Introduction
Atherosclerosis (AS) is a chronic multi-factorial
vascular disease and an underlying cause of cardiovascular disease,
which accounts for 16.7 million deaths each year worldwide
(1). AS is a major risk factor for
cardiovascular and cerebrovascular diseases (2) and is characterized by lipid
accumulation and the formation of fat-laden plaque in vessels
(3). Despite advances in
treatment, the morbidity and mortality of AS remain at high levels.
Therefore, it is urgent to uncover new mechanisms underlying
atherogenesis and to find potent therapeutic targets and methods
for AS.
The pathogenesis of AS is a complex process, in
which the dysfunction of vascular smooth muscle cells (VSMCs) and
endothelial cells, as well as the excessive secretion of
pro-inflammatory cytokines by macrophages serve important roles
(4,5). Particularly, VSMCs serve a crucial
role in this process owing to their capacities to promote plaque
growth in early stages and to contribute to plaque stability in
advanced stages (6), which
suggested that VSMCs may be an efficient therapeutic target for
AS.
Although ~90% of the human genes are transcribed,
only 2% of the human genome possesses the ability to encode
proteins, the remaining being non-coding genes (7,8).
Long non-coding (lnc)RNAs are a class of ncRNAs that have been
demonstrated to regulate gene expression at transcription and
post-transcription levels (9).
Accumulated evidence has shown that lncRNAs are involved in a
number of pathological processes, including carcinogenesis
(10) and chronic diseases such as
AS (11–13). lncRNA H19 (H19) is transcribed from
the H19/insulin-like growth factor 2 gene cluster that is located
in human chromosome 11p15.5 (14).
Recently, H19 was reported to be highly expressed in patients with
AS and upregulation of H19 promoted the proliferation and repressed
the apoptosis of VSMCs and human umbilical vein endothelial cells
(HUVECs) (15). However, the
detailed mechanisms underlying H19 in AS remains to be
elucidated.
p53 is an essential molecule in the regulation of
cell growth, apoptosis and cell cycle, and it is thought to be
strongly implicated in the pathogenesis of AS (16,17).
The inactivation of p53 promotes the development of AS (18–20).
Wu et al (21) demonstrated
that long intergenic non-coding RNA-p21 (lincRNA-p21) represses
proliferation and promotes apoptosis of VSMCs by increasing p53
expression. However, whether p53 is involved in H19-regulated VMSC
proliferation and apoptosis remains unclear. Therefore, the present
study aimed to explore the effects of H19/p53 in regulating VMSCs
apoptosis and proliferation in vitro and in vivo,
thereby elucidating its role in the pathogenesis of AS.
Materials and methods
Ethics approval
Animal experiments were approved by the Committee of
Jining First People's Hospital (Jining, China) and was performed in
accordance with the National Institutes of Health Guide for the
Care and Use of Laboratory Animals.
Animal experiments
Male specific pathogen free (SPF) grade
apolipoprotein (Apo)E−/− mice (n=25; age, 8 weeks;
weight, 20–22 g) were purchased from Genomeditech and were used to
establish AS models. Wild-type C57BL/6J mice without ApoE knockdown
(n=5; age, 8 weeks; weight, 20–22 g) were used as the control
(control group). All mice were raised under SPF conditions in a
controlled temperature (23±2°C) and humidity (55–60%) environment
under a 12 h light/dark cycle. After 1 week of acclimation, the
ApoE−/− mice (n=25) were divided into five groups (n=5
mice/group): i) AS group; ii) AS + short hairpin RNA (sh)-negative
control (NC) group; iii) AS + sh-H19 group; iv) AS + sh-NC +
simvastatin group; and v) AS + sh-H19 + simvastatin group. To
induce AS models, the ApoE−/− mice were given a high-fat
diet comprising 21% fat and 0.15% cholesterol on the basis of a
chow diet for 8 weeks as in a previous study (22). Following 8 weeks of receiving the
high-fat diet, the mice were subsequently fed a standard laboratory
diet (10% fat, 15% protein and 75% carbohydrate) for the remaining
10 weeks. Mice in the control group were fed a standard laboratory
diet for 18 weeks. Mice in the AS + sh-H19 group were given
intraperitoneal injection of 100 µl shRNA targeting mouse H19
(sh-H19) once/week for 10 consecutive weeks after high-fat diet
administration, and the lentiviral vector sh-NC was used as a
negative control of sh-H19. Additionally, the AS + sh-NC +
simvastatin group and the AS + sh-H19 + simvastatin group were
given an oral administration of 5 mg/kg of simvastatin (Merck KGaA)
every day for 10 weeks following 8 weeks of high-fat diet
administration (23), whereas mice
in other groups were orally administrated with vehicle (80%
Tween).
Thoracic aorta collection and
histopathological analysis
At the end of sh-H19 and simvastatin administration,
mice were sacrificed by cervical dislocation and the entire
thoracic aortas (from the branches of the abdominal aorta to the
aortic arch) were removed and stored in liquid nitrogen for
pathological examination or RNA extraction. For histopathological
analysis, the aortic arch was fixed with 10% formaldehyde at room
temperature for 24 h. Subsequently, samples were dehydrated with
different concentrations (80, 95 and 100%) of ethyl alcohol,
cleared with xylene, embedded in paraffin and sectioned (5 µm).
Sections were stained with 100% hematoxylin (Beyotime Institute of
Biotechnology) for 3 min and 0.5% eosin (Beyotime Institute of
Biotechnology) for 30 sec at room temperature. The staining was
visualized using an inverted light microscope at a magnification of
×100.
Immunohistochemical staining
Tissue sections were deparaffinized and rehydrated
with xylene and ethanol, as aforementioned. Subsequently, the
antigen was retrieved using citrate antigen retrieval solution
(Beyotime Institute of Biotechnology) and blocked with 5% goat
serum (AmyJet Scientific, Inc.) diluted in TBS + 0.5% Tween 20
(TBST), the sections were probed with the primary antibody against
cleaved caspase3 (c-caspase3; 1:100; cat. no. 9661; Cell Signaling
Technology, Inc.) at 4°C overnight. Following the primary antibody
incubation, sections were incubated with a horseradish
peroxidase-conjugated secondary antibody (1:500; cat. no. 18653;
Cell Signaling Technology, Inc.) at room temperature for 1 h,
followed by incubation with the chromogen 3,30-diaminobenzidine
tetrachloride (R&D Systems, Inc.) for 2–3 sec at room
temperature. Cell nuclei were stained with 1% Harris hematoxylin
solution for 30 sec at room temperature. The staining of c-caspase3
was evaluated by two pathologists on the basis of the positive
staining proportion and the staining intensity (24). The positive staining percentage was
scored as 0 for ≤5%, 1 for 6–25%, 2 for 26–50%, 3 for 51–75% and 4
for ≥75%, respectively. Intensity was marked as follows: 0, no
staining; 1 weak staining; 2, moderate staining; and 3, strong
staining. The final score was obtained by multiplying the
percentage score and intensity score.
