Introduction
Sepsis is a life-threatening organ dysfunction
caused by a complex host response to an infection, and the hospital
mortality rate of septic shock was >40% in 2016, worldwide
(1). Cardiac dysfunction is a
series complication of sepsis and it is associated with the
prognosis of patients (2).
Lipopolysaccharide (LPS) is a principal component of the cell wall
in Gram-negative bacteria and it can trigger an inflammatory
cascade reaction, which results in the synthesis and release of
inflammatory mediators, such as tumor necrosis factor (TNF)-α,
interleukin (IL)-1β and IL-6 (3).
The overexpression of inflammatory factors could lead to
endothelial injury and the disruption of vascular homeostasis,
which could subsequently promote irreversible cardiac dysfunction
(4). Therefore, the effective
treatment of septic cardiac dysfunction may be beneficial for
improving the prognosis of patients.
Astaxanthin (ATX) is a major carotenoid in marine
organisms, which is ubiquitously present in the biological world,
particularly in the feathers of shrimp, crab, fish, algae, yeast
and birds (5). Previous studies
have demonstrated that ATX serves a variety of beneficial
physiological effects, including antioxidative, anti-inflammatory
and antiapoptotic functions (6,7). In
addition, it has been suggested that ATX may reduce the myocardial
infarct area and improve the myocardial mitochondrial membrane
potential and contractility index (7). Notably, it has also been reported
that ATX may also protect against LPS-induced cardiac dysfunction
by reducing the levels of inflammatory mediators and oxidative
stress (8). However, the specific
mechanisms by which ATX protects against LPS-induced cardiac
dysfunction remain poorly understood.
The PI3K/AKT signaling pathway is involved in cell
proliferation, differentiation and apoptosis (9), and it was revealed to serve an
important role in cardiac dysfunction caused by sepsis (10). In a previous study, the mitogen
activated protein kinase (MAPK) signaling pathway was also observed
to participate in the regulation of cell growth and
differentiation, in addition to modulating the cell's response to
cytokines and various stresses, such as TNF-α and LPS (11). Numerous studies have revealed that
MAPK served a major role in promoting the production of
inflammatory cytokines during sepsis, such as TNF-α, IL-6 (12,13).
To investigate whether ATX exhibited protective effects during
cardiac dysfunction the present study established an in vivo
LPS-induced sepsis model using C57BL/6 mice to identify the
possible underlying mechanisms of action of ATX. We hypothesized
that ATX may be used for the treatment of LPS-induced cardiac
dysfunction through the regulation of the PI3K/AKT and the MAPK
signaling pathways. Understanding the mechanisms underlying the
action of ATX may assist in the development of a novel treatment
for patients with septic cardiomyopathy to improve the prognosis of
patients.
Materials and methods
Animal studies
A total of 45 specific pathogen-free male C57BL/6
mice (age, 8–10 weeks; weight, 24–26 g) were obtained from The
Institute of Laboratory of Animal Sciences, Chinese Academy of
Medical Sciences and Peking Union Medical College. Mice were
randomly divided into three groups (n=15 per group): Control, LPS
and LPS + ATX. Animals were housed at a controlled temperature of
25°C and a humidity of 45–50%, with a 12-h light/dark cycle and
free access to standard laboratory water and chow; the animals were
allowed to adapt to the laboratory conditions for 7 days prior to
use for subsequent experiments. The animal experiments were
approved by the Ethics Committee of Renmin Hospital of Wuhan
University (Wuhan, China).
Induction of sepsis-associated cardiac
dysfunction models
Cardiac dysfunction was induced by an
intraperitoneal injection of LPS (10 mg/kg; cat. no. L2880;
Sigma-Aldrich; Merck KGaA) for 24 h, as previously described
(14). ATX (40 mg/kg; cat. no.
SML0982; Sigma-Aldrich; Merck KGaA) dissolved in polyethylene
glycol 400-N, N-dimethylacetamide (PEG400; 1:1 v/v; cat. no. 91863;
Sigma-Aldrich; Merck KGaA) was administered to the mice in the LPS
+ ATX group 30 min after LPS administration for 24 h. The mice in
the control and LPS groups were administered an equivalent volume
of the vehicle (PEG400; 1:1 v/v; cat. no. 91863; Sigma-Aldrich;
Merck KGaA). The health, behavior and death of mice were monitored
every 6 h following LPS administration. In the LPS and LPS + ATX
groups, 8 and 3 mice died, respectively, of severe infection
following LPS treatment. All the surviving animals were
anesthetized with pentobarbital solution (50 mg/kg) via an
intraperitoneal injection 24 h after LPS treatment. Following
anesthesia, 0.5 ml blood was collected from the mice by eyeball
enucleation; which prompted two mice to die from severe anemia. To
isolate myocardial tissue, mice were sacrificed by cervical
dislocation. Following a 2 min observation, no respiration and
corneal reflex in the mice confirmed death. The heart tissue was
isolated post-mortem and stored in 4% paraformaldehyde or at
−80°C.
Mortality observation
The point at which LPS was administered was
considered as the 0 h time point. The number of surviving mice was
observed at 6, 12, 18 and 24 h after LPS injection and the
mortality of each group was calculated using the following method:
Mortality=dead mice/total number of mice ×100%.
Analysis of blood samples using
ELISA
Blood samples from the mice were collected from the
posterior orbital plexus venous and were centrifuged at 4,200 × g
for 10 min at room temperature. The supernatants were subsequently
collected and stored at −20°C. ELISA kits (cat. nos. EL-M0049c and
E-EL-M0044c; Elabscience Biotechnology Co., Ltd.) were used to
analyze the levels of TNF-α and IL-6 in the blood samples using an
enzyme-labeled instrument (Multiskan MK3; Thermo Fisher Scientific,
Inc.), according to the manufacturer's protocols.
Histological examination
Left ventricular and whole heart tissues were
collected and fixed for 48 h in 4% paraformaldehyde at 4°C,
embedded in paraffin and sliced into 4-µm thick sections. The
tissue sections were subsequently stained with hematoxylin &
eosin (H&E) for 70 min at room temperature. Stained sections
were visualized using a light microscope, at X100 and X200
magnification.
Reverse transcription-quantitative
(RT-q)PCR
mRNA expression levels of B-type natriuretic peptide
(BNP) were analyzed using RT-qPCR. The fresh frozen tissue stored
at −80°C was weighed to 100 mg, following which 1 ml
TRIzol® reagent (cat. no. 15596026; Invitrogen; Thermo
Fisher Scientific, Inc.) was added, and the tissue ground into a
slurry using a homogenizer, according to the manufacturer's
protocol. Total RNA was reverse transcribed into cDNA using a
HiScript RT reagent kit (cat. no. R101-01/02; Vazyme Biotech Co.,
Ltd.), using the following conditions: 25°C for 5 min, 50°C for 15
min, 85°C for 5 min then 4°C for 10 min. qPCR was subsequently
performed using a SYBRGreen master mix (cat. no. Q111-02; Vazyme
Biotech Co., Ltd.), according to the manufacturer's protocol, on a
QuantStudio 6 Flex Real-Time PCR system (Applied Biosystems; Thermo
Fisher Scientific, Inc.), using the following conditions 50°C for 2
min, 95°C for 10 min, then 40 cycles of 95°C for 30 sec and 60°C
for 30 sec. The data was analyzed with the 2−ΔΔCq method
(15). The primer sequences and
the amplicon sizes of the target genes are presented in Table I. β-actin was used as the
endogenous loading control.
 | Table I.Primer sequences and the amplicon
sizes of the target genes used for the reverse
transcription-quantitative PCR. |
Table I.
