Introduction
Alström syndrome (AS; OMIM 203800) was first
reported in 1959 (1). AS is a
multisystem disease with cone-rod retinal dystrophy, which can lead
to visual impairment, sensorineural hearing loss, obesity and
hyperinsulinemia (2). Other
disease phenotypes that may seriously affect prognosis and survival
include endocrine disorders, dilated cardiomyopathy and restrictive
pulmonary disease (1–3).
AS is caused by mutations in the Alström syndrome
protein 1 (ALMS1) gene that is located on chromosome 2p13
(4). The total length of
ALMS1 is 224 kb, comprising 23 exons, which were identified
in 2002 (5). The ALMS1
consists of 4,169 amino acids (NM_015120.2). ALMS1 has been
reported to be widely expressed and localized to the centrosomes
and basal bodies of ciliated cells of tissues including the central
nervous, photoreceptor, cardiopulmonary, reproductive, endocrine
and urological system (6–8). Although the exact biological function
of ALMS1 remains obscure, current evidence suggests that the
functions include maintaining ciliary function, intracellular
transport and adipocyte differentiation (6–8).
Early diagnosis of AS is usually based on the phenotype and by
direct sequencing analysis of the ALMS1 gene (8), but is complicated by the progressive
and non-simultaneous onset of the principal symptoms (2,6,8).
In the present study, two compound heterozygous
mutations were identified in the proband, which presented symptoms
of low vision, hearing loss and childhood obesity. Based on these
results, prenatal diagnosis for the fetus was provided for this
family and at the same time, relevant literature to explore the
main clinical characteristics, diagnosis and treatments for AS was
reviewed.
Patients and methods
Patient
A Chinese woman who was 4 months pregnant, visited
the Prenatal Diagnosis Center of West China Second University
Hospital of Sichuan University (Chengdu, China) for genetic
consultation in May 2018. The patient reported that her first child
was a 10-year-old male, who was diagnosed with retinitis pigmentosa
at another hospital. The proband was obese since he was 6 months
old, his height was 140 cm and his weight was 65 kg (body mass
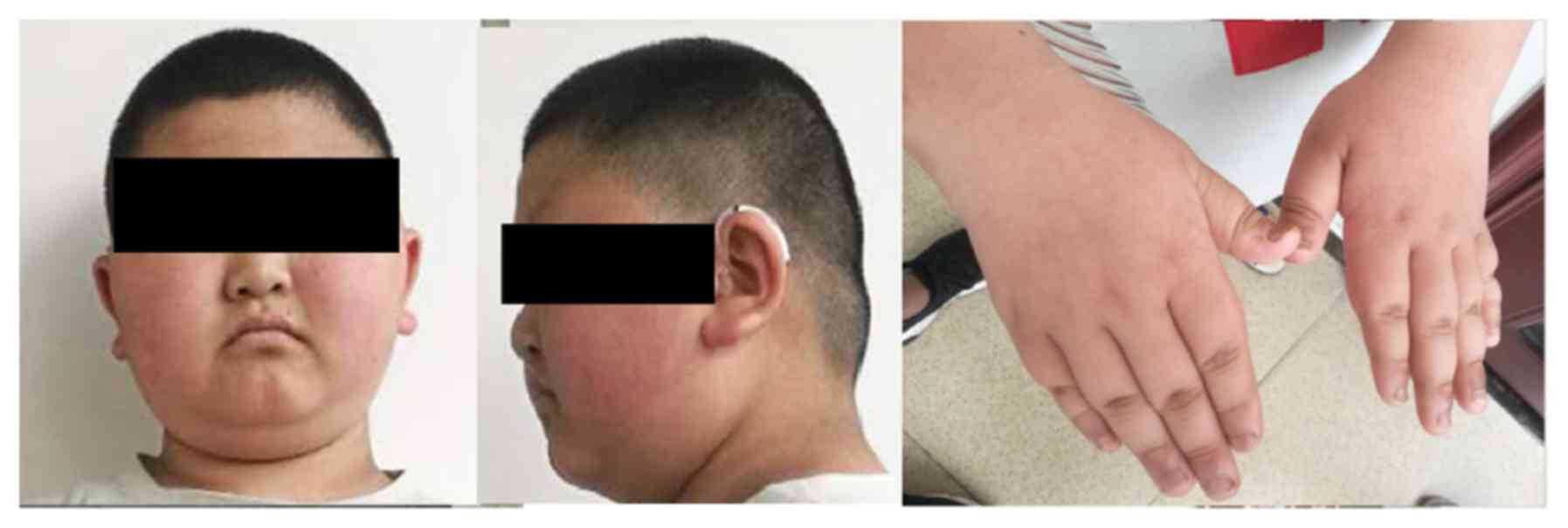
index was 33 kg/m2). He had unique facial features,
including narrow blepharoplasty, round face and sparse hair
(Fig. 1). Additionally, he had
wide and thick feet with short, thick fingers and toes. He
displayed photophobia and poor vision in the first year of life,
which eventually led to partial blindness at the age of 7–8 years.
