Introduction
Acute pancreatitis (SAP) is a prevalent and serious
disease caused by activation of digestive proteases in the
pancreas, which may result in pancreatic tissue autodigestion,
tissue edema, hemorrhage or acinar necrosis, although most are mild
patients (mild acute pancreatitis, MAP), ~25% of patients worldwide
are severe (severe acute pancreatitis, SAP), which is clinically
dangerous and often complicated by systemic inflammatory response
syndrome (SIRS) and multiple organ dysfunction syndrome (MODS)
(1). Although gallstones are known
to be the main cause of acute pancreatitis, its diagnosis and
management remain challenging, and the pathogenesis of this disease
is not fully understood despite the proposal of several explanatory
theories (2).
The exocrine pancreas is one of the main organs
responsible for digestion; thus, acinar cells have abundant
endoplasmic reticulum (ER) to support their central role in the
synthesis of digestive enzymes (3). Therefore, acinar cells are extremely
sensitive to external stimuli in response to ER perturbations
(4). A previous study reported
that ER stress serves an important role during the development of
AP (5). Furthermore, Hartley et
al (6) identified
morphological changes in the ER caused by ER stress. Pancreatic
acinar cell injury, characterized by the formation of vesiculation
in the ER a few minutes after a retrograde injection of
taurocholate into the biliopancreatic duct in rats, has also been
reported by Bhatia et al (7). Moreover, the ER is one of the largest
cell organelles and is recognized as a vital site in the regulation
of protein synthesis, folding and assembly, in addition to
regulation of calcium ion levels and cellular stress response
(8). Various external stimuli,
such as calcium homeostasis imbalance, insufficient energy supply
or the production of reactive oxygen species can all lead to
excessive accumulation of unfolded or misfolded proteins in the ER,
causing ER stress (6). To overcome
ER stress, cells activate a series of specific signal
transductionpathways, which are collectively known as the unfolded
protein response (UPR) (9). The
UPR in the ER is regulated by three ER membrane-associated proteins
[PKR-like ER kinase (PERK), inositol-requiring enzyme 1 (IRE1) and
activating transcription factor 6 (ATF6)] (10), with the initial intention to alter
cellular transcriptional and translational programs, enhance
protein folding ability and accelerate protein degradation, in
order to restore homeostasis and normal ER function (8,11).
However, when ER injury is irreversible, the UPR pathways initiate
apoptosis (11). Moreover,
induction of the CCAAT/enhancer binding protein (C/EBP)-homologous
protein (CHOP), a member of the C/EBP family of transcription
factors, is a signaling event involved in ER stress-induced
apoptosis (12–14).
CHOP plays a crucial role in ER stress-induced
apoptosis, and was first revealed to be one of the main mediating
factors in growth arrest and damage response (15). It has been reported that CHOP
expression is significantly increased in severe ER stress, and thus
promotes cell cycle arrest and/or apoptosis (16,17).
However, knockdown of CHOP was revealed to confer resistance to ER
stress-induced apoptosis in various experimental models (14). The involvement of CHOP-mediated
apoptosis has also been revealed in numerous diseases, such as
brain ischemia, diabetes, neurodegenerative abnormalities and some
cardiovascular diseases (14,18,19).
Previous studies have suggested that ER stress can
initiate an inflammatory response via NF-κB activation in several
cells and diseases, including Alzheimer's disease and pancreatic
acinar cells (20–24). Allagnat et al (25) reported that CHOP plays a pivotal
role in the pathogenesis of the inflammatory response by directly
contributing to NF-κB pathway activation and subsequent cytokine
and chemokine expression, leading to ER stress and CHOP expression,
and in turn, CHOP facilitates and amplifies NF-κB pathway activity.
Furthermore, NF-κB is a key transcriptional factor that has an
important role in the initial stage of inflammation (26), which controls the expression of
numerous inflammatory mediators and cell apoptosis genes (27,28).
However, continued NF-κB activity and excessive inflammatory
responses lead to the development of AP (29,30).
Thus, it was speculated that CHOP may have a central role in the
pathogenesis of inflammatory responses.
Melatonin, which is mainly secreted by the pineal
gland, plays a key role in inflammation and immune defense
(31). In addition, melatonin is
able to attenuate oxidative stress damage and inhibit inflammatory
activities (32), and has
exhibited anti-inflammatory effects via the NF-κB signaling pathway
(33). As early as 1999, studies
have reported the beneficial effects of melatonin, which attenuates
tissue edema and lipid peroxidation in cerulein (Cer)-induced AP
(34). Moreover, previous studies
revealed that melatonin can reduce liver fibrosis and cirrhosis by
inhibiting ER stress (35,36). However, it remains unknown whether
melatonin exhibits its anti-inflammatory effect via the ER
stress-induced CHOP-mediated pathway.
The present study investigated the potential role of
melatonin in the CHOP-mediated signaling pathway to decrease
inflammation and apoptosis in AR42J cell models of AP.
Materials and methods
Lentivirus (LV)-mediated stable RNA
interference (RNAi) of CHOP in AR42J cells
AR42J cells (American Type Culture Collection) were
cultured in DMEM with 20% FBS (Sigma-Aldrich; Merck KGaA) and
antibiotics (100 U/ml penicillin and 100 µl/ml streptomycin) at
37°C in a humidified incubator with 5% CO2. The LV
(vehicle information: hU6-MCS-Ubiquitin-EGFP-IRES-puromycin;
Shanghai GeneChem Co., Ltd.), which carried short hairpin RNA
(shRNA) that targeted the CHOP gene (LV-shCHOP; GenBank NM_024134)
or that did not have an RNAi effect (LV-control; product no.
GCNL89264) were both purchased from Shanghai GeneChem Co., Ltd.,
and designated as LV-shCHOP and LV-control cells, respectively.
AR42J wild-type cells (3-5×104
cells/well) were seeded in 12-well plates and incubated for 24 h at
37°C in a humidified incubator with 5% CO2. The medium
in each well was subsequently replaced with 100 µl viral suspension
(1×108 TU/ml) and 400 µl DMEM without FBS and
antibiotics in the presence of 10 µg/ml polybrene (Shanghai
GeneChem Co., Ltd.). The multiplicity of infection was ~100. This
was followed by 8 h of culture under standard conditions and then
replacement with 1 ml fresh medium. During this period, cell
passaging and medium refreshment were routinely performed. After
72–96 h of virus infection, green fluorescent protein
(GFP)-positive cells were observed under a Nikon inverted
fluorescence microscope (Nikon Corporation) at ×40 magnification.
The effects of knockdown of CHOP expression in AR42J cells were
analyzed by western blotting and reverse transcription-quantitative
PCR (RT-qPCR).
Cell treatment and groups
In total, three types of AR42J cell lines
(wild-type, LV-control and LV-shCHOP cells) were seeded in 6-well
plates (5×105 cells/well). Following incubation for 24 h
at 37°C in a humidified incubator with 5% CO2, the
groups were defined as follows: Group I, three types of AR42J cells
were added to PBS alone and used as the negative control (control);
Group II, three types of AR42J cells were treated with Cer (10 mM,
Sigma-Aldrich; Merck KGaA) + LPS (10 mg/l, Sigma-Aldrich; Merck
KGaA) to induce AP (Cer + LPS) for 8 h at 37°C in a humidified
incubator with 5% CO2; Group III, LV-control cells were
treated with a low dose of melatonin (0.5 mmol/l, Sigma-Aldrich;
Merck KGaA) 30 min before AP was induced (0.5 mM) at 37°C in a
humidified incubator with 5% CO2; and Group IV,
LV-control cells were treated with a high dose of melatonin (2
mmol/l) 30 min before AP was induced (2 mM) at 37°C in a humidified
incubator with 5% CO2. Then 9 h later, cell extracts
were collected for western blotting and RT-qPCR.
Furthermore, to assess the effects of Cer + LSP
treatment on the protein expression levels of ER stress markers,
apoptosis and inflammatory-related molecules, wild-type cells were
treated with Cer + LPS for 0 (control), 3, 6, 9 and 12 h at 37°C in
a humidified incubator with 5% CO2, and then analyzed by
western blotting. Each experiment was performed in triplicate.
