Introduction
Posterior capsular opacification (PCO) is the main
complication following cataract surgery and it is a leading cause
of visual impairment worldwide (1,2).
While there has been an improvement in surgical techniques and
intraocular lens material, the incidence of PCO remains high in
15–50% of patients within 2–5 years following cataract surgery
(3,4). The proliferation of residual lens
epithelial cells (LECs) serves an important role in PCO formation;
residual LECs have been discovered to regenerate within a few hours
following cataract surgery, before migrating across the posterior
capsule (5,6). Thus, inhibiting the proliferation of
LECs may be an important therapeutic strategy for PCO prevention in
clinical practice.
High levels of vitamin C intake have been revealed
to serve beneficial effects in preventing age-related cataracts or
PCO formation following cataract surgery (7–10).
In addition, the long-term supplement use of vitamin C has been
inversely associated with the occurrence of cataracts or PCO risk
(11,12). Hypoxia-inducible factor-1α (HIF-1α)
and vascular endothelial growth factor (VEGF) have also been
discovered to serve important roles in the stimulation of cell
proliferation and migration; VEGF is a target gene of the HIF-1α
and the upregulation of the HIF-1α/VEGF signaling axis was
identified to promote cell proliferation and migration (13,14).
The proline hydroxylation of HIF-1α by prolyl hydroxylases (PHDs)
is responsible for the rapid degradation of HIF-1α (15,16).
Notably, vitamin C has been identified to serve as a cofactor of
PHDs (17). The authors' previous
study demonstrated that vitamin C inhibited the proliferation of
human LECs by enhancing the rapid degradation of HIF-1α via proline
hydroxylation and thus, inhibited the expression levels of VEGF
(10).
The present study aimed to investigate the molecular
mechanisms of vitamin C on the expression levels of VEGF in more
detail. The findings of the present study demonstrated that the
HIF-1 inhibitor BAY 87–2243 significantly inhibited cell
proliferation and VEGF expression levels in LECs. Moreover, vitamin
C further inhibited the proliferation and expression levels of VEGF
in LECs following the treatment with the HIF-1 inhibitor. These
findings suggested that vitamin C may inhibit VEGF expression
levels via both HIF-1α-dependent and -independent pathways in
LECs.
Materials and methods
Cell culture
The human LEC line HLE-B3 was purchased from the
American Type Culture Collection and the 293T cell line was
obtained from the Cell Bank of Type Culture Collection of the
Chinese Academy of Sciences. Both cells were cultured in DMEM
(Gibco; Thermo Fisher Scientific, Inc.), supplemented with 10% FBS
(Gibco; Thermo Fisher Scientific, Inc.), and maintained in a
humidified atmosphere at 37°C and 5% CO2.
Reagents
Primary antibodies against glucose transporter 1
(GLUT1; 1:2,000; cat. no. 12939), lysine-specific demethylase 1
(LSD1; 1:2,000; cat. no. 2184), HIF-1α (1:1,1000; cat. no. 36169),
PHD2 (1:3,000; cat. no. 4835), AKT (1:2,000; cat. no. 2938) and
phosphorylated (p)-T308-AKT (1:1,000; cat. no. 13038) were
purchased from Cell Signaling Technology, Inc. Primary antibodies
against β-actin (1:5,000; cat. no. ab16039), VEGF (1:1,000; cat.
no. ab46154) and hydroxyproline (1:500; cat. no. ab37067; for
detecting prolyl hydroxylated AKT) were purchased from Abcam. The
horseradish peroxidase-conjugated secondary antibody (1:5,000; cat.
no. ab205718) was obtained from Abcam and normal rabbit IgG (1:500;
cat. no. 2729) was purchased from Cell Signaling Technology, Inc.
Vitamin C (100 µM; cat. no. 95209) and dimethyloxaloylglycine
(DMOG; 1 mM; cat. no. D3695) were obtained from Sigma-Aldrich;
Merck KGaA. The PHD2 selective inhibitor IOX2 (10 µM; cat. no.
9451) and AKT inhibitor A-674563 (5 µM; cat. no. B1761) were
purchased from BioVision, Inc. The HIF-1 inhibitor BAY 87–2243 (100
nM; cat. no. B1115) was from APeXBIO Technology LLC and the PHD
inhibitor Molidustat (5 µM; cat. no. S8138) was obtained from
Selleck Chemicals. The above drugs were used to treat HLE-B3 cells
at 37°C.
Lentiviral transfection
PHD2 short hairpin RNA (shRNA/sh) targeting
sequences (sh1, 5′-CGCAATAACTGTTTGGTATTT-3′; and sh2,
5′-CTGTTATCTAGCTGAGTTCAT-3′) and a scramble control
(5′-CCTAAGGTTAAGTCGCCCTCGCTCGAGCGAGGGCGACTTAACCTTAGG-3′) were
cloned into a pLKO.1 lentiviral knockdown vector (Open Biosystems,
Inc.). Lentiviral expression vectors (pCDH-CMV-MCS-EF1-Puro; System
Biosciences, LLC) expressing wild-type (WT) AKT and
hydroxylation-deficient mutants of AKT (AKT-P125/313A) were
purchased from GrowHealthy. A total of 5×106 293T cells
were transfected with 12 µg plasmids using
Lipofectamine® 3000 reagent (Thermo Fisher Scientific,
Inc.). Lentiviral vectors were produced in 293T cells with
packaging plasmids according to our previous study (14). Following incubation for 48 h at
37°C, the lentivirus supernatants were harvested and transfected
into 1×106 HLE-B3 cells in the presence of 8 µg/ml
polybrene [cat. no. 28728-55-4; Sigma-Aldrich (Merck KGaA)].
Following incubation with puromycin-containing media for 72 h, the
expression levels of target proteins were analyzed using western
blotting.
Reverse transcription-quantitative PCR
(RT-qPCR)
Total RNA was extracted from cells using the RNeasy
Mini kit (cat. no. 74106; Qiagen, Inc.), according to the
manufacturer's protocol. Total RNA was reverse transcribed into
cDNA using a PrimeScript™ RT reagent kit (Takara Bio, Inc.),
according to the manufacturer's protocol, for 1 h at 37°C. qPCR was
subsequently performed using SYBR Green I dye (Takara Bio, Inc.),
according to the manufacturer's protocol. The following
thermocycling conditions were used for the qPCR: 95°C for 15 min;
followed by 40 cycles of 94°C for 15 sec, 55°C for 30 sec and 70°C
for 30 sec; and a final extension step at 72°C for 5 min. The
following primer pairs were used for the qPCR: VEGF forward,
5′-TGCAGATTATGCGGATCAAACC-3′ and reverse,
5′-TGCATTCACATTTGTTGTGCTGTAG-3′; and β-actin forward,
5′-GAGCACAGAGCCTCGCCTTT-3′ and reverse, 5′-AGAGGCGTACAGGGATAGCA-3′.
