zhao M, Liu
C, Liu L, Li H, Feng F and Sun C: Prognostic value of aberrantly
expressed methylation gene profiles in lung squamous cell
carcinoma: A study based on The Cancer Genome Atlas. J Cell
Physiol. 234:6519–6528. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Shi YX, Wang Y, Li X, Zhang W, Zhou HH,
Yin JY and Liu ZQ: Genome-wide DNA methylation profiling reveals
novel epigenetic signatures in squamous cell lung cancer. BMC
Genomics. 18:9012017. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Gao C, Zhuang J, Li H, Liu C, Zhou C, Liu
L and Sun C: Exploration of methylation-driven genes for monitoring
and prognosis of patients with lung adenocarcinoma. Cancer Cell
Int. 18:1942018. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Xiao-Jie L, Ai-Mei G, Li-Juan J and Jiang
X: Pseudogene in cancer: Real functions and promising signature. J
Med Genet. 52:17–24. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Poliseno L, Salmena L, Zhang J, Carver B,
Haveman WJ and Pandolfi PP: A coding-independent function of gene
and pseudogene mRNAs regulates tumour biology. Nature.
465:1033–1038. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Grandér D and Johnsson P:
Pseudogene-expressed RNAs: Emerging roles in gene regulation and
disease. Curr Top Microbiol Immunol. 394:111–126. 2016.PubMed/NCBI
|
|
40
|
Johnsson P, Ackley A, Vidarsdottir L, Lui
WO, Corcoran M, Grandér D and Morris KV: A pseudogene
long-noncoding-RNA network regulates PTEN transcription and
translation in human cells. Nat Struct Mol Biol. 20:440–446. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Yang Z, Liu A, Xiong Q, Xue Y, Liu F, Zeng
S, Zhang Z, Li Y, Sun Y and Xu C: Prognostic value of
differentially methylated gene profiles in bladder cancer. J Cell
Physiol. 234:18763–18772. 2019.PubMed/NCBI
|
|
42
|
Takahashi A, Seike M, Chiba M, Takahashi
S, Nakamichi S, Matsumoto M, Takeuchi S, Minegishi Y, Noro R,
Kunugi S, et al: Ankyrin repeat domain 1 overexpression is
associated with common resistance to afatinib and osimertinib in
EGFR-mutant lung cancer. Sci Rep. 8:148962018. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Lei Y, Henderson BR, Emmanuel C, Harnett
PR and DeFazio A: Inhibition of ANKRD1 sensitizes human ovarian
cancer cells to endoplasmic reticulum stress-induced apoptosis.
Oncogene. 34:485–495. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Jen J and Wang YC: Zinc finger proteins in
cancer progression. J Biomed Sci. 23:532016. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Xiang S, Xiang T, Xiao Q, Li Y, Shao B and
Luo T: Zinc-finger protein 545 is inactivated due to promoter
methylation and functions as a tumor suppressor through the
Wnt/β-catenin, PI3K/AKT and MAPK/ERK signaling pathways in
colorectal cancer. Int J Oncol. 51:801–811. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Weigel C, Chaisaingmongkol J, Assenov Y,
Kuhmann C, Winkler V, Santi I, Bogatyrova O, Kaucher S, Bermejo JL,
Leung SY, et al: DNA methylation at an enhancer of the three prime
repair exonuclease 2 gene (TREX2) is linked to gene expression and
survival in laryngeal cancer. Clin Epigenetics. 11:672019.
View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Pogribny IP, Pogribna M, Christman JK and
James SJ: Single-site methylation within the p53 promoter region
reduces gene expression in a reporter gene construct: Possible in
vivo relevance during tumorigenesis. Cancer Res. 60:588–594.
2000.PubMed/NCBI
|