Cell viability assay
The effects of melatonin on the viability of AR42J
wild-type cells after Cer + LPS treatment was assessed using Cell
Counting Kit-8 (CCK-8; Dojindo Molecular Technologies, Inc.)
according to the manufacturer's instructions. Wild-type cells
(4×103 cells/well) were seeded into 96-well (100
µl/well) plates. After 24 h, the cells were treated with various
concentrations of melatonin (0.5, 1, 2 and 4 mM) 30 min before AP
was induced (melatonin treatment groups; 0.5–4 mM), 0.01 M PBS
(negative control group) and Cer + LPS (AP group) at 37°C in a
humidified incubator with 5% CO2. Each group consisted
of five parallel wells. Following incubation for 0, 3, 6, 9 and 12
h at 37°C in a humidified incubator with 5% CO2, 100 µl
of culture medium containing 10 µl CCK-8 was then added to the
culture media. An enzyme standard instrument (Infinite®
200 PRO NanoQuant; Tecan Austria GmbH) was used to measure the
supernatant of each well at a wavelength of 450 nm. Cell viability
was analyzed in each group by the optical density value. Each
experiment was performed in triplicate.
Western blot analysis
Western blotting was performed to assess the protein
expression levels of β-actin, 78 kDa glucose-regulated protein
(GRP78), CHOP, tumor necrosis factor-α (TNF-α), Bcl-2, Bax,
caspase-3, phospho-NF-κB inhibitor α (P-IκBα) and phospho-NF-κB p65
(p-p65).
Total proteins from pancreatic acinar cells were
extracted and homogenized in ice-cold RIPA buffer (Shanghai
Biyuntian Bio-Technology Co., Ltd.) supplemented with protease and
phosphatase inhibitors for 30 min on ice. The extracts were then
transferred to a microcentrifuge tube and centrifuged at
1.2×104 × g for 20 min at 4°C, and the protein
concentrations were determined using a bicinchoninic acid assay kit
(Thermo Fisher Scientific, Inc.). Subsequently, 45 µg total protein
per lane was separated by 10% SDS-PAGE and then transferred to PVDF
membranes (EMD Millipore) at 300 mA for 0.5–1 h. The membranes were
blocked at 20–25°C for 2 h with 5% non-fat milk and then
immunoblotted with specific primary antibodies overnight at 4°C.
The following primary antibodies were used: GRP78 (product code
ab108615), CHOP (also known as DDIT3; product code ab179823), TNF-α
(product code ab6671), Bcl-2 (product code ab32124), Bax (product
code Ab182733) and caspase-3 (product code Ab32351; all from
Abcam), p-IκBα (Ser 32; product no. 2859), IκBα (44D4; product code
4812), p-p65 (Ser 536; product code 3033), p65 (D14E12; product
code 8242) and β-actin (D6A8; product code 8457) all from Cell
Signaling Technology, Inc.) at 1:1,000 dilution. β-actin was used
as an internal control. The following day, the membranes were
washed three times with TBST containing 1% Tween-20 for 10 min each
time and incubated for 1 h at 20–25°C with a goat anti-rabbit IgG
secondary antibody conjugated to horseradish peroxidase (1:5,000;
product code BS10003; Bioworld Technology, Inc.), and then washed
with TBST as before. Protein bands were visualized using a Western
Bright ECL detection kit (Advansta, Inc.). Images of the protein
bands were captured using the ChemiDoc MP imaging densitometer
(Bio-Rad Laboratories, Inc.) and the density of the bands was
quantified using Image Lab software 4.1 (Bio-Rad Laboratories,
Inc.). Each experiment was performed in triplicate.
RT-qPCR
Total RNA from pancreatic acinar cells was extracted
using TRIzol® reagent (Invitrogen; Thermo Fisher
Scientific, Inc.) and then synthesized to cDNA using a RT kit
(product code K1691; Invitrogen; Thermo Fisher Scientific, Inc.) at
25°C for 5 min, 42°C for 60 min and 70°C for 5 min. PCR was
subsequently performed in the presence of specific primers to the
cDNA of rat genes. The PCR primers were synthesized by Sangon
Biotech Co., Ltd., and the sequences were as follows: GRP78
forward, 5′-CAAGAACCAACTCACGTCCA-3′ and reverse,
5′-ACCACCTTGAATGGCAAGAA-3′; CHOP forward,
5′-CCAGGAAACGAAGAGGAAGA-3′ and reverse, 5′-CTTTGGGAGGTGCTTGTGA-3′;
TNF-α forward, 5′-TGATCCGAGATGTGGAACTG-3′ and reverse,
5′-CGAGCAGGAATGAGAAGAGG-3′; interleukin (IL)-6, forward
5′-TACCCCAACTTCCAATGCTC-3′ and reverse, 5′-GGTTTGCCGAGTAGACCTCA-3′;
Bcl-2 forward, 5′-AGGATTGTGGCCTTCTTTGA-3′ and reverse,
5′-CAGATGCCGGTTCAGGTACT-3′; Bax forward, 5′-CAGGATCGAGCAGAGAGGAT-3′
and reverse, 5′-GTCCAGTTCATCGCCAATTC-3′; caspase-3 forward,
5′-ACTGGACTGTGGCATTGAGA-3′ and reverse,
5′-AATTTCGCCAGGAATAGTAACC-3′; and β-actin, forward
5′-CGTGAAAAGATGACCCAGAT-3′ and reverse, 5′-ACCCTCATAGATGGGCACA-3′.
The qPCR procedure was conducted using a RT qPCR system (Bio-Rad
Laboratories, Inc.) with the following: Initial denaturation at
50°C for 2 min; 40 cycles of 95°C for 30 sec, 95°C for 5 sec and
60°C for 34 sec) and the Takara Power SYBR-Green PCR Master mix
(cat. no. DRR820A; Takara Bio, Inc.). β-actin was used as the
internal standard, and quantified relative gene expression levels
were calculated using the 2−ΔΔCq method (37). Each experiment was performed in
triplicate.
Statistical analysis
The results were analyzed using SPSS software 20.0
(SPSS, Inc.), and data are presented as the mean ± SEM. One-way
ANOVA followed by a Tukey's post-hoc test was performed for
comparisons of ≥3 groups. P<0.05 was considered to indicate a
statistically significant difference.
Results
Effects of melatonin on the viability
of wild-type cells after Cer + LPS treatment
Cell viability of the Cer + LPS group was
significantly reduced compared with the control group after 12 h of
treatment (70% viability; Fig. 1).
However, when the concentration of melatonin was increased, cell
viability was gradually enhanced (Fig.
1), with a maximum effect observed in the 4-mM group compared
with the Cer + LPS group at the same time period (Fig. 1). Thus, it was speculated that
melatonin may have a dose-dependent effect on cell viability.
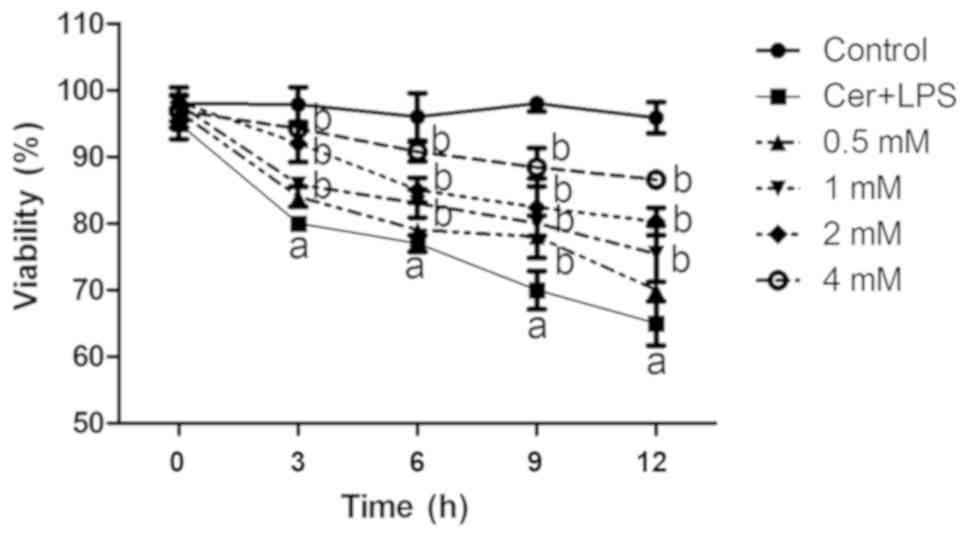 | Figure 1.Effects of melatonin on the viability
of AR42J cells after Cer + LPS treatment. Wild-type cells were
treated with various concentrations of melatonin (0.5, 1, 2 and 4
mM) for 30 min before Cer + LPS treatment (melatonin treatment
groups, 0.5–4 mM), 0.01 M PBS (negative control) and Cer + LPS (AP
group). Cell viability was examined at 0, 3, 6, 9 and 12 h with a
Cell Counting Kit-8 assay. Data are presented as the mean ± SEM of
≥3 independent experiments. aP<0.05 vs. the control
group. bP<0.05 vs. the Cer + LPS group at the same
time period. Cer + LPS, cerulein plus LPS; AP, acute pancreatitis;
LPS, lipopolysaccharide. |
Effects of Cer + LPS on the
CHOP-mediated pathway at different treatment times
The expression levels of factors related to the
CHOP-mediated pathway were examined by western blotting in cells
cultured with Cer + LPS for 0, 3, 6, 9 and 12 h. It was
demonstrated that expression levels of the inflammatory markers,
p-p65, P-IκBα, and TNF-α, and ER stress markers, GRP78 and CHOP,
were increased over time (Fig. 2).