Cell culture and simvastatin
treatment
Human VSMCs (ATCC CRL-1999) were obtained from the
American Type Culture Collection. VSMCs were cultured in DMEM
(HyClone; GE Healthcare Life Sciences), supplemented with 1%
penicillin (100 U/ml)/streptomycin (100 mg/ml) (Beyotime Institute
of Biotechnology) and 10% FBS (Gibco; Thermo Fisher Scientific,
Inc.) and maintained in a 37°C constant humidified incubator with
5% CO2.
For simvastatin treatment, a total of
5×105 VSMCs/ml were incubated with 2 µM simvastatin
(Xingqiong Co., Ltd.) for 24 h following transduction for 24 h with
the lentiviral vectors. H19 and p53 expression levels were reduced
by transfecting cells with 2 mg shRNAs (lentiviral vectors)
targeting H19 (sh-H19) and p53 (sh-p53), respectively, and control
cells were transduced with 2 µg negative control vector (sh-NC);
all shRNAs were purchased from Shanghai GenePharma Co., Ltd. VSMCs
were transduced with sh-p53, sh-H19 or sh-NC in a pGLVH1/GFP+ Puro
lentiviral vector (Shanghai GenePharma Co., Ltd.) using Polybrene
(4 µg/ml; Merck KGaA).
Reverse transcription-quantitative PCR
(RT-qPCR)
The expression levels of H19/p53 in mouse aorta and
VSMCs were tested by RT-qPCR; expression levels were normalized to
GAPDH. Briefly, total RNA was acquired from cells (1×106
cells/sample) and aorta tissues (100 g/sample) by using
TRIzol® reagent (Invitrogen; Thermo Fisher Scientific,
Inc.), according to the manufacturer's protocol. Subsequently, 1 µg
of the total RNA was reversed transcribed using a cDNA Synthesis
Kit (CoWin Biosciences), according to the manufacturers' protocol.
qPCR was performed using the SYBR® Green PCR Master mix
(CoWin Biosciences), according to the manufacturer's protocol using
the following primers: H19, forward 5′-ATCGGTGCCTCAGCGTTCGG-3′,
reverse 5′-CTGTCCTCGCCGTCACACCG-3′; p53, forward
5′-CCTGGATTGGCCAGACTGC-3′, reverse 5′-TTTTCAGGAAGTAGTTTCCATAGGT-3′;
GAPDH, forward 5′-CACTAGGCGCTCACTGTTCTCT-3′, reverse
5′-CGTTCTCAGCCTTGACGGT-3′. The following thermocycling conditions
for the qPCR were used: Initial denaturation for 3 min at 95°C;
followed by 35 cycles of denaturation at 95°C for 10 sec and
annealing/extension at 55°C for 30. Expression levels were
quantified using the 2−∆∆Cq method (25) and normalized to GAPDH.
Western blot analysis
VMSCs were collected and lysed with RIPA lysis
buffer (cat. no. R0010; Beijing Solarbio Science & Technology
Co., Ltd.) containing protease inhibitor. After being centrifuged
at 20,000 × g for 30 min at 4°C and quantified with a BCA kit
(Thermo Fisher Scientific, Inc.), 30 µg protein from each sample
was separated by 10% SDS-PAGE and transferred onto PVDF membranes
(EMD Millipore). The membranes were blocked with 5% nonfat milk
diluted in TBST at room temperature for 1 h and then incubated with
anti-p53 (1:1,000; cat. no. 9282; Cell Signaling Technology, Inc.),
anti-bcl-2 (1:1,000; cat. no. 3498; Cell Signaling Technology,
Inc.), anti-p53 upregulated modulator of apoptosis (PUMA; 1:1,000;
cat no. 4976; Cell Signaling Technology, Inc.), anti-c-caspase3
(1:2,000; cat. no. 9661; Cell Signaling Technology, Inc.) and
anti-GAPDH monoclonal antibody (1:5,000; cat. no. 5174; Cell
Signaling Technology, Inc.) at 4°C overnight. After washing with
TBST three times, the membranes were probed with the horseradish
peroxidase-conjugated goat anti-mouse and goat anti-rabbit
secondary antibodies (1:10,000; cat. nos. SA00001-1 and SA00001-2,
respectively; ProteinTech Group, Inc.) for 1 h at room temperature
and visualized with ECL detection reagents (EMD Millipore). Protein
expression was quantified using ImageJ version 1.48 software
(National Institutes of Health). GAPDH was used as a loading
control to normalize the data.
Co-immunoprecipitation analysis of the
interaction of p53 and Bax proteins
VSMCs transfected with sh-H19 or sh-NC were
collected and rinsed with cold PBS and then lysed in IP Lysis
Buffer (Thermo Fisher Scientific, Inc.), followed by centrifugation
at 20,000 × g for 30 min at 4°C. Next, total protein was quantified
using a BCA assay and the lysate containing 200 µg protein were
incubated with Dynabeads® protein G for 1 h and
incubated with 2 µg anti-p53 (cat. no. 9282; Cell Signaling
Technology, Inc.) or anti-Bax (cat. no. ab216494; Abcam) antibodies
overnight at 4°C, followed by incubation with Dynabeads®
protein G for another 1 h. The whole cell lysate was incubated
overnight at 4°C with 2 µg anti-rabbit IgG antibody (cat. no. 3900;
Cell Signaling Technology, Inc.) as the negative control for the
anti-p53 antibody or 2 µg anti-mouse IgG antibody (cat. no. ab6709;
Abcam) as the negative control for the anti-Bax antibody. The
immunocomplex were washed with IP Lysis Buffer and analyzed by the
aforementioned western blot procedure using anti-Bax (1:1,000; cat.
no. 2774; Cell Signaling Technology, Inc.) or anti-p53 (1:1,000;
cat. no. 2524; Cell Signaling Technology, Inc.) antibodies.
