Introduction
Fibroblast growth factor receptors (FGFRs), which
belong to the receptor tyrosine kinase (RTK) family, are known to
signal from the cell membrane as well as from endosomal
compartments (1). There are four
FGFRs: FGFR1, FGFR2, FGFR3 and FGFR4; these FGFs bind their
receptors and >20 known ligands to these receptors, resulting in
diverse effects in many different target cells (2). FGFR signaling plays an important role
in cell proliferation, angiogenesis and many normal biological
processes (3); however, FGFR
signaling dysregulation has been implicated in aberrant pathologies
associated with tumor growth, including ovarian, colon, breast,
prostate, soft tissue sarcomas, melanoma and lung cancer (4–9).
Despite advances in treatment over the past decades,
ovarian cancer has the highest mortality among gynecologic
malignancies (10). Limited
prognosis remains a key obstacle for the treatment of patients with
advanced ovarian cancer (11).
Upregulation of all four members of the FGFR family and other
various fibroblast growth factors has been found in epithelial
ovarian carcinoma tissue (10,12),
suggesting that dysregulated FGFR signaling contributes to ovarian
carcinogenesis and may represent a suitable therapeutic target
(13). The FGFR4 GlyArg388
polymorphism has been shown to predict prolonged survival and
platinum sensitivity in advanced ovarian cancer (14). FGFR1 and FGFR2 mutations have also
been demonstrated to promote ovarian cancer progression and
invasion (15,16). The mechanisms of FGFR1 in other
cancer types have been studied; for example, the upregulation of
FGFR1 in carcinoma cells is critical for prostate cancer
progression and invasion (17).
Furthermore, the FGFR1 pathway recruits macrophages to the mammary
epithelium and promotes paracrine interactions between tumor cells
and macrophages, thus inducing tumor growth (18,19).
However, to the best of the authors' knowledge, not many studies on
the role of FGFR1 in ovarian cancer exist, and how FGFR1 functions
in ovarian cancer is unclear.
Genetic evidence and structure analysis indicated
that the N-glycosylation of FGFR may constitute an important
regulatory input (20). The
disruption of N-glycosylation can cause the mutation of an
asparagine residue in the extracellular domain of FGFR2 and FGFR3,
and result in skeletal growth defects. Abnormal cellular
glycosylation has been shown to play a key role in cancer
progression and malignancy (21–23).
Therefore, understanding the regulation of FGFR glycosylation may
provide novel insight into cancer biology and result in developing
possible therapeutic strategies. Glycosylation is regulated by
various glycosyltransferases, such as fucosyl-, sialyl- and
galactosyltransferases (24). The
β galactoside α2,6-sialyltransferase, CMP-NeuAc: Galβ (1,4)
GlcNAc: α2,6-sialyltransferase (ST6Gal-I) is a vital
sialyltransferase that adds sialic acid residues to N-linked
oligosaccharides (25). ST6Gal-I
has been reported to induce adhesion and migration, and promote
drug resistance in various cancer cells (26–29).
However, the possible biological effect of ST6Gal-I on FGFR1 in
ovarian cancer has not been clearly established.
In the present study, ST6Gal-I knockdown or
overexpression OVCAR3 ovarian cell lines were prepared and
characterized, to investigate the sialylation of FGFR1 and its
effects on cancer cell proliferation and migration, and sensitivity
to anticancer drugs. It was identified that ST6Gal-I overexpression
induced high sialylation levels of FGFR1, and activated ERK and
focal adhesion kinase (FAK) signaling in cells. ST6Gal-I
overexpression decreased the effects of anticancer drugs, but
ST6Gal-I knockdown resulted in the opposite effect. Collectively,
these data suggested that FGFR1 sialylation affects FGFR1-mediated
cell growth and chemotherapeutic drug sensitivity in human ovarian
cancer cells. FGFR1 sialylation levels are hypothesized to be a
reliable biomarker for anti-FGFR1 therapy.
Materials and methods
Cell culture and transfection
OVCAR3 ovarian cancer cells, purchased from The
American Type Culture Collection, were cultured in DMEM (Gibco;
Thermo Fisher Scientific, Inc.) with 10% FBS (Gibco; Thermo Fisher
Scientific, Inc.) and 1% penicillin/streptomycin (Gibco; Thermo
Fisher Scientific, Inc.) at 37°C in a 5% CO2-humidified
atmosphere. Stable ST6Gal-I overexpression (oe-ST6Gal-I), knockdown
small hairpin-ST6Gal-I (sh-ST6Gal-I) or empty vector cell lines
were established, as previously described (30). In brief, pcDNA3.1(−)/ST6Gal-I,
small hairpin (sh)-ST6Gal-I and empty vector plasmids (10 µg/ml)
were purchased from Invitrogen; Thermo Fisher Scientific, Inc., and
transfected into OVCAR3 ovarian cancer cells with
Lipofectamine® 2000 (Thermo Fisher Scientific, Inc.). A
limiting dilution was applied to obtain subcell line clones after
24 h of transfection. Blasticidin S Hcl (1.5 µg/ml, Invitrogen;
Thermo Fisher Scientific, Inc.) was used to select the sh-ST6Gal-I
clone, and Geneticin® Selective Antibiotic G418 (350
µg/ml, Invitrogen; Thermo Fisher Scientific, Inc.) was utilized to
select the ST6Gal-I overexpression clone. ST6Gal-I overexpression
or knockdown cell lines were verified by reverse transcription
(RT)-semi-quantitative (q)PCR and immunoblotting.
RT-qPCR
Total RNA was isolated from the cells using
TRIzol® reagent (Invitrogen; Thermo Fisher Scientific,
Inc.), and cDNA was synthesized using a PrimeScript® RT
Master Mix kit (Takara Bio) according to the manufacturer's
protocol: 37°C for 60 min, 85°C for 5 min and hold at 4°C. RT-qPCR
was performed on a Real-Time PCR Detection System (Bio-Rad
Laboratories). PCR cycles were: Pretreatment at 95°C for 10 min,
93°C for 15 sec, 67°C for 45 sec (45 cycles), then 93°C for 15 sec,
67°C for 1 min, 95°C for 15 sec, 75°C for 10 min and hold at 4°C.
The primer sequences used for the real-time PCR assays were as
follows: Forward, 5′-CCTCTGGGATGCTTGGTATC-3′; and reverse,
5′-GTGCAGGCACTATCGAAGAA-3′ for ST6Gal-I; forward,
5′-AGCCTCAAGATCATCAGC-3′ and reverse, 5′-GAGTCCTTCCACGATACC-3′ for
GAPDH (BGI, Inc.). The gene expression was determined using the
2−ΔΔCq method (31).
ST6Gal-I activity assay
Lectin staining was conducted to measure ST6Gal-I
activity. Cells were stained with FITC-conjugated SNA lectin (EY
Laboratories, Inc.), which is specific for 2–6 sialic acids. Cells
were stained for 40 min at 4°C with SNA-FITC (1:200) and analyzed
by fluorescence-activated cell sorting (FACS; Becton Dickinson). In
addition, cells were stained with SNA-FITC (1:100) for 4 h at room
temperature for an immunofluorescence assay using a Leica DM2500
LED microscope (Leica Microsystems GmbH) at 200× magnification.
Scratch wound healing assay
After the oe-ST6Gal-I and sh-ST6Gal-I subcell line
clones were verified, a wound healing assay was used to assess cell
migration (32). Cells
(105 cells/well) were seeded in a 6-well plate, and the
tip of a 200-µl micropipette was used to make a straight scratch on
a confluent monolayer of cells to create a wound. The detached
cells were rinsed with PBS twice, and serum-free DMEM was then
added. The wound closure in the area was imaged in brightfield
using a microscope (Olympus Corporation; magnification, ×100) after
incubation for 0, 12 and 24 h. The wound closure areas were
selected randomly and the width of the wound was quantified in
ImageJ (v1.8, National Institutes of Health) to show the wound
closure at each time point. The results of four independent
experiments were imaged under a microscope and quantified.