Bilateral hearing loss occurred at the age of 7 years. According to
the aforementioned clinical phenotype, the doctor's clinical
diagnosis was suspected AS.
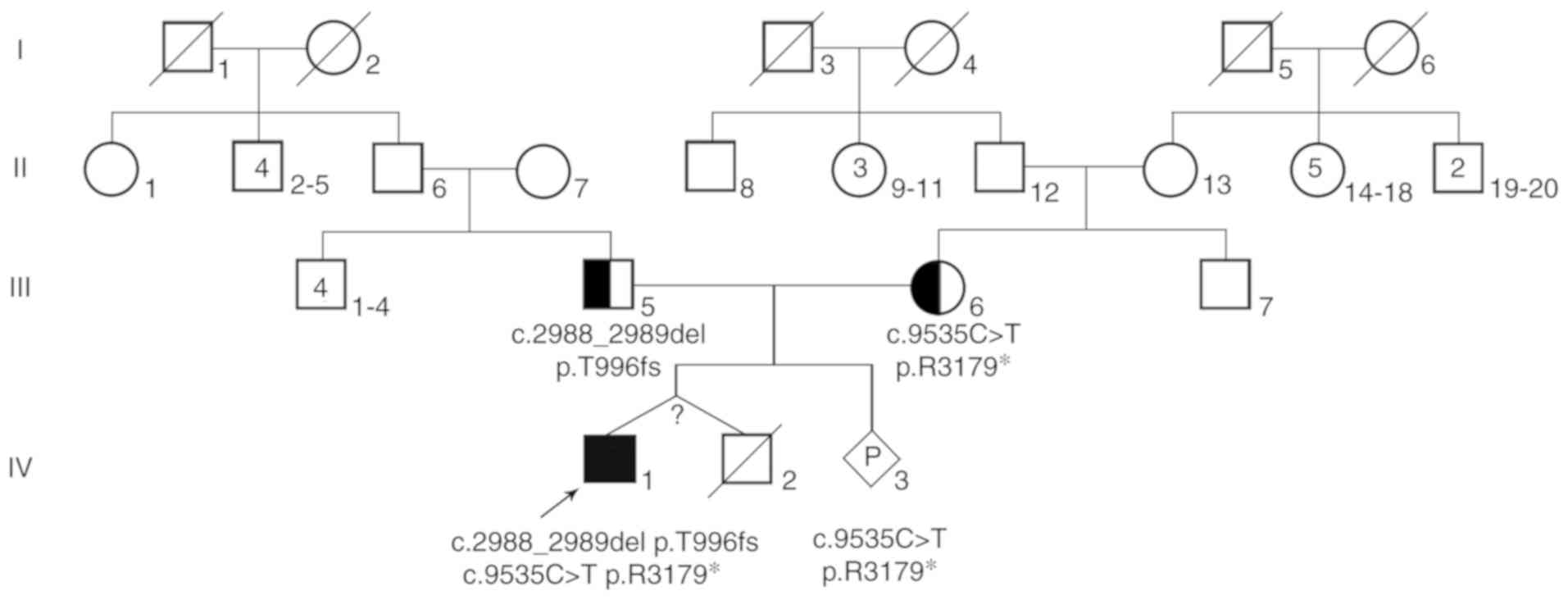
In addition, the proband had a twin brother. The
twins were born prematurely by caesarean section, so they were born
at the same time. However, the mother could not provide
characteristics at birth, including their birth length, weight and
head circumference. Unfortunately, the proband's twin brother died
of fever and diarrhea 50 days after birth, leaving no specimen for
further laboratory testing. The parents of the proband were
healthy, unrelated adults and the mother denied any existence of
family history of congenital malformation and genetic diseases, and
also denied the exposure to teratogenic environment during
pregnancy. The pedigree of the patient's family is described in
Fig. 2. The current study was
approved by the Medical Ethics Committee of West China Second
University Hospital of Sichuan University (Chengdu, China) and
written informed consent was obtained from the parents of the
patient.
