Introduction
Scalp-ear-nipple (SEN) syndrome is a rare,
autosomal-dominant disorder that commonly presents as cutis aplasia
of the scalp, minor anomalies of the external ears, digits and
nails, and malformation of the breast (1). In mice, homozygous deficiency of the
lymphoid enhancer-binding factor 1 (Lef-1) gene results in the lack
of whiskers and the arrest of hair-follicle development, lack of
mammary glands and edentulism, indicating that Lef-1 may serve as a
potential candidate gene for treating SEN syndrome (2). Ten mutations in the broad-complex,
tramtrack and bric a brac (BTB) domain of potassium-channel
tetramerization-domain-containing 1 (KCTD1) have been identified in
ten families with SEN syndrome of European, Brazilian and North
African background (3). This
conserved domain regulates transcriptional activity, suggesting it
may serve a key role in KCTD1 protein function during ectodermal
development (3).
Transcription factor AP-2α-knockout mice exhibit a
number of severe developmental phenotypes, including neural tube,
body wall and craniofacial abnormality (4). Dominant AP-2α mutant Doarad results
in a missense mutation that affects the
Y58F59P60P61P62Y63
(PY) motif in the transactivation domain of AP-2α, causing a
misshapen malleus, incus and stapes (5). This P59L mutation in AP-2α leads to
increased transcriptional activation of AP-2α (5). Patients with Char syndrome have three
mutations in the basic DNA-binding domain and a PY motif in the
activation domain of the AP-2β gene, causing dominant-negative
effects. In the USA, individuals with Char syndrome who have a PY
motif P62R mutation in AP-2β have a higher incidence rate (10 of
14) of patent ductus arteriosus, but milder hand and facial
abnormal phenotypes compared with individuals with DNA-binding
domain mutations in AP-2α (6,7).
Point mutations or insertions/deletions in the conserved
DNA-binding domain in AP-2α, encoded by exons 4 and 5, cause the
majority of branchio-oculo-facial syndrome (BOFS) cases (8). BOFS is associated with >25
different mutations in AP-2α (9–12).
Therefore, AP-2α mutants have been associated with certain
developmental abnormalities.
Our previous study (13) demonstrated that KCTD1 binds with
AP-2α and decreases its transcriptional activity. KCTD15 is
associated with the KCTD1 protein in the KCTD family. It is likely
that KCTD15 functions during embryogenesis by interacting with the
activation domain of AP-2α to inhibit neural crest induction
(14). Individuals with KCTD15
mutations exhibit developmental defects in neural crest derivatives
and the torus lateralis (TLa) in the central nervous system,
resulting in a significant decrease in normal growth rates
(15,16). Moreover, both AP-2α and KCTD1
regulate the Wnt/β-catenin pathway. AP-2α directly interacts with
adenomatous polyposis coli (APC), stabilizing interactions between
APC and β-catenin, attenuating interactions between β-catenin and
transcription factor-4 in the nucleus and suppressing TOPFLASH
luciferase reporter activity in human colorectal cancer cells
(17). AP-2α-interacting protein
KCTD1 directly binds to β-catenin, enhances β-catenin
ubiquitination and degradation, which is dependent on casein kinase
1/glycogen synthase kinase-3β-mediated phosphorylation of
β-catenin, and inhibits the canonical Wnt/β-catenin signaling
pathway (18).
The present study aimed to investigate whether KCTD1
and AP-2α protein mutants reciprocally regulate their interactions
and inhibit the Wnt/β-catenin signaling pathway. The results
demonstrated that BTB domain mutations in KCTD1 were not associated
with AP-2α. The P59A mutation in the PY motif of AP-2α prevented
binding with KCTD1, but the majority of BTB domain mutations in
KCTD1 and PY motif mutations in AP-2α resulted in inhibited Wnt
pathway-responsive TOPFLASH reporter gene expression levels.
Wild-type KCTD1 suppressed the transcriptional activity of AP-2α
P60A and P61R to repress TOPFLASH reporter gene expression levels.
Moreover, wild-type KCTD1 and AP-2α downregulated β-catenin
expression levels and decreased the viability of SW480 cells. These
findings highlighted the importance of AP-2α and KCTD1 regulation
in maintaining a normal tissue phenotype.