Relative mRNA expression levels were quantified using the
2−ΔΔCq method (18) and
normalized to β-actin as the endogenous control.
Immunoprecipitation assay
A total of 1×107 cells were lysed with 1
ml NP40 lysis buffer (Beyotime Institute of Biotechnology).
Following centrifugation at 16,500 × g for 20 min at 4°C, cell
lysates were pre-cleared with 40 µl Protein A/G beads alone (cat.
no. sc-2003; Santa Cruz Biotechnology, Inc.) for 1 h at 4°C to
reduce the non-specific protein binding. The pre-cleared cell
lysates were subsequently incubated with an anti-AKT antibody
(1:500) to isolate total AKT protein or normal rabbit IgG (1:500)
as the negative control for 3 h at 4°C and then with 40 µl protein
A/G agarose beads overnight at 4°C. Following the incubations,
protein A/G agarose beads were washed with NP40 buffer five times
and the immunoprecipitated samples were eluted from protein A/G
agarose beads with 40 µl 1X SDS sample buffer by heating at 100°C
for 5 min. Following centrifugation at 3,650 × g for 1 min at 4°C,
the proteins (20 µl from the total 40 µl sample) were analyzed
using western blotting. The expression levels of immunoprecipitated
endogenous prolyl hydroxylated AKT (pro-AKT) were detected using an
anti-hydroxyproline antibody, as described below.
Western blotting
Total protein was extracted from cells using RIPA
lysis buffer containing a protease/phosphatase inhibitors mixture
(Beyotime Institute of Biotechnology). Nuclear proteins were
extracted using a nucleoprotein extraction kit (cat. no. BB-3102-1;
BestBio Co. Ltd.), according to the manufacturer's protocol. Total
protein was quantified using the Bradford method and 50 µg
protein/lane was separated via 10% SDS-PAGE. The separated proteins
were subsequently transferred onto PVDF membranes (EMD Millipore)
and blocked with 5% BSA (Beijing Solarbio Science & Technology
Co., Ltd.) in TBS-0.1% Tween (TBST) buffer (Thermo Fisher
Scientific, Inc.) at 37°C for 1 h. The membranes were then
incubated with the following primary antibodies at 4°C overnight:
Anti-GLUT1, anti-LSD1, anti-HIF-1α, anti-PHD2, anti-AKT,
anti-p-T308-AKT, anti-β-actin, anti-VEGF and anti-hydroxyproline.
Following the primary antibody incubation, the membranes were
washed thrice with PBST buffer and incubated with the horseradish
peroxidase-conjugated secondary antibody at 37°C for 1 h. Protein
bands were detected using an ECL™ Western Blotting Detection
reagent (GE Healthcare Life Sciences) and the expression levels
were quantified using ImageJ version 1.47v software (National
Institutes of Health). Each experiment was independently repeated
three times.
Colony formation assay
A total of 2.5×102 HLE-B3 cells/well were
plated into 6-cm well plates and incubated for 24 h. Cells were
subsequently treated with the DMSO vehicle control [equal volume to
the inhibitor; Sigma-Aldrich (Merck KGaA)] or different inhibitors
(100 nM BAY 87-2243100; 100 µM vitamin C; 5 µM A-674563) at 37°C
for 12 days. Following the incubation, cells were fixed with 75%
ethanol at 37°C for 15 min and then stained with 0.01% crystal
violet at 37°C for 30 min and visualized. The relative colony
formation rate (%) was determined using the following equation:
(Number of colonies formed from drug-treated cells/number of
colonies formed from untreated cells) ×100.
ELISA
An ELISA was used to detect the secretory levels of
VEGF in HLE-B3 cells. Briefly, a total of 1×106 HLE-B3
cells were treated with DMSO vehicle, 100 nM BAY 87-2243100 or 100
nM BAY 87-2243100 and 100 µM vitamin C at 37°C for 12 h. Following
centrifugation of 2,000 × g for 20 min at room temperature, cell