Primer sequences and the amplicon
sizes of the target genes used for the reverse
transcription-quantitative PCR.
| Gene | Primer sequence
(5′→3′) | Size (bp) |
|---|
| β-actin | F:
CACGATGGAGGGGCCGGACTCATC | 240 |
|
| R:
TAAAGACCTCTATGCCAACACAGT |
|
| B-type natriuretic
peptide | F:
GAGGTCACTCCTATCCTCTGG | 202 |
|
| R:
GCCATTTCCTCCGACTTTTCTC |
|
Western blot analysis
To assess the expression levels and activation state
of proteins in the MAPK and PI3K/AKT signaling pathways, total
protein was extracted from the cardiac tissues. Cardiac tissues
were collected and homogenized with a homogenizer using
radioimmunoprecipitation assay lysis buffer (cat. no. P0013B),
containing phenylmethanesulfonyl fluoride (cat. no. ST506) and
phosphoesterase inhibitors (cat. no. S1873) (all Beyotime Institute
of Biotechnology). Total protein was quantified using a
bicinchoninic acid kit (cat. no. P0010; Beyotime Institute of
Biotechnology) and 40 µg protein/lane was separated using 10%
SDS-PAGE. The separated proteins were subsequently transferred onto
a PVDF membrane (cat. no. IPVH00010; EMD Millipore) and blocked
using 5% skimmed milk in Tris buffered saline containing 0.1%
Tween-20 (TBST) for 2 h at room temperature. The membranes were
incubated overnight at 4°C in TBST containing 1% skimmed milk with
the following primary antibodies: Anti-p38 (1:1,000; cat. no. 8690;
Cell Signaling Technology, Inc.), anti-phosphorylated (p)-p38
(1:1,000; cat. no. 9211; Cell Signaling Technology, Inc.),
anti-ERK1/2 (1:1,000; cat. no. ab17942; Abcam), anti-p-ERK1/2
(1:2,000; cat. no. 4370S; Cell Signaling Technology, Inc.),
anti-JNK (1:1,000; cat. no. 9252S; Cell Signaling Technology,
Inc.), anti-p-JNK (1:1,000; cat. no. 9251S; Cell Signaling
Technology, Inc.), anti-PI3K (1:1,000; cat. no. 4257S; Cell
Signaling Technology, Inc.), anti-p-PI3K (1:1,000; cat. no.
ab182651; Abcam), anti-AKT (1:1,000; cat. no. 4691S; Cell Signaling
Technology, Inc.), anti-p-AKT (1:1,000; cat. no. 9271T; Cell
Signaling Technology, Inc.), anti-mTOR (1:500; cat. no. ab87540;
Abcam), anti-p-mTOR (1:1,000; cat. no. 5536T; Cell Signaling
Technology, Inc.), anti-glycogen synthase kinase-3 (1:500; GSK3) β
(cat. no. ab93926; Abcam), anti-p-GSK3β (1:500; cat. no. ab75745;
Abcam), anti-Bax (1:1,000; cat. no. ab32503; Abcam), anti-Bcl-2
(1:800; cat. no. 26593-1-AP; ProteinTech Group, Inc.) and
anti-GAPDH (1:1,000; cat. no. AB-P-R 001; Hangzhou Goodhere Biotech
Co., Ltd.). Following the incubation with the primary antibodies,
the membranes were incubated with the following secondary
antibodies for 2 h at 37°C: Horseradish peroxidase (HRP) conjugated
AffiniPure goat anti-mouse IgG (dilution 1:50,000; cat. no. BA1051)
and HRP conjugated AffiniPure goat anti-rabbit IgG (dilution
1:50,000; cat. no. BA1054) (both Boster Biological Technology Co.,
Ltd). The protein bands were visualized using an ECL kit (Pierce;
Thermo Fisher Scientific, Inc.) and densitometry analysis was
performed using Bandscan v5.0 software (Glyko Biomedical Ltd.). The
protein expression levels were normalized to GAPDH and
phosphorylated proteins were normalized to the respective total
protein.
TUNEL staining
A TUNEL assay was used to assess cardiomyocyte
apoptosis. Briefly, heart tissues were collected, fixed and
embedded as aforementioned, then the paraffin-embedded tissues were
cut into 4-µm thick sections placed flat in 42°C water bath and
incubated at 60°C. The tissue sections were deparaffinized, rinsed
with PBS and incubated with proteinase K (20 µg/ml; cat. no.
40308ES20; Yeasen Biotech Co., Ltd) for 20 min at room temperature.
A total of 50 µl TUNEL reaction mixture was subsequently added to
each sample and the slices were placed in a wet box for 60 min at
37°C in the dark. Following the incubation, the slides were rinsed
with PBS (pH 7.4) three times for 5 min each and the cell nuclei
were stained with DAPI (10 µg/ml) for 5 min at room temperature in
the dark, and then washed with PBS four times for 5 min each time.
Antifade mounting medium (cat. no. 0100-01; Southern Biotechnology
Associates, Inc.) was used for sealing. TUNEL-positive cells were
observed using a fluorescence microscope with 22 fields of view and
X200 magnification; the apoptotic cells in the tissue sections
fluoresced red and the nuclei fluoresced blue. The number of
apoptotic and total cells were counted and the percentage of
apoptotic cells were calculated using the following equation:
Apoptosis index=apoptotic cells/total cells ×100%.
Statistical analysis
Data are presented as the mean ± SD. Statistical
differences among multiple groups were determined using an one-way
ANOVA, followed by Tukey's post-hoc test. Kaplan-Meier survival
curves were used to plot the mortality rates of the three groups
and the differences in the survival between the groups were
compared using a log-rank test. The experiments were repeated three
times. All data were analyzed using SPSS version 17.0 (SPSS, Inc.).
P<0.05 was considered to indicate a statistically significant
difference.