Moreover, the expression levels of pro-apoptosis-related molecules
caspase-3 and Bax increased, while the anti-apoptotic protein Bcl-2
decreased in the early stage of inflammation. However, at the 12-h
time-point, Bax expression was decreased (Fig. 2).
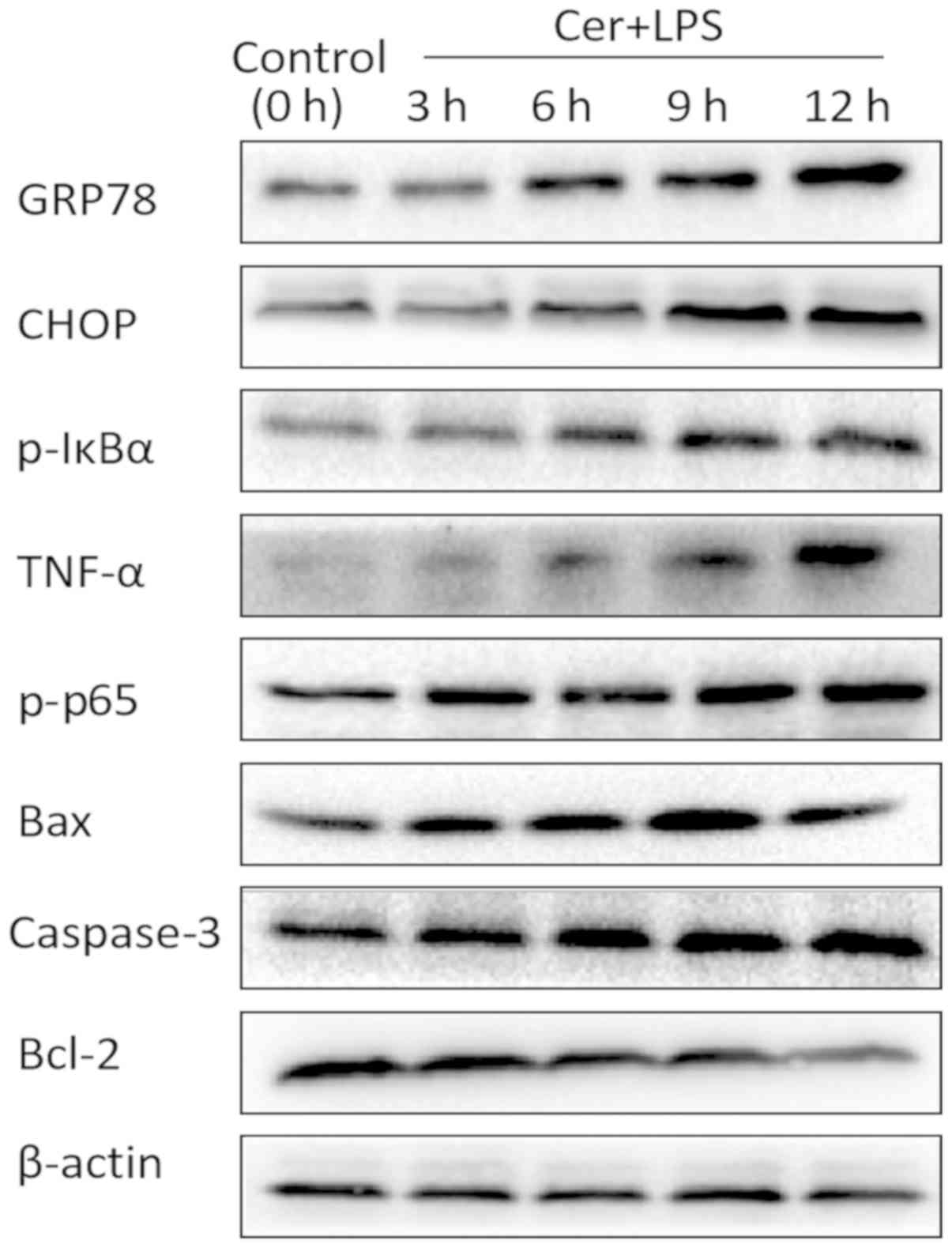 | Figure 2.Effects of Cer + LPS treatment on the
protein expression levels of ER stress markers, apoptosis and
inflammation-related molecules in AR42J cells. Wild-type cells were
treated with Cer + LPS for 0, 3, 6, 9 and 12 h. The protein
expression levels of GRP78, CHOP, p-p65, TNF-α, p-IκBα, Bcl-2, Bax
and caspase-3 were assessed by western blotting, and β-actin was
used as an internal control. Cer + LPS, cerulein + LPS; LPS,
lipopolysaccharide; GRP78, 78 kDa glucose-regulated protein; CHOP,
CCAAT/enhancer binding protein (C/EBP)-homologous protein; p-IκBα,
phospho-NF-κB inhibitor α; TNF-α, tumor necrosis factor-α; p-p65,
phospho-NF-κB p65. |
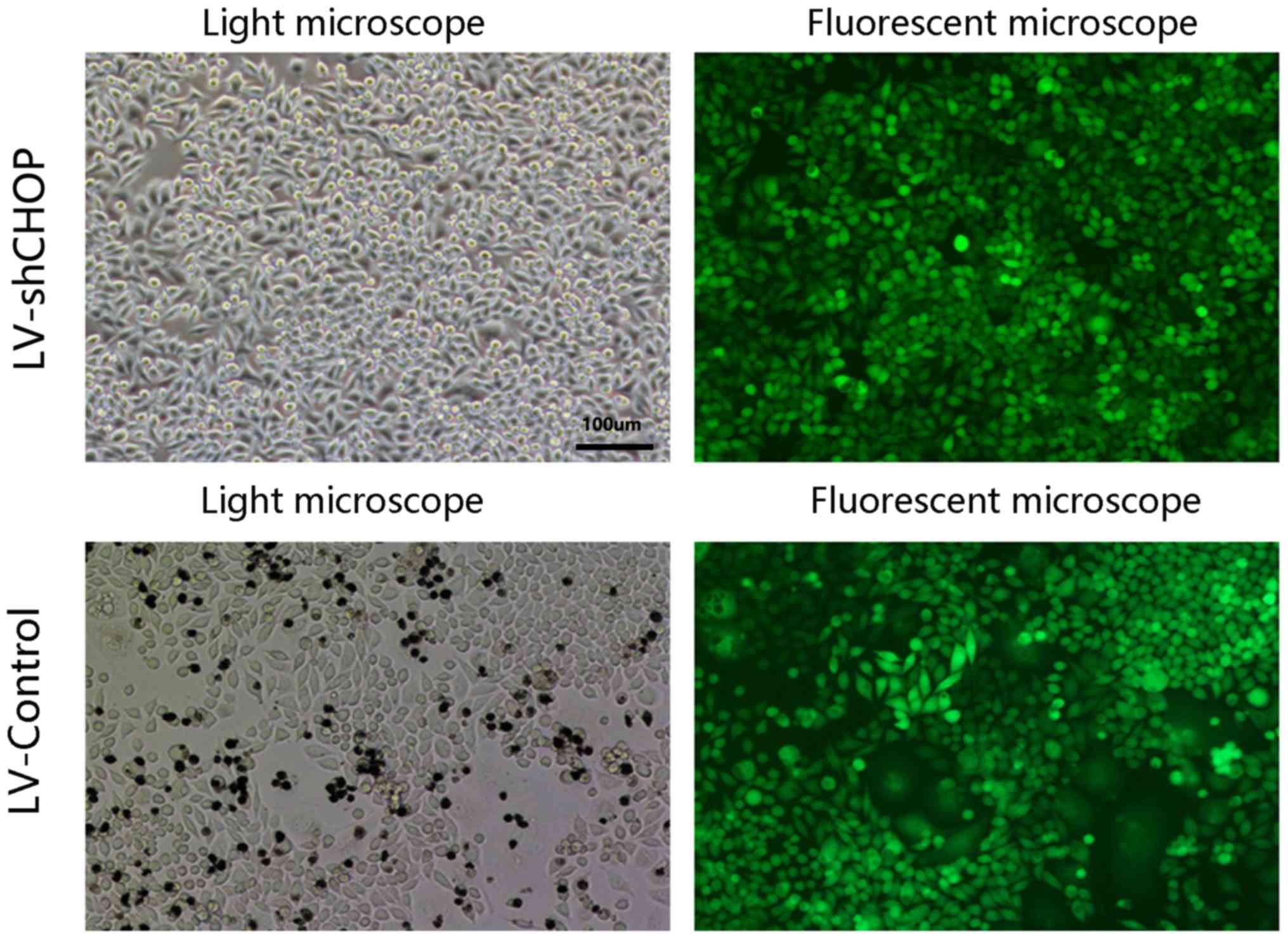
Transduction efficacy of
LV-shCHOP-mediated RNAi in AR42J cells
At 72–96 h after infection, highly efficient
transduction (>95%) of LV-shCHOP was identified in AR42J cells,
as GFP expression was observed under a Nikon inverted fluorescence
microscope (Fig. 3), and the
LV-control and LV-shCHOP cells had similar transduction
efficiencies (Fig. 3).