Cell proliferation detection
Cell proliferation was measured by Cell Counting
Kit-8 (CCK-8; Dojindo Molecular Technologies, Inc.) according to
the manufacturer's protocol. Briefly, 100 µl cell suspension
containing 2×103 cells was placed into a 96-well plate
and incubated at 37°C overnight. Then, the cells were transfected
with sh-NC, sh-H19, sh-p53 or sh-H19 + sh-p53 for 1, 2, 3, 4 or 5
days, with the medium changed every day. At each time point, 10 µl
CCK-8 solution was added to the wells and the absorbance at 450 nm
was detected using a spectrophotometer (Shjingmi Co., Ltd.).
Cell apoptosis
Cell apoptosis was assessed by using Annexin
V-FITC/PI Double-Staining kit (YESEN Co., Ltd; http://www.yeasen.com), according to the
manufacturer's protocol. After staining, cells were analyzed using
a BD FACSCalibur flow cytometer (BD Biosciences) at 488 nm
excitation and 630 nm emission to determine apoptotic rates with
FlowJo version 10 software (FlowJo LLC).
Statistical analysis
Data from three independent experiments are
presented as the mean ± SD. Comparisons were made using Student's
t-test or with one-way ANOVA followed by Tukey's test if >2
groups using SPSS 17.0 software (SPSS, Inc.). P<0.05 was
considered to indicate a statistically significant difference.
Results
H19 is highly expressed in AS model
mice and H19 downregulation promotes VSMC apoptosis
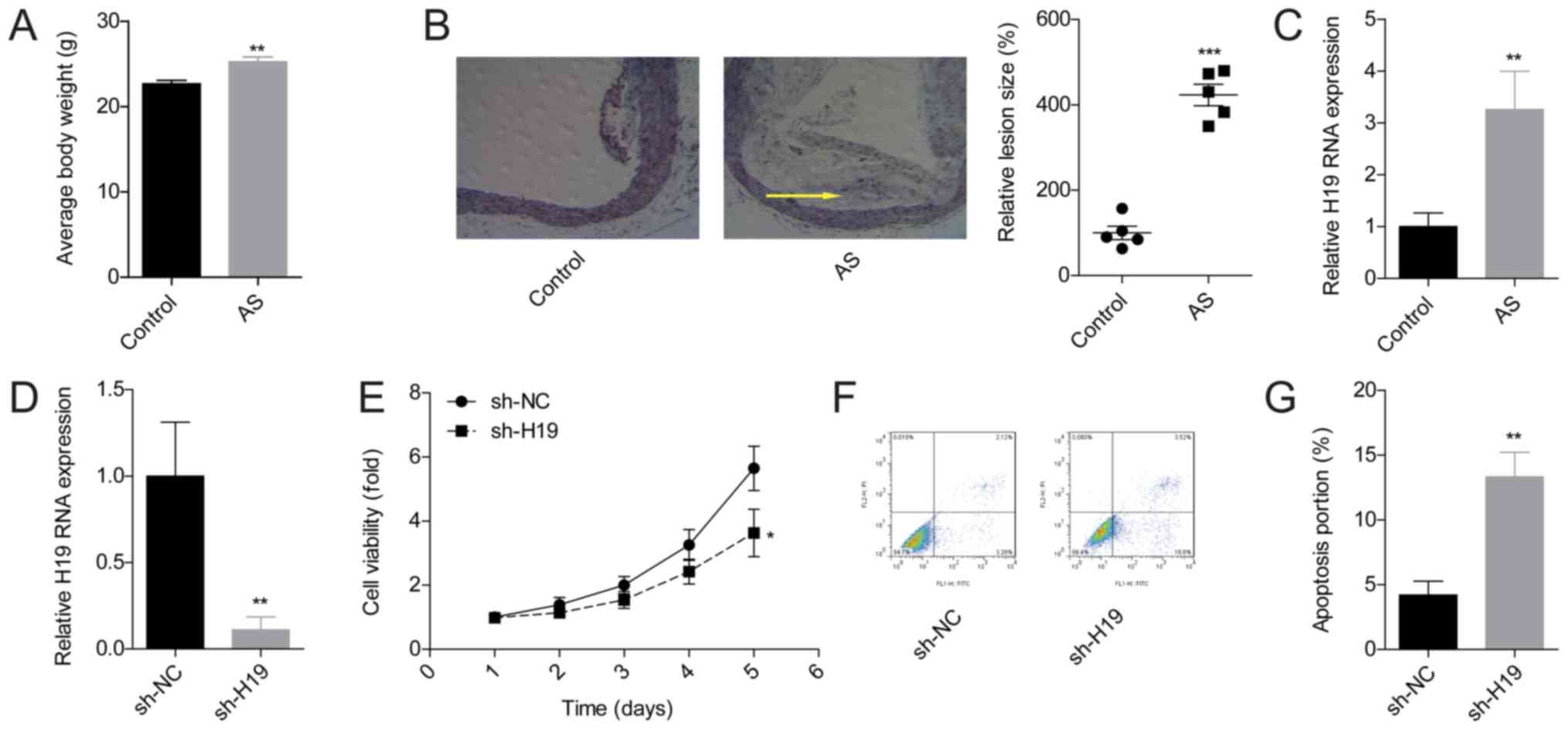
To further explore the effect of H19 in the
pathogenesis of AS, the expression levels of H19 in the mouse AS
model were assessed. The average mouse body weights following 8
weeks of high-fat diet were clearly increased in the AS model group
compared with the control group (Fig.
1A). In addition, compared with the control group, plaque
formation was visible in the aortic roots of mice in the AS group
and demonstrated significantly increased lesion sizes (Fig. 1B), which indicated that the AS
model had been constructed successfully. H19 expression patterns in
mouse aorta tissues from the control and the AS group were compared
using RT-qPCR. The results demonstrated that H19 expression levels
were significantly higher in AS group mice compared with the
control group (Fig. 1C).
The effects of H19 on the proliferation and
apoptosis of VSMCs were investigated through loss-of-function
experiments. The mRNA level of H19 was reduced by approximately 80%
after VSMCs were infected with sh-H19 (Fig. 1D). Silencing of H19 significantly
inhibited the viability (Fig. 1E)
and promoted the apoptosis of VSMCs (Fig. 1F and G). These results suggested
that H19 may serve an important role in the regulation of VSMC
growth.