Results
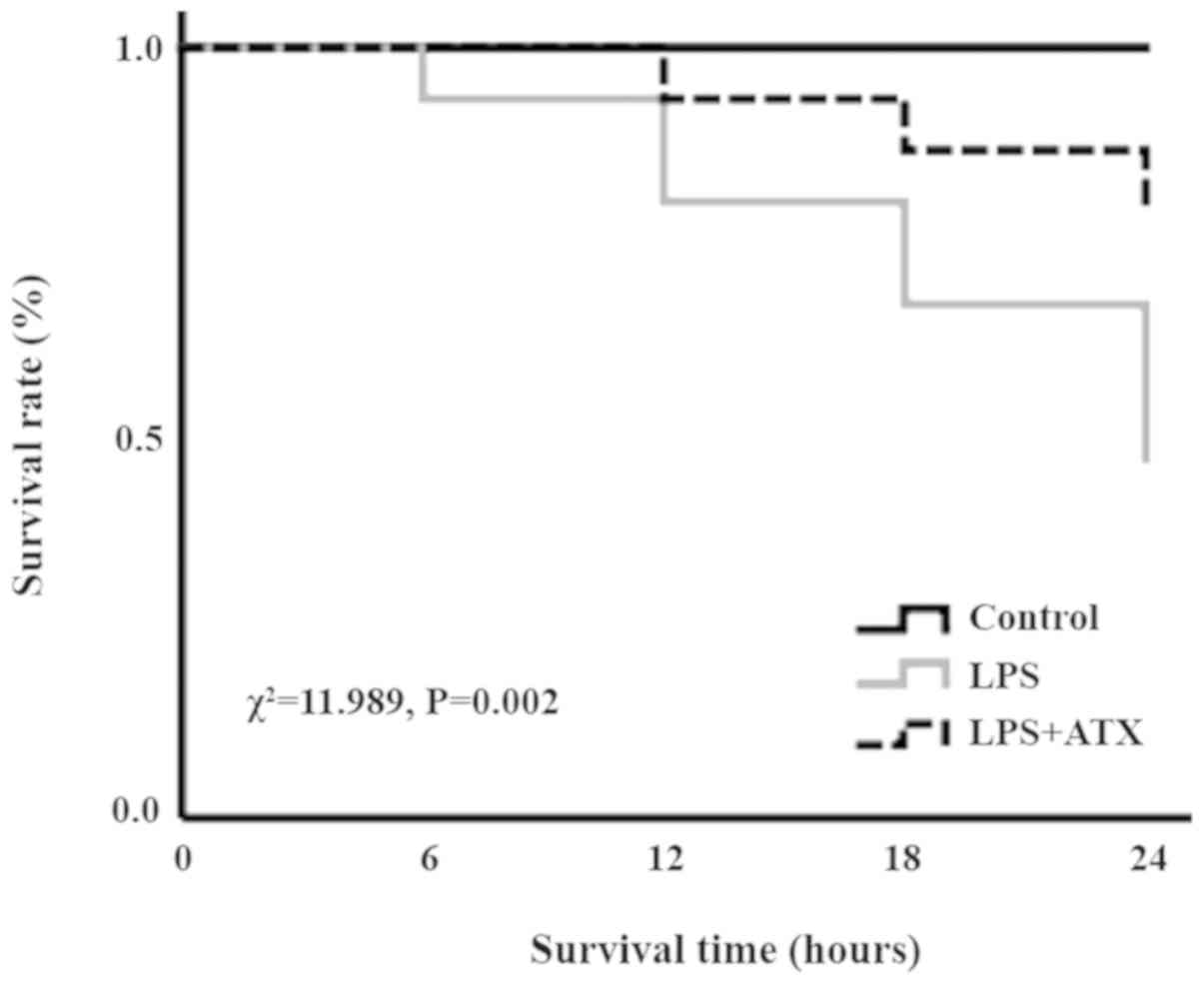
ATX improves the survival rate in
septic mice
In the LPS group, following 6, 12, 18 and 24 h of
LPS treatment, the number of deaths was 1, 2, 2 and 3,
respectively, whereas in the LPS + ATX group, the number of deaths
was 0, 1, 1 and 1, following 6, 12, 18 and 24 h of LPS treatment,
respectively. The Kaplan-Meier survival curves revealed that the 24
h mortality rate of septic mice, induced by LPS without ATX
administration, was 53.3%; however, the 24 h mortality rate
decreased to 20% when treated with ATX, and this difference was
significant (χ2=11.989; P=0.002; Fig. 1).
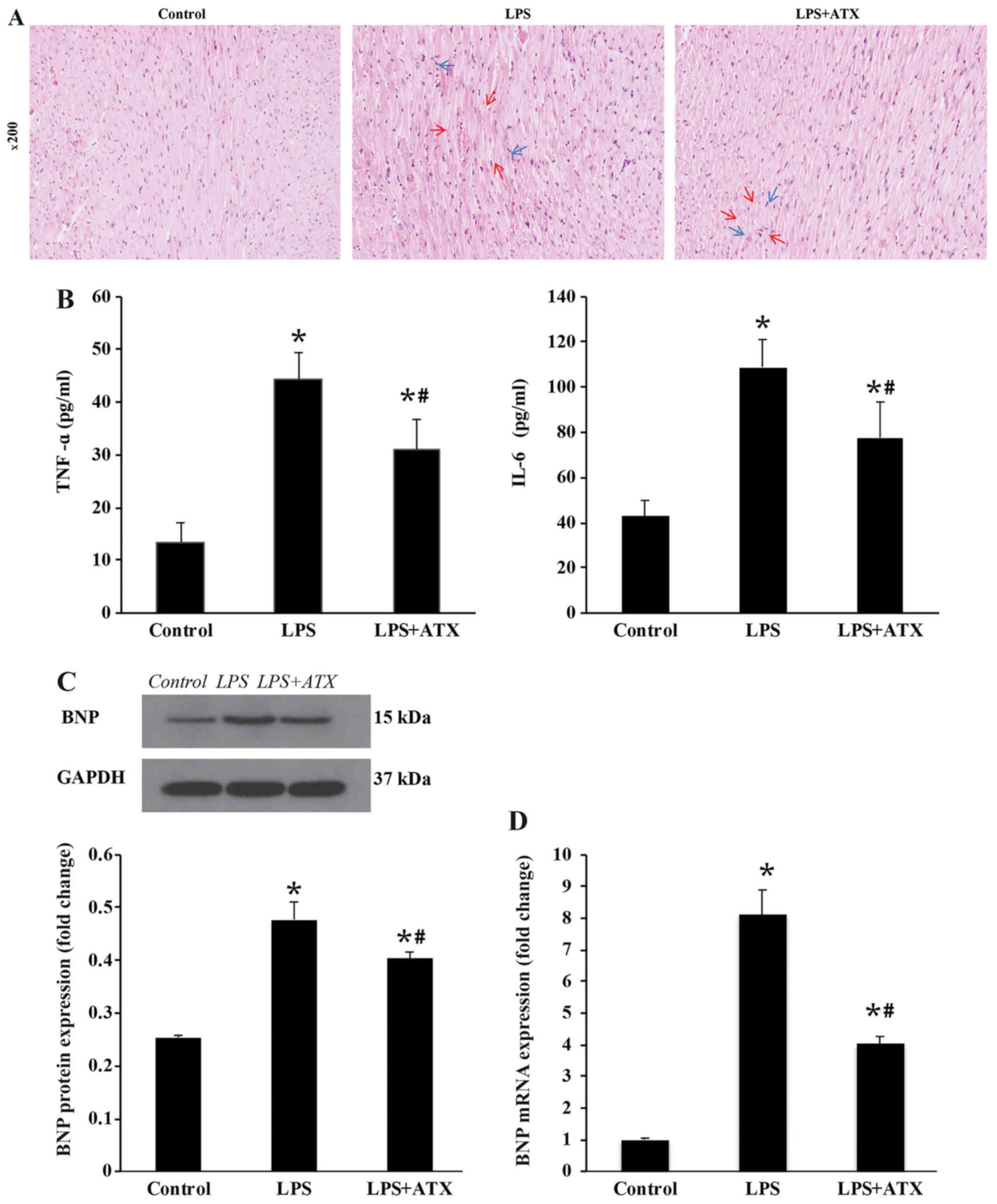
ATX alleviates the inflammatory
response and downregulates the expression levels of BNP
H&E staining demonstrated that LPS caused
cardiomyocyte necrosis (indicated by the red arrows) and neutrophil
infiltration (indicated by the blue arrows) in the LPS only group,
while ATX treatment attenuated the LPS-induced necrosis (indicated
by the red arrows) and neutrophil granulocyte infiltration
(indicated by the blue arrows) in the myocardium (Fig. 2A). LPS treatment also significantly
increased the protein and mRNA expression levels of BNP and the
protein expression levels of the inflammatory cytokines, TNF-α and
IL-6, compared with that in the control group, whereas ATX
treatment significantly reversed these changes (Fig. 2B-D).