Materials and methods
Plasmid generation
Recombinant overexpressing plasmids pCMV-Myc-AP-2α,
pCMV-HA-KCTD1, pCMV-Myc-KCTD1 and pEGFP-C1-AP-2α were generated as
previously described (13,18). The mutants AP-2α P61R and KCTD1
P20S, A30E, P31R, H33P and H33Q were also generated as previously
described (13,18). The KCTD1 mutants P31L, P31H, G62D,
D69E and H74P and AP-2α mutants P59A, P60A and 4A
(P59AP60AP61AY62A) were modified by PCR-based site-directed
mutagenesis and cloned into pCMV-Myc plasmids. Mutated primers are
presented in Table I. Reporter
vectors A2 and TOPFLASH were generated as previously described
(18,19). Eight recombinant plasmids were
sequenced using the Sanger method, in order to verify mutant site
points (Sangon Biotech, Co., Ltd.) (Fig. S1).
 | Table I.Mutants of KCTD1 and AP-2α and their
PCR primers. |
Table I.
Mutants of KCTD1 and AP-2α and their
PCR primers.
| Gene | Mutant | Primer sequence
(5′→3′) |
|---|
| KCTD1 | P20S | F: TCTACTCCAGCACAACTCACA |
|
|
| R:
TGTGAGTTGTGCTGGAGTAGA |
|
| A30E | F:
ATCCAATGAGCCTGTCCACAT |
|
|
| R:
ATGTGGACAGGCTCATTGGAT |
|
| P31R | F:
CAATGCGCGTGTCCACATTGA |
|
|
| R:
TCAATGTGGACACGCGCATTG |
|
| P31L | F:
CAATGCGCTTGTCCACATTGA |
|
|
| R:
TCAATGTGGACAAGCGCATTG |
|
| P31H | F:
CAATGCGCATGTCCACATTGA |
|
|
| R:
TCAATGTGGACATGCGCATTG |
|
| H33P | F: CCATTGATGTGGGCGGCCAC |
|
|
| R:
GTGGCCGCCCACATCAATGG |
|
| H33Q | F:
CGCCTGTCCAAATTGATGTGG |
|
|
| R:
CCACATCAATTTGGACAGGCG |
|
| G62D | F:
TTTGATGATACAGAGCCCATTG |
|
|
| R:
CAATGGGCTCTGTATCATCAAA |
|
| D69E | F: AAGTCTCAAACAGCACTATTTC |
|
|
| R:
GAAATAGTGCTGTTTGAGACTT |
|
| H74P | F: CCTATTTCATTGACAGAGATGG |
|
|
| R:
CCATCTCTGTCAATGAAATAGG |
| AP-2α | P61R | F:
TTCCCCCCAAGATACCAGCCT |
|
|
| R:
AGGCTGGTATCTTGGGGGGAA |
|
| P59A | F:
ATACTTCGCCCCACCCTACC |
|
|
| R:
GGTAGGGTGGGGCGAAGTAT |
|
| P60A | F:
ACTTCCCCGCACCCTACCAG |
|
|
| R:
CTGGTAGGGTGCGGGGAAGT |
|
| 4A | F:
ATACTTCGCCGCAGCCGCCCAGCCT |
|
|
| R:
AGGCTGGGCGGCTGCGGCGAAGTAT |
Cell culture and transfection
SW480, 293 and HeLa cells (American Type Culture
Collection) were cultured in complete DMEM (Sigma-Aldrich; Merck
KGaA). All cells were maintained with 10% fetal calf serum
(HyClone; GE Healthcare Life Sciences), 2 mM glutamine, 10 U/ml
penicillin and 100 µg/ml streptomycin at 37°C with 5%
CO2. The cells were transiently transfected with
different amounts of plasmid DNA (1 µg for luciferase reporter
assays; 6 µg for immunofluorescence analysis; 20 µg for
co-immunoprecipitation; Clontech Laboratories, Inc.) using
Lipofectamine® 2000 reagent (Invitrogen; Thermo Fisher
Scientific, Inc.) for 6 h. The cells were collected 24–30 h
following transfection for subsequent experimentation.