Downregulation of H19 induces the
apoptosis of VSMCs through enhancing p53 expression
The role of p53 in H19 downregulation-induced VMSC
apoptosis promotion was next investigated. The expression of p53
was significantly increased when H19 expression was reduced in
VSMCs at both mRNA and protein levels (Fig. 2A and B), which was evidently
rescued when p53 expression was silenced (Fig. 2C and D). p53 downregulation also
impaired sh-H19 roles in inhibiting VSMC viability (Fig. 2E) and promoting VSMC apoptosis
(Fig. 2F and G).
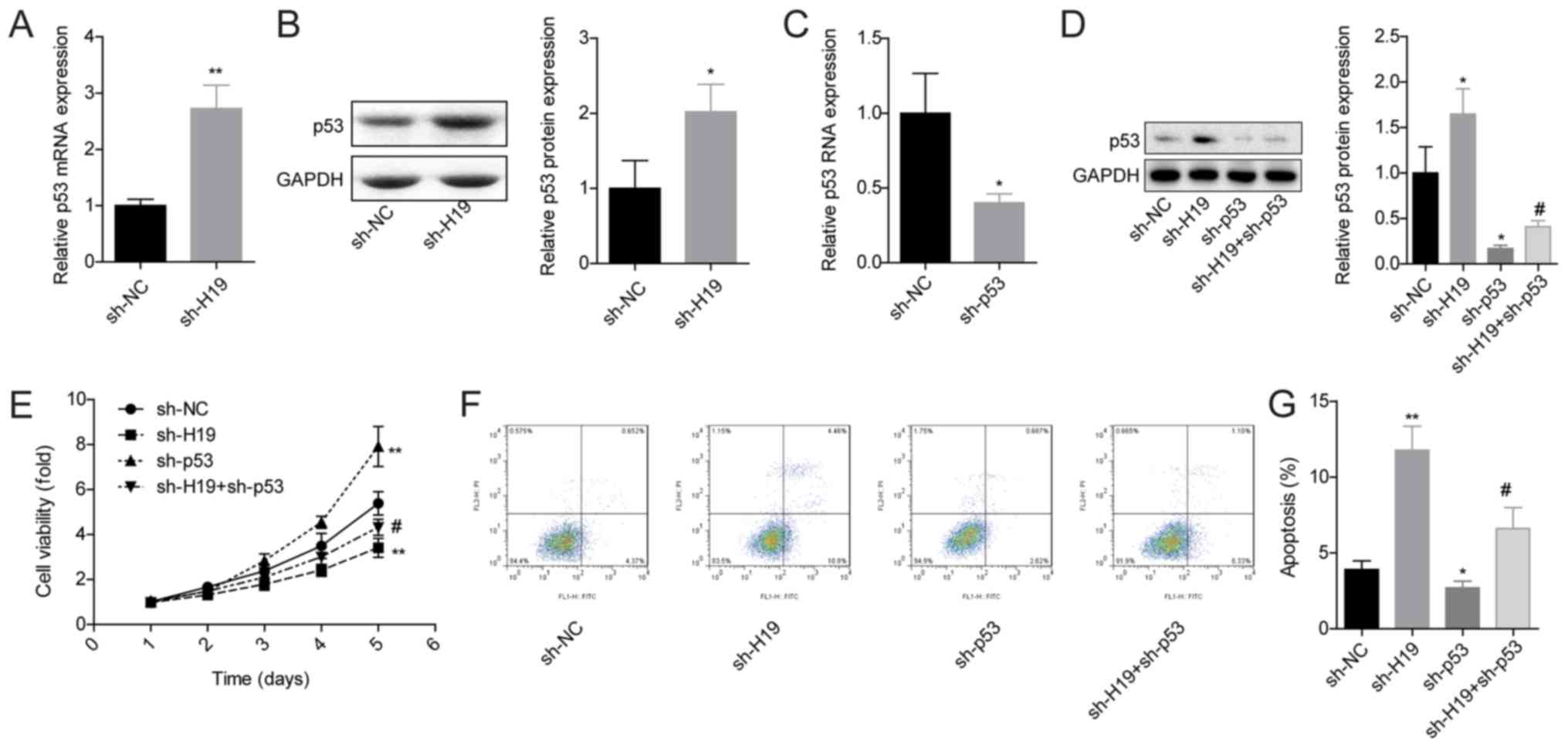 | Figure 2.H19 downregulation promotes cell
apoptosis through upregulating p53 expression in VSMCs. (A) mRNA
and (B) protein expression levels of p53 were assessed by RT-qPCR
and western blotting, respectively. (C) p53 mRNA expression levels
after VSMCs were infected with sh-NC or sh-p53. (D) p53 protein
expression of p53 was detected after VSMCs were infected with
sh-NC, sh-H19, sh-53 or sh-H19 + sh-p53. (E) Cell proliferation was
detected by Cell Counting Kit-8 assay after VSMCs were infected
with sh-NC, sh-H19, sh-p53 and sh-H19 + sh-p53. (F and G) Cell
apoptosis was measured by flow cytometry after VSMCs were infected
with sh-NC, sh-H19, sh-p53 and sh-H19 + sh-p53. *P<0.05 and
**P<0.01 vs. sh-NC; #P<0.05 vs. sh-H19. NC,
negative control; RT-qPCR, reverse transcription-qPCR; sh, short
hairpin RNA; VSMCs, vascular smooth muscle cells. |
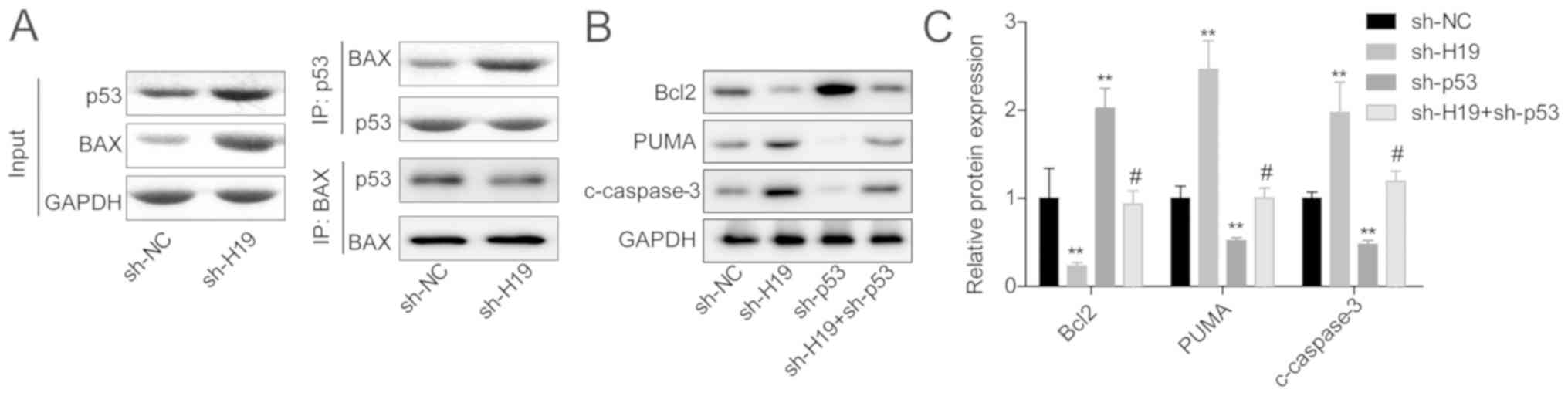
Downregulation of H19 in VSMCs enhanced the
interaction between p53 protein and Bax protein (Fig. 3A). In addition, the expression
levels of PUMA and c-caspase3 were increased, whereas Bcl-2
expression was reduced in the sh-H19 group compared with the sh-NC
group, and this effect was neutralized when p53 was also
downregulated in VSMCs (Fig. 3B and
C). Together, these findings indicated that H19 downregulation
promoted VSMC apoptosis in a p53-dependent manner.