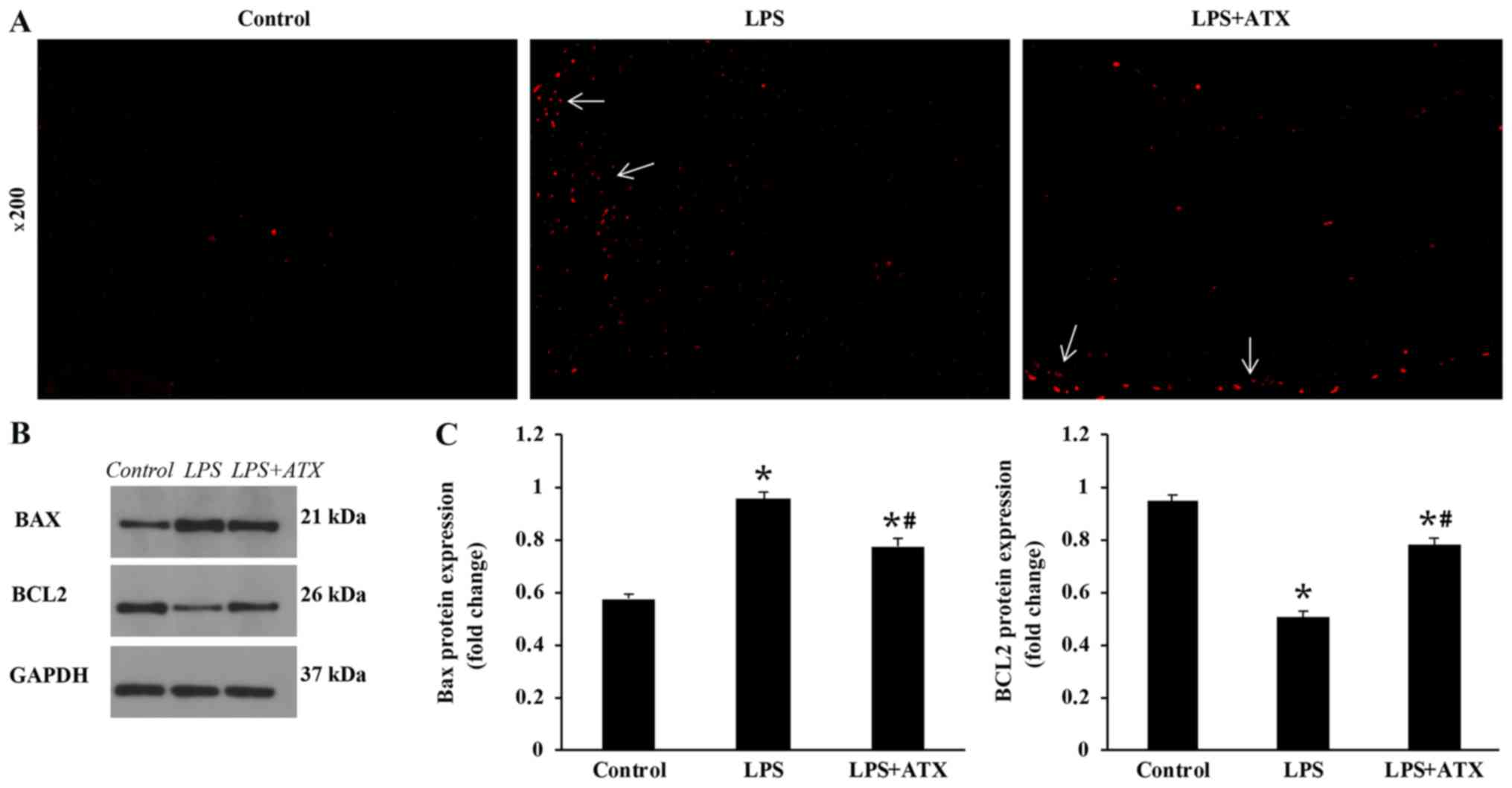
ATX inhibits the levels of
cardiomyocyte apoptosis in response to LPS
Compared with that in the control group, there was a
marked increase in the levels of cardiomyocyte apoptosis in the LPS
group; conversely, ATX treatment was observed to reduce the
LPS-induced increased levels in cardiomyocyte apoptosis (Fig. 3A). Furthermore, LPS increased the
protein expression levels of Bax and decreased the Bcl-2 protein
expression levels in the LPS group, compared with that in the
control group. ATX treatment decreased the protein expression
levels of Bax and increased the Bcl-2 protein expression levels in
the mice hearts following LPS treatment, compared with that in the
LPS group (Fig. 3B and C).
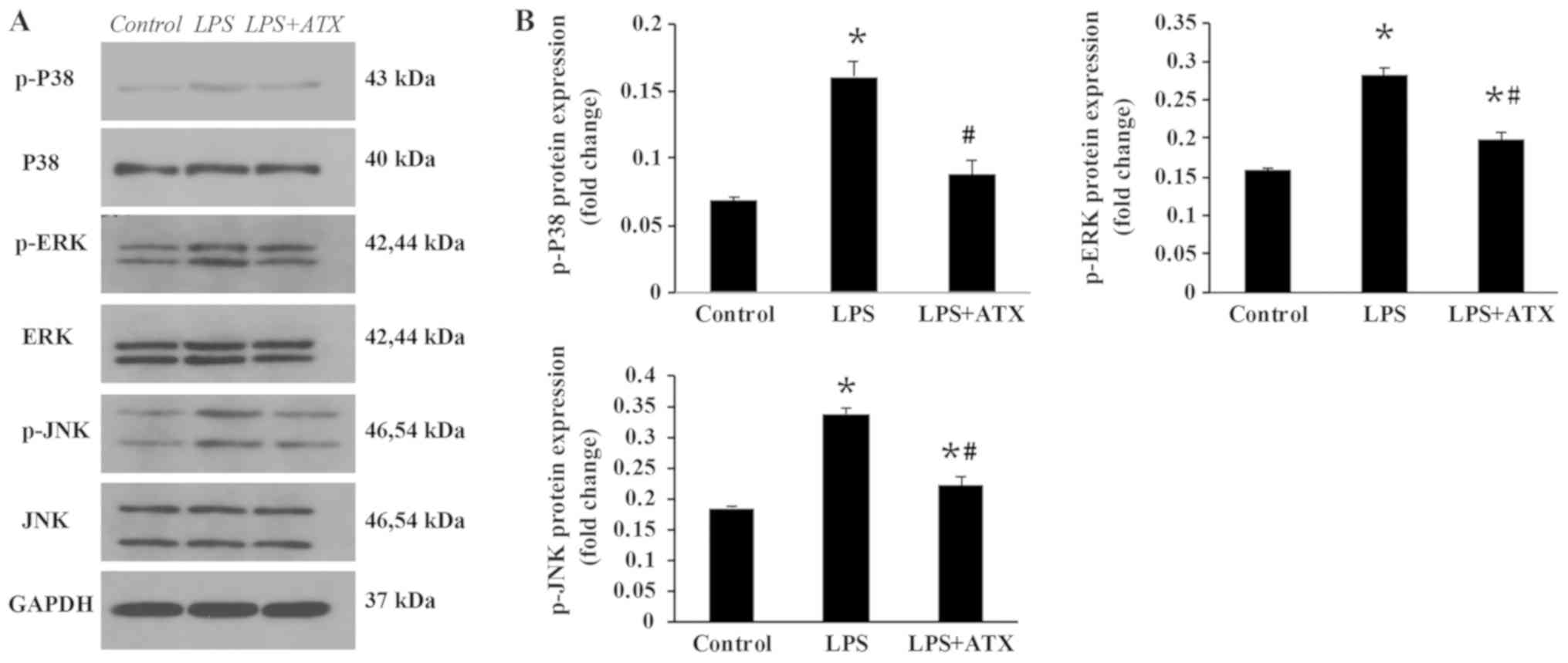
ATX inhibits the activation of the
MAPK signaling pathway
To investigate the anti-inflammatory molecular
mechanisms of ATX, the activation state of proteins in the MAPK
signaling pathway were analyzed. The administration of LPS was
found to significantly increase the protein expression levels of
p-p38, p-ERK and p-JNK compared with that in the control group
(Fig. 4A and B); however, ATX
treatment significantly decreased the LPS-induced expression levels
of these proteins.