Luciferase reporter assays
The 293 cells were cultured and transiently
transfected with 0.3 µg reporter plasmids A2 (generous gift of
Professor Trevor Williams) or pTOPFLASH (Clontech Laboratories,
Inc.) (18) and the indicated
plasmids (0.3 µg pCMV-Myc-KCTD1 and/or 0.3 µg of pCMV-Myc-AP-2α) in
12-well plates using Lipofectamine 2000 reagent as previously
described (20). Briefly, 0.1 µg
pCMV-LacZ plasmid (19) was
co-transfected in each well to measure transfection efficiency as
an internal β-galactosidase control. The total amount of 1 µg
plasmid DNA in each well was maintained by adding empty vector
pCMV-Myc (Clontech Laboratories, Inc.) to each transfection.
β-galactosidase and luciferase activities were assessed 24 h after
transfection using a Promega Luciferase Assay system (Promega
Corporation) and a Turner TD20/20 luminometer (Turner Designs). The
luciferase activity is normalized relative to the β-galactosidase
control. A total of three experimental repeats were performed.
Immunofluorescence localization
analysis
HeLa cells were grown to ~70% confluence on glass
coverslips and transiently transfected with3 µg GFP-AP-2α and 3 µg
Myc-KCTD1 mutants or 3 µg Myc-AP-2α mutants and 3 µg HA-KCTD1. At
30 h post-transfection, cells were collected for immunofluorescence
staining, as previously described (19). Primary antibodies included Anti-Myc
mouse monoclonal antibody (clone. no. 9E10; Santa Cruz
Biotechnology, Inc.) and anti-HA rabbit polyclonal antibody (clone
no. Y-11) (Santa Cruz Biotechnology, Inc.) at 1:200 dilution for 1
h at 4°C. Alexa Fluor 488 goat anti-rabbit IgG (cat. no. A27034)
and Alexa Fluor 594 goat anti-mouse IgG (cat. no. A-11005)
secondary antibodies were purchased from Molecular Probes; Thermo
Fisher Scientific, Inc. and used at 1:500 dilution for 45 min at
4°C. Hoechst 33258 (1 µg/ml) was used to stain the nuclei
(Sigma-Aldrich; Merck KGaA) for 5 min at 4°C. The fluorescence
signals were observed with a fluorescence microscope (Carl Zeiss
AG).
Co-immunoprecipitation
The 293 cells were grown to ~80% confluence and
transiently transfected with 10 µg Myc-AP-2α and 10 µg Myc-KCTD1 in
10-cm dishes. At 30 h after transfection, cells were collected and
lysed as previously described (13). The cells were lysed in RIPA buffer
[50 mM Tris-HCl pH 7.2; 150 mM NaCl, 1% (v/v) Triton X-100; 1%
(w/v) sodium deoxycholate; 0.1% (w/v) SDS] supplemented with
protease inhibitors. The cellular lysates were precleared with
protein A/G plus agarose (Santa Cruz Biotechnology, Inc.) at 4°C
for 1 h, and then immunoprecipitated using rabbit anti-KCTD1
polyclonal antibodies (21) and
protein A/G plus agarose. The immunoprecipitates were washed four
times for 15 min each in 1 ml 1% Triton buffer by keeping gentle
agitation and then centrifuged at 500 × g for 3 min at 4°C. The
immunocomplexes were separated by SDS-PAGE on 13% gels and detected
with mouse anti-Myc tag monoclonal antibody at 1:2,000 dilution
overnight at 4°C as described in the following Western blot
section. Pre-immune rabbit IgG (cat. no. sc-2027; Santa Cruz
Biotechnology, Inc.) was used as a negative control. The
quantification of immunoprecipitated bands was analyzed using
ImageJ software (ImageJ 1.48v, http://imagej.nih.gov/ij/).