culture supernatants were collected. VEGF levels were measured
using a Human VEGF Quantikine ELISA kit (cat. no. DVE00; R&D
Systems, Inc.) according to the manufacturer's protocol.
Statistical analysis
Statistical analysis was performed using GraphPad
Prism 7.0 software (GraphPad Software, Inc.) and data are expressed
as the mean ± SD of three independent experiments. Statistical
differences between two groups were analyzed by a two-tailed
Student's t-test, whereas a one-way ANOVA followed by a Tukey's
multiple comparison test was used for multiple groups. P<0.05
were considered to indicate a statistically significant
difference.
Results
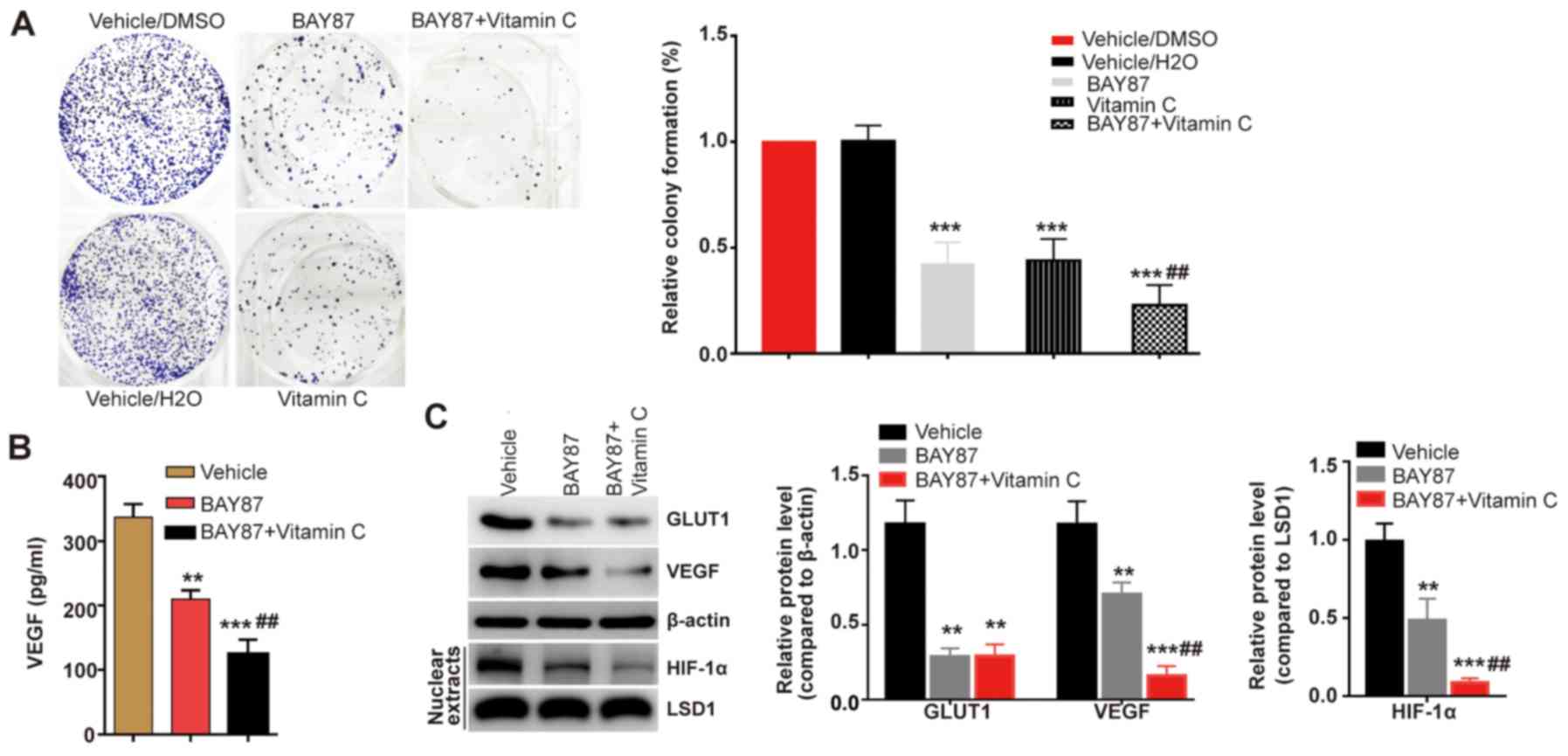
Vitamin C inhibits cell proliferation
and VEGF expression levels following HIF-1 inhibition
Our previous study revealed that vitamin C inhibited
cell proliferation and VEGF expression levels (10). Vitamin C, an essential cofactor of
PHDs, has been discovered to increase the proline hydroxylation of
HIF-1α, thus promoting the degradation of HIF-1α via the
ubiquitin-proteasome pathway (17). Therefore, it was hypothesized that
HIF-1α may serve a crucial role in vitamin C-inhibited LEC
proliferation. The present study used a HIF-1 inhibitor (BAY
87-2243) to investigate the effect of vitamin C in HLE-B3 LECs.
While the proliferation of HLE-B3 cells was significantly inhibited
following the treatment with BAY 87-2243 compared with the vehicle
group, the combined treatment of BAY 87-2243 with vitamin C was
found to further inhibit the proliferation of LECs compared with
the BAY 87-2243 group (Fig.
1A).
GLUT1 and VEGF, which both possess hormone response
elements, are well-known targets of HIF-1α (19). Thus, the present study analyzed the
expression levels of GLUT1 and the secretion of VEGF in the
presence of BAY 87-2243 and vitamin C. It was found that BAY
87-2243 significantly inhibited the expression levels of GLUT1 and
the secretion of VEGF compared with the vehicle group (Fig. 1B and C). However, whilst the
combined treatment of BAY 87-2243 and vitamin C further inhibited
VEGF expression levels and secretion in HLE-B3 cells, the
expression levels of GLUT1 were not further affected by vitamin C
(Fig. 1B and C). Although the
HIF-1α protein is rapidly degraded by the ubiquitin-proteasome
pathway under normoxic conditions (20), HIF-1α can be detected from isolated
nuclear extracts under normoxic conditions (21,22).
In the present study, it was revealed that vitamin C demonstrated a
synergistic effect with BAY 87-2243 by decreasing the expression
levels of HIF-1α compared with the vehicle and to a further extent
compared with the BAY 87-2243 group (Fig. 1C). Therefore, the present results
suggested that vitamin C may inhibit VEGF expression levels via
HIF-1α dependent and independent pathways in LECs.
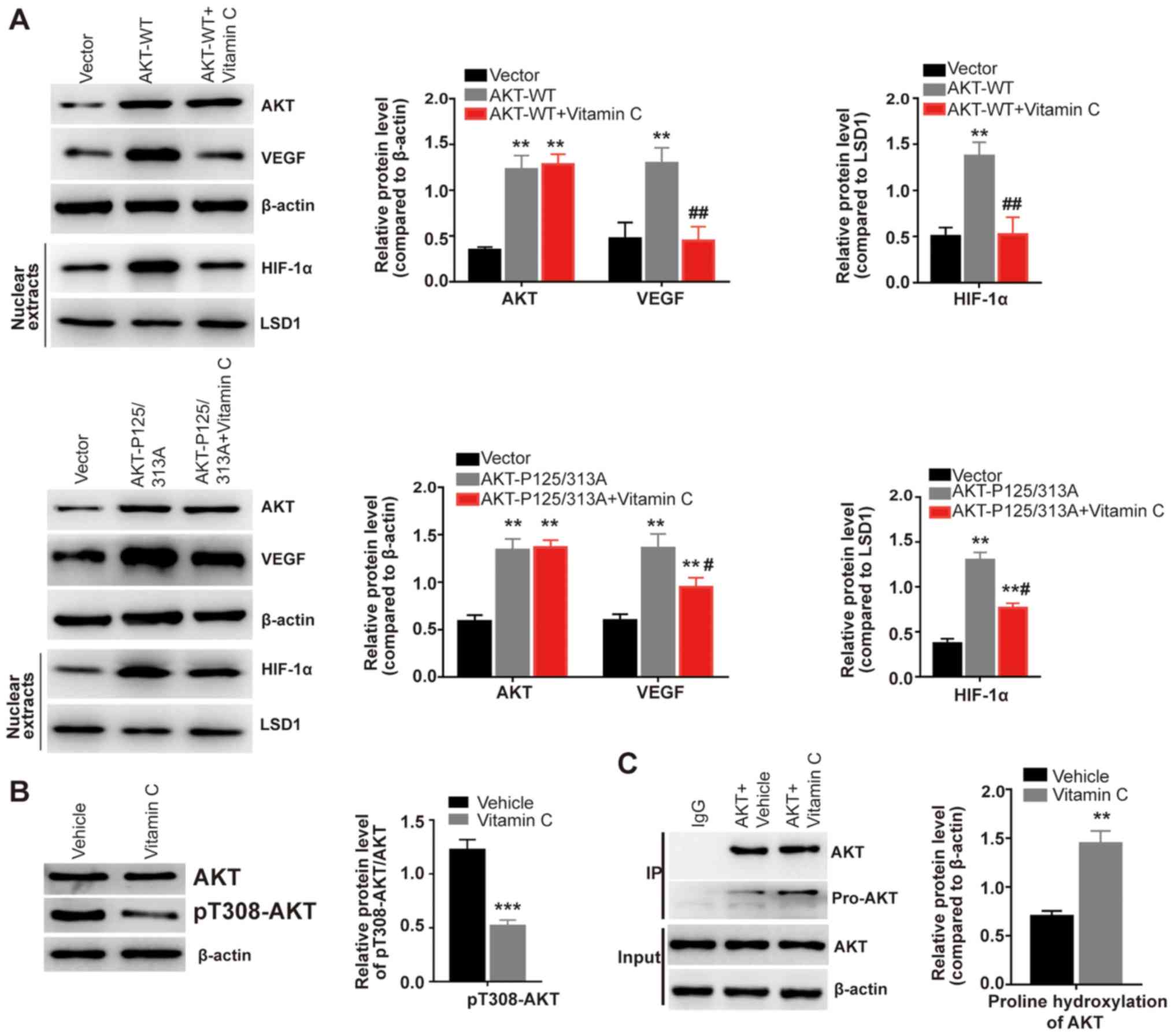
Vitamin C increases proline
hydroxylation and inhibits the activity of AKT
The activation of the serine/threonine-specific
protein kinase AKT has been found to promote the proliferation of
various types of cancer (23). AKT
activation is involved in mediating VEGF upregulation (24). Furthermore, AKT is
prolyl-hydroxylated by the oxygen-dependent prolyl hydroxylase PHD2
(25). The present results
observed that the overexpression of AKT-WT significantly increased
the expression levels of VEGF compared with the vector group
(Fig. 2A). Notably, following the
treatment with vitamin C, the increased expression levels of VEGF
observed in the AKT-WT group were significantly decreased compared
with the AKT-WT group (Fig. 2A).