ATX inhibits the activation of the
PI3K/AKT signaling pathway
To determine the mechanisms by which ATX reduced the
apoptosis levels, the activation state of proteins in the PI3K/AKT
signaling pathway was detected. The administration of LPS
significantly increased the protein expression levels of p-PI3K,
p-AKT, p-mTOR and p-GSK3β compared with that in the control group
(Fig. 5A and B). In contrast, the
treatment with ATX significantly reversed these trends observed in
the LPS group (Fig. 5A and B).
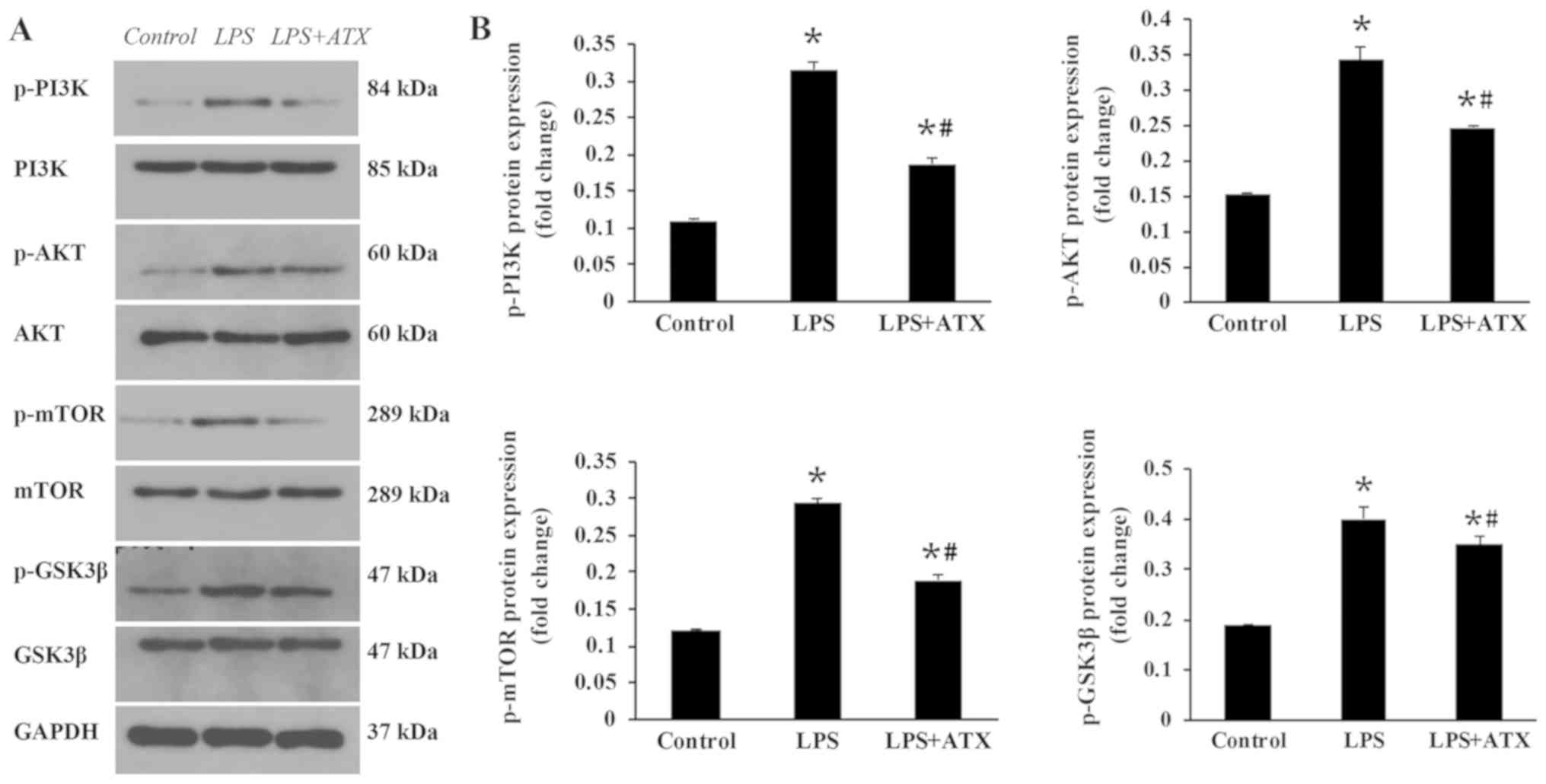 | Figure 5.Effect of ATX on the PI3K/AKT
signaling pathway. The protein expression levels of p-PI3K, PI3K,
p-AKT, AKT, p-mTOR, mTOR, p-GSK3β and GSK3β were determined using
(A) western blot analysis and the results were subsequently (B)
semi-quantified. Data are presented as the mean ± SD from three
independent experiments. *P<0.05 vs. control;
#P<0.05 vs. LPS. ATX, Astaxanthin; LPS,
lipopolysaccharide; GSK3β, glycogen synthase kinase-3β; p-,
phosphorylated. |
Discussion
Sepsis is a significant public health concern and
patients with septic shock had a high rate of mortality worldwide
in 2016 (1). Septic shock
frequently results in the dysfunction of multiple organs such as
reduced hepatic clearance, elevated creatinine levels, and impaired
aerobic respiration (1). The
ventricular myocardium is depressed during sepsis and it has been
discovered to exhibit features associated with diastolic
dysfunction (16) and systolic
dysfunction (17). Furthermore,
the pathogenesis of septic cardiomyopathy has been associated with
oxidative stress, increases in the production of inflammatory
cytokines and apoptosis (17).
However, the current therapeutic options available for cardiac
dysfunction in sepsis are limited, and include hemofiltration
techniques, statins, mesenchymal stem cells, and phosphodiesterase
inhibitors (17). As a carotenoid,
ATX has exhibited a variety of biological properties, including
antioxidant, anti-inflammatory, antithrombotic and antiapoptotic
functions (6,7).
LPS is highly pathogenic and can induce severe
sepsis (18–21). In different studies, the mortality
rate of mice was different following 24 h of intraperitoneal
injection of LPS (10 mg/kg), such as 25, 36, 40 and 50% (18–21).
In the present study, the mortality of mice after 24 h of LPS
administration was 53.3%. As the principal component of the cell
wall of Gram-negative bacteria, LPS can promote the synthesis and
release of inflammatory mediators, such as TNF-α, IL-1β and IL-6
(3). Numerous studies have
reported that proinflammatory mediators, including TNF-α, IL-1β and
IL-6, were involved in the occurrence and development of myocardial
dysfunction (4,22). For example, TNF-α was discovered to
induce myocardial depression through the modulation of the
inflammatory response (23,24).
Furthermore TNF-α was identified to serve as a modulator of
secondary factors (25), such as
nitric oxide and caspase activation, which induced myocardial
apoptosis leading to cardiac dysfunction (26). IL-6 is an important mediator of
cardiac inflammation and dysfunction; in a previous study, IL-6
knockout mice were found to have a reduced inflammatory response
and the levels of apoptosis, and an improved systolic function
following sepsis, compared with that in the wild-type mice
(27). The present study revealed
that following 24 h of LPS administration, the levels of the
inflammatory factors in the serum of mice were significantly
increased, whereas ATX treatment decreased the synthesis and
release of TNF-α and IL-6, and prevented LPS-induced cardiac
function.