Cell Counting Kit-8 (CCK-8) assay
To measure the proliferation of different
transfected cloned cell lines, a CCK-8 detection kit (Dojindo
Molecular Technologies, Inc.) was used, according to the
manufacturer's protocol. In total, ~3,000 cells were seeded into a
96-well plate in quintuplicate for 6 h, and complete medium was
then changed to DMEM with different concentrations (0, 10, 100,
1,000 and 10,000 nM) of paclitaxel, Adriamycin or PD173074
(Sigma-Aldrich; Merck KGaA) for 24 h or the cells were cultured
without FBS for 0, 24, 48, 72, 96, 120 and 144 h at 37°C. Next, 10
µl CCK-8 reagent was added to each well, and the absorbance value
was measured at 490 nm using a Multiskan Spectrum spectrophotometer
(BioTek Instruments, Inc.).
Apoptosis analysis by flow
cytometry
Cells were incubated with different concentrations
of paclitaxel, Adriamycin or PD173074 (Sigma-Aldrich; Merck KGaA)
for 24 h and collected for staining. After centrifugation at 200 ×
g at 4°C, the cell pellets were resuspended and placed in 100 µl
Annexin V binding buffer containing 5 µl Annexin V-Phycoerythrin
(PE) and 2 µl 7-aminoactinomycin D (7-AAD; BD Biosciences). The
cells were incubated with PE-labeled Annexin V binding buffer in
the dark at room temperature for 20 min. Staining controls were
prepared and were single-stained or unstained. A positive apoptotic
control was obtained by incubating cells for 15 h with 1 mM
hydrogen peroxide (Sigma-Aldrich; Merck KGaA). The stained cell
populations were analyzed using a FACSCalibur flow cytometer (BD
Biosciences), and the cell cycle distributions were analyzed using
FlowJo software v10 (FlowJo LLC).
Immunoprecipitation and
immunoblotting
Cells were lysed using cell lysis buffer (PBS, 1%
NP40, 1% sodium deoxycholate and 0.1% SDS, 100 µg/ml PMSF, 1 mmol/l
sodium orthovanadate and 1 protease inhibitor tablet/10 ml), and
the protein concentration was measured using a bicinchoninic acid
assay kit (Beyotime Institute of Biotechnology). Equal amounts of
denatured proteins (20 µg) were separated by SDS-PAGE on 10% gels
and transferred onto PVDF membranes. Antibodies against ST6Gal-I
(1:200, cat. no. AF5924, R&D Systems), FGFR1 (1:200, cat. no.
ab156031, Abcam), phosphorylated (p)-FGFR1 (1:200, cat. no.
ab59194, Abcam), ERK (1:200, cat. no. sc514302, Santa Cruz
Biotechnology), p-ERK(1:200, cat. no. sc156521, Santa Cruz
Biotechnology), FAK (1:200, cat. no. ab72140, Abcam), p-FAK (1:200,
cat. no. ab4792, Abcam), cleaved caspase-3 (1:200, cat. no.
ab49822, Abcam) and GAPDH (1:1,000, cat. no. sc32233, Santa Cruz
Biotechnology, Inc.) were used as the primary antibodies. For
SNA-FGFR1, 100 µl SNA-conjugated agarose (EY Laboratories, Inc.)
was added to the lysed protein for 1 h, and the beads were
collected. α2–6 sialylated proteins bound to SNA-agarose beads were
precipitated by centrifugation (200 × g) at 4°C for 10 min and
washed extensively with lysis buffer. Sialylated proteins were
released from the complexes and then boiled in SDS-PAGE sample
buffer and immunoblotted for FGFR1 (Santa Cruz Biotechnology,
Inc.). The membranes were blocked with 5% non-fat milk at room
temperature, and then incubated with a primary antibody and
horseradish peroxidase-conjugated secondary antibody (Goat IgG
Horseradish Peroxidase-conjugated Antibody, cat. no. HAF019,
R&D Systems, Oakville, Canada; Mouse IgG Horseradish
Peroxidase-conjugated Antibody, cat. no. sc516132, Santa Cruz
Biotechnology, Inc.; Rabbit IgG Horseradish Peroxidase-conjugated
Antibody, cat. no. HAF008, R&D Systems Oakville, Canada), and
detected using an ECL kit (GE Healthcare) according to the
manufacturer's protocol. The relative amount of protein was
determined by densitometry using ImageJ software (version v1.8.0,
National Institutes of Health).
Statistical analysis
All the experiments were repeated 3 times. The data
are presented as the mean ± SD and analyzed by GraphPad Prism 6
(GraphPad Software Inc.). One-way ANOVA with the Least Significant
Difference post hoc test was performed to determine statistical
significance between groups. P<0.05 was considered to indicate a
statistically significant difference.
Results
Establishment of oe-ST6Gal-I or
sh-ST6Gal-I subcell clones of OVCAR3 human ovarian cancer
cells
The majority of previous FGFR1 studies have focused
on FGFR1 amplification and activating mutations (33–35),
whereas regulation of FGFR1 activity via post-translational
modifications, such as glycosylation, fucosylation and sialylation,
has been studied considerably less. To the best of the authors'
knowledge, there are no studies that have investigated the
relevance of FGFR1 α2,6-sialylation in the poor prognosis and
treatment of ovarian cancer. Therefore, the present study aimed to
evaluate the effects of ST6Gal-I on FGFR1 and ovarian cancer
progression. An overexpressing plasmid and shRNA vector of ST6Gal-I
were constructed and transfected into OVCAR3 cells. After limiting
dilution and persistent culture, stable subcell line clones were
established. The expression of ST6Gal-I was further confirmed by
PCR and western blotting assays. The endogenous ST6Gal-I gene and
protein were stably overexpressed in the oe-ST6Gal-I clone, whereas
the ST6Gal-I gene and protein were decreased in the sh-ST6Gal-I
cell line compared with their expression in the empty vector cell
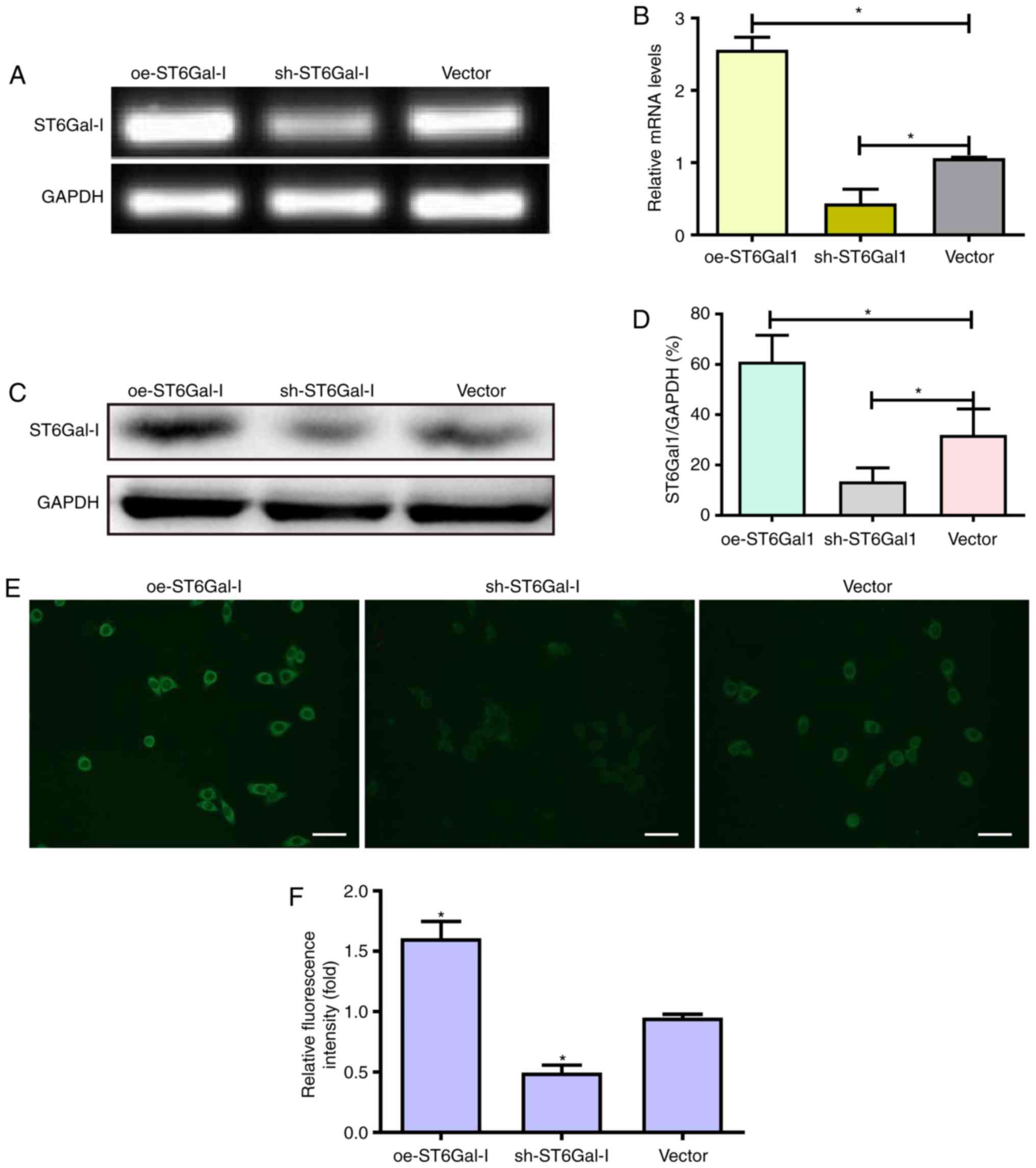
line (Fig. 1A-D). These data
indicated that ST6Gal-I was stably overexpressed or knocked down in
OVCAR3 cells.