Western blotting
SW480 cells were transiently transfected with
pCMV-Myc-AP-2α and/or pCMV-Myc-KCTD1 expression plasmids. At 24 h
after post-transfection, cells were collected and lysed in RIPA
buffer. The protein concentration of whole-cell extracts was
measured using a BCA assay kit (Pierce; Thermo Fisher Scientific,
Inc.). Total protein (10 µg/lane) was resolved by SDS-PAGE on 12%
gels, then transferred to PVDF membranes (Bio-Rad Laboratories,
Inc.) at 4°C for 2 h at 100 V. The membrane was blocked with TBS
buffer [100 mM Tris-HCl pH7.5; 0.9% (w/v) NaCl] with 1% BSA
(Sigma-Aldrich; Merck KGaA) for 1 h at 4°C. After blocking, the
membrane was incubated with primary antibodies against Myc-tag
(clone. no. 9E10; 1:2,000), β-catenin (E-5, 1:1,000), cyclin D1
(CCND1; clone. no. HD11; 1:1,000) and GAPDH (clone. no. 6C5;
1:5,000) (Santa Cruz Biotechnology, Inc.) overnight at 4°C and
washed with TBS and 0.1% (v/v) Tween-20 for 1 h. After washing, the
membrane was then incubated with goat anti-mouse IgG-horseradish
peroxidase-conjugated secondary antibody (cat. no. sc-2005,
1:7,500; Santa Cruz Biotechnology, Inc.) at room temperature for 45
min followed by washing five times for 1 h. The signal was detected
with SuperSignal West Pico chemiluminescent Substrate (Pierce;
Thermo Fisher Scientific, Inc.) and visualized with tanon-5200
system (Tanon Science and Technology Co., Ltd.).
Cell viability assays
A total of 3×103 transfected SW480 cells
were grown at 37°C in 96-well plates in octuplicate. Cell viability
was analyzed 24 h after transfection by adding 1 mg/ml MTT to each
well, followed by incubation for 4 h at 37°C. Subsequently, the
formazan crystals were dissolved in 100 µl DMSO. The absorbance was
measured at 490 nm using a spectrophotometer. To detect the effects
of AP-2α and KCTD1 proteins on the sensitivity of SW480 cells to
sorafenib, SW480 cells were transfected and subsequently treated
with 20 µM cisplatin (DDP) for 24 h; viability was assessed by MTT,