Genetic testing
The peripheral blood samples of the patient and his
parents were collected and the genomic (g)DNA was extracted using a
QIAamp DNA Blood Mini kit (Qiagen GmbH) according to the
manufacturer's protocol. The quality and concentration of gDNA were
assessed with the NanoDrop 1000 (Thermo Fisher Scientific, Inc.)
and 1% agarose gel electrophoresis. For the proband, a
disease-targeted gene panel (selected genes sequenced by a
next-generation sequencing method) was employed using the MGP011
Targeted Exome Capture kit (MyGenostics, Inc.) according to the
manufacturer's instructions; the kit targets 171 genes known to
cause metabolic diseases (including ALMS1 gene). AmpliSeq™
Library PLUS (96 Reactions) for Illumina® reagent (cat.
no. 20019102; Illumina, Inc.) was used for enrichment libraries.
The purified and enriched DNA libraries were sequenced using the
Illumina Nextseq 500/550 medium flux V2 kit (300 cycles; cat. no.
FC-404-2004; Illumina, Inc.) and NextSeq 500 platform (Illumina,
Inc.) to generate paired-end reads for 150 cycles per read. The
loading concentration of DNA libraries was 1.8 pM, which was
measured using qPCR analysis (7500 Fast Dx Real-Time PCR
Instrument, Thermo Fisher Scientific, Inc.) according to the
manufacturer's protocol. DNA libraries quantification was performed
by amplifying the set of six pre-diluted DNA Standards and diluted
library samples by qPCR, using the KAPA SYBR FAST qPCR MasterMix
(cat. no. KK4824; Kapa Biosystems; Roche Diagnostics) and primers
targeting the Illumina P5 and P7 flow cell oligo sequences. Library
Quantification Primer Premix (10X), contained the following
primers: Primer 1, 5′-AATGATACGGCGACCACCGA-3′ and Primer 2,
5′-CAAGCAGAAGACGGCATACGA-3′. The PCR protocol included initial
denaturation at 95°C for 5 min and then 35 cycles of 95°C for 30
sec followed by 60°C for 30 sec. The average Cq score for each DNA
Standard was plotted against log10 (concentration in pM)
to generate a standard curve. The concentrations of diluted library
samples were then calculated against the standard curve using the
2−ΔΔCq method (9). The
sequencing data were analyzed by BWA (version 0.7.10; http://www.plob.org/tag/bwa), ANNOVAR (version 1;
http://annovar.openbioinformatics.org/en/latest/)
and GATK (version 4.0.8.1; http://www.broadinstitute.org/gatk/) software as well
as the public databases 1000G (http://browser.1000genomes.org), ExAC (http://exac.broadinstitute.org/), gnomAD
(http://gnomad.broadinstitute.org/),
Esp6500 (http://evs.gs.washington.edu/EVS) and human genome
mutation database (HGMD; https://portal.biobase-international.com/cgi-bin/portal/login.cgi.
Additionally, the American College of Medical Genetics and Genomics
(ACMG) guidelines (10) were used
for the interpretation of sequence variants. Furthermore, Sanger
sequencing was used to verify the novel compound heterozygous
mutations with two primer pairs to amplify c.2988_2989 and c.9535
of the ALMS1 gene (reference genome version: GRCh37/hg19,
reference transcript: NM_015120.2). The following primers were
used: c.2988_2989 forward, 5′-ACCCTGCCAGACTTTCTTTT-3′ and reverse,
5′-GCATAACTATCTGGCCACTCC-3′; c.9535 forward,
5′-TCCAAGTCGTACAGCCTTCT-3′ and reverse, 5′- TCAGAG
AAATCACACGGCCT-3′. Finally, Chromas software (version 2.4.1;
Biosoft) was used to analyze the Sanger sequencing data.
Amniocentesis and prenatal
diagnosis
Amniocentesis was performed at 21±2 weeks of
gestation. A total of 5 ml amniotic fluid sample was taken for
prenatal diagnosis of ALMS1. Fetal exfoliated cells in the
amniotic fluid were collected by centrifugation (400 × g; 10 min,
room temperature) and washed with phosphate buffer saline. gDNA was
extracted with the DNeasy Blood and Tissue kit (Qiagen GmbH)
according to the manufacturer's protocol. Short tandem repeat
analysis (11) of the amniotic
fluid cells and parental peripheral blood was used to confirm the
relationship between the samples and also to confirm that there was
no maternal cell pollution in the amniotic fluid. Sanger sequencing
(12) was used to analyze the
nucleotide sequences of the ALMS1 gene c.9535 and
c.2988_2989.