To assess the effect of ST6Gal-I upregulation or
downregulation on the FGFR1 receptor in tumor cells,
FITC-conjugated SNA lectin was used to recognize α2,6-linked sialic
acids by immunofluorescence microscopy. The immunofluorescence
results showed that oe-ST6Gal-I cells expressed significantly
higher levels of α2,6-linked sialic acids than vector cells;
conversely, sh-ST6Gal-I cells expressed lower levels of α2,6-linked
sialic acids than vector control cells (Fig. 1E and F).
Cells with high ST6Gal-I expression
enhance tumor cell viability and migratory ability
To test whether ST6Gal-I expression affected ovarian
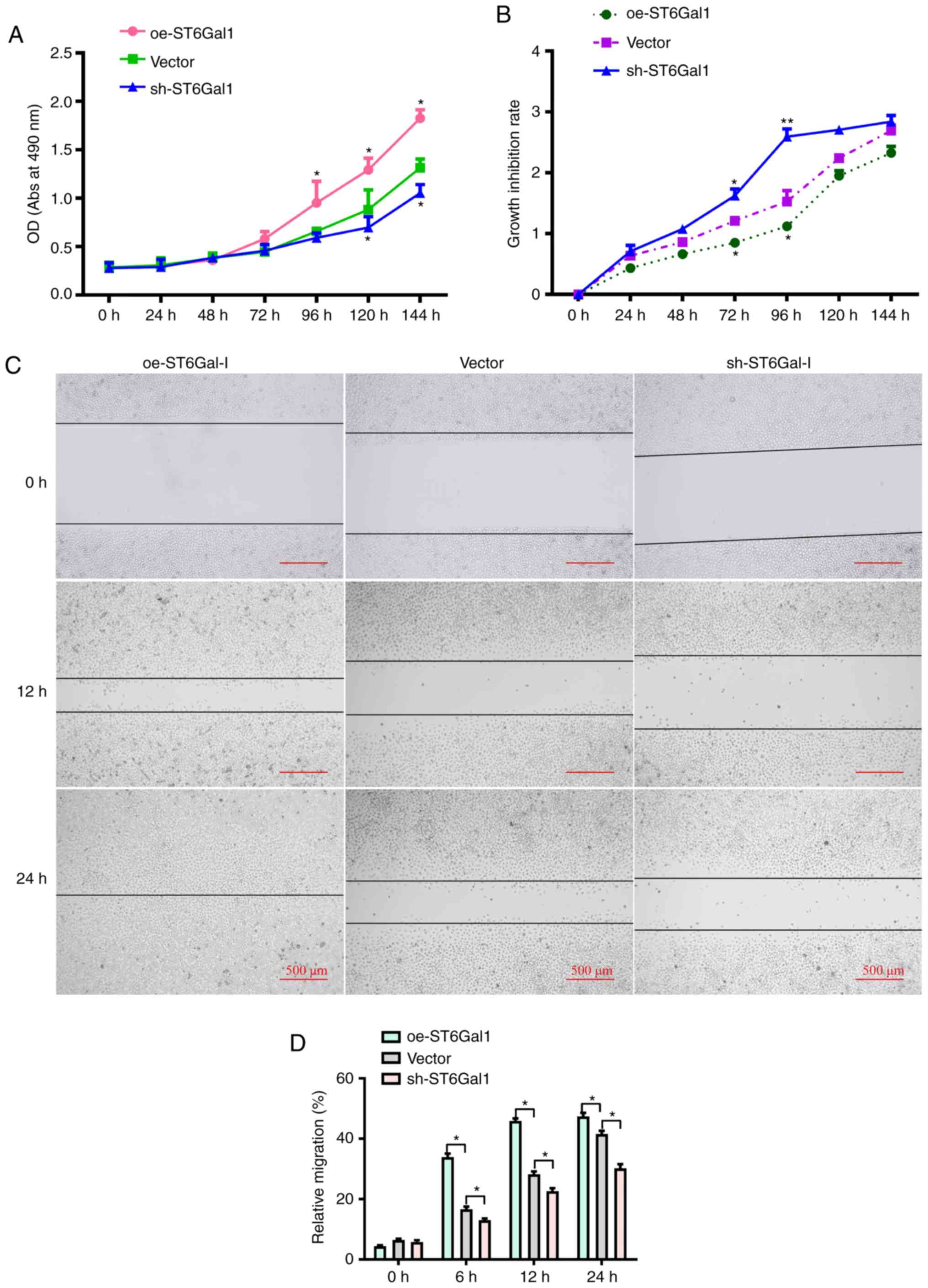
cancer cell viability, a CCK-8 assay was conducted. It was observed
that the growth rate and cell viability were markedly higher in
oe-ST6Gal-I cells than in sh-ST6Gal-I cells and vector cells
incubated with complete culture medium (Fig. 2A). To determine the role of
ST6Gal-I in protecting against serum withdrawal, oe-ST6Gal-I,
sh-ST6Gal-I and vector cells were cultured under serum starvation
conditions for 0, 24, 48, 72, 96, 120 or 144 h. As shown in
Fig. 2B, the CCK-8 results
indicated that the growth inhibition rate and cytotoxicity were
lower in oe-ST6Gal-I cells than in sh-ST6Gal-I cells. Additionally,
scratch-wound healing assays were conducted to detect cell
migration. The results showed that compared with vector cells, the
increased ST6Gal-I expression in oe-ST6Gal-I cells significantly
promoted cell migration, and the decreased ST6Gal-I expression in
sh-ST6Gal-I cells significantly attenuated cell migration at 6, 12
and 24 h (Fig. 2C and D).
Collectively, these results suggested that ST6Gal-I overexpression
promoted the proliferation and migration of ovarian cancer
cells.