The MAPK signaling pathway serves an important role
in inflammation and MAPKs were discovered to be important mediators
that drive the production of inflammatory cytokines in sepsis
(12). For example, the activation
of p38 was observed to stimulate monocyte and macrophages to
produce TNF-α and IL-6, while the inhibition of p38 exerted a
significant protective effect on lung tissue and cardiomyocytes
(28,29). In addition, the JNK signaling
pathway was reported to serve an active role in the inflammatory
response by promoting the release of TNF-α, IL-1β and IL-6
(30). The activation of
inflammation requires two distinct steps: Priming and activation.
The priming or licensing of the inflammasome are independent of
transcription and translation, and they are centered upon the ERK
signaling pathway (31). In a
previous study using the standard model of LPS priming followed by
ATP, an ERK inhibitor (U0126) was found to significantly block
inflammasome priming and activation (31). However, the inhibition of ERK
following priming was unable to block LPS/ATP-mediated human
monocyte inflammasome activation (32). Consistent with the findings of the
previous studies, the present study also demonstrated that ATX
inhibited the production of inflammation by inhibiting the
p38/JNK/ERK signaling pathway. PI3K/AKT signaling is also involved
in the inflammatory response; for example, Stark et al
(33) reported that PI3K/AKT
signaling served a complex role in the coordination of both
proinflammatory and anti-inflammatory pathways, which promoted the
production of proinflammatory cytokines through NF-κB activation
downstream of AKT, and exerted an inhibitory effect on toll-like
receptor (TLR)2, TLR3 and TLR4-mediated inflammation through the
AKT-dependent inhibition of GSK3β and forkhead box O1,
respectively. Jope et al (34) also demonstrated that the activation
of TLR4 induced the production of cytokines through myeloid
differentiation factor 88 (MyD88)-dependent and MyD88-independent
pathways, and GSK3 enhanced the inflammatory signaling via both
pathways. In addition, numerous studies have reported that the
TLR4/MAPK signaling pathway is an important mechanism of
inflammatory activation in sepsis (35,36).
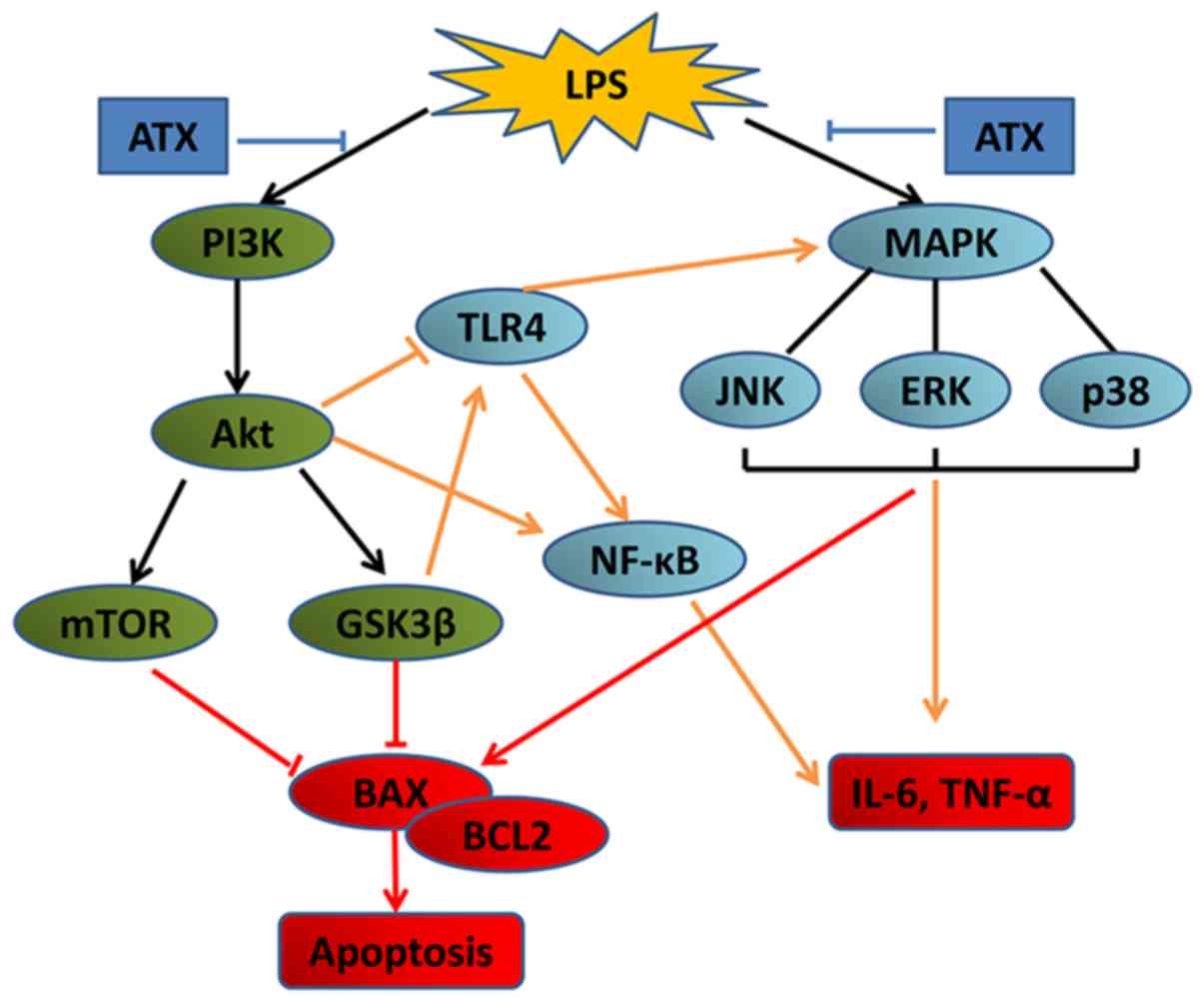
Therefore, the PI3K/AKT pathway may also promote or inhibit the
MAPK pathway, which stimulates the production of inflammatory
factors, and ATX may mitigate the septic myocardial injury by
partially blocking the activation of the MAPK and PI3K/AKT
signaling pathways.
The PI3K/AKT signaling pathway serves an important
role in cell proliferation, differentiation, apoptosis and survival
(9). mTOR and GSK3β, downstream
members of the AKT signaling pathway, were discovered to be
involved in the regulation of cell apoptosis (37,38).
The PI3K/AKT signaling pathway has also been associated with septic
myocardial injury, and the inhibition of the PI3K/AKT signaling
pathway was observed to mitigate myocardial injury in sepsis
(10). In the present study, the
protein expression levels of p-PI3K, p-AKT, p-mTOR and p-GSK3β were
increased in the LPS-treated mice, whereas ATX reversed these
increased expression levels of p-PI3K, p-AKT, p-mTOR and p-GSK3β,
suggesting an antiapoptotic effect. However, the PI3K/AKT signaling
pathway is known to inhibit apoptosis (9). MAPK signaling is involved in
apoptosis and JNK signaling was discovered to induce activator
protein-1 dependent Bax and caspase activation, which resulted in
neuronal apoptosis (39). In a
review by Lu and Xu (40) the
authors found that the activation of ERK1/2 typically promoted cell
survival; however, under certain conditions, ERK1/2 exhibited
proapoptotic functions. Therefore, sepsis-induced myocardial
apoptosis may be associated with the p38/JNK/ERK signaling pathway.
In addition, the reactivation of AKT, following hypoxia or
ischemia, was revealed to be regulated by JNKs (41). Chaanine et al (42) discovered that both JNK and AKT
activities increased with pressure overload; however, JNK signaling
was dominant over AKT signaling for the activation of the
transcription factor, FOXO3a and for the transcription of its
effector, BNIP3, promoting mitochondrial apoptosis. Therefore, we
hypothesized that both the MAPK and the PI3K/AKT signaling pathways
were activated by LPS, and signaling via the MAPK pathway was more
prominent compared with signaling via the PI3K/AKT pathway, which
thus increased apoptosis. In the present study, ATX inhibited the
activation of the MAPK and PI3K/AKT signaling pathways and reduced
the protein expression levels of Bax/ Bcl-2, which protected the
myocardium from apoptosis in sepsis.