Effects of Cer + LPS and treatment
with melatonin on the expression of ER stress-related factors
Western blotting results identified increased GRP78
and CHOP protein expression levels in LV-control cells treated with
Cer + LPS compared with the respective controls, and there were no
differences between wild-type cells and LV-control cells. However,
LV-shCHOP cells after Cer + LPS treatment and LV-control cells in
the melatonin treatment groups both had reduced GRP78 and CHOP
expression levels, compared with LV-control cells treated with Cer
+ LPS (Fig. 4A). These findings
were also demonstrated by RT-qPCR (Fig. 4B). LV-shCHOP cells had a greater
effect of silencing on CHOP expression in AR42J cells and
significantly attenuated ER stress after Cer + LPS treatment.
Moreover, pre-treatment of LV-control cells with melatonin also
downregulated CHOP expression and inhibited ER stress to protect
the cells. Therefore, the extent of inhibition by melatonin was
dose dependent.
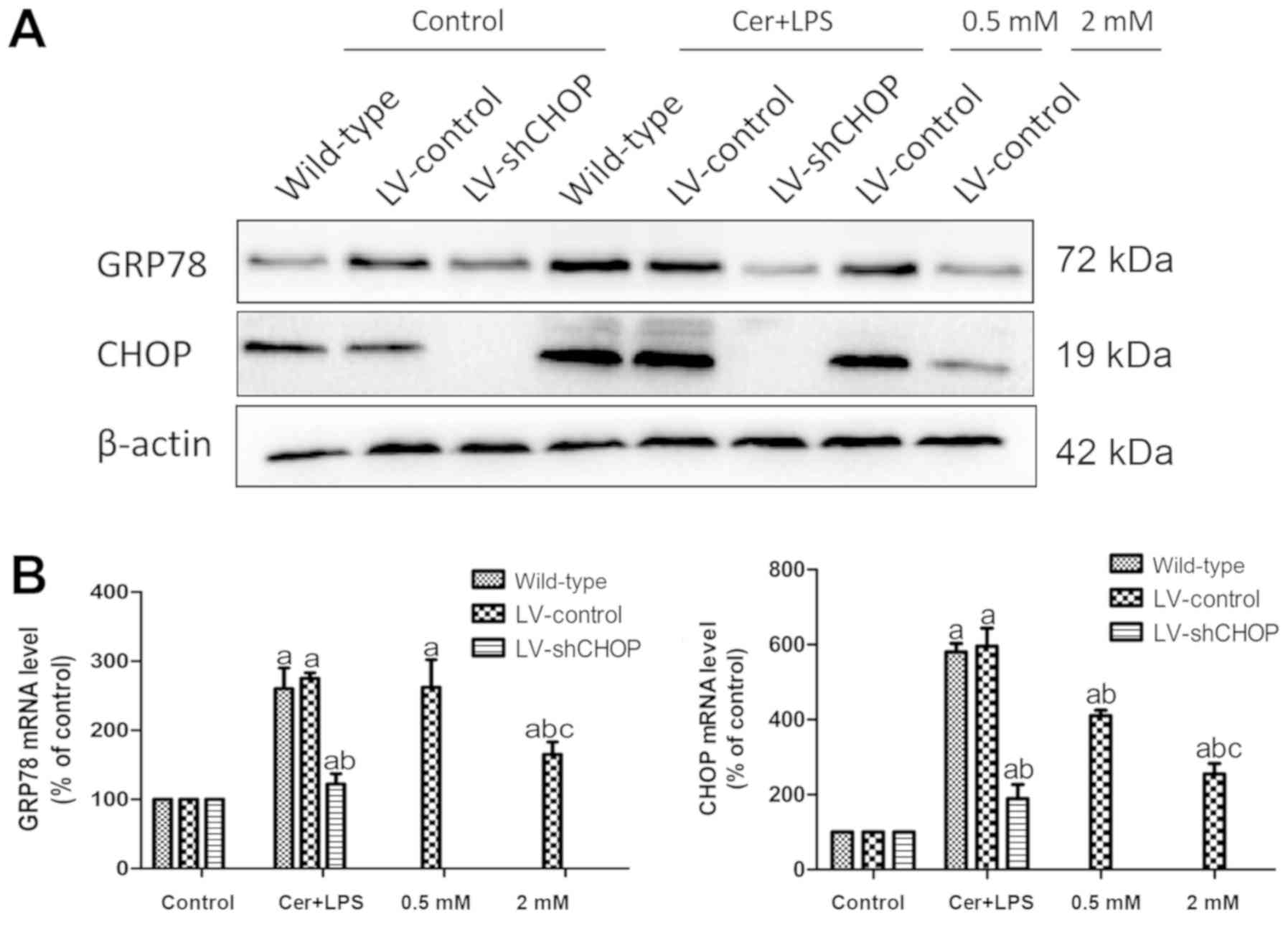 | Figure 4.Effects of Cer + LPS and treatment
with melatonin on the expression of ER stress-related molecules in
AR42J cells. (A) Protein expression levels of GRP78 and CHOP at 9 h
after Cer + LPS and melatonin treatment were assessed by western
blotting, and β-actin was used as the internal control. (B) mRNA
expression levels of GRP78 and CHOP were quantified by reverse
transcription-quantitative PCR at 9 h. Data are presented as the
mean ± SEM of ≥3 independent experiments. Control, treated with
PBS; 0.5 mM, treated with 0.5 mM melatonin 30 min before CER + LPS
treatment; 2 mM, treated with 2 mM melatonin 30 min before CER +
LPS treatment. aP<0.05 vs. the respective controls.
bP<0.05 vs. the LV-control cells treated with Cer +
LPS. cP<0.05 vs. the 0.5 mM group. Cer + LPS,
cerulein + LPS; LV, lentivirus; sh, short hairpin RNA; CHOP,
CCAAT/enhancer binding protein (C/EBP)-homologous protein; LPS,
lipopolysaccharide; GRP78, 78 kDa glucose-regulated protein. |
Effects of Cer + LPS and treatment
with melatonin on inflammation and apoptosis-associated
molecules
The expression levels of NF-κB pathway and
pro-inflammatory molecules were increased following Cer + LPS
treatment, compared with the respective controls. However, the
expression levels of these molecules in LV-shCHOP cells after Cer +
LPS treatment and LV-control cells in the melatonin treatment
groups were lower compared with the LV-control cells, after Cer +
LPS treatment (Fig. 5A and B).