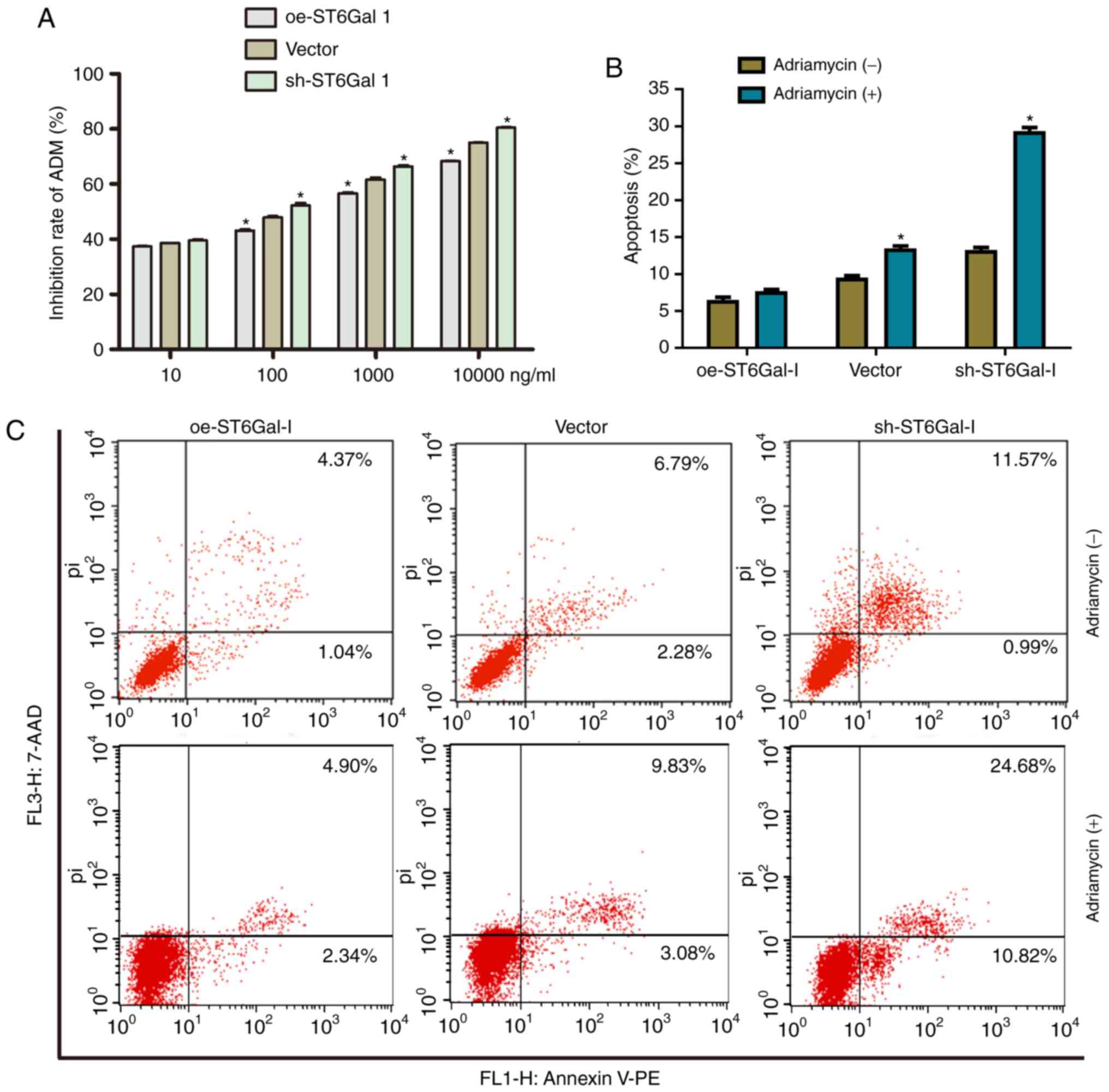
Cells with high ST6Gal-I expression
have reduced cell apoptosis and increased chemoresistance
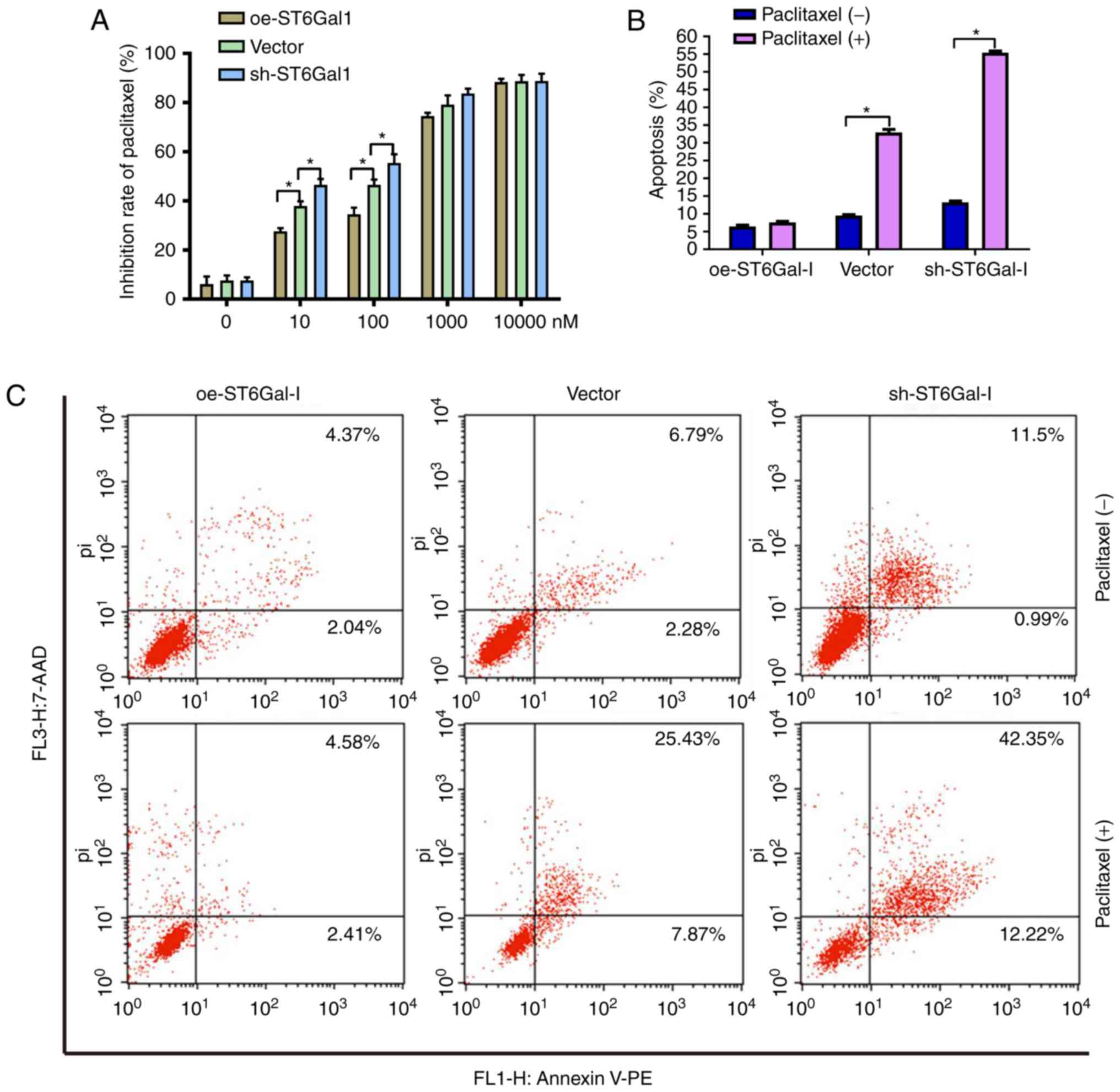
To test the effect of ST6Gal-I expression status on
the anticancer efficacy of paclitaxel, a cell viability assay and
FACS analysis were performed in ovarian cancer cells. The data
showed that the growth inhibition of paclitaxel was dose-dependent
in each group. The growth inhibition was higher in sh-ST6Gal-I
stable clone cells than in oe-ST6Gal-I cells at drug concentrations
of 10–1,000 nM, and most of the cells died at the high
concentration of 10 µM (Fig. 3A).