At present, the most common methods for modeling
sepsis include intraperitoneal injection or intravenous injection
with LPS; however, cecal ligation and puncture have been found to
improve the development of sepsis and clinical infection (43). This is achieved by necrotizing the
end of the cecum following surgery and enabling the contents to
enter the abdominal cavity following intestinal puncture; however,
this method is affected by the bacterial composition of the
intestinal contents and differences between operators (44). The model of sepsis induced with LPS
by tail vein injection is acute, and acute cardiac dysfunction can
be induced within 4–6 h (45). LPS
can also be injected intraperitoneally to induce septic myocardial
injury after 6 h; the dosage of LPS is easy to control and the
surgery is simple; however, the model of LPS administration does
not reflect all the complex physiological human responses, such as
proinflammatory cytokines production, hemodynamic response
(46,47). A previous study used PEG400 as an
ATX solvent and confirmed that PEG400 exerted no significant effect
on the experiment (48). Thus, ATX
dissolved in PEG400 was administered to the mice in the LPS + ATX
group in the present study.
Nonetheless, the results of the current study are
limited. BNP is an important indicator of cardiac dysfunction,
which has been discovered to be significantly associated with
sepsis induced myocardial dysfunction and mortality in patients
with septic shock (49). Thus, due
to the limited conditions in the present study, only BNP was
selected to evaluate cardiac function without ultrasonic,
hemodynamic and electrocardiography methods. Our future studies aim
to improve the experimental method.
In conclusion, the present study demonstrated that
compared with that in the LPS group, ATX treatment significantly
reduced the levels of IL-6 and TNF-α in the serum, reversed the
histopathological alterations and inhibited the LPS-induced
apoptosis of mouse cardiomyocytes. Furthermore, the increased mRNA
and protein expression levels of BNP induced by LPS were reversed
by ATX, and ATX treatment reduced the mortality rates in mice with
sepsis. Together, these data indicate that ATX may exhibit a
protective effect on LPS-induced cardiac dysfunction in septic
mice. Thus, ATX treatment may protect the heart from sepsis and
lower the mortality rates in mice. The mechanisms of ATX may be
related to the inhibition of the MAPK and PI3K/AKT/mTOR/GSK3β
signaling pathways (Fig. 6).
Therefore, ATX may serve as a potentially effective intervention
for the treatment of cardiac dysfunction in patients with
sepsis.
Acknowledgements
Not applicable.
Funding
This study was supported by the National Natural
Science Foundation of China (grant nos. 81870939 and 81571147).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
WJX and GH designed the experiments. LW and SSW
performed the experiments. GH performed the statistical analysis.
WJX prepared the manuscript. XXX designed the experiments,
interpreted and analyzed the data, and revised the manuscript. All
authors read and approved the final manuscript.
Ethics approval and consent to
participate
All animal experiments were conducted in accordance
with the institutional guidelines of the Animal Care and Use
Committee of Renmin Hospital of Wuhan University (Wuhan,
China).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Singer M, Deutschman CS, Seymour CW,
Shankar-Hari M, Annane D, Bauer M, Bellomo R, Bernard GR, Chiche
JD, Coopersmith CM, et al: The third international consensus
definitions for sepsis and septic shock (sepsis-3). JAMA.
315:3338–810. 2016. View Article : Google Scholar
|
|
2
|
Merx MW and Weber C: Sepsis and the heart.
Circulation. 116:793–802. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Matsuda N and Hattori Y: Systemic
inflammatory response syndrome (SIRS): Molecular pathophysiology
and gene therapy. J Pharmacol Sci. 101:189–198. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Siti HN, Kamisah Y and Kamsiah J: The role
of oxidative stress, antioxidants and vascular inflammation in
cardiovascular disease (a review). Vascul Pharmacol. 71:40–56.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Hussein G, Nakamura M, Zhao Q, Iguchi T,
Goto H, Sankawa U and Watanabe H: Antihypertensive and
neuroprotective effects of astaxanthin in experimental animals.
Biol Pharm Bull. 28:47–52. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Wu H, Niu H, Shao A, Wu C, Dixon BJ, Zhang
J, Yang S and Wang Y: Astaxanthin as a potential neuroprotective
agent for neurological diseases. Mar Drugs. 13:5750–5766. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Fassett RG and Coombes JS: Astaxanthin: A
potential therapeutic agent in cardiovascular disease. Mar Drugs.
9:447–465. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Zhou L, Gao M, Xiao Z, Zhang J, Li X and
Wang A: Protective effect of astaxanthin against multiple organ
injury in a rat model of sepsis. J Surg Res. 195:559–567. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Kandel ES and Hay N: The regulation and
activities of the multifunctional serine/threonine kinase Akt/PKB.
Exp Cell Res. 253:210–229. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Chen L, Liu P, Feng X and Ma C:
Salidroside suppressing LPS-induced myocardial injury by inhibiting
ROS-mediated PI3K/Akt/mTOR pathway in vitro and in vivo. J Cell Mol
Med. 21:3178–3189. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Joh EH, Gu W and Kim DH: Echinocystic acid
ameliorates lung inflammation in mice and alveolar macrophages by
inhibiting the binding of LPS to TLR4 in NF-κB and MAPK pathways.
Biochem Pharmacol. 84:331–340. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Frazier WJ, Wang X, Wancket LM, Li XA,
Meng X, Nelin LD, Cato AC and Liu Y: Increased inflammation,
impaired bacterial clearance, and metabolic disruption after
gram-negative sepsis in Mkp-1-deficient mice. J Immunol.
183:7411–7419. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Wang X, Meng X, Kuhlman JR, Nelin LD,
Nicol KK, English BK and Liu Y: Knockout of Mkp-1 enhances the host
inflammatory responses to gram-positive bacteria. J Immunol.
178:5312–5320. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Lee SJ, Bai SK, Lee KS, Namkoong S, Na HJ,
Ha KS, Han JA, Yim SV, Chang K, Kwon YG, et al: Astaxanthin
inhibits nitric oxide production and inflammatory gene expression
by suppressing I(kappa)B kinase-dependent NF-kappaB activation. Mol
Cells. 16:97–105. 2003.PubMed/NCBI
|
|
15
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Kakihana Y, Ito T, Nakahara M, Yamaguchi K
and Yasuda T: Sepsis-induced myocardial dysfunction:
Pathophysiology and management. J Intensive Care. 4:222016.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Balija TM and Lowry SF: Lipopolysaccharide
and sepsis-associated myocardial dysfunction. Curr Opin Infect Dis.
24:248–253. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Honda T, He Q, Wang F and Redington AN:
Acute and chronic remote ischemic conditioning attenuate septic
cardiomyopathy, improve cardiac output, protect systemic organs,
and improve mortality in a lipopolysaccharide-induced sepsis model.
Basic Res Cardiol. 114:152019. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
de Pádua Lúcio K, Rabelo ACS, Araújo CM,
Brandão GC, de Souza GHB, da Silva RG, de Souza DMS, Talvani A,
Bezerra FS, Calsavara AJC and Costa DC: Anti-inflammatory and
antioxidant properties of black mulberry (Morus nigra L.) in a
model of LPS-induced sepsis. Oxid Med Cell Longev.