aforementioned.
Statistical analysis
All statistical analysis was performed using SPSS
statistical software (version 16.0; SPSS, Inc.). Data are presented
as the mean ± SD of three independent experiments. Significant
differences were determined using Student's t-test to compare two
groups, and two-way ANOVA to compare three or more groups.
Multi-group comparisons were analyzed using Tukey's post hoc test.
P<0.05 was considered to indicate a statistically significant
difference.
Results
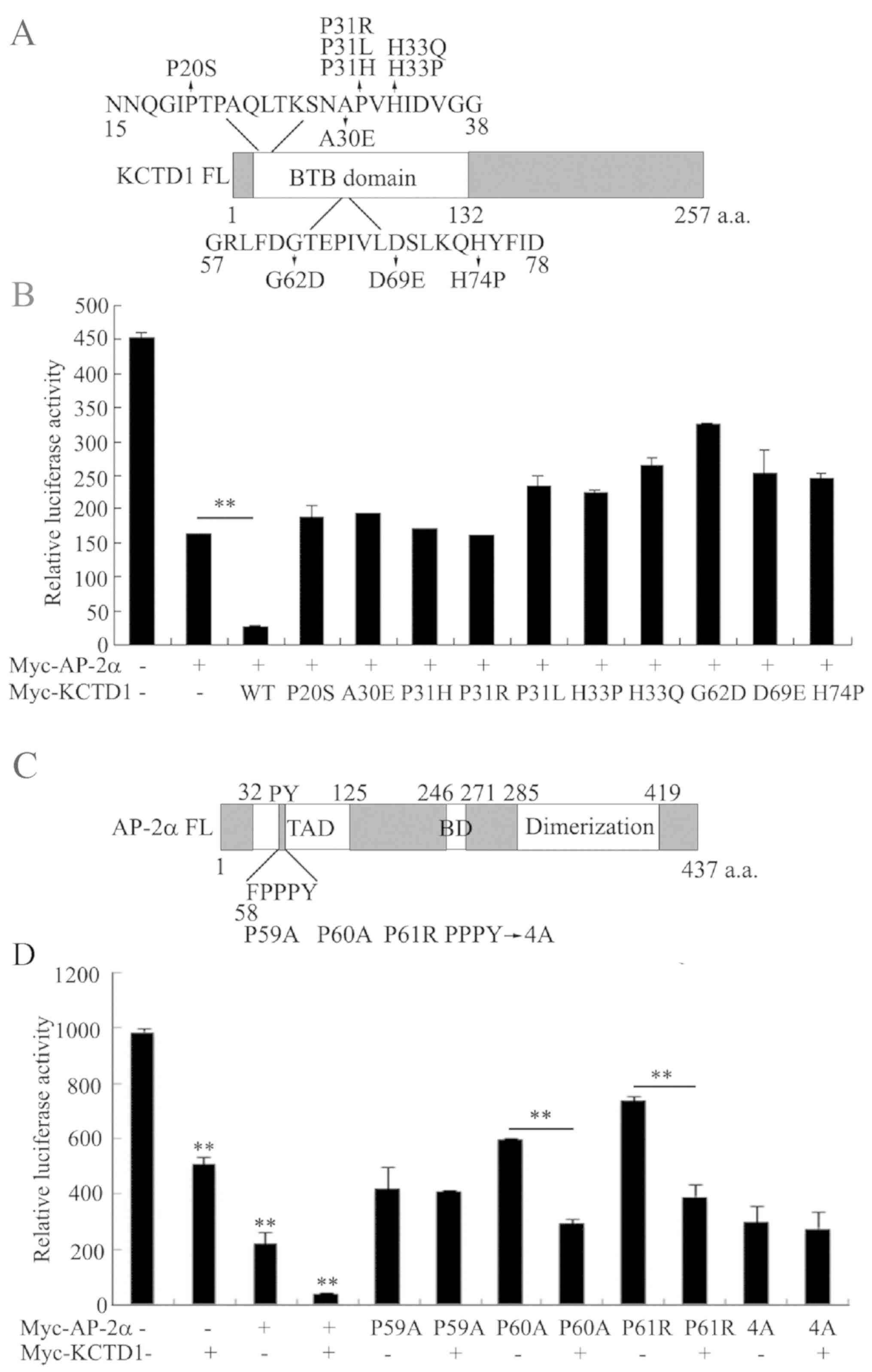
Known KCTD1 mutants have no inhibitory
effect on the transcriptional activity of AP-2α, and KCTD1 does not
repress the transcriptional activity of the AP-2αP59A mutation
All the known KCTD mutations in the BTB-domain lead
to SEN syndrome, whereas mutations in AP-2α possibly cause Char
syndrome. As the BTB domain of KCTD1 binds to the activation domain
of AP-2α and suppresses its transcriptional activity (13), it was investigated whether these
KCTD1 mutations affect AP-2α transcriptional activity (Fig. 1A). An A2 reporter plasmid with
three AP-2 binding sites was transiently transfected in the
presence or absence of pCMV-Myc-KCTD1 wild-type or KCTD1 mutants
into 293 cells. Consistent with previous results (13) wild-type KCTD1 overexpression
significantly decreased A2 luciferase activity, whereas 10 KCTD1
mutants had no effect on the transcriptional inhibition of AP-2α,
compared with wild-type KCTD1 (Fig.
1B).
The present study also investigated the effect of
KCTD1 on the transcriptional activity of AP-2α with mutations in
the PY motif (Fig. 1C), which is
required for a normal developmental phenotype (5). KCTD1 markedly repressed A2 luciferase
activity of AP-2α P60A and P61R, but not of AP-2α P59A or 4A,
indicating that the AP-2α mutation of amino acid 59 abrogated KCTD1
regulation (Fig. 1D). Taken
together, these data revealed that pathogenic mutations in KCTD1 or
AP-2α affect their regulatory mechanisms.