Knockdown of H19 represses plaque
formation in ApoE−/− mice
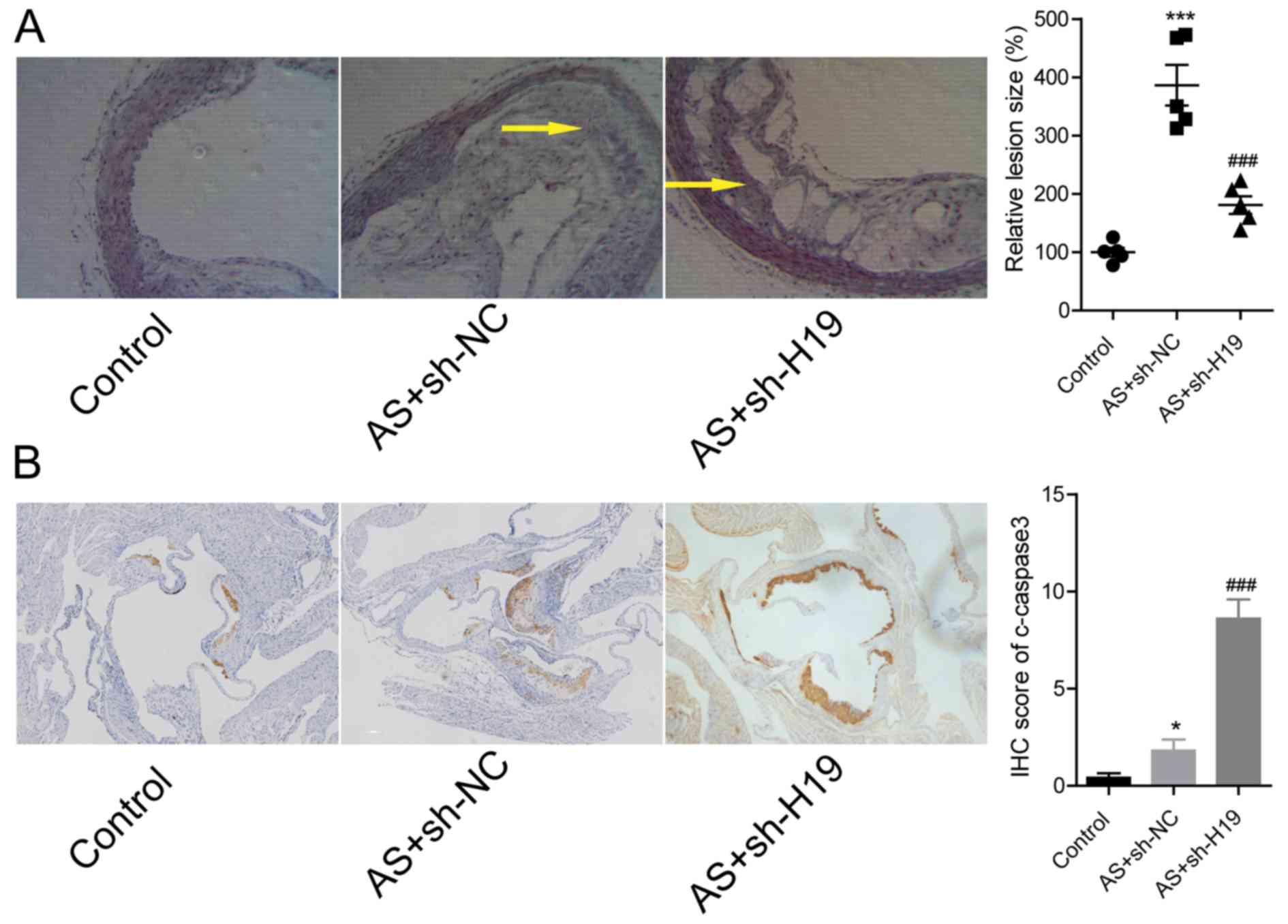
Next, the effects of H19 downregulation in the
pathogenesis of AS in ApoE−/− mice was explored.
Compared with the AS group, the lesion size in the aortic root was
significantly reduced in AS + sh-H19 group (Fig. 4A). The downregulation of H19 in the
AS + sh-H19 group significantly increased the expression of
c-caspase3, an apoptotic marker in atherosclerotic plaque tissues,
compared with the AS + sh-NC group (Fig. 4B). These findings suggested that
knockdown of H19 could alleviate high-fat diet-induced plaque
formation in ApoE−/− mice.
Knockdown of H19 enhances the role of
simvastatin in alleviating AS
Finally, the effects of H19 downregulation on the
progression of AS under simvastatin administration was explored
in vivo. Compared with the sh-NC group, VSMC apoptosis was
significantly increased in both the sh-H19 and sh-NC + simvastatin
groups (Fig. 5A and B). In
addition, the apoptotic rate of VSMCs in the sh-H19 + simvastatin
group was significantly higher compared with the sh-NC +
simvastatin group (Fig. 5A and B).