Furthermore, PE-labeled Annexin V and 7-AAD staining were analyzed
by FACS to determine cell apoptosis. The Annexin V receptor is a
phosphatidylserine, which is normally an asymmetric resident of the
inner membrane. Only when its asymmetric distribution is lost can a
population with increased Annexin V staining be detected. As shown
in Fig. 3B and C, FACS analysis of
oe-ST6Gal-I cells showed only low surface staining for Annexin V,
but Annexin V staining was distinctly higher in sh-ST6Gal-I and
vector cells after treatment with paclitaxel. A small population of
oe-ST6Gal-I cells showed apoptosis (both early- and late-stage),
whereas sh-ST6Gal-I cells had a large population undergoing
apoptosis. Collectively, these results suggested that increased
α2,6 sialylation in cancer cells reduced cell apoptosis and
increased paclitaxel resistance.
α2,6 sialylation of FGFR1 affects the
ERK and FAK signaling pathways
FGFR1 signaling was significantly correlated with
tumorigenesis and metastasis in different types of cancer (10,17).
A previous study demonstrated that FGFR1 phosphorylation can
activate downstream ERK signaling cascades, which play a vital role
in the proliferation and survival of cancer cells (36). Prior findings strongly suggested
that FGFR1 and β3 integrin work in a complex, leading to the
activation of FAK signaling to drive tumor metastasis. To
investigate whether α2,6 sialylation of FGFR1 affected FGFR1
phosphorylation and the downstream ERK and FAK signaling pathways,
a western blotting assay was used to detect the protein levels of
FGFR1, ERK and FAK in oe-ST6Gal-I and sh-ST6Gal-I cells with or
without paclitaxel treatment. As shown in Fig. 4A and B, α2,6-sialylated proteins in
oe-ST6Gal-I and sh-ST6Gal-I cells bound by SNA-agarose were
isolated by SDS-PAGE and immunoblotted for FGFR1. The SNA
precipitation results demonstrated that SNA-FGFR1 expression was
notably higher in oe-ST6Gal-I cells than in sh-ST6Gal-I cells.
ST6Gal-I overexpression in hypersialylated FGFR1 cells, and
paclitaxel treatment attenuated this effect. p-FGFR1 expression was
notably lower in oe-ST6Gal-I cells than in sh-ST6Gal-I cells, and
p-FGFR1 expression was enhanced after 10 µM paclitaxel treatment.
The total FGFR1 expression was similar in each group (Fig. 4A and C). ERK1/2 and FAK
phosphorylation levels were higher in oe-ST6Gal-I cells than in
sh-ST6Gal-I cells, and paclitaxel decreased ERK and FAK
phosphorylation levels in cancer cells, especially in sh-ST6Gal-I
cells (Fig. 4A, D and E). The
present results showed that ST6Gal-I overexpression in cancer cells
increased the α2,6 sialylation of proteins and decreased the
phosphorylation of FGFR1. Both α2,6 sialylation and phosphorylation
of FGFR1 can activate downstream ERK and FAK signaling. Therefore,
it was suggested that the decreased FGFR1 phosphorylation did not
attenuate the effect of high α2,6 sialylation on downstream cascade
activation.
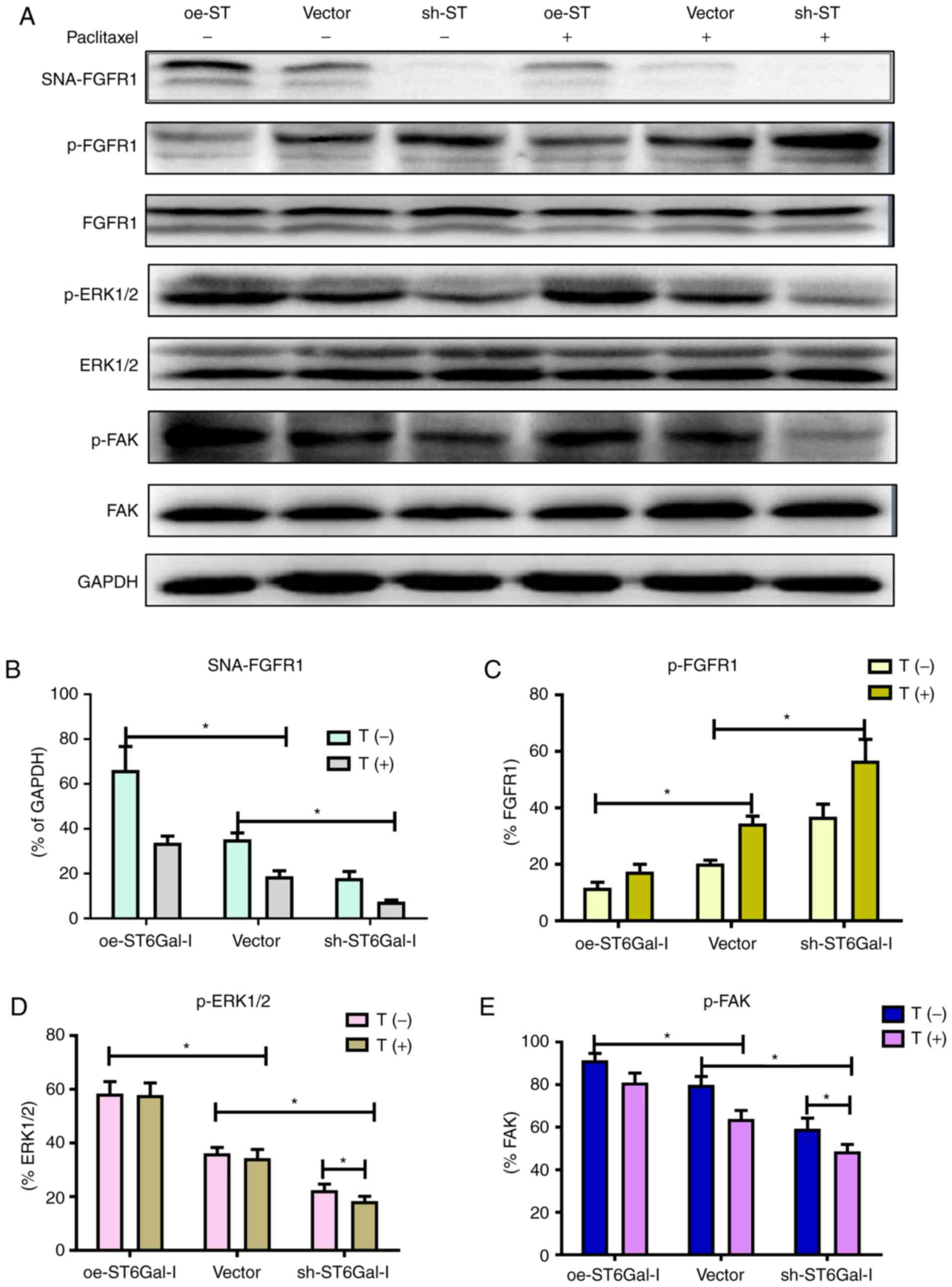 | Figure 4.α2,6 sialylation of FGFR1 affects the
ERK and FAK signaling pathways. (A) oe-ST6Gal-I and sh-ST6Gal-I
cells were treated with or without paclitaxel for 24 h, and
SNA-FGFR1, p-FGFR1, FGFR1, p-ERK1/2, ERK1/2, p-FAK, FAK and GAPDH
protein levels were then measured by western blotting. Relative
protein intensities of (B) SNA-FGFR1, (C) p-FGFR1, (D) p-ERK1/2 and
(E) p-FAK were detected using ImageJ. *P<0.05. FGFR1, fibroblast
growth factor receptor 1; FAK, focal adhesion kinase; ST6Gal-I/ST,
α2,6-sialyltransferase; oe, overexpression; sh, small hairpin; p,
phosphorylated; T, paclitaxel. |
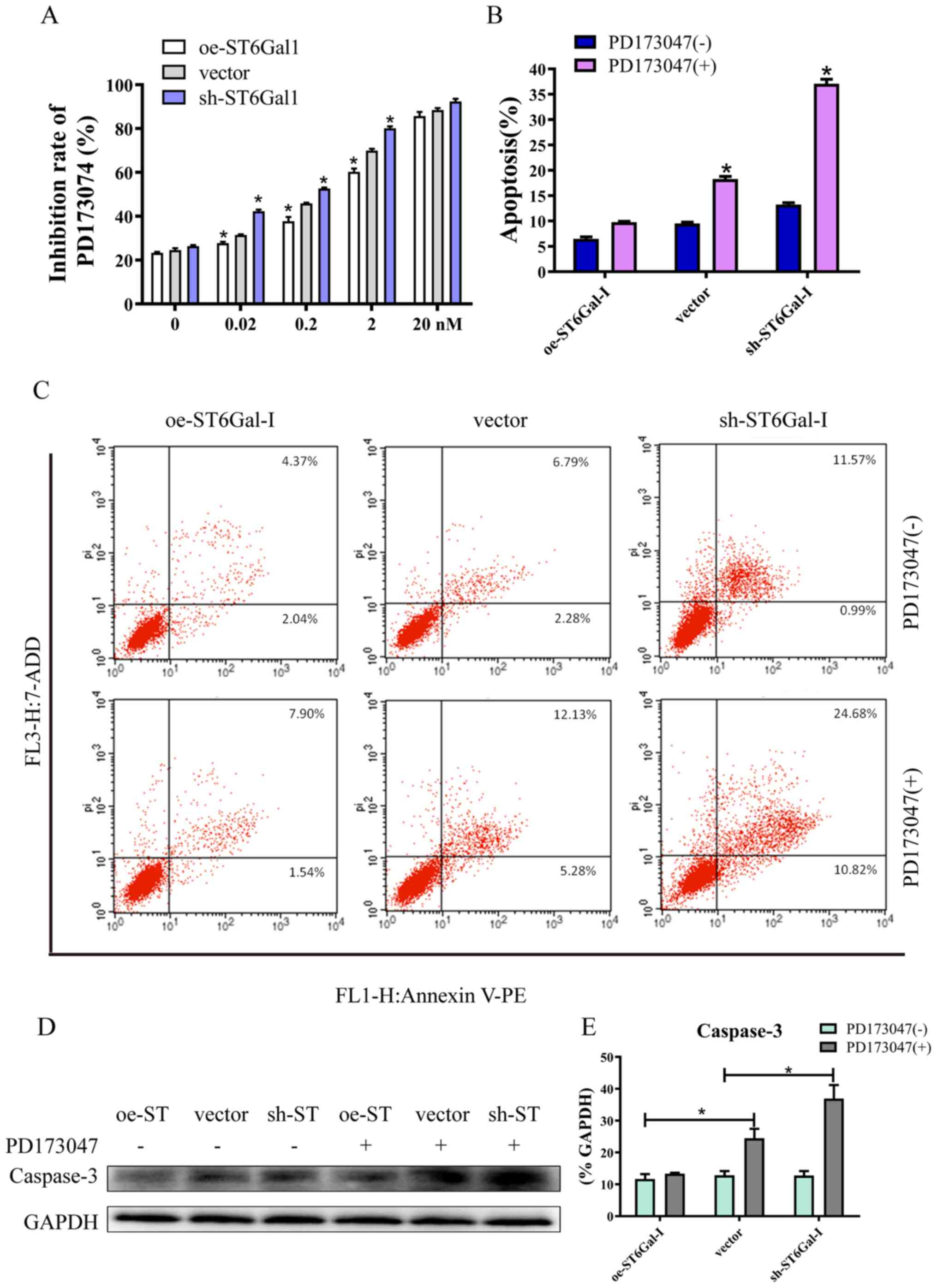
High ST6Gal-I expression attenuates
FGFR1 inhibitor-induced cell apoptosis
Since aberrant FGFR activity has been implicated in
various cancer types, several FGFR inhibitors are currently in the
early phases of clinical development (37). PD173074 has reportedly shown both
high affinity and selectivity for the FGFR family, and is being
used as an FGFR inhibitor in the clinical settings and in
experiments (38,39). The CCK-8 assay results showed that
PD173074 inhibited cancer cell growth in a dose-dependent manner,