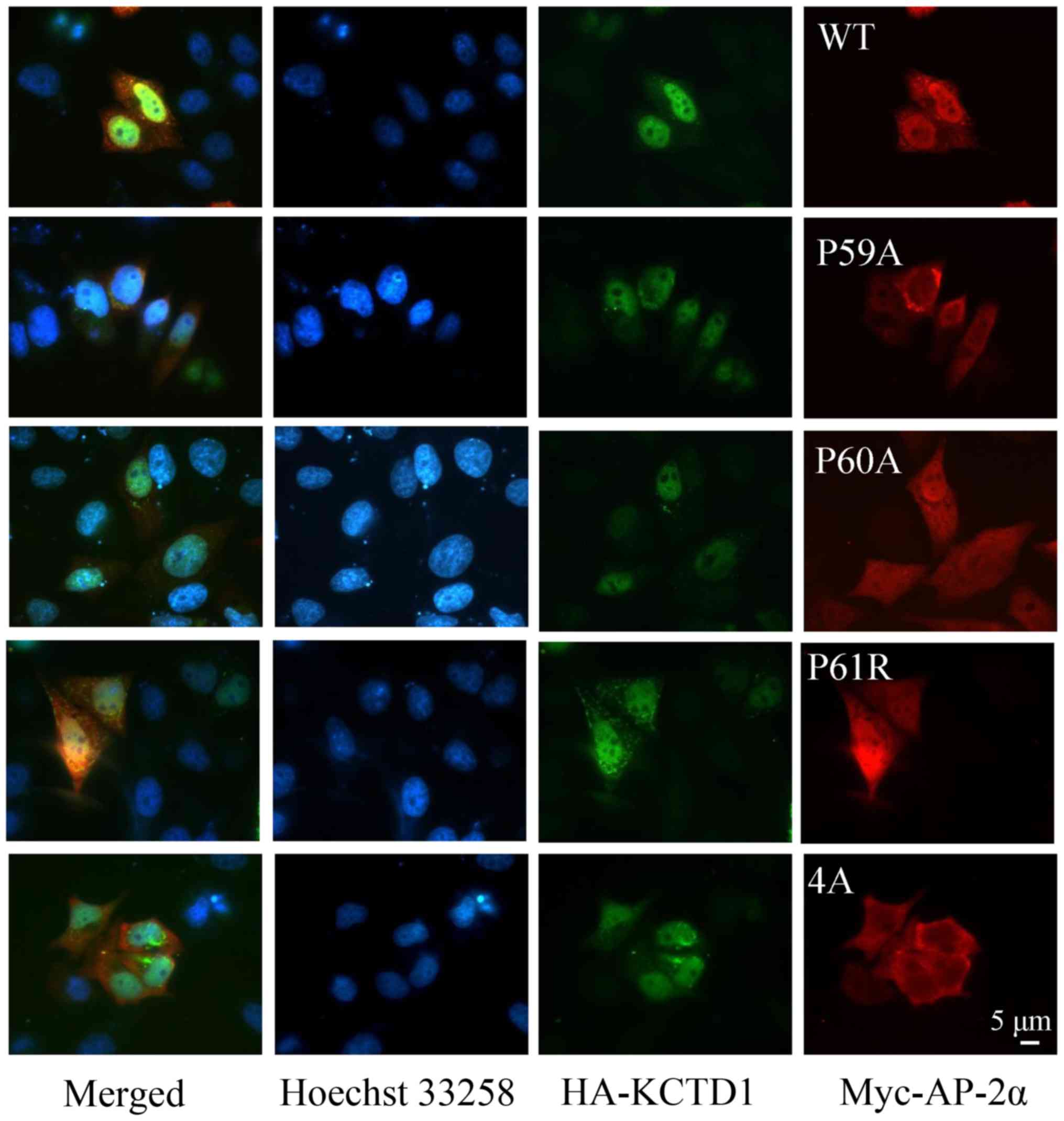
Altered cellular localization of KCTD1
and AP-2α mutants, compared with wild-type
Plasmids encoding mutant KCTD1 and AP-2α proteins
were transfected into 293 cells to investigate the localization of
both mutant proteins using immunofluorescence (Figs. 2 and 3, respectively). It was observed that
wild-type KCTD1 localized with wild-type AP-2α, consistent with
previously reported results (13).
The KCTD1 mutants (A30E, P31R, P31L, H33Q, G62D, D69E and H74P)
were distributed throughout the whole cell, predominantly exhibited
strong fluorescence in the nucleus, and co-localized with AP-2α
(Fig. 2). However, localization of
KCTD1 P20S was detected in the nuclear membrane. KCTD1 P31H was
localized in the nuclear membrane and the cytoplasm (Fig. 2A). KCTD1 H33P clustered in an
aggregated pattern in a significant portion of the cells (Fig. 2B). These data suggested that
wild-type KCTD1 and its mutant proteins primarily localized in the
nucleus, but KCTD1 mutants with mutated amino acids 20 and 31
localized to the nuclear membrane. In addition, wild-type AP-2α and
mutant P60A and P61R protein appeared to demonstrate strong
fluorescence in the nucleus and co-localized with KCTD1 protein.
However, the AP-2α P59A and 4A proteins were predominantly detected
in the cytoplasm and showed no overlap with KCTD1 protein (Fig. 3). These observations were
indicative of altered localization of KCTD1 and AP-2α.