Similarly, the in vivo experiments demonstrated that
simvastatin treatment significantly reduced the lesion size in the
aortic root in the AS + sh-NC + simvastatin group compared with the
AS group, and this effect was significantly enhanced when H19 was
downregulated in the AS + sh-H19 + simvastatin group (Fig. 5C). In addition, c-caspase3
expression was increased in mice atherosclerotic plaque tissues in
the AS + simvastatin + sh-H19 group compared with the AS +
simvastatin group (Fig. 5D and E),
as well as increased expression levels of Bax/Bcl-2 and PUMA
(Fig. 5E). These results indicated
that H19 downregulation may cooperate with simvastatin to promote
VSMCs apoptosis, thereby alleviating AS.
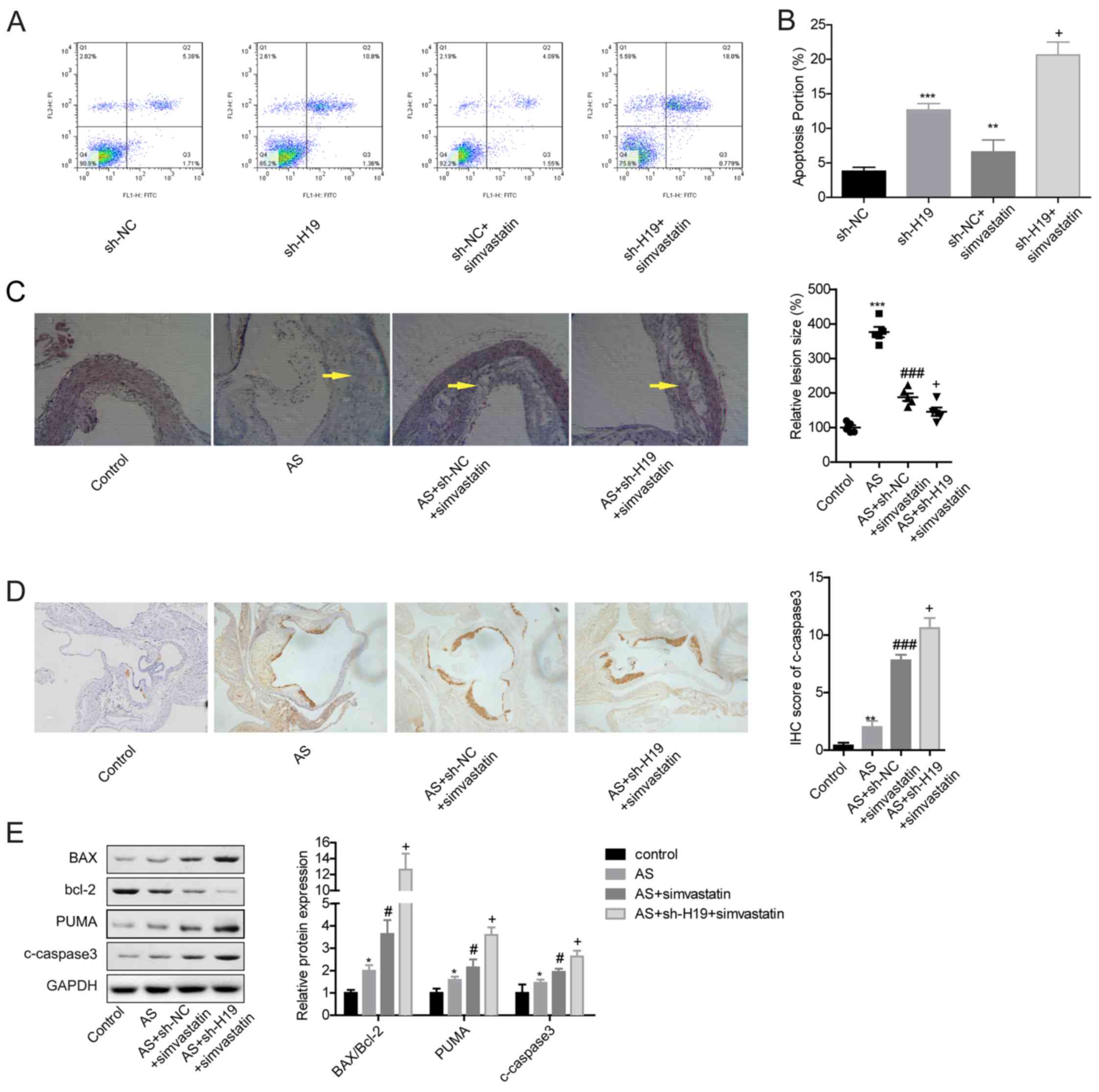 | Figure 5.H19 downregulation cooperates with
simvastatin to alleviate AS. (A and B) Apoptosis was detected with
flow cytometry after VSMCs were infected with sh-NC, sh-H19, sh-NC
+ simvastatin or sh-H19 + simvastatin. **P<0.01, ***P<0.001
vs. sh-NC; +P<0.05 vs. sh-NC + simvastatin. (C)
Hematoxylin and eosin staining analysis of the lesions in mice
aortic roots from different groups. Arrows indicate the
atherosclerotic lesions. Magnification, ×200. (D) C-caspase3
expression levels in the atherosclerotic plaque tissues from mice
in control, AS + sh-NC, AS + sh-NC + simvastatin or AS + sh-H19 +
simvastatin groups were analyzed by IHC. Magnification, ×100. (E)
Western blot analysis of the expression levels of Bcl2, PUMA,
c-caspase3 and Bax in atherosclerotic plaque tissues of mice from
control, AS, AS + simvastatin or AS + simvastatin + sh-H19 groups.
*P<0.05 and **P<0.01, ***P<0.001 vs. Control;
#P<0.05 and ###P<0.001 vs. AS;
+P<0.05 vs. AS + sh-NC + simvastatin. AS,
atherosclerosis; c-, cleaved; IHC, immunohistochemistry; NC,
negative control; sh, short hairpin RNA; PUMA, p53 upregulated
modulator of apoptosis; VSMCs, vascular smooth muscle cells. |
Discussion
AS is a clinical symptom that can induce narrowing
inside the artery interior due to plaque accumulation. Several risk
factors, including excessive drinking, unbalanced diet, smoking and
obesity, may facilitate the progression of AS (2). The dysfunction of VSMCs serves a
vital role in the formation and eventual rupture of atherosclerotic
plaques due to their hyperproliferative properties (26). The excessive proliferation of VSMCs
can trigger plaque formation, which accounts for an important step
in the initiation of AS, indicating that effective control of VSMC
proliferation may be a promising method for AS prevention and
treatment (27). However, VSMC
apoptosis in advanced plaques increase plaque vulnerability,
stenosis and medial degeneration (28,29).