Results
The average sequencing depth was 1247.02X on the
targeted regions, and 1,546 variants in the disease-targeted gene
panel of the proband were identified. The summary of the
disease-targeted next generation sequencing (NGS) were listed in
Table I. A total of two
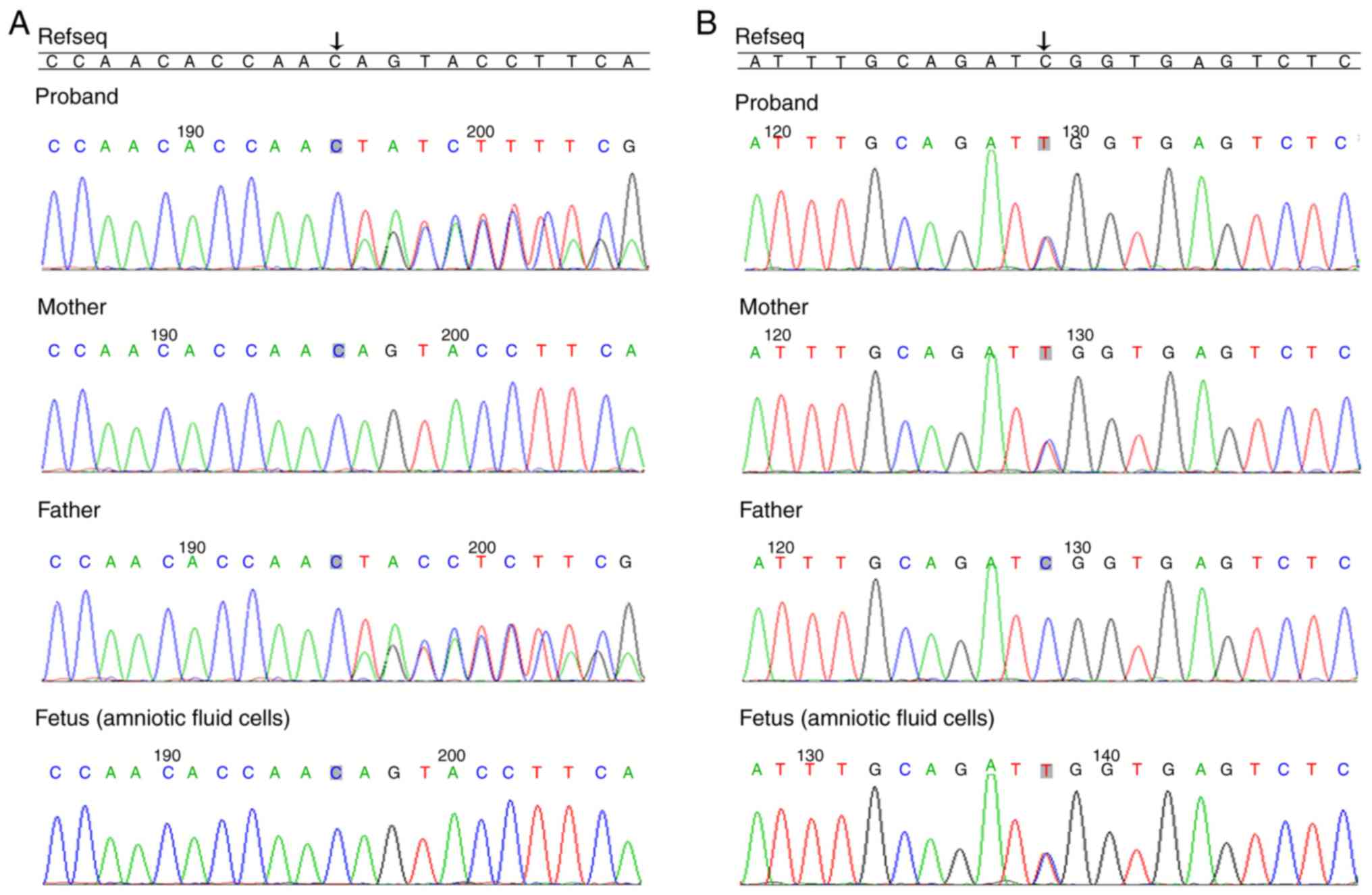
pathological variants in ALMS1 were observed:
c.2988_2989delAG (chr2-73676644 73676646) in exon 8 and
c.9535C>T (chr2-73718624) in exon 10. These variants were
further verified by Sanger sequencing and the results demonstrated
that c.2988_2989delAG (p.T996fs) was paternally inherited and
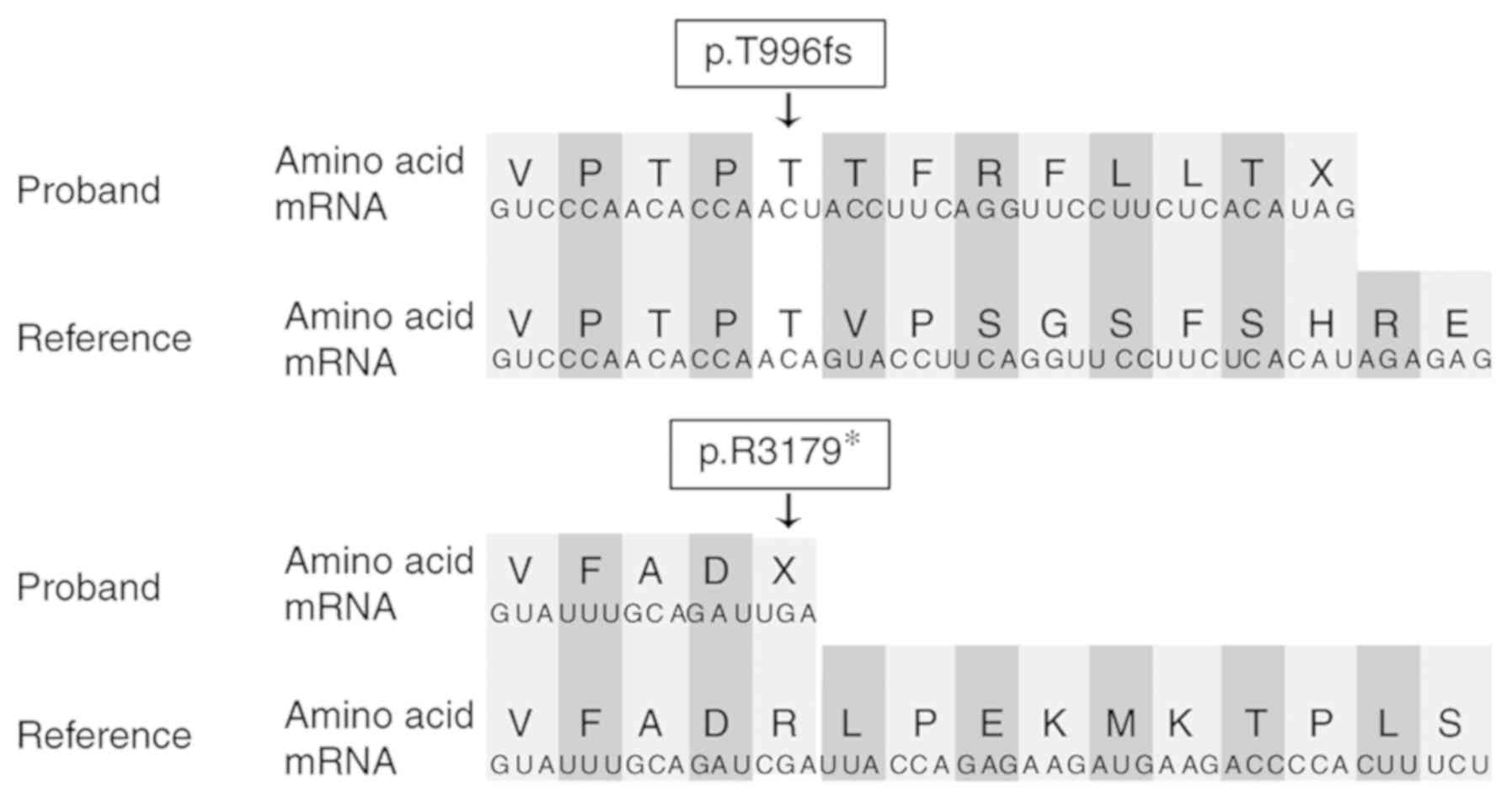
c.9535C>T (p.R3179*) was maternally inherited (Fig. 3). These mutations lead to premature
termination codons (Fig. 4).
 | Table I.Summary of targeted next-generation
sequencing on the patient. |
Table I.
Summary of targeted next-generation
sequencing on the patient.
| Statistics data
indicators | Statistical
results |
|---|
| Raw databases,
Mb | 1,463.69155 |
| Clean databases,
Mb | 1,411.56 |
| Aligned bases,
Mb | 1,392.32 |
| Aligned, % | 98.64 |
| Initial bases on
target, n | 564,206 |
| Base covered on
target, n | 564,150 |
| Coverage of target
region, % | 99.99 |
| Effective bases on
target, n | 703,575,596 |
| Fraction of effective
bases on target, % | 50.53 |
| Average sequencing
depth on target, n | 1,247.02 |
| Fraction of target
covered with at least 20X, % | 99.66 |
The present study also observed that there was a
heterozygous variation at c.9535C>T of the ALMS1 gene in
amniotic fluid cells, and there was no mutation at c.2988_2989
(Fig. 3). In addition, the fetus
was followed-up for >2 years. The mother of the proband
delivered a 3.5 kg male baby at 40±3 weeks of gestation. The baby
was examined by the children's health care department on a monthly
basis, and had no abnormal phenotypes until 15 months of age. The
mother of the proband did not wish for blood samples to be taken
from the baby after birth to confirm the prenatal test results.