and the inhibition rate was lower in oe-ST6Gal-I cells than in
sh-ST6Gal-I cells. Moreover, there was no significant difference
between oe-ST6Gal-I cells and sh-ST6Gal-I cells when the
concentration of PD173074 reached 20 nM, and most of the cells died
(Fig. 5A). The FACS results
demonstrated that the number of apoptotic cells (including early
and late apoptotic cells) was increased with PD173074 treatment in
oe-ST6Gal-I cells. The number of apoptotic sh-ST6Gal-I cells was
notably higher with PD173074 treatment than the oe-ST6Gal-I cells
(Fig. 5B and C). In agreement with
the flow cytometry results, the western blotting results indicated
that the apoptotic marker caspase-3 was significantly increased in
sh-ST6Gal-I cells treated with PD173074, whereas caspase-3 levels
in oe-ST6Gal-I cells were slightly increased after PD173074
treatment compared with vector cells (Fig. 5D and E). Taken together, these data
suggested that FGFR1 inhibitors can effectively induce cell death
in vector cells and sh-ST6Gal-I cells; however, ST6Gal-I
overexpression reduced this anticancer effect.
High ST6Gal-I expression protects
cancer cells from Adriamycin
To further investigate the drug-resistant effects of
ST6Gal-I on another chemotherapy drug, the inhibition rate of cells
and cell apoptosis after treatment with Adriamycin were assessed.
Similar to the paclitaxel results, the growth inhibition of
Adriamycin was dose-dependent in each group. The growth inhibition
was higher in sh-ST6Gal-I stable clone cells than in oe-ST6Gal-I
cells at drug concentrations of 10–10000 nM (Fig. 6A). Furthermore, the FACS results
showed that only a small population of oe-ST6Gal-I cells showed
apoptosis (7.24%), whereas sh-ST6Gal-I cells had a large population
undergoing apoptosis (29.34%) with Adriamycin treatment (Fig. 6B and C). In conclusion, ST6Gal-I
overexpression enhances the chemoresistance of ovarian cancer
cells; conversely, ST6Gal-I knockdown decreases
chemoresistance.
Discussion
Ovarian cancer is characterized by a lack of early
symptoms or screening methods, which often lead to late diagnosis
in advanced stages and a high mortality rate (40). ST6Gal-I has been demonstrated to
confer radiation resistance in colon cancer cell lines (41). However, the functional contribution
of ST6Gal-I to ovarian cancer has yet to be elucidated.
Accumulating evidence suggests that ST6Gal-I is a major inhibitor
of cell death pathways initiated by Fas, TNFR1 and galectins
(27,42). RTKs, such as epidermal growth
factor receptor and FGFR, are highly expressed or activated in
ovarian cancer (10,43,44).
Previous studies have demonstrated that the amplification and
mutation of FGFR1 are associated with poor ovarian cancer prognosis
and malignancy (45,46). In the present study, it was
verified that ST6Gal-I overexpression leads to high sialylation of
FGFR1 in ovarian cancer and the subcellular mechanism was
specifically investigated; FGFR1 signaling regulates cancer cell
behavior. The present results provide novel insight for the role of
α2,6 sialylated FGFR1 in ovarian cancer drug resistance.
In the present study, it was observed that ST6Gal-I
overexpression induced high FGFR1 sialylation, leading to high
proliferation and a low growth inhibition rate under serum
deprivation conditions. Moreover, compared with sh-ST6Gal-I cells,
ST6Gal-I overexpression promoted cancer cell migration after
scratch wounding. In addition, it was identified that ST6Gal-I
overexpression attenuated paclitaxel-induced apoptosis. These
findings are consistent with the hypothesis that ST6Gal-I activity
might underlie the survival or drug resistance of cancer cell
populations (29,47).
FGFR1 is a member of the FGFR family of RTKs; FGFR1
activation leads to downstream signaling via the ERK and FAK
pathways, which are central to growth, survival migration and
angiogenesis in many cancer types (48,49).
ERK activation might be associated with the progression of a wide
variety of neoplasias, as well as poor prognosis and
chemotherapeutic resistance in cancer cells (50,51).
Accumulating evidence supports that FAK also plays a vital role in
tumor cell proliferation, survival and migration (52,53).