It has been reported that proline at position 125 or 313 of AKT can
be hydroxylated by PHD2, which leads to aberrant AKT activation
(25). The present study used a
hydroxylation-deficient mutant of AKT by replacing proline at
position 125 and 313 with alanine (P125/313A). While AKT-P125/313A
overexpression significantly increased the expression levels of
VEGF compared with the vector group, vitamin C treatment
significantly reduced the increased expression levels of VEGF
induced by AKT-P125/313A overexpression (Fig. 2A). The expression and activity of
HIF-1α is also reportedly enhanced by AKT signaling under both
normoxic and hypoxic conditions (26,27).
In the present study, the overexpression of AKT-WT or AKT-P125/313A
significantly increased the expression levels of HIF-1α from
isolated nuclear extracts compared with the vector group (Fig. 2A). Moreover, vitamin C
significantly weakened the increased expression levels of HIF-1α
induced by AKT-WT or AKT-P125/313A overexpression (Fig. 2A). Moreover, vitamin C was
demonstrated to significantly decrease the expression levels of
pT308-AKT compared with the vehicle-treated HLE-B3 cells (Fig. 2B). An immunoprecipitation assay was
subsequently performed using a human AKT antibody to isolate
endogenous AKT in HLE-B3 cells. It was observed that vitamin C
treatment significantly increased the prolyl hydroxylation of AKT
compared with the vehicle treated cells (Fig. 2C). Collectively, these findings
suggested that vitamin C may suppress AKT kinase activity by
enhancing the prolyl hydroxylation of AKT.
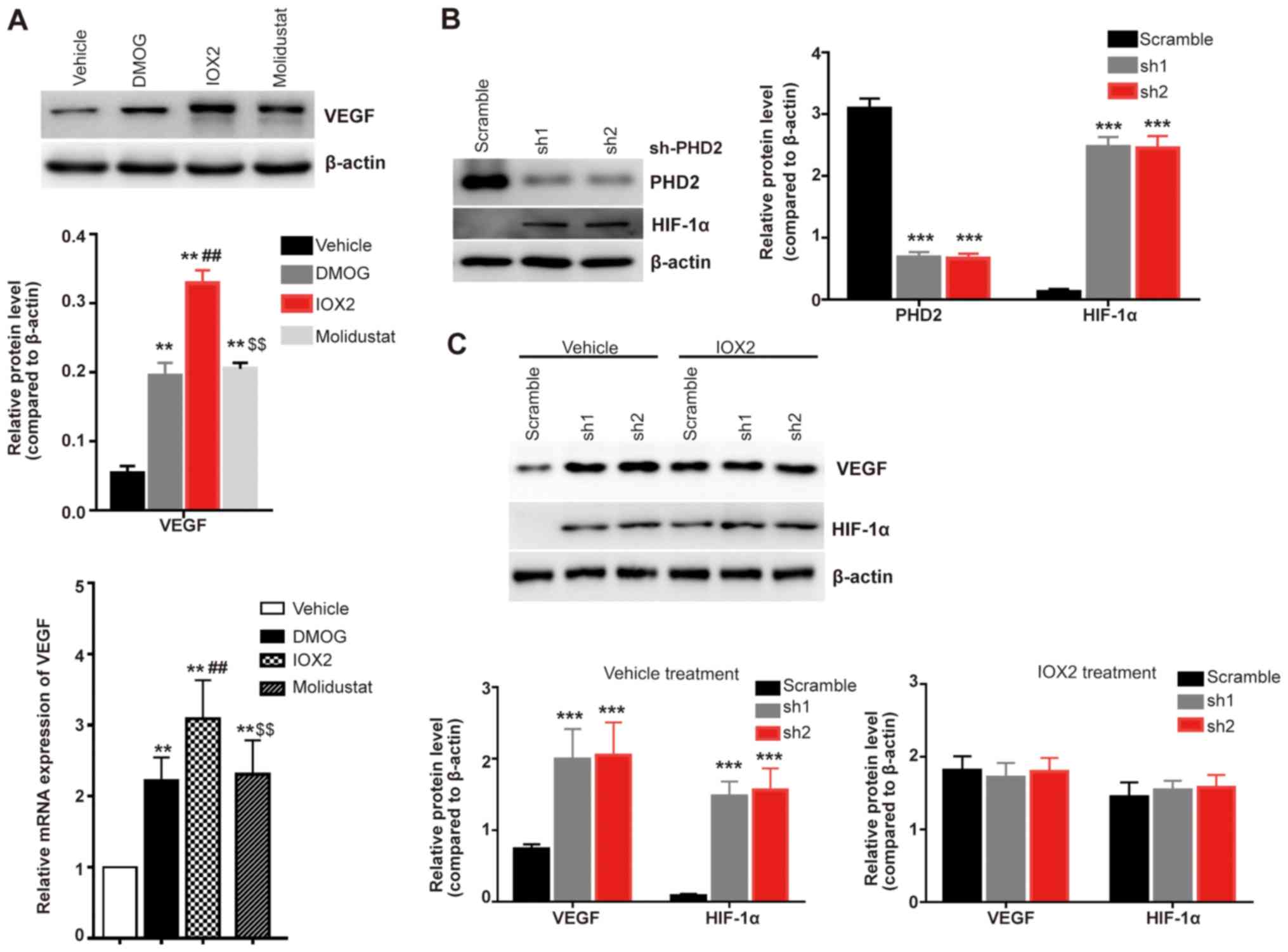
Inhibition of PHD2 increases VEGF
expression levels
To further investigate the mechanism via which
vitamin C regulates VEGF expression levels, the present study used
three PHDs inhibitors (DMOG, IOX2 and Molidustat) to investigate
their effects on the expression levels of VEGF. All of these
inhibitors were found to significantly increase the expression
levels of VEGF compared with the vehicle treated cells (Fig. 3A); the PHD2 specific inhibitor IOX2
exhibited the most significant effect at both the mRNA and protein
expression level compared with the non-selective inhibitors DMOG
and Molidustat (Fig. 3A). A PHD2
stable genetic knockdown model was subsequently established in
HLE-B3 cells (Fig. 3B) and
increased expression levels of HIF-1α were observed in the stable
PHD2 knockdown HLE-B3 cells (both sh1 and sh2) compared with the
scramble control-transfected cells (Fig. 3B). Furthermore, the genetic
knockdown of PHD2 significantly increased the expression levels of
VEGF compared with the scramble group (Fig. 3C); however, there were no
significant differences in the expression levels of VEGF and HIF-1α
in PHD2 knockdown HLE-B3 cells treated with the PHD2 specific
inhibitor IOX2 compared with the scramble IOX2-treated cells
(Fig. 3C). PHD2 is responsible for
the proline hydroxylation of AKT and hence inhibits the activation
of AKT (25). These findings were
consistent with the hypothesis that vitamin C inhibits AKT kinase
activity by enhancing the prolyl hydroxylation of AKT.