Discussion
The most common symptoms of AS are cone-rod
dystrophy, sensorineural hearing loss, short stature, obesity,
cardiomyopathy and metabolic disorders (13–16).
The clinical symptoms, which are age of onset and severity, vary
greatly among patients (2).
Currently, clinical diagnostic standard for AS are
based on the major and minor criteria. The criteria include a
pathogenic mutation in one ALMS1 allele, and/or family
history of AS and vision problems. The minor criteria include
obesity and/or insulin resistance and/or type 2 diabetes mellitus,
dilated cardiomyopathy with congestive heart failure and hearing
loss (2). However, diagnosis of AS
is difficult because some symptoms (such as nystagmus and
photophobia) start at birth whereas others (such as hypertension
and renal failure) occur as the child grows (2,17).
Genetic testing should be performed when the criteria fail to allow
for a clear clinical diagnosis (17). Due to the size and mutation
spectrum of ALMS1, the mutation analysis of ALMS1 by
conventional Sanger sequencing is usually complicated (2,6).
This difficulty can be resolved by the application of NGS.
Disease-targeted gene panel is often used for
specific suspected disease or a group of diseases (18). Disease-targeted gene panels often
have higher diagnostic rates compared with those of exome
sequencing or genome sequencing, being designed to maximize
coverage, sensitivity and specificity for the included genes
(19). The cost of
disease-targeted gene panels is variable but is often lower
compared with that of exome sequencing and genome sequencing
(19). In the present study, the
patient was diagnosed with a high clinical suspicion of AS, and two
compound heterozygous mutations in ALMS1 were detected using
a disease-targeted gene panel.
Mutations are highly heterogeneous within
ALMS1 (17). According to
the statistics of HGMD (https://portal.biobase-international.com/cgi-bin/portal/login.cgi),
a total of 306 disease-causing variants of ALMS1 have been
reported, 285 of which are related to the syndrome phenotype. The
majority of the variants are nonsense mutations (39.87%) and
frameshift mutations (48.37%; Table
II), and most of the mutations occur in exon 8, 10 and 16
(6,20). In this case, an unreported
mutation: c.2988_2989del (p.T996fs), which was a frameshift
mutation located in exon 8, was identified. The c.9535C>T
(p.R3179*), a nonsense mutation located in exon 10, was previously
reported as a disease mutation variant (6,21) in
the HGMD database. Flintoff and Boute-Benejean (21) reported the variant p.R3179* as a
novel pathological mutation without any details. Marshall et
al (6) evaluated a large
cohort of patients (including 250 affected individuals) with AS for
variants in the ALMS1 gene and reported 55 novel variants
which included p.R3179*. However, Marshall et al (6) only stated that p.R3179* was a novel
variant, but did not indicate whether the variant was a homozygous
or compound heterozygous, and whether the variants were from the
proband's parents. This is very important according to the
diagnostic criteria for AS (2). In
addition, the authors did not specifically describe the clinical
phenotype of the patient carrying p.R3179*. In the present study,
the specific phenotypes of the patient was described, the variant
(p.R3179*) was verified by NGS (the sequencing depth was 373X) and
Sanger sequencing was used to verify the authenticity of the
variant and whether the parents were also carriers. Combined with
the clinical phenotype and gene test results of the patient, the
patient was diagnosed with AS. The diagnosis of AS was very
important as the patient was not diagnosed by molecular and genetic
techniques until the age of 10 years. The diagnosis was crucial at
that time due to his mother's pregnancy and desire for a prenatal
diagnosis, which was made possible. The present study demonstrated
that p.R3179* is a pathogenic variant in patients with AS, and
prenatal diagnosis was performed for this variant, to the best of
our knowledge, for the first time. Sequence analysis performed in
the current study demonstrated that the mutations resulted in code
shifting and nonsense mutations, which lead to premature
termination codons. According to the criteria of the ACMG (10), the mutations were pathogenic, and
the evidence for this was as follows: i) Very strong evidence of
pathogenicity: Nonsense, frameshift; ii) moderate evidence of
pathogenicity: Absent from controls in 1000G; and iii) supporting
evidence of pathogenicity: Patient's phenotype is highly specific
for a disease with a single genetic etiology (10).
| Table II.Disease mutation of Alström syndrome
protein 1 gene. |
Table II.