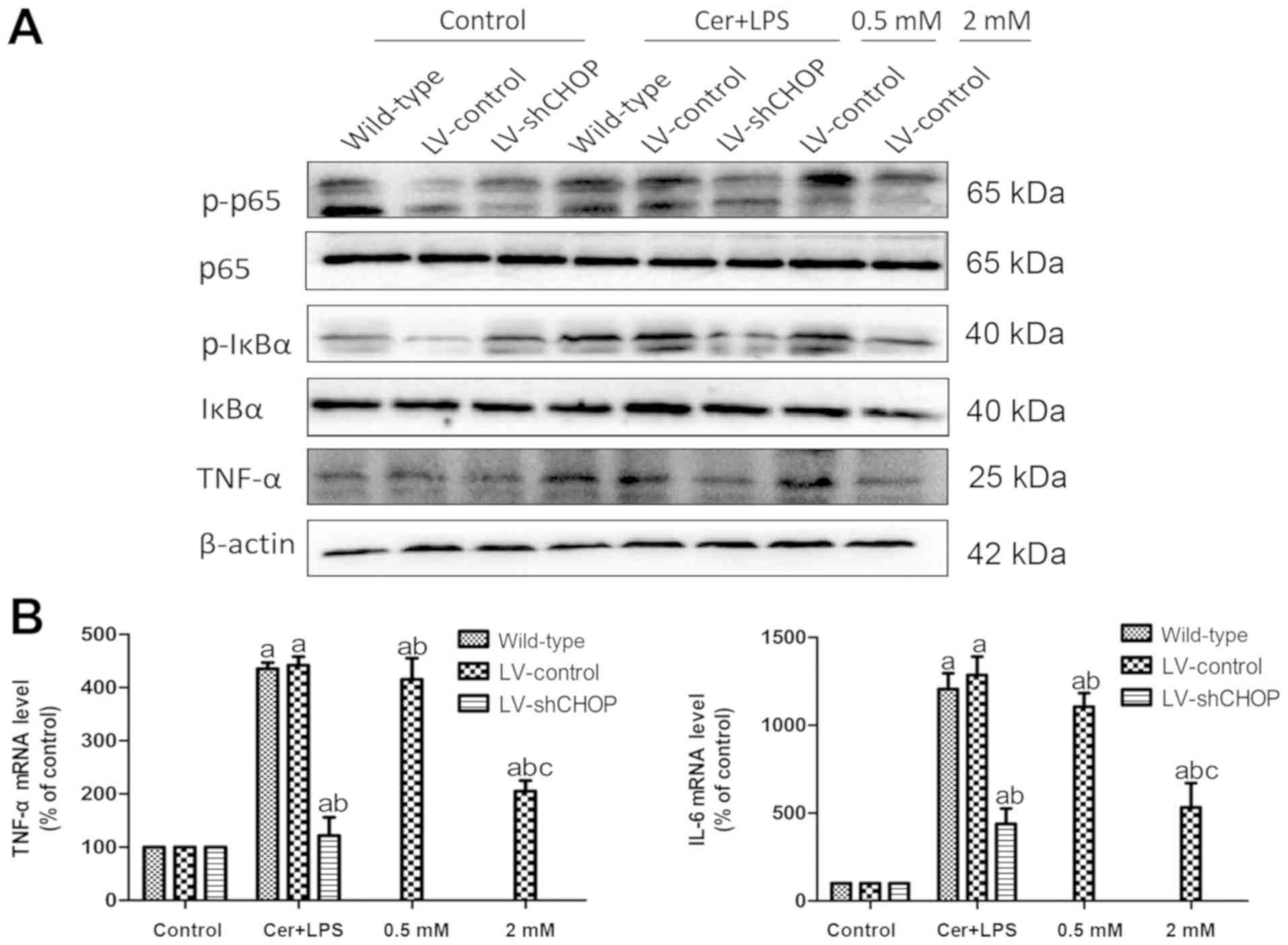 | Figure 5.Effects of Cer + LPS and treatment
with melatonin on the expression of inflammation-related molecules
in AR42J cells. (A) Protein expression levels of p-p65, p65,
p-IκBα, IκBα and TNF-α at 9 h after Cer + LPS and melatonin
treatment were assessed by western blotting, and β-actin was used
as the internal control. (B) mRNA expression levels of IL-6 and
TNF-α were quantified by reverse transcription-quantitative PCR at
9 h. Data are presented as the mean ± SEM of ≥3 independent
experiments. Control, treated with PBS; 0.5 mM, treated with 0.5 mM
melatonin 30 min before Cer + LPS treatment; 2 mM, treated with 2
mM melatonin 30 min before Cer + LPS treatment.
aP<0.05 vs. the respective controls.
bP<0.05 vs. the LV-control cells treated with Cer +
LPS. cP<0.05 vs. the 0.5 mM group. Cer + LPS,
cerulein + LPS; LV, lentivirus; sh, short hairpin RNA; CHOP,
CCAAT/enhancer binding protein (C/EBP)-homologous protein; LPS,
lipopolysaccharide; TNF-α, tumor necrosis factor-α; p-IκBα,
phospho-NF-κB inhibitor α; p-p65, phospho-NF-κB p65. |
Furthermore, there were significantly enhanced Bax
and caspase-3 expression levels and reduced Bcl-2 expression in
LV-control cells after Cer + LPS treatment, compared with the
respective controls (Fig. 6A and
B). Moreover, LV-shCHOP cells treated with Cer + LPS and
LV-control cells in the melatonin treatment groups had
significantly reduced expression levels of Bax and caspase-3 and
enhanced expression of Bcl-2 compared with the LV-control cells
treated with Cer + LPS. Collectively, the results indicated that
knockdown of CHOP expression reduced activation of the NF-κB
pathway, inflammation and the apoptotic response after Cer + LPS
treatment, while pre-treatment with melatonin also attenuated
inflammation and apoptosis. Furthermore, the extent of inhibition
by melatonin exhibited a dose-dependent effect.
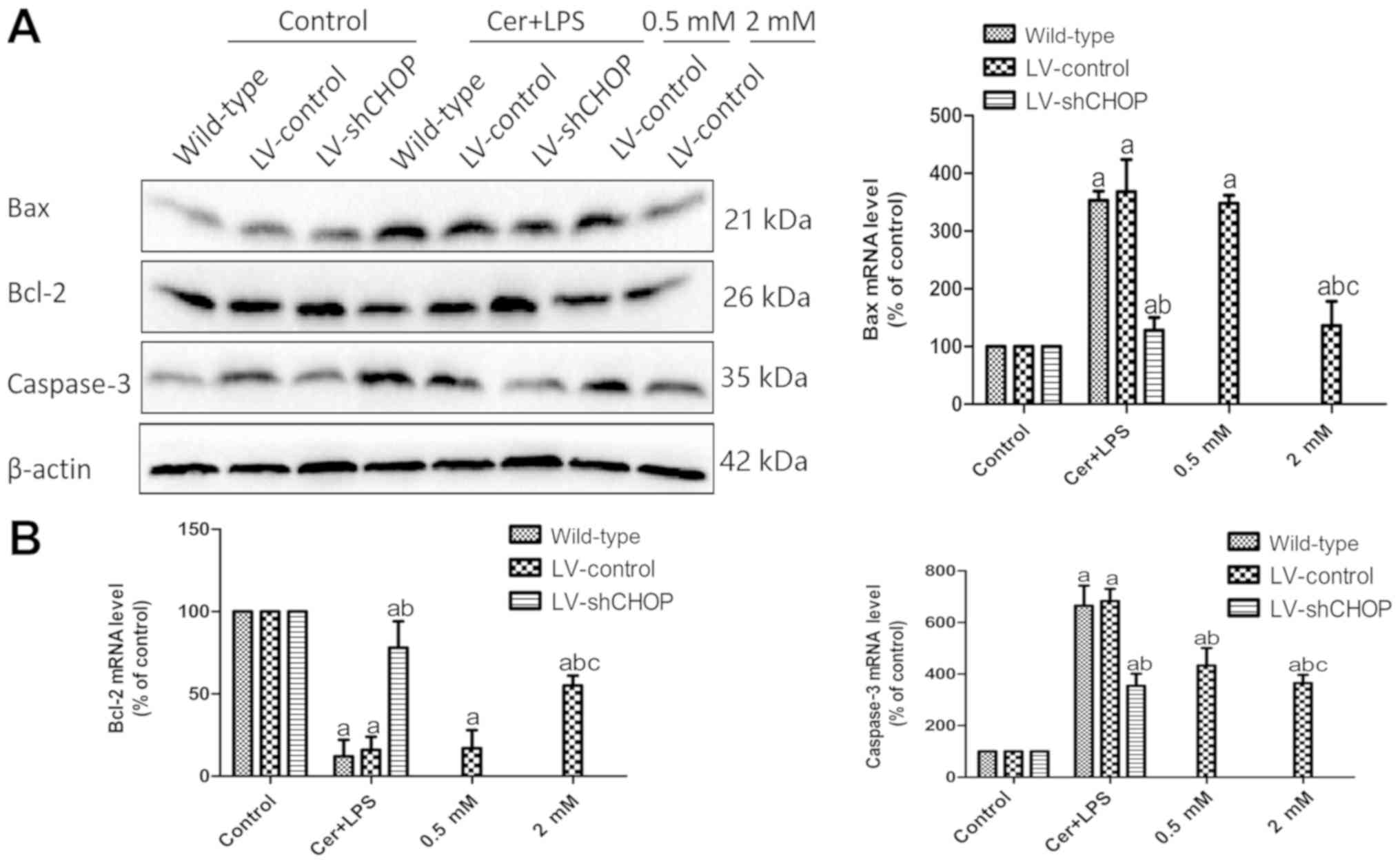 | Figure 6.Effects of Cer + LPS and treatment
with melatonin on the expression of apoptosis-related molecules in
AR42J cells. (A) Protein expression levels of Bax, Bcl-2 and
caspase-3 at 9 h after Cer + LPS and melatonin treatment were
assessed by western blotting, and β-actin was used as the internal
control. (B) mRNA expression levels of Bcl-2, Bax and caspase-3
were quantified by reverse transcription-quantitative PCR at 9 h.