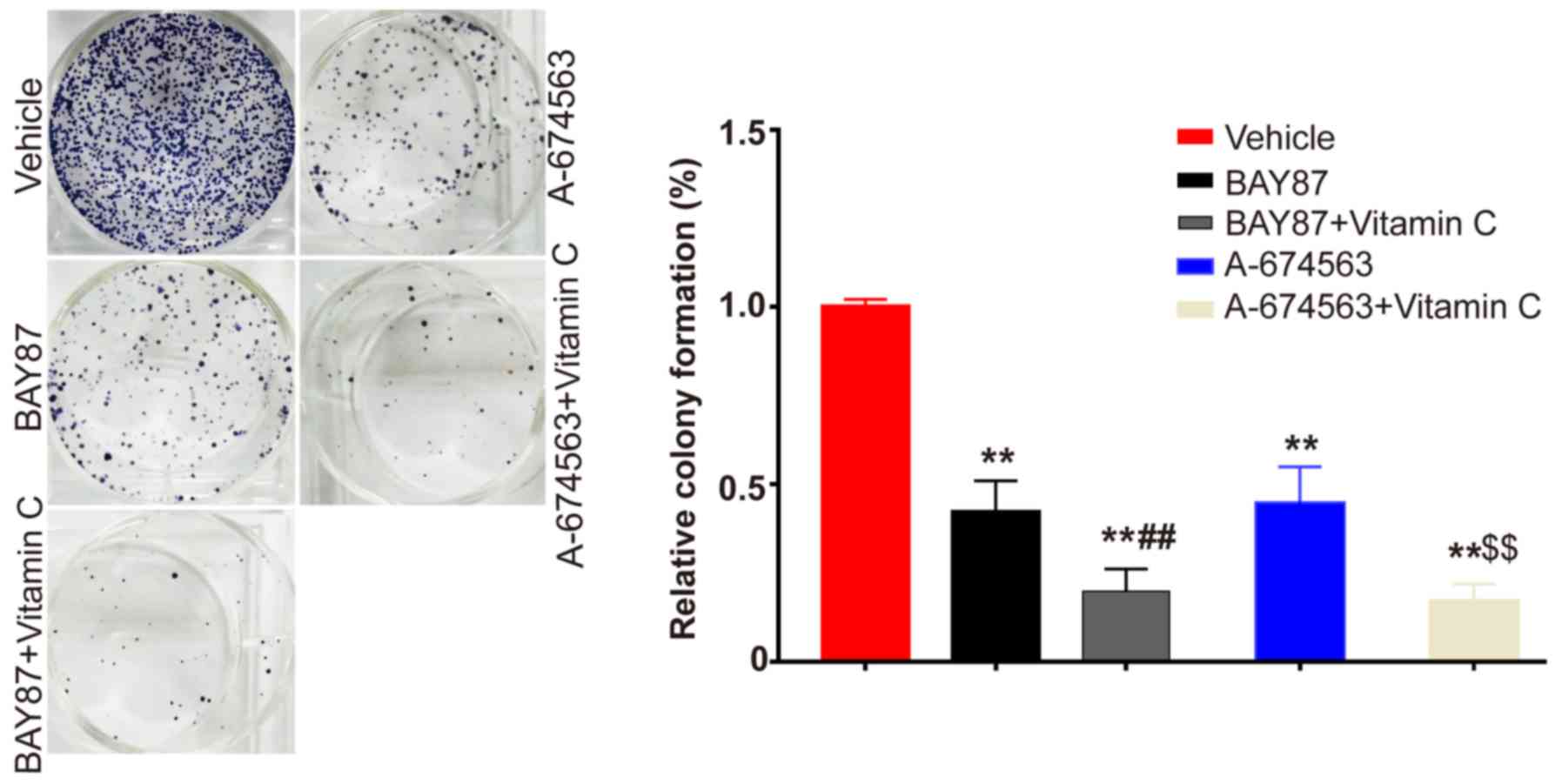
Synergistic effects of vitamin C with
AKT inhibition or HIF-1 inhibition
The present study indicated that vitamin C may
enhance the prolyl hydroxylation of HIF-1α to promote its
degradation and the prolyl hydroxylation of AKT to suppress its
activity. Thus, it was subsequently investigated whether vitamin C
had synergistic effects with HIF-1α or AKT inhibition. The
selective AKT inhibitor A-674563 was used to compare the effects
with vitamin C and HIF-1 inhibition. The colony formation assay
results demonstrated that vitamin C had synergistic effects with a
HIF-1 inhibitor (BAY 87-2243) and AKT inhibitor (A-674563); the
combined treatment of each inhibitor with vitamin C significantly
inhibited the proliferation of HLE-B3 cells to a greater extent
compared with the inhibitor treatment alone (Fig. 4). To further demonstrate that
vitamin C functions via HIF-1α dependent and AKT dependent pathways
to inhibit VEGF expression levels in LECs, western blotting was
used to determine the effect of the AKT inhibitor on the expression
levels of the HIF-1α target protein GLUT1. It was discovered that
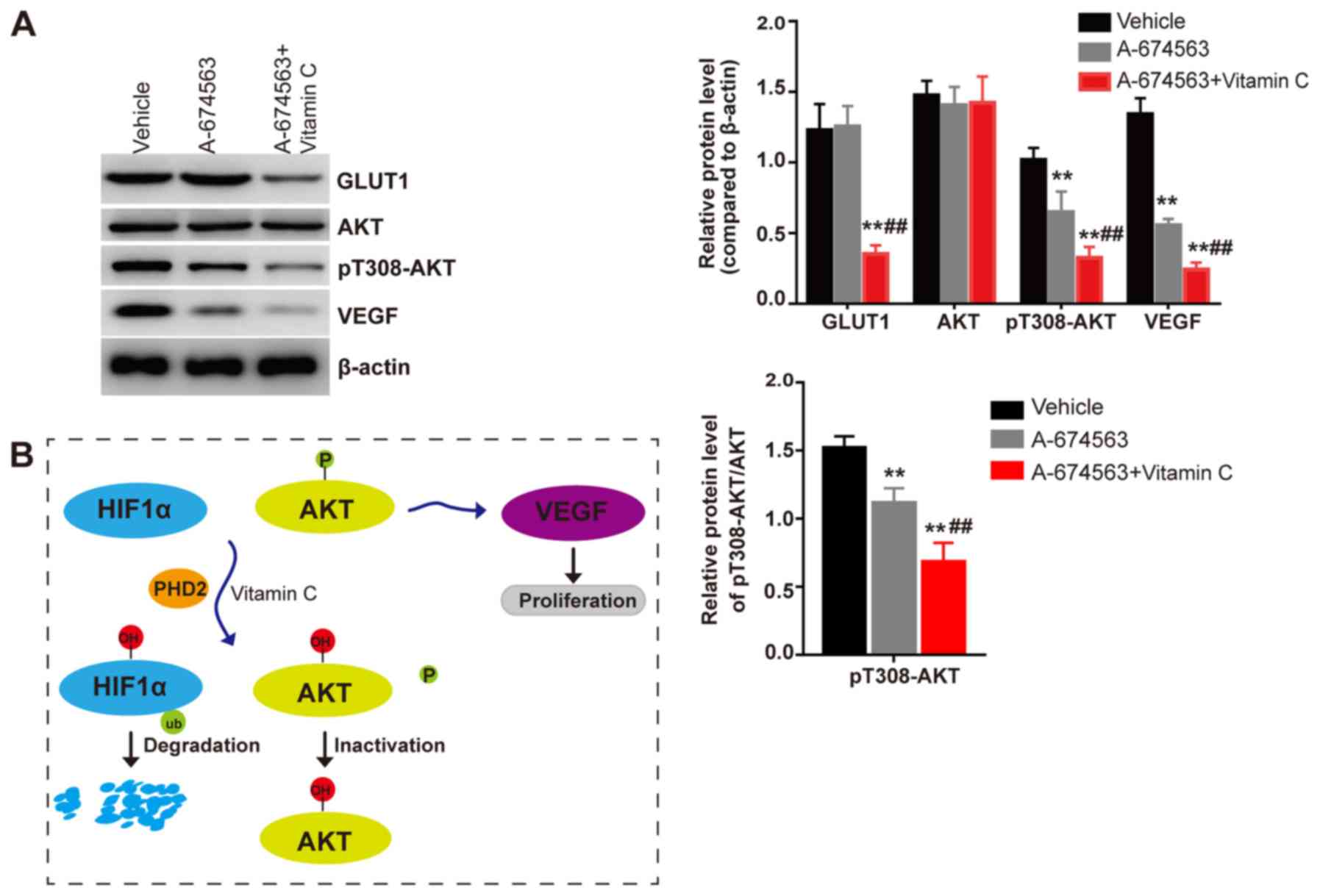
the AKT inhibitor significantly inhibited the phosphorylation
status of AKT and the expression levels of VEGF compared with the
vehicle treated cells (Fig. 5A).
Meanwhile, no significant differences were observed in the
expression levels of the HIF-1α target protein GLUT1 between the
cells treated with the AKT inhibitor or the vehicle control.
Notably, it was demonstrated that vitamin C treatment significantly
inhibited the phosphorylation status of AKT and the expression
levels of VEGF and GLUT1 in the presence of the AKT inhibitor
compared with AKT inhibitor treated group (Fig. 5A). In conclusion, these findings
suggested that vitamin C may inhibit the expression levels of VEGF
via HIF-1α and AKT dependent pathways in LECs. Hence, vitamin C was
found to act synergistically with the HIF-1 or AKT inhibitor to
inhibit the proliferation of LECs (Fig. 5B).
Discussion
PCO, the main complication after cataract surgery,