Disease mutation of Alström syndrome
protein 1 gene.
| Mutation type | No. of mutation | Ratio, % |
|---|
| Missense | 18 | 5.88 |
| Nonsense | 122 |
39.87 |
| Splicing
substitutions | 7 | 2.29 |
| Small
deletions/insertions/duplications | 148 |
48.37 |
| Gross
deletions/insertions/duplications | 10 | 3.27 |
| Complex
rearrangements | 1 | 0.33 |
| Total | 306 | 100.0 |
According to the recommended clinical diagnostic
criteria for AS, patients aged 3–14 years should fulfill two major
criteria or one major and three minor criteria (2). The patient in the current study was a
10-year-old male and fulfilled two major criteria (compound
heterozygous mutations in ALMS1 and vision problems
including photophobia and visual impairment) in addition to some
minor criteria (obesity and hearing loss). Therefore, the diagnosis
of AS was determined in the proband. However, due to the rarity of
AS, delay in onset of some of the cardinal features, inter and
intra familial clinical heterogeneity, some minor criteria clinical
findings were not observed in the proband, such as renal failure
(2). Marshall et al
(6), demonstrated that a
significant association between alterations in exon 8 and absent,
mild or delayed renal disease may be due to the tissue-specific
expression of different splicing subtypes or the lack of another
subtype of exon 8 in the kidney, which may protect patients with
exon 8 mutations from serious renal complications. This was
consistent with the phenotype of the proband; the frameshift
variant in the current study was in exon 8, and the renal function
(creatinine and blood urea nitrogen) of the patient was normal,
without any renal disease. The case is still in the follow-up of
the genetic counseling clinic. However, although the renal function
was normal, renal ultrasound was not performed. Thus, the renal
function of the patient should be actively monitored in future.
Patients with AS rarely live >40 years (22). At present, there is no specific
treatment for AS, but early diagnosis and intervention can
alleviate the progress of the disease phenotypes and improve the
survival period and quality of life of patients (2). Early diagnosis can be treated by
multi-disciplinary combination therapy (2). For patients with retinopathy, heart
disease, obesity, deafness and diabetes, the possibility of AS
should be considered and gene detection should be performed as
early as possible for a clear diagnosis, which is conducive to the
early assessment of the disease in order to improve the prognosis
and long-term survival. For pregnant women with a positive AS
family history, genetic counseling and prenatal diagnosis should be
performed.
In the present study, a Chinese patient presenting
clinical features compatible with AS was described. Using a
disease-targeted gene panel, two compound heterozygous variants in
the exons 8 and 10 were identified. Both the mutations were
predicted to lead to premature termination codons, which may result
in truncated ALMS1 protein formation. In addition, the specific DNA
sequences of the two mutation sites were tested for in the fetus of
the proband's mother.
This study identified two compound heterozygous
ALMS1 mutations in a patient with the symptoms of AS and reported a
novel ALMS1 variant which expands the spectrum of ALMS1 variants in
AS.
Acknowledgements
Not applicable.
Funding
The current study was supported by the National Key
Research and Development Program of China (grant no.
2018YFC1002200) and the Program of Science and Technology
Department of Sichuan Province (grant no. 2020YFS0095).
Availability of data and materials
All data generated or analyzed during this study are
included in this published article.
Authors'contributions
CZ, SL and JW designed the study. CZ, YX and HX
performed the experiments. CZ and JW conducted data analysis. All
authors read and approved the final manuscript.
Ethics approval and consent to
participate
The present study was approved by the Medical Ethics
Committee of West China Second University Hospital of Sichuan
University. Written informed consent to participate was obtained
from the parents of the patient.
Patient consent for publication
Written informed consent for publication was
obtained from the parents of the patient.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Alström CH, Hallgren B, Nilsson LB and
Asander H: Retinal degeneration combined with obesity, diabetes
mellitus and neurogenous deafness: a specific syndrome (not
hitherto described) distinct from the Laurence-Moon-Bardet-Biedl
syndrome: a clinical, endocrinological and genetic examination
based on a large pedigree. Acta Psychiatr Neurol Scand Suppl.