Data are presented as the mean ± SEM of ≥3 independent experiments.
Control, treated with PBS; 0.5 mM, treated with 0.5 mM melatonin 30
min before cerulein + LPS treatment; 2 mM, treated with 2 mM
melatonin 30 min before cerulein + LPS treatment.
aP<0.05 vs. the respective controls.
bP<0.05 vs. the LV-control cells treated with Cer +
LPS. cP<0.05 vs. the 0.5 mM group. Cer + LPS,
cerulein + LPS; LV, lentivirus; sh, short hairpin RNA; CHOP,
CCAAT/enhancer binding protein (C/EBP)-homologous protein; LPS,
lipopolysaccharide. |
Discussion
The present results indicated that melatonin had an
anti-inflammatory effect via the ER stress-induced CHOP-mediated
pathway in AR42J cells. Moreover, ER stress was significantly
activated in the early stage of AP induced by Cer + LPS. The
CHOP-mediated pathway also aggravated acinar cell damage by
inducing apoptosis and NF-κB activation. Furthermore, it was
indicated that treatment with melatonin significantly attenuated
the expression of pro-inflammatory cytokines and ER stress-related
molecules, and exerted a protective effect by inhibiting apoptosis
of acinar cells.
In cells, heat shock proteins (HSPs) promote the
modification of newly synthesized proteins in the ER and their
translocation to the cellular membrane (38), and this is essential in the process
of recovery to reduce stimulation and injury (39). In addition, under inflammatory
conditions, the regulation of HSPs may attenuate the inflammatory
response (40). GRP78, which
belongs to the HSP family, is important in the response to ER
stress (41). When excess unfolded
or misfolded proteins accumulate in the ER causing ER stress, GRP78
separates from the three major triggering molecules (ATF6, PERK and
IRE1) of the UPR, thus initiating the downstream UPR signaling
pathways (42). The present
results revealed that GRP78 was upregulated early after Cer + LPS
treatment and exhibited a time-dependent increase, which suggested
that ER stress was activated early in AP and became increasingly
severe over time.
Apoptosis is the process of programmed cell death.
Numerous studies have reported that apoptosis and necrosis occur in
clinical and experimental AP (43–45),
and some studies have indicated that these are involved in ER
stress-induced apoptosis (46,47).
Furthermore, the apoptotic signal can be induced when cells are
subjected to excessive and prolonged ER stress (11).
Previous studies have revealed that ER stress can
exacerbate inflammatory damage in pancreatic tissue by regulating
cell death, and thus promote the development of pancreatitis
(48–50). ER stress-induced apoptosis includes
the following three signaling pathways: i) CHOP-mediated apoptosis
via the downregulation of Bcl-2; ii) caspase-12-mediated pathway,
which plays a role in apoptosis by activating caspase-9 and
caspase-3; and iii) kinase 1-c-Jun N-terminal kinase pathway
activation (13,14). Therefore, it is important to
identify the extent to which signaling pathways contribute to
acinar cell apoptosis after Cer + LPS treatment. CHOP is a
pro-apoptotic molecule and can be activated by all three branches
of the ER stress pathway, and in turn has been targeted for the
modulation of ER stress (51). In
the present study, the in vitro models of AP demonstrated
that CHOP expression was significantly increased after Cer + LPS
treatment, indicating that ER stress was activated early in AR42J
cells under the condition of Cer + LPS-induced inflammation. Thus,
the LV-mediated RNAi method was used to determine the potential
role of CHOP in the regulation of cell apoptosis during AP. The
present results indicated that the mRNA and protein expression
levels of CHOP could be specifically silenced, while this did not
occur using a non-target LV that did not have an RNAi effect on
CHOP expression in AR42J cells. Furthermore, knockdown of CHOP led
to an almost complete suppression of apoptosis after Cer + LPS
treatment, which significantly decreased the expression levels of
Bax and caspase-3, and increased the expression of Bcl-2 in the
experimental pancreatitis model. This pro-apoptotic potential may
be due to CHOP, as it is one of the highest inducible genes
underlying ER stress. Moreover, excessively activated IRE1α may
result from the recruitment of TNF receptor-associated factor 2
(TRAF2) and its combining with apoptosis signal-regulated kinase 1
(ASK1) to form the IRE-1-TRAF2-ASK1 complex on the outer membrane
of ER, which activates the c-Jun amino-terminal kinase and p38
mitogen-activated protein kinase (52). In addition, phosphorylation of p38
mitogen-activated protein kinase can induce the expression of CHOP
after transduction of the activation domain serine 78/81 (14,52,53).
ER stress-induced CHOP-mediated apoptosis is speculated to
upregulate the pro-apoptotic molecule Bax and translocation of Bax
to the mitochondria, downregulate anti-apoptotic molecules such as
Bcl-2 and Bcl-xl (14,54,55),
and activate growth arrest and DNA damage inducible 34 and ER
oxidoreductin-1a (51).
Collectively, these findings indicated that CHOP plays a central
role in pancreatitis acinar cell apoptosis and leads to acinar cell
injury in AP. Therefore, in the future, CHOP inhibitors may be
developed as novel therapeutic drugs for AP.
In recent years, the ER stress-induced,
CHOP-mediated pathway has been reported in various inflammatory
diseases (8,56–58),
but to the best of our knowledge, it has not been demonstrated in a
cell model of pancreatic injury. The present results revealed that
the CHOP-mediated pathway enhances pancreatitis inflammation,
possibly via the transcriptional regulation of NF-κB, and this was
consistent with the findings from Allagnat et al (25). NF-κB plays an important role in the
inflammatory response (29). When
cells are subjected to pathological stimulation such as
inflammation or viral infection, IκB kinase is activated and
phosphorylates IκB, which is subsequently degraded and exposes a
nuclear-localization signal for NF-κB, allowing NF-κB to rapidly
migrate into the nucleus, where it initiates the transcription and
activation of numerous inflammation-associated genes (12). In addition, the association between
ER stress and the inflammatory response are interconnected, and
inflammation can regulate and activate ER stress in cells (12). Zhang et al (59) revealed that ER stress can activate
the cAMP-responsive element-binding protein H (CREBH) to induce an
acute inflammatory response, and pro-inflammatory cytokines such as
IL-6, IL-1β or TNF-α can also exacerbate ER stress and promote the
production of CREBH in hepatoma cells in vivo. Previous
studies have also reported that inflammatory factors can lead to
insulin resistance, promote the occurrence and development of
metabolic syndrome, and aggravate tissue and cell stress and damage
(60,61). In line with these previous
findings, the present results demonstrated that the NF-κB pathway
and pro-inflammatory cytokines were significantly reduced by CHOP
knockdown after Cer + LPS treatment. Therefore, it was speculated
that CHOP knockdown inhibited NF-κB signaling pathway activation
and attenuated pancreatitis inflammation and acinar cell injury.