is a leading cause of visual impairment (1). The proliferation of residual LECs has
been found to serve a critical role in PCO formation (28). Therefore, the inhibition of the
proliferation of LECs may provide an important strategy for PCO
prevention and treatment in clinical practice (29). It has been previously reported that
high levels of vitamin C exert beneficial effects, such as
preventing age-related cataracts or PCO formation following
cataract surgery (11,12).
The HIF-1α/VEGF axis has been demonstrated to serve
an important role in the stimulation of cell proliferation and
migration (30,31). The authors' previous study revealed
that vitamin C inhibited the proliferation of human LECs by
promoting the degradation of HIF-1α via proline hydroxylation,
which subsequently inhibited the expression of the HIF-1α target
gene VEGF (10). In the present
study, it was found that vitamin C could further inhibit the
proliferation of LECs and VEGF expression levels in LECs following
HIF-1 inhibition. Moreover, the HIF-1α target gene GLUT1 could not
be regulated by vitamin C treatment following the treatment with
the HIF-1 inhibitor BAY 87-2243. Thus, the present results
suggested that vitamin C may suppress VEGF expression levels via
both HIF-1α dependent and independent manners. Indeed, it has been
reported that AKT and HIF-1α can independently regulate the
expression levels of VEGF (32,33).
Moreover, a previous study also demonstrated that AKT and HIF-1α
independently enhanced cell proliferation (34). In the present study, vitamin C also
demonstrated synergistic inhibitory effects with the HIF-1 or AKT
inhibitor over the proliferation of LECs.
Vitamin C serves as an electron-donor, maintaining
the Fe iron in the ferrous (Fe2+) state, which is
necessary to achieve the full activity of prolyl-hydroxylase
(17,35). Vitamin C has been revealed to
increase the degradation of HIF-1α via proline hydroxylation
(10,36). Furthermore, PHDs were found to be
responsible for the proline hydroxylation of HIF-1α at proline
residues 402 and 564 within the oxygen-dependent degradation domain
(37,38). AKT kinase activity is an important
mediator of the upregulation of VEGF (24,39).