129:3271–35. 1959.
|
|
2
|
Marshall JD, Beck S, Maffei P and Naggert
JK: Alström syndrome. Eur J Hum Genet. 15:1193–1202. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Marshall JD, Bronson RT, Collin GB,
Nordstrom AD, Maffei P, Paisey RB, Carey C, Macdermott S,
Russell-Eggitt I, Shea SE, et al: New Alström syndrome phenotypes
based on the evaluation of 182 cases. Arch Intern Med. 165:675–683.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Hearn T, Renforth GL, Spalluto C, Hanley
NA, Piper K, Brickwood S, White C, Connolly V, Taylor JF,
Russell-Eggitt I, et al: Mutation of ALMS1, a large gene with a
tandem repeat encoding 47 amino acids, causes Alström syndrome. Nat
Genet. 31:79–83. 2002. View
Article : Google Scholar : PubMed/NCBI
|
|
5
|
Collin GB, Marshall JD, Cardon LR and
Nishina PM: Homozygosity mapping at Alström syndrome to chromosome
2p. Hum Mol Genet. 6:213–219. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Marshall JD, Hinman EG, Collin GB, Beck S,
Cerqueira R, Maffei P, Milan G, Zhang W, Wilson DI, Hearn T, et al:
Spectrum of ALMS1 variants and evaluation of genotype-phenotype
correlations in Alström syndrome. Hum Mutat. 28:1114–1123. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Collin GB, Marshall JD, King BL, Milan G,
Maffei P, Jagger DJ and Naggert JK: The Alström syndrome protein,
ALMS1, interacts with α-actinin and components of the endosome
recycling pathway. PLoS One. 7:e379252012. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Hearn T: ALMS1 and Alström syndrome: A
recessive form of metabolic, neurosensory and cardiac deficits. J
Mol Med (Berl). 97:1–17. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(−Δ Δ C(T)) Method. Methods. 25:402–408. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Richards S, Aziz N, Bale S, Bick D, Das S,
Gastier-Foster J, Grody WW, Hegde M, Lyon E, Spector E, et al ACMG
laboratory quality assurance committee, : Standards and guidelines
for the interpretation of sequence variants: A joint consensus
recommendation of the American College of Medical Genetics and
Genomics and the Association for Molecular Pathology. Genet Med.
17:405–424. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Wang J, Chen L, Zhou C, Wang L, Xie H,
Xiao Y, Zhu H, Hu T, Zhang Z, Zhu Q, et al: Prospective chromosome
analysis of 3429 amniocentesis samples in China using copy number
variation sequencing. Am J Obstet Gynecol. 219:287.e1–287.e18.
2018. View Article : Google Scholar
|
|
12
|
Beck TF, Mullikin JC; NISC Comparative
Sequencing Program, ; Biesecker LG: Systematic evaluation of Sanger
validation of next-generation sequencing variants. Clin Chem.
62:647–654. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Casey J, McGettigan P, Brosnahan D, Curtis
E, Treacy E, Ennis S and Lynch SA: Atypical Alstrom syndrome with
novel ALMS1 mutations precluded by current diagnostic criteria. Eur
J Med Genet. 57:55–59. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Russell-Eggitt IM, Clayton PT, Coffey R,
Kriss A, Taylor DS and Taylor JF: Alström syndrome. Report of 22
cases and literature review. Ophthalmology. 105:1274–1280. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Waldman M, Han JC, Reyes-Capo DP, Bryant
J, Carson KA, Turkbey B, Choyke P, Naggert JK, Gahl WA, Marshall
JD, et al: Alström syndrome: Renal findings in correlation with
obesity, insulin resistance, dyslipidemia and cardiomyopathy in 38
patients prospectively evaluated at the NIH clinical center. Mol
Genet Metab. 125:181–191. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Collin GB, Marshall JD, Ikeda A, So WV,
Russell-Eggitt I, Maffei P, Beck S, Boerkoel CF, Sicolo N, Martin
M, et al: Mutations in ALMS1 cause obesity, type 2 diabetes and
neurosensory degeneration in Alström syndrome. Nat Genet. 31:74–78.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Marshall JD, Maffei P, Collin GB and
Naggert JK: Alström syndrome: Genetics and clinical overview. Curr
Genomics. 12:225–235. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Pulignani S, Vecoli C, Borghini A, Foffa
I, Ait-Alì L and Andreassi MG: Targeted next-generation sequencing
in patients with non-syndromic congenital heart disease. Pediatr
Cardiol. 39:682–689. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Adams DR and Eng CM: Next-generation
sequencing to diagnose suspected genetic disorders. N Engl J Med.
379:1353–1362. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Marshall JD, Muller J, Collin GB, Milan G,
Kingsmore SF, Dinwiddie D, Farrow EG, Miller NA, Favaretto F,
Maffei P, et al: Alström syndrome: Mutation spectrum of ALMS1. Hum
Mutat. 36:660–668. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Flintoff KJ and Boute-Benejean O: Novel
human pathological mutations. Gene symbol: ALMS1. Disease: Alstrom
syndrome. Hum Genet. 121:645–652. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Nerakh G and Ranganath P: Alström Syndrome
presenting as isolated dilated cardiomyopathy. Indian J Pediatr.
86:296–298. 2019. View Article : Google Scholar : PubMed/NCBI
|