Thus, the present results suggested that ER stress-induced,
CHOP-mediated pathway had a detrimental role in the pathogenesis of
AP.
Other pro-inflammatory mechanisms of CHOP have also
been investigated in previous studies. For example, one study using
a CHOP-deficient mouse model of experimental pancreatitis
identified a reduction in pancreatic tissue inflammation and IL-1β
activity by inhibiting the induction of inflammation-associated
caspases, caspase-11 and caspase-1 (56). It has also been revealed that in a
mouse model of myocardial ischemia/reperfusion injury, knockdown of
CHOP led to reduced myocardial inflammation, possibly via the
regulation of the transcription of specific pro-inflammatory
cytokine genes, such as IL-6 (8).
However, the underlying molecular mechanisms of the CHOP-mediated
pathway and pro-inflammatory activity are not fully understood and
require further investigation.
The anti-inflammatory effects of melatonin on
pancreatitis have been reported in numerous cellular and animal
studies, which also functions as a protectant and antioxidant;
however, the potential mechanisms are largely unknown (32). It has also been revealed that
melatonin could reduce cell injury in different models via the
inhibition of ER stress (58).
Moreover, the present results revealed that treatment with
melatonin significantly decreased the protein expression of CHOP,
inhibited the activation of the NF-κB signaling pathway and the
release of pro-inflammatory factors, as well as attenuated the ER
stress and suppressed apoptosis of AR42J cells. Thus, it was
speculated that melatonin protected acinar cells against AP, and
alleviated acinar cell injury and inflammatory response by
inhibiting the ER stress-induced, CHOP-mediated pathway.
However, the present study had some limitations. The
clinical application of melatonin is still not universal, and
further clinical trials are required. Moreover, the underlying
anti-inflammatory molecular mechanism and the optimal concentration
of melatonin need to be identified, in order to further reduce
pancreatitis mortality and improve survival prognosis.
Acknowledgements
Not applicable.
Funding
This study was supported by the Public Projects of
Zhejiang Province (grant no. 2016C33215).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
QZ and HZ performed the majority of the research. JH
and XL designed the study. XJ and JW analyzed the data. XL, JW and
HZ collected the information. QZ wrote the manuscript. All authors
read and approved the final manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Bhatia M, Wong FL, Cao Y, Lau HY, Huang J,
Puneet P and Chevali L: Pathophysiology of acute pancreatitis.
Pancreatology. 5:132–144. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Walczak-Galezewska MK, Skrypnik D,
Szulinska M, Skrypnik K and Bogdanski P: Conservative management of
acute calculous cholecystitis complicated by pancreatitis in an
elderly woman: A case report. Medicine (Baltimore). 97:e112002018.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Berridge MJ: The endoplasmic reticulum: A
multifunctional signaling organelle. Cell Calcium. 32:235–249.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Kubisch CH and Logsdon CD: Endoplasmic
reticulum stress and the pancreatic acinar cell. Expert Rev
Gastroenterol Hepatol. 2:249–260. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Thrower EC, Gorelick FS and Husain SZ:
Molecular and cellular mechanisms of pancreatic injury. Curr Opin
Gastroenterol. 26:484–489. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Hartley T, Siva M, Lai E, Teodoro T, Zhang
L and Volchuk A: Endoplasmic reticulum stress response in an INS-1
pancreatic beta-cell line with inducible expression of a
folding-deficient proinsulin. BMC Cell Biol. 11:592010. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Bhatia M, Neoptolemos JP and Slavin J:
Inflammatory mediators as therapeutic targets in acute
pancreatitis. Curr Opin Investig Drugs. 2:496–501. 2001.PubMed/NCBI
|
|
8
|
Miyazaki Y, Kaikita K, Endo M, Horio E,
Miura M, Tsujita K, Hokimoto S, Yamamuro M, Iwawaki T, Gotoh T, et
al: C/EBP homologous protein deficiency attenuates myocardial
reperfusion injury by inhibiting myocardial apoptosis and
inflammation. Arterioscler Thromb Vasc Biol. 31:1124–1132. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Guo G, Meng Y, Tan W, Xia Y, Cheng C, Chen
X and Gu Z: Induction of apoptosis coupled to endoplasmic reticulum
stress through regulation of CHOP and JNK in bone marrow
mesenchymal stem cells from patients with systemic lupus
erythematosus. J Immunol Res. 2015:1837382015. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Boyce M and Yuan J: Cellular response to
endoplasmic reticulum stress: A matter of life or death. Cell Death
Differ. 13:363–373. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Wu JS, Li WM, Chen YN, Zhao Q and Chen QF:
Endoplasmic reticulum stress is activated in acute pancreatitis. J
Dig Dis. 17:295–303. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Zhang K and Kaufman RJ: From
endoplasmic-reticulum stress to the inflammatory response. Nature.
454:455–462. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Xu C, Bailly-Maitre B and Reed JC:
Endoplasmic reticulum stress: Cell life and death decisions. J Clin
Invest. 115:2656–2664. 2005. View
Article : Google Scholar : PubMed/NCBI
|
|
14
|
Oyadomari S and Mori M: Roles of
CHOP/GADD153 in endoplasmic reticulum stress. Cell Death Differ.
11:381–389. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Park JS, Luethy JD, Wang MG, Fargnoli J,
Fornace AJ Jr, McBride OW and Holbrook NJ: Isolation,
characterization and chromosomal localization of the human GADD153
gene. Gene. 116:259–267. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Wang XZ, Lawson B, Brewer JW, Zinszner H,
Sanjay A, Mi LJ, Boorstein R, Kreibich G, Hendershot LM and Ron D:
Signals from the stressed endoplasmic reticulum induce
C/EBP-homologous protein (CHOP/GADD153). Mol Cell Biol.
16:4273–4280. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Matsumoto M, Minami M, Takeda K, Sakao Y
and Akira S: Ectopic expression of CHOP (GADD153) induces apoptosis
in M1 myeloblastic leukemia cells. FEBS Lett. 395:143–147. 1996.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Myoishi M, Hao H, Minamino T, Watanabe K,
Nishihira K, Hatakeyama K, Asada Y, Okada K, Ishibashi-Ueda H,
Gabbiani G, et al: Increased endoplasmic reticulum stress in
atherosclerotic plaques associated with acute coronary syndrome.
Circulation. 116:1226–1233. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Fu HY, Okada K, Liao Y, Tsukamoto O,
Isomura T, Asai M, Sawada T, Okuda K, Asano Y, Sanada S, et al:
Ablation of C/EBP homologous protein attenuates endoplasmic
reticulum-mediated apoptosis and cardiac dysfunction induced by
pressure overload. Circulation. 122:361–369. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Walter P and Ron D: The unfolded protein
response: From stress pathway to homeostatic regulation. Science.
334:1081–1086. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Kitamura M: Control of NF-kB and
inflammation by the unfolded protein response. Int Rev Immunol.
30:4–15. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Kitamura M: Biphasic, bidirectional
regulation of NF-kappaB by endoplasmic reticulum stress. Antioxid
Redox Signal. 11:2353–2364. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Sah RP, Dudeja V, Dawra RK and Saluja AK:
Cerulein-induced chronic pancreatitis does not require intra-acinar
activation of trypsinogen in mice. Gastroenterology.
144:1076–1085.e1072. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Sah RP, Garg SK, Dixit AK, Dudeja V, Dawra
RK and Saluja AK: Endoplasmic reticulum stress is chronically
activated in chronic pancreatitis. J Biol Chem. 289:27551–27561.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Allagnat F, Fukaya M, Nogueira T,
Delaroche D, Welsh N, Marselli L, Marchetti P, Haefliger JA,
Eizirik DL and Cardozo AK: C/EBP homologous protein contributes to
cytokine-induced pro-inflammatory responses and apoptosis in
β-cells. Cell Death Differ. 19:1836–1846. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Rius J, Guma M, Schachtrup C, Akassoglou
K, Zinkernagel AS, Nizet V, Johnson RS, Haddad GG and Karin M:
NF-kappaB links innate immunity to the hypoxic response through
transcriptional regulation of HIF-1alpha. Nature. 453:807–811.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Sun XF and Zhang H: NF-κB and NF-κB1
polymorphisms in relation to susceptibility of tumor and other
diseases. Histol Histopatol. 22:1387–1398. 2007.
|
|
28
|
Czyz M: Specificity and selectivity of the
NFkappaB response. Postepy Biochem. 51:60–68. 2005.(In Polish).