It has since been discovered that AKT can be prolyl hydroxylated by
PHD2 and its activity was also found to be inhibited by
proline-hydroxylation (25). Other
previous studies have also supported our results and suggested that
vitamin C enhanced PHD2 hydroxylase activity (17,40–42).
In the present study, it was demonstrated that the overexpression
of AKT-WT increased the expression levels of VEGF. Moreover,
vitamin C was revealed to prevent the AKT-WT-induced increases in
VEGF expression levels. Furthermore, whilst the overexpression of
hydroxylation-deficient mutants of AKT increased the expression
levels of VEGF, vitamin C weakened its regulatory effect over
AKT-induced VEGF expression. It was demonstrated that vitamin C
inhibited AKT activity through decreasing the phosphorylation
levels, which was followed by increased levels of proline
hydroxylation of AKT. Moreover, the present results discovered that
the PHD2 specific inhibitor IOX2 increased the protein and mRNA
expression levels of VEGF to a higher extent compared with the
non-selective inhibitors. It was also observed that the PHD2
knockdown reversed the regulation of IOX2 over VEGF expression
levels in human LECs.
In conclusion, the findings of the present study
demonstrated that the AKT inhibitor A-674653 inhibited the activity
of AKT and the expression levels of VEGF but did not affect the
expression levels of the HIF-1α target protein GLUT1. Notably, the
cotreatment of cells with vitamin C and the AKT inhibitor A-674563
reduced the phosphorylation status of AKT and the expression levels
of VEGF compared with the A-674563 treatment alone. In addition,
vitamin C exhibited synergistic effects with the HIF-1 inhibitor or
AKT inhibitor to suppress the proliferation of human LECs.
Therefore, the present study provided evidence to suggest that
vitamin C may inhibit the expression levels of VEGF via HIF-1α
dependent and AKT dependent pathways in LECs. Thus, it is possible
that vitamin C treatment may facilitate PCO prevention in clinical
practice by inhibiting the proliferation of LECs.
Acknowledgements
Not applicable.
Funding
The present study was supported by the Key Program
of Research and Development of Shaanxi, China (grant no.
2017SF-266).
Availability of data and materials
All data generated or analyzed during the present
study are included in this published article.
Authors' contributions
FW and LZ conceived and designed the study, acquired
and analyzed the data and drafted the manuscript. LZ, YZ and LW
contributed to the data analysis and experimental materials. JW and
MY contributed to the design of the study and revised the
manuscript. All authors read and approved the final manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Raj SM, Vasavada AR, Johar SR and Vasavada
VA and Vasavada VA: Post-operative capsular opacification: A
review. Int J Biomed Sci. 3:237–250. 2007.PubMed/NCBI
|
|
2
|
Moulick PS, Rodrigues F and Shyamsundar K:
Evaluation of posterior capsular opacification following
phacoemulsification, extracapsular and small incision cataract
surgery. Med J Armed Forces India. 65:225–228. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Duman R, Karel F, Özyol P and Ates C:
Effect of four different intraocular lenses on posterior capsule
opacification. Int J Ophthalmol. 8:118–121. 2015.PubMed/NCBI
|
|
4
|
Zukin LM, Pedler MG, Groman-Lupa S,
Pantcheva M, Ammar DA and Petrash JM: Aldose reductase inhibition
prevents development of posterior capsular opacification in an in
vivo model of cataract surgery. Invest Ophthalmol Vis Sci.
59:3591–3598. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Zhang C, Liu J, Jin N, Zhang G, Xi Y and
Liu H: SiRNA targeting mTOR effectively prevents the proliferation
and migration of human lens epithelial cells. PLoS One.
11:e01673492016. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Andjelić S, Drašlar K, Lumi X, Yan X, Graw
J, Facskó A, Hawlina M and Petrovski G: Morphological and
proliferative studies on ex vivo cultured human anterior lens
epithelial cells-relevance to capsular opacification. Acta
Ophthalmol. 93:e499–e506. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Abdelkader H, Alany RG and Pierscionek B:
Age-related cataract and drug therapy: Opportunities and challenges
for topical antioxidant delivery to the lens. J Pharm Pharmacol.
67:537–550. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Chang JR, Koo E, Agrón E, Hallak J,
Clemons T, Azar D, Sperduto RD, Ferris FL III and Chew EY;
Age-Related Eye Disease Study Group, : Risk factors associated with
incident cataracts and cataract surgery in the age-related eye
disease study (AREDS): AREDS report number 32. Ophthalmology.
118:2113–2119. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Thiagarajan R and Manikandan R:
Antioxidants and cataract. Free Radic Res. 47:337–345. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Zhao L, Quan Y, Wang J, Wang F, Zheng Y
and Zhou A: Vitamin C inhibit the proliferation, migration and
epithelial-mesenchymal- transition of lens epithelial cells by
destabilizing HIF-1α. Int J Clin Exp Med. 8:15155–15163.
2015.PubMed/NCBI
|
|
11
|
Kaur A, Gupta V, Christopher AF, Malik MA
and Bansal P: Nutraceuticals in prevention of cataract-An evidence
based approach. Saudi J Ophthalmol. 31:30–37. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Valero MP, Fletcher AE, De Stavola BL,
Vioque J and Alepuz VC: Vitamin C is associated with reduced risk
of cataract in a mediterranean population. J Nutr. 132:1299–1306.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Yum S, Jeong S, Kim D, Lee S, Kim W, Yoo
JW, Kim JA, Kwon OS, Kim DD, Min DS and Jung Y: Minoxidil induction
of VEGF is mediated by inhibition of HIF-prolyl hydroxylase. Int J
Mol Sci. 19(pii): E532017. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Zhang Y, Xu Y, Ma J, Pang X and Dong M:
Adrenomedullin promotes angiogenesis in epithelial ovarian cancer
through upregulating hypoxia-inducible factor-1α and vascular
endothelial growth factor. Sci Rep. 7:405242017. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Takei A, Ekström M, Mammadzada P, Aronsson
M, Yu M, Kvanta A and André H: Gene transfer of prolyl hydroxylase
domain 2 inhibits hypoxia-inducible angiogenesis in a model of
choroidal neovascularization. Sci Rep. 7:425462017. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Xiong GF, Stewart RL, Chen J, Gao T, Scott
TL, Samayoa LM, O'Connor K, Lane AN and Xu R: Collagen prolyl
4-hydroxylase 1 is essential for HIF-1α stabilization and TNBC
chemoresistance. Nat Commun. 9:44562018. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Kuiper C and Vissers MC: Ascorbate as a
co-factor for fe- and 2-oxoglutarate dependent dioxygenases:
Physiological activity in tumor growth and progression. Front
Oncol. 4:3592014. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Zhou L, Wang Y, Zhou M, Zhang Y, Wang P,
Li X, Yang J, Wang H and Ding Z: HOXA9 inhibits HIF-1α-mediated
glycolysis through interacting with CRIP2 to repress cutaneous
squamous cell carcinoma development. Nat Commun. 9:14802018.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Carmeliet P, Dor Y, Herbert JM, Fukumura
D, Brusselmans K, Dewerchin M, Neeman M, Bono F, Abramovitch R,
Maxwell P, et al: Role of HIF-1alpha in hypoxia-mediated apoptosis,
cell proliferation and tumour angiogenesis. Nature. 394:485–490.