In agreement with this evidence, it was observed that ST6Gal-I
overexpression increased the α2,6-linked sialic acids of FGFR1 and
enhanced ERK- and FAK-mediated cell signaling pathways. Although
ST6Gal-I overexpression decreased FGFR1 phosphorylation, the high
FGFR1 sialylation levels could activate the ERK- and FAK-mediated
signaling pathways through other mechanisms. Therefore, further
investigation is needed to thoroughly explore the underlying
mechanism.
Multiple FGFR inhibitors are in development. Many of
these are multi-targeted tyrosine kinase inhibitors against
targeted receptors, including FGFR1 (54). In the present study, it was
identified that the FGFR1 inhibitor PD173074 suppressed cancer cell
proliferation and induced cell apoptosis; however, ST6Gal-I
overexpression attenuated the effects of PD173074. Consistent with
these results, ST6Gal-I overexpression also weakened the effect of
Adriamycin on cancer cells. A hypothesis is that ST6Gal-I
overexpression in cancer cells may protect cells from multiple
apoptotic stimuli, thus promoting cell proliferation and
migration.
In conclusion, the present data suggested that
ST6Gal-I overexpression induces high levels of protein
α2,6-sialylation and that FGFR1 is one of the targeted molecules.
Sialylated FGFR1 activated the ERK and FAK pathways, thus promoting
cell proliferation and migration. Overall, the present study
provides new insight into how ST6Gal-I and FGFR1 signaling regulate
cancer progression and drug resistance in ovarian cancer cells.
Acknowledgements
Not applicable.
Funding
This study was supported by The National Natural
Science Foundation of China (grant nos. 81071751 and 81703120),
Guangdong Provincial Medical Research Foundation (grant no.
A2016360) and Natural Science Foundation of Guangdong Province
(grant no. 2017A030310365).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
LO and XH performed the experiments. SL and XW
secured the funding for the present study. SL designed the present
study, and made final corrections to the manuscript; XW provided
ovarian cancer cell lines and analyzed the chemical resistant data.
XH and ZD sourced and prepared the equipment and reagents for the
experiments. NL and YS performed data collection. JL and LG
analyzed and interpreted the data. LO wrote the manuscript. LO and
LG revised the manuscript. LO responded to the comments and
questions and revised the paper accordingly and LG edited the
manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Glossary
Abbreviations
Abbreviations:
|
FGFR1
|
fibroblast growth factor receptor
1
|
|
ST6Gal-I
|
α2,6-sialyltransferase
|
|
FAK
|
focal adhesion kinase
|
References
|
1
|
Eswarakumar VP, Lax I and Schlessinger J:
Cellular signaling by fibroblast growth factor receptors. Cytokine
Growth Factor Rev. 16:139–149. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Cotton LM, O'Bryan MK and Hinton BT:
Cellular signaling by fibroblast growth factors (FGFs) and their
receptors (FGFRs) in male reproduction. Endocr Rev. 29:193–216.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Touat M, Ileana E, Postel-Vinay S, André F
and Soria JC: Targeting FGFR signaling in cancer. Clin Cancer Res.
21:2684–2694. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Manchado E, Weissmueller S, Morris JP IV,
Chen CC, Wullenkord R, Lujambio A, de Stanchina E, Poirier JT,
Gainor JF, Corcoran RB, et al: A combinatorial strategy for
treating KRAS-mutant lung cancer. Nature. 534:647–651. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Turkington RC, Longley DB, Allen WL,
Stevenson L, McLaughlin K, Dunne PD, Blayney JK, Salto-Tellez M,
Van Schaeybroeck S and Johnston PG: Fibroblast growth factor
receptor 4 (FGFR4): A targetable regulator of drug resistance in
colorectal cancer. Cell Death Dis. 5:e10462014. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Ye T, Wei X, Yin T, Xia Y, Li D, Shao B,
Song X, He S, Luo M, Gao X, et al: Inhibition of FGFR signaling by
PD173074 improves antitumor immunity and impairs breast cancer
metastasis. Breast Cancer Res Treat. 143:435–446. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Wan X, Corn PG, Yang J, Palanisamy N,
Starbuck MW, Efstathiou E, Li Ning Tapia EM, Zurita AJ, Aparicio A,
Ravoori MK, et al: Prostate cancer cell-stromal cell crosstalk via
FGFR1 mediates antitumor activity of dovitinib in bone metastases.
Sci Transl Med. 6:252ra1222014. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Schweiger N, Hauck M, Steinhoff H, Sampl
S, Reifinger M, Walter I, Kreilmeier T, Marian B, Grusch M, Berger
W, et al: Canine and human sarcomas exhibit predominant FGFR1
expression and impaired viability after inhibition of signaling.
Mol Carcinog. 54:841–852. 2015. View
Article : Google Scholar : PubMed/NCBI
|
|
9
|
Garay T, Molnár E, Juhász É, László V,
Barbai T, Dobos J, Schelch K, Pirker C, Grusch M, Berger W, et al:
Sensitivity of melanoma cells to EGFR and FGFR activation but not
inhibition is influenced by oncogenic BRAF and NRAS mutations.
Pathol Oncol Res. 21:957–968. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Cole C, Lau S, Backen A, Clamp A, Rushton
G, Dive C, Hodgkinson C, McVey R, Kitchener H and Jayson GC:
Inhibition of FGFR2 and FGFR1 increases cisplatin sensitivity in
ovarian cancer. Cancer Biol Ther. 10:495–504. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Meng QH, Xu E, Hildebrandt MA, Liang D, Lu
K, Ye Y, Wagar EA and Wu X: Genetic variants in the fibroblast
growth factor pathway as potential markers of ovarian cancer risk,
therapeutic response, and clinical outcome. Clin Chem. 60:222–232.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Birrer MJ, Johnson ME, Hao K, Wong KK,
Park DC, Bell A, Welch WR, Berkowitz RS and Mok SC: Whole genome
oligonucleotide-based array comparative genomic hybridization
analysis identified fibroblast growth factor 1 as a prognostic
marker for advanced-stage serous ovarian adenocarcinomas. J Clin
Oncol. 25:2281–2287. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Ivan M and Matei D: Blockade of FGF
signaling: Therapeutic promise for ovarian cancer. Cancer Biol
Ther. 10:505–508. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Marmé F, Hielscher T, Hug S, Bondong S,
Zeillinger R, Castillo-Tong DC, Sehouli J, Braicu I, Vergote I,
Isabella C, et al: Fibroblast growth factor receptor 4 gene (FGFR4)
388Arg allele predicts prolonged survival and platinum sensitivity
in advanced ovarian cancer. Int J Cancer. 131:E586–E591. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Zhao X, Zhou Y, Chen YU and Yu F: miR-494
inhibits ovarian cancer cell proliferation and promotes apoptosis
by targeting FGFR2. Oncol Lett. 11:4245–4251. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Zecchini S, Bombardelli L, Decio A,
Bianchi M, Mazzarol G, Sanguineti F, Aletti G, Maddaluno L, Berezin
V, Bock E, et al: The adhesion molecule NCAM promotes ovarian
cancer progression via FGFR signalling. EMBO Mol Med. 3:480–494.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Yang F, Zhang Y, Ressler SJ, Ittmann MM,
Ayala GE, Dang TD, Wang F and Rowley DR: FGFR1 is essential for
prostate cancer progression and metastasis. Cancer Res.
73:3716–3724. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Welm BE, Freeman KW, Chen M, Contreras A,
Spencer DM and Rosen JM: Inducible dimerization of FGFR1:
Development of a mouse model to analyze progressive transformation
of the mammary gland. J Cell Biol. 157:703–714. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Bohrer LR and Schwertfeger KL: Macrophages
promote fibroblast growth factor receptor-driven tumor cell
migration and invasion in a CXCR2-dependent manner. Mol Cancer Res.
10:1294–1305. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Duchesne L, Tissot B, Rudd TR, Dell A and
Fernig DG: N-glycosylation of fibroblast growth factor receptor 1
regulates ligand and heparan sulfate co-receptor binding. J Biol
Chem. 281:27178–27189. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Taniguchi N and Korekane H: Branched
N-glycans and their implications for cell adhesion, signaling and
clinical applications for cancer biomarkers and in therapeutics.