2018:50480312018. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Kawaguchi S, Okada M, Ijiri E, Koga D,
Watanabe T, Hayashi K, Kashiwagi Y, Fujita S and Hasebe N:
β3-Adrenergic receptor blockade reduces mortality in
endotoxin-induced heart failure by suppressing induced nitric oxide
synthase and saving cardiac metabolism. Am J Physiol Heart Circ
Physiol. 318:H283–H294. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Kumari A, Dash D and Singh R: Curcumin
inhibits lipopolysaccharide (LPS)-induced endotoxemia and airway
inflammation through modulation of sequential release of
inflammatory mediators (TNF-α and TGF-β1) in murine model.
Inflammopharmacology. 25:329–341. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Romero-Bermejo FJ, Ruiz-Bailen M,
Gil-Cebrian J and Huertos-Ranchal MJ: Sepsis-induced
cardiomyopathy. Curr Cardiol Rev. 7:163–183. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Kapadia S, Lee J, Torre-Amione G, Birdsall
HH, Ma TS and Mann DL: Tumor necrosis factor-alpha gene and protein
expression in adult feline myocardium after endotoxin
administration. J Clin Invest. 96:1042–1052. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Haudek SB, Bryant DD and Giroir BP:
Differential regulation of myocardial NF kappa B following acute or
chronic TNF-alpha exposure. J Mol Cell Cardiol. 33:1263–1271. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Meldrum DR: Tumor necrosis factor in the
heart. Am J Physiol. 274:R577–R595. 1998.PubMed/NCBI
|
|
26
|
Carlson DL, Willis MS, White DJ, Horton JW
and Giroir BP: Tumor necrosis factor-alpha-induced caspase
activation mediates endotoxin-related cardiac dysfunction. Crit
Care Med. 33:1021–1028. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Zhang H, Wang HY, Bassel-Duby R, Maass DL,
Johnston WE, Horton JW and Tao W: Role of interleukin-6 in cardiac
inflammation and dysfunction after burn complicated by sepsis. Am J
Physiol Heart Circ Physiol. 292:H2408–H2416. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Nick JA, Young SK, Arndt PG, Lieber JG,
Suratt BT, Poch KR, Avdi NJ, Malcolm KC, Taube C, Henson PM and
Worthen GS: Selective suppression of neutrophil accumulation in
ongoing pulmonary inflammation by systemic inhibition of p38
mitogen-activated protein kinase. J Immunol. 169:5260–5269. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Frazier WJ, Xue J, Luce WA and Liu Y: MAPK
signaling drives inflammation in LPS-stimulated cardiomyocytes: The
route of crosstalk to G-protein-coupled receptors. PLoS One.
7:e500712012. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Li ST, Dai Q, Zhang SX, Liu YJ, Yu QQ, Tan
F, Lu SH, Wang Q, Chen JW, Huang HQ and Li M: Ulinastatin
attenuates LPS-induced inflammation in mouse macrophage RAW264.7
cells by inhibiting the JNK/NF-κB signaling pathway and activating
the PI3K/Akt/Nrf2 pathway. Acta Pharmacol Sin. 39:1294–1304. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Ghonime MG, Shamaa OR, Das S, Eldomany RA,
Fernandes-Alnemri T, Alnemri ES, Gavrilin MA and Wewers MD:
Inflammasome priming by lipopolysaccharide is dependent upon ERK
signaling and proteasome function. J Immunol. 192:3881–3888. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Mehta VB, Hart J and Wewers MD:
ATP-stimulated Release of interleukin (IL)-1beta and IL-18 requires
priming by lipopolysaccharide and is independent of caspase-1
cleavage. J Biol Chem. 276:3820–3826. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Stark AK, Sriskantharajah S, Hessel EM and
Okkenhaug K: PI3K inhibitors in inflammation, autoimmunity and
cancer. Curr Opin Pharmacol. 23:82–91. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Jope RS, Cheng Y, Lowell JA, Worthen RJ,
Sitbon YH and Beurel E: Stressed and inflamed, can gsk3 be blamed?
Trends Biochem Sci. 42:180–192. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Liu C, Tang X, Zhang W, Li G, Chen Y, Guo
A and Hu C: 6-bromoindirubin-3′-oxime suppresses LPS-induced
inflammation via inhibition of the TLR4/NF-κB and TLR4/MAPK
signaling pathways. Inflammation. 42:2192–2204. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Park J, Ha SH, Abekura F, Lim H, Magae J,
Ha KT, Chung TW, Chang YC, Lee YC, Chung E, et al:
4-O-carboxymethylascochlorin inhibits expression levels of on
inflammation-related cytokines and matrix metalloproteinase-9
through NF-κB/MAPK/TLR4 signaling pathway in LPS-activated RAW264.7
cells. Front Pharmacol. 10:3042019. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Kumar D, Shankar S and Srivastava RK:
Rottlerin induces autophagy and apoptosis in prostate cancer stem
cells via PI3K/Akt/mTOR signaling pathway. Cancer Lett.
343:179–189. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Guo B, Zhang W, Xu S, Lou J, Wang S and
Men X: GSK-3β mediates dexamethasone-induced pancreatic β cell
apoptosis. Life Sci. 144:1–7. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Putcha GV, Le S, Frank S, Besirli CG,
Clark K, Chu B, Alix S, Youle RJ, Lamarche A, Maroney AC and
Johnson EM Jr: JNK-mediated BIM phosphorylation potentiates
BAX-dependent apoptosis. Neuron. 38:899–914. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Lu Z and Xu S: ERK1/2 MAP kinases in cell
survival and apoptosis. IUBMB Life. 58:621–631. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Shao Z, Bhattacharya K, Hsich E, Park L,
Walters B, Germann U, Wang YM, Kyriakis J, Mohanlal R, Kuida K, et
al: c-jun N-terminal kinases mediate reactivation of Akt and
cardiomyocyte survival after hypoxic injury in vitro and in vivo.
Circ Res. 98:111–118. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Chaanine AH, Jeong D, Liang L, Chemaly ER,
Fish K, Gordon RE and Hajjar RJ: JNK modulates FOXO3a for the
expression of the mitochondrial death and mitophagy marker BNIP3 in
pathological hypertrophy and in heart failure. Cell Death Dis.
3:2652012. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Fink MP: Animal models of sepsis.
Virulence. 5:143–153. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Hubbard WJ, Choudhry M, Schwacha MG, Kerby
JD, Rue LW III, Bland KI and Chaudry IH: Cecal ligation and
puncture. Shock. 24 (Supp 1):S52–S57. 2005. View Article : Google Scholar
|
|
45
|
Wang X, Su L, Yang R, Zhang H and Liu D:
Myocardial strain/stress changes identified by echocardiography may
reveal early sepsis-induced myocardial dysfunction. J Int Med Res.
46:1439–1454. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Dejager L, Pinheiro I, Dejonckheere E and
Libert C: Cecal ligation and puncture: The gold standard model for
polymicrobial sepsis? Trends Microbiol. 19:198–208. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Diao X and Sun S: PMicroRNA-124a regulates
LPS-induced septic cardiac dysfunction by targeting STX2.
Biotechnol Lett. 39:1335–1342. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Guo SX, Zhou HL, Huang CL, You CG, Fang Q,
Wu P, Wang XG and Han CM: Astaxanthin attenuates early acute kidney
injury following severe burns in rats by ameliorating oxidative
stress and mitochondrial-related apoptosis. Marine Drugs.
13:2105–2123. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Kakoullis L, Giannopoulou E,
Papachristodoulou E, Pantzaris ND, Karamouzos V, Kounis NG, Koniari
I and Velissaris D: The utility of brain natriuretic peptides in
septic shock as markers for mortality and cardiac dysfunction: A
systematic review. Int J Clin Pract. 73:e133742019. View Article : Google Scholar : PubMed/NCBI
|