Collectively, these findings indicate that VSMC apoptosis and
proliferation serve dual roles in the pathogenesis of AS. The
present study demonstrated that knockdown of lncRNA H19 could
inhibit plaque formation through inducing the apoptosis and
repressing the proliferation of VSMCs partly in a p53-dependent
way, indicating a protective role of H19 downregulation in AS.
Recently, studies have reported that lncRNAs are
strongly implicated in the pathogenesis of AS. For instance, Chen
et al (30) found that
tanshinol administration could reduce the aortic atherosclerotic
lesion area by downregulating lncRNA TUG1. Yao et al
(31) demonstrated that lncRNA
ENST00113 significantly promotes the proliferation and migration of
VSMCs and HUVECs. lncRNA H19 was confirmed to be overexpressed in
patients with AS, and H19 overexpression significantly promoted the
proliferation and repressed the apoptosis of HUVECs and VSMCs in
vitro (15). Consistently, the
present study clarified that H19 was highly expressed in AS mice,
and H19 knockdown significantly inhibited the progression of AS by
inducing the apoptosis of VSMCs.
p53 serves an important role in the pathogenesis of
AS. For example, Guevara et al (16) reported that deficiency of p53 in
ApoE−/− mice accelerated the formation of aortic plaque
and increased cell proliferation in the atherosclerotic lesions.
van Vlijmen et al (18)
demonstrated that p53 expression was increased in human
atherosclerotic plaques, such as VSMCs and macrophages, and
depletion of it in macrophages can accelerate AS progression in
ApoE−/− mice. Wassmann et al (32) reported that upregulation of p53
induced by Krüppel-like factors (such as Krüppel-like Factor 4) can
significantly promote VSMCs apoptosis. These data demonstrated that
the downregulation of p53 may promote the progression of AS through
repressing VSMC apoptosis. The present study demonstrated that
knockdown of H19 could increase p53 expression and increase its
interaction with Bax, an apoptotic marker. In addition, it was
observed that the increase in cell apoptosis caused by H19
downregulation was rescued when p53 was downregulated in VSMCs,
which suggested that H19 may promote the apoptosis of VSMCs partly
in a p53-dependent manner. This result was congruent to a previous
study by Wu et al (21),
which demonstrated that upregulation of p53 induces an increase in
VSMC apoptosis.
Simvastatin, a cholesterol-lowering drug, is widely
used as an atheroprotective drug mainly due to its inhibitory role
in cholesterol syntheses (33,34).
Increasing evidence has demonstrated that simvastatin stimulation
can inhibit the proliferation of VSMCs (35,36).
Notably, Chan et al (36)
found that p53 upregulation is strongly implicated in
simvastatin-mediated VSMC growth repression. Based on this, the
present study used simvastatin as a drug for AS treatment in
vivo and in vitro to further reveal the roles of H19 in
AS progression. The results demonstrated that knockdown of H19
enhanced the role of simvastatin in promoting VSMC apoptosis and
increasing c-caspase3 expression in atherosclerotic plaque tissues.
In conclusion, the present study identified that the knockdown of
lncRNA H19 may inhibit AS progression by inducing VSMC apoptosis in
a p53-dependent manner.
Acknowledgements
Not applicable.
Funding
No funding was received.
Availability of data and materials
All data generated or analyzed during this study are
included in this published article.
Authors' contributions
KC conceived the study and revised the manuscript.
HS and QJ performed all the experiments, performed data analysis
and were major contributors to the writing of the manuscript. LS
performed some of the experiments. All authors read and approved
the final manuscript.
Ethics approval and consent to
participate
Animal experiment was approved by The Committee of
Jining First People's Hospital (Jining, China) and were performed
in accordance with the National Institutes of Health Guide for the
Care and Use of Laboratory Animals.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Schaftenaar F, Frodermann V, Kuiper J and
Lutgens E: Atherosclerosis: The interplay between lipids and immune
cells. Curr Opin Lipdol. 27:3095–215. 2016.
|
|
2
|
Weber C and Noels H: Atherosclerosis:
Current pathogenesis and therapeutic options. Nat Med.
17:1410–1422. 2011. View
Article : Google Scholar : PubMed/NCBI
|
|
3
|
Moore KJ, Sheedy FJ and Fisher EA:
Macrophages in atherosclerosis: A dynamic balance. Nat Rev Immunol.
13:709–721. 2013. View
Article : Google Scholar : PubMed/NCBI
|
|
4
|
Galkina E and Ley K: Immune and
inflammatory mechanisms of atherosclerosis. Ann Rev Immunol.
27:165–197. 2009. View Article : Google Scholar
|
|
5
|
Leischik R, Foshag P, Straub M, Garg P,
Dworrak B, Littwitz H, Lazic JS and Horlitz M: Physical activity,
cardiorespiratory fitness and carotid intima thickness: Sedentary
occupation as risk factor for atherosclerosis and obesity. Eur Rev
Med Pharmacol Sci. 19:3157–3168. 2015.PubMed/NCBI
|
|
6
|
Bennett MR, Sinha S and Owens GK: Vascular
smooth muscle cells in atherosclerosis. Circ Res. 118:692–702.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Kapranov P, Willingham AT and Gingeras TR:
Genome-wide transcription and the implications for genomic
organization. Nat Rev Genet. 8:413–423. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Yang JX, Rastetter RH and Wilhelm D:
Non-coding RNAs: An introduction. Adv Exp Med Biol. 886:13–32.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Rinn JL and Chang HY: Genome regulation by
long noncoding RNAs. Ann Rev Biochem. 81:145–166. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Huarte M: The emerging role of lncRNAs in
cancer. Nat Med. 21:1253–1261. 2015. View
Article : Google Scholar : PubMed/NCBI
|
|
11
|
Lorenzen JM and Thum T: Long noncoding
RNAs in kidney and cardiovascular diseases. Nat Rev Nephrol.