1998. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Finley LW, Carracedo A, Lee J, Souza A,
Egia A, Zhang J, Teruya-Feldstein J, Moreira PI, Cardoso SM, Clish
CB, et al: SIRT3 opposes reprogramming of cancer cell metabolism
through HIF1α destabilization. Cancer Cell. 19:416–428. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Groulx I and Lee S: Oxygen-dependent
ubiquitination and degradation of hypoxia-inducible factor requires
nuclear-cytoplasmic trafficking of the von Hippel-Lindau tumor
suppressor protein. Mol Cell Biol. 22:5319–5336. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Shiozaki A, Shen-Tu G, Bai X, Iitaka D, De
Falco V, Santoro M, Keshavjee S and Liu M: XB130 mediates cancer
cell proliferation and survival through multiple signaling events
downstream of Akt. PLoS One. 7:e436462012. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Karar J and Maity A: PI3K/AKT/mTOR pathway
in angiogenesis. Front Mol Neurosci. 4:512011. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Guo J, Chakraborty AA, Liu P, Gan W, Zheng
X, Inuzuka H, Wang B, Zhang J, Zhang L, Yuan M, et al: pVHL
suppresses kinase activity of Akt in a
proline-hydroxylation-dependent manner. Science. 353:929–932. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Woo YM, Shin Y, Lee EJ, Lee S, Jeong SH,
Kong HK, Park EY, Kim HK, Han J, Chang M and Park JH: Inhibition of
aerobic glycolysis represses Akt/mTOR/HIF-1α axis and restores
tamoxifen sensitivity in antiestrogen-resistant breast cancer
cells. PLoS One. 10:e01322852015. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Pore N, Jiang Z, Shu HK, Bernhard E, Kao
GD and Maity A: Akt1 activation can augment hypoxia-inducible
factor-1alpha expression by increasing protein translation through
a mammalian target of rapamycin-independent pathway. Mol Cancer
Res. 4:471–479. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Li JH, Wang NL and Wang JJ: Expression of
matrix metalloproteinases of human lens epithelial cells in the
cultured lens capsule bag. Eye (Lond). 22:439–444. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Chandler HL, Barden CA, Lu P, Kusewitt DF
and Colitz CM: Prevention of posterior capsular opacification
through cyclooxygenase-2 inhibition. Mol Vis. 13:677–691.
2007.PubMed/NCBI
|
|
30
|
Shi GH and Zhou L: Emodin suppresses
angiogenesis and metastasis in anaplastic thyroid cancer by
affecting TRAF6-mediated pathways in vivo and in
vitro. Mol Med Rep. 18:5191–5197. 2018.PubMed/NCBI
|
|
31
|
Palazon A, Tyrakis PA, Macias D, Veliça P,
Rundqvist H, Fitzpatrick S, Vojnovic N, Phan AT, Loman N, Hedenfalk
I, et al: An HIF-1α/VEGF-A axis in cytotoxic T cells regulates
tumor progression. Cancer Cell. 32:669–683 e665.e5. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Zhang Z, Yao L, Yang J, Wang Z and Du G:
PI3K/Akt and HIF-1 signaling pathway in hypoxia-ischemia (Review).
Mol Med Rep. 18:3547–3554. 2018.PubMed/NCBI
|
|
33
|
Choi SB, Park JB, Song TJ and Choi SY:
Molecular mechanism of HIF-1-independent VEGF expression in a
hepatocellular carcinoma cell line. Int J Mol Med. 28:449–454.
2011.PubMed/NCBI
|
|
34
|
Arsham AM, Plas DR, Thompson CB and Simon
MC: Akt and hypoxia-inducible factor-1 independently enhance tumor
growth and angiogenesis. Cancer Res. 64:3500–3507. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Satheesh NJ, Samuel SM and Busselberg D:
Combination therapy with vitamin c could eradicate cancer stem
cells. Biomolecules. 10(pii): E792020. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Fischer AP and Miles SL: Ascorbic acid,
but not dehydroascorbic acid increases intracellular vitamin C
content to decrease hypoxia inducible factor-1 alpha activity and
reduce malignant potential in human melanoma. Biomed Pharmacother.
86:502–513. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Jaakkola P, Mole DR, Tian YM, Wilson M,
Gielbert J, Gaskell SJ, von Kriegsheim A, Hebestreit HF, Mukherji
M, Schofield CJ, et al: Targeting of HIF-alpha to the von
Hippel-Lindau ubiquitylation complex by O2-regulated prolyl
hydroxylation. Science. 292:468–472. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Marxsen JH, Stengel P, Doege K, Heikkinen
P, Jokilehto T, Wagner T, Jelkmann W, Jaakkola P and Metzen E:
Hypoxia-inducible factor-1 (HIF-1) promotes its degradation by
induction of HIF-alpha-prolyl-4-hydroxylases. Biochem J.
381:761–767. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Kawasaki Y, Fujiki M, Uchida S, Morishige
M, Momii Y and Ishii K: A single oral dose of geranylgeranylacetone
upregulates vascular endothelial growth factor and protects against
kainic acid-induced neuronal cell death: Involvement of the
phosphatidylinositol-3 kinase/akt pathway. Pathobiology.
84:184–191. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Lindsey RC, Cheng S and Mohan S: Vitamin C
effects on 5-hydroxymethylcytosine and gene expression in
osteoblasts and chondrocytes: Potential involvement of PHD2. PLoS
One. 14:e02206532019. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Xing W, Pourteymoor S and Mohan S:
Ascorbic acid regulates osterix expression in osteoblasts by
activation of prolyl hydroxylase and ubiquitination-mediated
proteosomal degradation pathway. Physiol Genomics. 43:749–757.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Lee HY, Lee T, Lee N, Yang EG, Lee C, Lee
J, Moon EY, Ha J and Park H: Src activates HIF-1α not through
direct phosphorylation of HIF-1α specific prolyl-4 hydroxylase 2
but through activation of the NADPH oxidase/Rac pathway.
Carcinogenesis. 32:703–712. 2011. View Article : Google Scholar : PubMed/NCBI
|