PubMed/NCBI
|
|
29
|
Huang H, Liu Y, Daniluk J, Gaiser S, Chu
J, Wang H, Li ZS, Logsdon CD and Ji B: Activation of nuclear
factor-kB in acinar cells increases the severity of pancreatitis in
mice. Gastroenterology. 144:202–210. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Sah RP, Garg P and Saluja AK: Pathogenic
mechanisms of acute pancreatitis. Curr Opin Gastroenterol.
28:507–515. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Wang H, Li L, Zhao M, Chen YH, Zhang ZH,
Zhang C, Ji YL, Meng XH and Xu DX: Melatonin alleviates
lipopolysaccharide-induced placental cellular stress response in
mice. J Pineal Res. 50:418–426. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Jaworek J, Leja-Szpak A, Kot M, Jaworek A,
Nawrot-Porbka K, Bonior J and Szklarczyk J: The role of melatonin
in pancreatic protection: Could melatonin be used in the treatment
of acute pancreatitis? Curr Pharm Des. 20:4834–4840. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Forman K, Vara E, Garcia C, Kireev R,
Cuesta S, Acuña-Castroviejo D and Tresguerres JA: Beneficial
effects of melatonin on cardiological alterations in a murine model
of accelerated aging. J Pineal Res. 49:312–320. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Qi W, Tan DX, Reiter RJ, Kim SJ,
Manchester LC, Cabrera J, Sainz RM and Mayo JC: Melatonin reduces
lipid peroxidation and tissue edema in cerulein-induced acute
pancreatitis in rats. Dig Dis Sci. 44:2257–2262. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Kim SJ, Kang HS, Lee JH, Park JH, Jung CH,
Bae JH, Oh BC, Song DK, Baek WK and Im SS: Melatonin ameliorates ER
stress-mediated hepatic steatosis through miR-23a in the liver.
Biochem Biophys Res Commun. 458:462–469. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
San-Miguel B, Crespo I, Sanchez DI,
González-Fernández B, Ortiz de Urbina JJ, Tuñón MJ and
González-Gallego J: Melatonin inhibits autophagy and endoplasmic
reticulum stress in mice with carbon tetrachloride-induced
fibrosis. J Pineal Res. 59:151–162. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Hartl FU and Hayer-Hartl M: Molecular
chaperones in the cytosol: From nascent chain to folded protein.
Science. 295:1852–1858. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Pfaffenbach KT and Lee AS: The critical
role of GRP78 in physiologic and pathologic stress. Curr Opin Cell
Biol. 23:150–156. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Ni M and Lee AS: ER chaperones in
mammalian development and human diseases. FEBS Lett. 581:3641–3651.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Lee AS: The glucose regulated proteins:
Stress induction and clinical applications. Trends Biochem Sci.
26:504–510. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Kimata Y and Kohno K: Endoplasmic
reticulum stress-sensing mechanisms in yeast and mammalian cells.
Curr Opin Cell Biol. 23:135–142. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Gukovskaya AS and Pandol SJ: Cell death
pathways in pancreatitis and pancreatic cancer. Pancreatology.
4:567–586. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Bhatia M: Apoptosis versus necrosis in
acute pancreatitis. Am J Physiol Gastrointest Liver Physiol.
286:189–196. 2004. View Article : Google Scholar
|
|
45
|
Mareninova OA, Sung KF, Hong P, Lugea A,
Pandol SJ, Gukovsky I and Gukovskaya AS: Cell death in
pancreatitis: Caspases protect from necrotizing pancreatitis. J
Biol Chem. 281:3370–3381. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Liu Y, Yang L, Chen KL, Zhou B, Yan H,
Zhou ZG and Li Y: Knockdown of GRP78 promotes apoptosis in
pancreatic acinar cells and attenuates the severity of cerulein and
LPS induced pancreatic inflammation. PLoS One. 9:e923892014.
View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Williams JA, Sans MD, Tashiro M, Schäfer
C, Bragado MJ and Dabrowski A: Cholecystokinin activates a variety
of intracellular signal transduction mechanisms in rodent
pancreatic acinar cells. Pharmacol Toxicol. 91:297–303. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Wu L, Cai B, Zheng S, Liu X, Cai H and Li
H: Effect of emodin on endoplasmic reticulum stress in rats with
severe acute pancreatitis. Inflammation. 36:1020–1029. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Jacob TG, Sreekumar VI, Roy TS and Garg
PK: Electron-microscopic evidence of mitochondriae containing
macroautophagy in experimental acute pancreatitis: Implications for
cell death. Pancreatology. 14:454–458. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Hall JC and Crawford HC: The conspiracy of
autophagy, stress and inflammation in acute pancreatitis. Curr Opin
Gastroenterol. 30:495–499. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Nashine S, Liu Y, Kim BJ, Clark AF and
Pang IH: Role of C/EBP homologous protein in retinal ganglion cell
death after ischemia/reperfusion injury. Invest Ophthalmol Vis Sci.
56:221–231. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Nishitoh H, Matsuzawa A, Tobiume K,
Saegusa K, Takeda K, Inoue K, Hori S, Kakizuka A and Ichijo H: ASK1
is essential for endoplasmic reticulum stress-induced neuronal cell
death triggered by expanded polyglutamine repeats. Genes Dev.
16:1345–1355. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Nagai H, Noguchi T, Takeda K and Ichijo H:
Pathophysiologica1 roles of ASK1-MAP kinase signaling pathways. J
Biochem Mol Biol. 40:1–6. 2007.PubMed/NCBI
|
|
54
|
McCullough KD, Martindale JL, Klotz LO, Aw
TY and Holbrook NJ: Gadd153 sensitizes cells to endoplasmic
reticulum stress by down-regulating Bcl2 and perturbing the
cellular redox state. Mol Cell Boil. 21:1249–1259. 2001. View Article : Google Scholar
|
|
55
|
Gotoh T, Terada K, Oyadomari S and Mori M:
hsp70-DnaJ chaperone pair prevents nitric oxide- and CHOP-induced
apoptosis by inhibiting translocation of Bax to mitochondria. Cell
Death Differ. 11:390–402. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Suyama K, Ohmuraya M, Hirota M, Ozaki N,
Ida S, Endo M, Araki K, Gotoh T, Baba H and Yamamura K: C/EBP
homologous protein is crucial for the acceleration of experimental
pancreatitis. Biochem Biophys Res Commun. 367:176–182. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Endo M, Mori M, Akira S and Gotoh T: C/EBP
homologous protein (CHOP) is crucial for the induction of
caspase-11 and the pathogenesis of lipopolysaccharide-induced
inflammation. J Immunol. 176:6245–6253. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Namba T, Tanaka K, Ito Y, Ishihara T,
Hoshino T, Gotoh T, Endo M, Sato K and Mizushima T: Positive role
of CCAAT/enhancer-binding protein homologous protein, a
transcription factor involved in the endoplasmic reticulum stress
response in the development of colitis. Am J Pathol. 174:1786–1798.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Zhang K, Shen X, Wu J, Sakaki K, Saunders
T, Rutkowski DT, Back SH and Kaufman RJ: Endoplasmic reticulum
stress activates cleavage of CREBH to induce a systemic
inflammatory response. Cell. 124:587–599. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Matulewicz N and Karczewska-Kupczewska M:
Insulin resistance and chronic inflammation. Postepy Hig Med Dosw
(Online). 70:1245–1258. 2016.PubMed/NCBI
|
|
61
|
Musialik K, Szulinska M, Hen K, Skrypnik D
and Bogdanski P: The relation between osteoprotegerin, inflammatory
processes, and atherosclerosis in patients with metabolic syndrome.
Eur Rev Med Pharmacol Sci. 21:4379–4385. 2017.PubMed/NCBI
|