12:360–373. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Zhou T, Ding JW, Wang XA and Zheng XX:
Long noncoding RNAs and atherosclerosis. Atherosclerosis.
248:51–61. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Jian L, Jian D, Chen Q and Zhang L: Long
noncoding RNAs in atherosclerosis. J Atheroscler Thromb.
23:376–384. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Kallen AN, Zhou XB, Xu J, Qiao C, Ma J,
Yan L, Lu L, Liu C, Yi JS, Zhang H, et al: The imprinted H19 lncRNA
antagonizes let-7 microRNAs. Mol Cell. 52:101–112. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Pan JX: LncRNA H19 promotes
atherosclerosis by regulating MAPK and NF-kB signaling pathway. Eur
Rev Med Pharmacol Sci. 21:322–328. 2017.PubMed/NCBI
|
|
16
|
Guevara NV, Kim HS, Antonova EI and Chan
L: The absence of p53 accelerates atherosclerosis by increasing
cell proliferation in vivo. Nat Med. 5:335–339. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Merched AJ, Williams E and Chan L:
Macrophage-specific p53 expression plays a crucial role in
atherosclerosis development and plaque remodeling. Arterioscler
Thromb Vasc Biol. 23:1608–1614. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
van Vlijmen BJ, Gerritsen G, Franken AL,
Boesten LS, Kockx MM, Gijbels MJ, Vierboom MP, van Eck M, van De
Water B, van Berkel TJ and Havekes LM: Macrophage p53 deficiency
leads to enhanced atherosclerosis in APOE*3-Leiden transgenic mice.
Circ Res. 88:780–786. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Mercer J and Bennett M: The role of p53 in
atherosclerosis. Cell Cycle. 5:1907–1909. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Mercer J, Figg N, Stoneman V, Braganza D
and Bennett MR: Endogenous p53 protects vascular smooth muscle
cells from apoptosis and reduces atherosclerosis in ApoE knockout
mice. Circ Res. 96:667–674. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Wu G, Cai J, Han Y, Chen J, Huang ZP, Chen
C, Cai Y, Huang H, Yang Y, Liu Y, et al: LincRNA-p21 regulates
neointima formation, vascular smooth muscle cell proliferation,
apoptosis, and atherosclerosis by enhancing p53 activity.
Circulation. 130:1452–1465. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Ma L, Qian L, Ying Q, Zhang Y, Zhou C and
Wu G: I4, a synthetic anti-diabetes agent, attenuates
atherosclerosis through its lipid-lowering, anti-inflammatory and
anti-apoptosis properties. Mol Cell Endocrinol. 440:80–92. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Yin M, Liu Q, Yu L, Yang Y, Lu M, Wang H,
Luo D, Rong X, Tang F and Guo J: Downregulations of CD36 and
calpain-1, inflammation, and atherosclerosis by simvastatin in
apolipoprotein E knockout mice. J Vasc Res. 54:123–130. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Xin B, He X, Wang J, Cai J, Wei W, Zhang T
and Shen X: Nerve growth factor regulates CD133 function to promote
tumor cell migration and invasion via activating ERK1/2 signaling
in pancreatic cancer. Pancreatology. 16:1005–1014. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Gorenne I, Kumar S, Gray K, Figg N, Yu H,
Mercer J and Bennett M: Vascular smooth muscle cell sirtuin 1
protects against DNA damage and inhibits atherosclerosis.
Circulation. 127:386–396. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Rudijanto A: The role of vascular smooth
muscle cells on the pathogenesis of atherosclerosis. Acta Med
Indones. 39:86–93. 2007.PubMed/NCBI
|
|
28
|
Clarke MC, Figg N, Maguire JJ, Davenport
AP, Goddard M, Littlewood TD and Bennett MR: Apoptosis of vascular
smooth muscle cells induces features of plaque vulnerability in
atherosclerosis. Nat Med. 12:1075–1080. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Clarke MC, Littlewood TD, Figg N, Maguire
JJ, Davenport AP, Goddard M and Bennett MR: Chronic apoptosis of
vascular smooth muscle cells accelerates atherosclerosis and
promotes calcification and medial degeneration. Circ Res.
102:1529–1538. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Chen C, Cheng G, Yang X, Li C, Shi R and
Zhao N: Tanshinol suppresses endothelial cells apoptosis in mice
with atherosclerosis via lncRNA TUG1 up-regulating the expression
of miR-26a. Am J Transl Res. 8:2981–2991. 2016.PubMed/NCBI
|
|
31
|
Yao X, Yan C, Zhang L, Li Y and Wan Q:
LncRNA ENST00113 promotes proliferation, survival, and migration by
activating PI3K/Akt/mTOR signaling pathway in atherosclerosis.
Medicine (Baltimore). 97:e04732018. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Wassmann S, Wassmann K, Jung A, Velten M,
Knuefermann P, Petoumenos V, Becher U, Werner C, Mueller C and
Nickenig G: Induction of p53 by GKLF is essential for inhibition of
proliferation of vascular smooth muscle cells. J Mol Cell Cardiol.
43:301–307. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Song G, Liu J, Zhao Z, Yu Y, Tian H, Yao
S, Li G and Qin S: Simvastatin reduces atherogenesis and promotes
the expression of hepatic genes associated with reverse cholesterol
transport in apoE-knockout mice fed high-fat diet. Lipids Health
Dis. 10:82011. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Sparrow CP, Burton CA, Hernandez M, Mundt
S, Hassing H, Patel S, Rosa R, Hermanowski-Vosatka A, Wang PR,
Zhang D, et al: Simvastatin has anti-inflammatory and
antiatherosclerotic activities independent of plasma cholesterol
lowering. Arterioscler Thromb Vasc Biol. 21:115–121. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Zhang Z, Zhang M, Li Y, Liu S, Ping S,
Wang J, Ning F, Xie F and Li C: Simvastatin inhibits the additive
activation of ERK1/2 and proliferation of rat vascular smooth
muscle cells induced by combined mechanical stress and oxLDL
through LOX-1 pathway. Cell Signal. 25:332–340. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Chan KC, Wang CJ, Ho HH, Chen HM and Huang
CN: Simvastatin inhibits cell cycle progression in
glucose-stimulated proliferation of aortic vascular smooth muscle
cells by up-regulating cyclin dependent kinase inhibitors and p53.
Pharmacol Res. 58:247–256. 2008. View Article : Google Scholar : PubMed/NCBI
|