BMB Rep. 44:772–781. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Dall'Olio F, Malagolini N, Trinchera M and
Chiricolo M: Mechanisms of cancer-associated glycosylation changes.
Front Biosci (Landmark Ed). 17:670–699. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Schultz MJ, Swindall AF and Bellis SL:
Regulation of the metastatic cell phenotype by sialylated glycans.
Cancer Metastasis Rev. 31:501–518. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Dall'Olio F, Malagolini N, Trinchera M and
Chiricolo M: Sialosignaling: Sialyltransferases as engines of
self-fueling loops in cancer progression. Biochim Biophys Acta.
1840:2752–2764. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Harduin-Lepers A, Vallejo-Ruiz V,
Krzewinski-Recchi MA, Samyn-Petit B, Julien S and Delannoy P: The
human sialyltransferase family. Biochimie. 83:727–737. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Lee M, Park JJ and Lee YS: Adhesion of
ST6Gal I-mediated human colon cancer cells to fibronectin
contributes to cell survival by integrin beta1-mediated paxillin
and AKT activation. Oncol Rep. 23:757–761. 2010.PubMed/NCBI
|
|
27
|
Swindall AF and Bellis SL: Sialylation of
the Fas death receptor by ST6Gal-I provides protection against
Fas-mediated apoptosis in colon carcinoma cells. J Biol Chem.
286:22982–22990. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Zhuo Y, Chammas R and Bellis SL:
Sialylation of beta1 integrins blocks cell adhesion to galectin-3
and protects cells against galectin-3-induced apoptosis. J Biol
Chem. 283:22177–22185. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Park JJ, Yi JY, Jin YB, Lee YJ, Lee JS,
Lee YS, Ko YG and Lee M: Sialylation of epidermal growth factor
receptor regulates receptor activity and chemosensitivity to
gefitinib in colon cancer cells. Biochem Pharmacol. 83:849–857.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Britain CM, Dorsett KA and Bellis SL: The
glycosyltransferase ST6Gal-I protects tumor cells against serum
growth factor withdrawal by enhancing survival signaling and
proliferative potential. J Biol Chem. 292:4663–4673. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) Method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Saporiti F, Piacentini L, Alfieri V, Bono
E, Ferrari F, Chiesa M and Colombo GI: Melanocortin-1 receptor
positively regulates human artery endothelial cell migration. Cell
Physiol Biochem. 52:1339–1360. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Park JS, Lee JS, Kim EY, Jung JY, Kim SK,
Chang J, Kim DJ, Lee CY, Jung I and Kim JH: The frequency and
impact of FGFR1 amplification on clinical outcomes in Korean
patients with small cell lung cancer. Lung Cancer. 88:325–331.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Preusser M, Berghoff AS, Berger W,
Ilhan-Mutlu A, Dinhof C, Widhalm G, Dieckmann K, Wöhrer A, Hackl M,
von Deimling A, et al: High rate of FGFR1 amplifications in brain
metastases of squamous and non-squamous lung cancer. Lung Cancer.
83:83–89. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Gessi M, Moneim YA, Hammes J, Goschzik T,
Scholz M, Denkhaus D, Waha A and Pietsch T: FGFR1 mutations in
Rosette-forming glioneuronal tumors of the fourth ventricle. J
Neuropathol Exp Neurol. 73:580–584. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Wu J, Wei T, Tang Q, Weng B, Li W, Jiang
X, Ding T, Li X, Liang G, Cai Y and Ji J: Discovery and anti-cancer
evaluation of two novel non-ATP-competitive FGFR1 inhibitors in
non-small-cell lung cancer. BMC Cancer. 15:2762015. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Turner N and Grose R: Fibroblast growth
factor signalling: From development to cancer. Nat Rev Cancer.
10:116–129. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Mohammadi M, Froum S, Hamby JM, Schroeder
MC, Panek RL, Lu GH, Eliseenkova AV, Green D, Schlessinger J and
Hubbard SR: Crystal structure of an angiogenesis inhibitor bound to
the FGF receptor tyrosine kinase domain. EMBO J. 17:5896–5904.
1998. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Nguyen PT, Tsunematsu T, Yanagisawa S,
Kudo Y, Miyauchi M, Kamata N and Takata T: The FGFR1 inhibitor
PD173074 induces mesenchymal-epithelial transition through the
transcription factor AP-1. Br J Cancer. 109:2248–2258. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Torre LA, Bray F, Siegel RL, Ferlay J,
Lortet-Tieulent J and Jemal A: Global cancer statistics, 2012. CA
Cancer J Clin. 65:87–108. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Lee M, Lee HJ, Bae S and Lee YS: Protein
sialylation by sialyltransferase involves radiation resistance. Mol
Cancer Res. 6:1316–1325. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Zhuo Y and Bellis SL: Emerging role of
alpha2,6-sialic acid as a negative regulator of galectin binding
and function. J Biol Chem. 286:5935–5941. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Gui T and Shen K: The epidermal growth
factor receptor as a therapeutic target in epithelial ovarian
cancer. Cancer Epidemiol. 36:490–496. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
McKie AB, Vaughan S, Zanini E, Okon IS,
Louis L, de Sousa C, Greene MI, Wang Q, Agarwal R, Shaposhnikov D,
et al: The OPCML tumor suppressor functions as a cell surface
repressor-adaptor, negatively regulating receptor tyrosine kinases
in epithelial ovarian cancer. Cancer Discov. 2:156–171. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Gorringe KL, Jacobs S, Thompson ER,
Sridhar A, Qiu W, Choong DY and Campbell IG: High-resolution single
nucleotide polymorphism array analysis of epithelial ovarian cancer
reveals numerous microdeletions and amplifications. Clin Cancer
Res. 13:4731–4739. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Whitworth MK, Backen AC, Clamp AR, Wilson
G, McVey R, Friedl A, Rapraeger AC, David G, McGown A, Slade RJ, et
al: Regulation of fibroblast growth factor-2 activity by human
ovarian cancer tumor endothelium. Clin Cancer Res. 11:4282–4288.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Chen X, Wang L, Zhao Y, Yuan S, Wu Q, Zhu
X, Niang B, Wang S and Zhang J: ST6Gal-I modulates docetaxel
sensitivity in human hepatocarcinoma cells via the p38 MAPK/caspase
pathway. Oncotarget. 7:51955–51964. 2016.PubMed/NCBI
|
|
48
|
Singleton KR, Hinz TK, Kleczko EK, Marek
LA, Kwak J, Harp T, Kim J, Tan AC and Heasley LE: Kinome RNAi
screens reveal synergistic targeting of MTOR and FGFR1 pathways for
treatment of lung cancer and HNSCC. Cancer Res. 75:4398–4406. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Anderson HJ and Galileo DS: Small-molecule
inhibitors of FGFR, integrins and FAK selectively decrease
L1CAM-stimulated glioblastoma cell motility and proliferation. Cell
Oncol (Dordr). 39:229–242. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Wales CT, Taylor FR, Higa AT, McAllister
HA and Jacobs AT: ERK-dependent phosphorylation of HSF1 mediates
chemotherapeutic resistance to benzimidazole carbamates in
colorectal cancer cells. Anticancer Drugs. 26:657–666.
2015.PubMed/NCBI
|
|
51
|
Xiao Z, Ding N, Xiao G, Wang S, Wu Y and
Tang L: Reversal of multidrug resistance by gefitinib via RAF1/ERK
pathway in pancreatic cancer cell line. Anat Rec (Hoboken).
295:2122–2128. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Zhang J and Hochwald SN: The role of FAK
in tumor metabolism and therapy. Pharmacol Ther. 142:154–163. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Lee BY, Timpson P, Horvath LG and Daly RJ:
FAK signaling in human cancer as a target for therapeutics.
Pharmacol Ther. 146:132–149. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Katoh M: FGFR inhibitors: Effects on
cancer cells, tumor microenvironment and whole-body homeostasis
(Review). Int J Mol Med. 38:3–15. 2016. View Article : Google Scholar : PubMed/NCBI
|