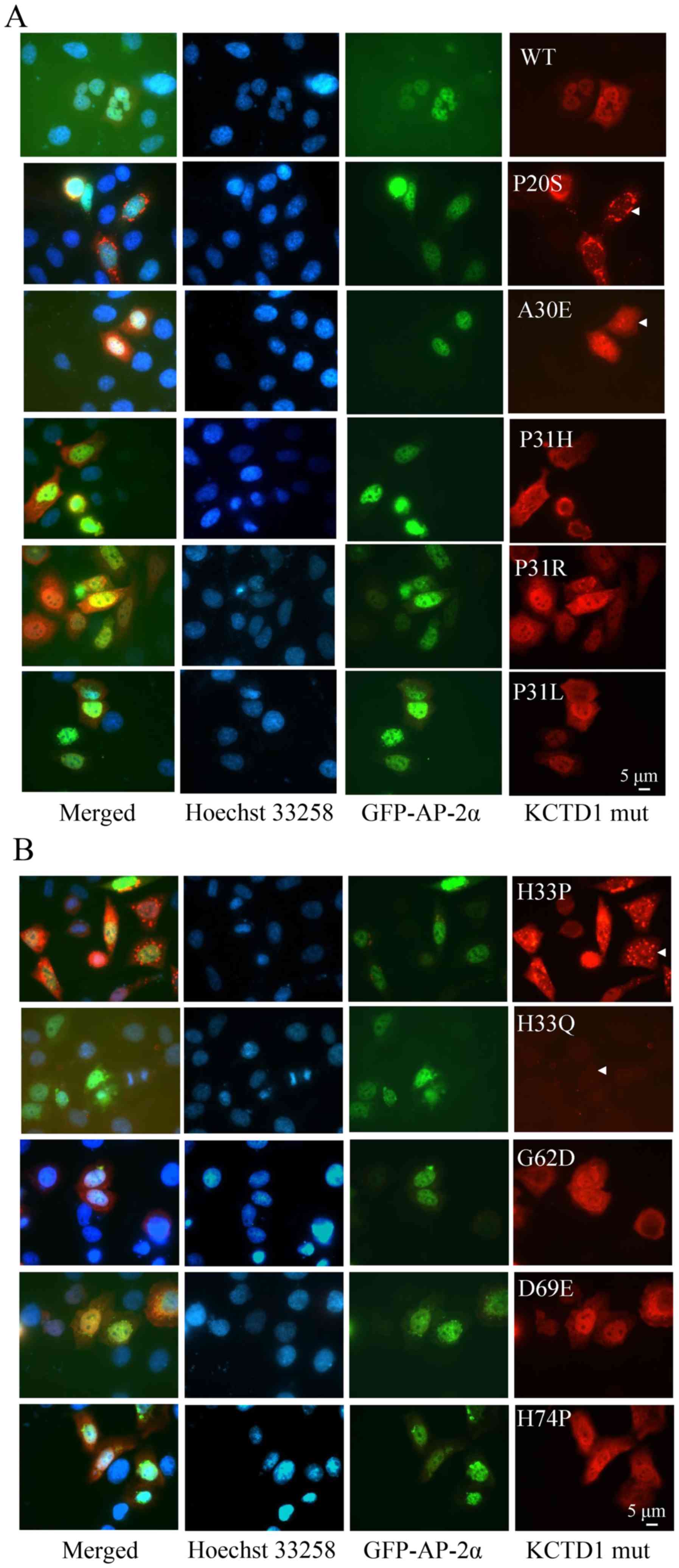 | Figure 2.Cellular localization of KCTD1
mutants. HeLa cells were transiently transfected with the
expression plasmids pEGFP-C1-AP-2α and pCMV-Myc-wild-type KCTD1 or
pCMV-Myc-KCTD1 mutants. (A) Localization of wild-type AP-2α and
wild-type KCTD1 or KCTD1 mutants including P20S, A30E, P31R, P31L,
P31H. (B) Localization of wild-type AP-2α and KCTD1 mutants
including H33P, H33Q, G62D, D69E and H74P. Images were captured at
36 h after transfection. Green, GFP-AP-2α expression; blue, Hoechst
33258 nuclear stain; red, KCTD1 WT and mutants; yellow indicates
overlapping expression. GFP, green fluorescent protein; KCTD1,
potassium-channel tetramerization-domain-containing 1; mut, mutant;
wt, wild-type. |
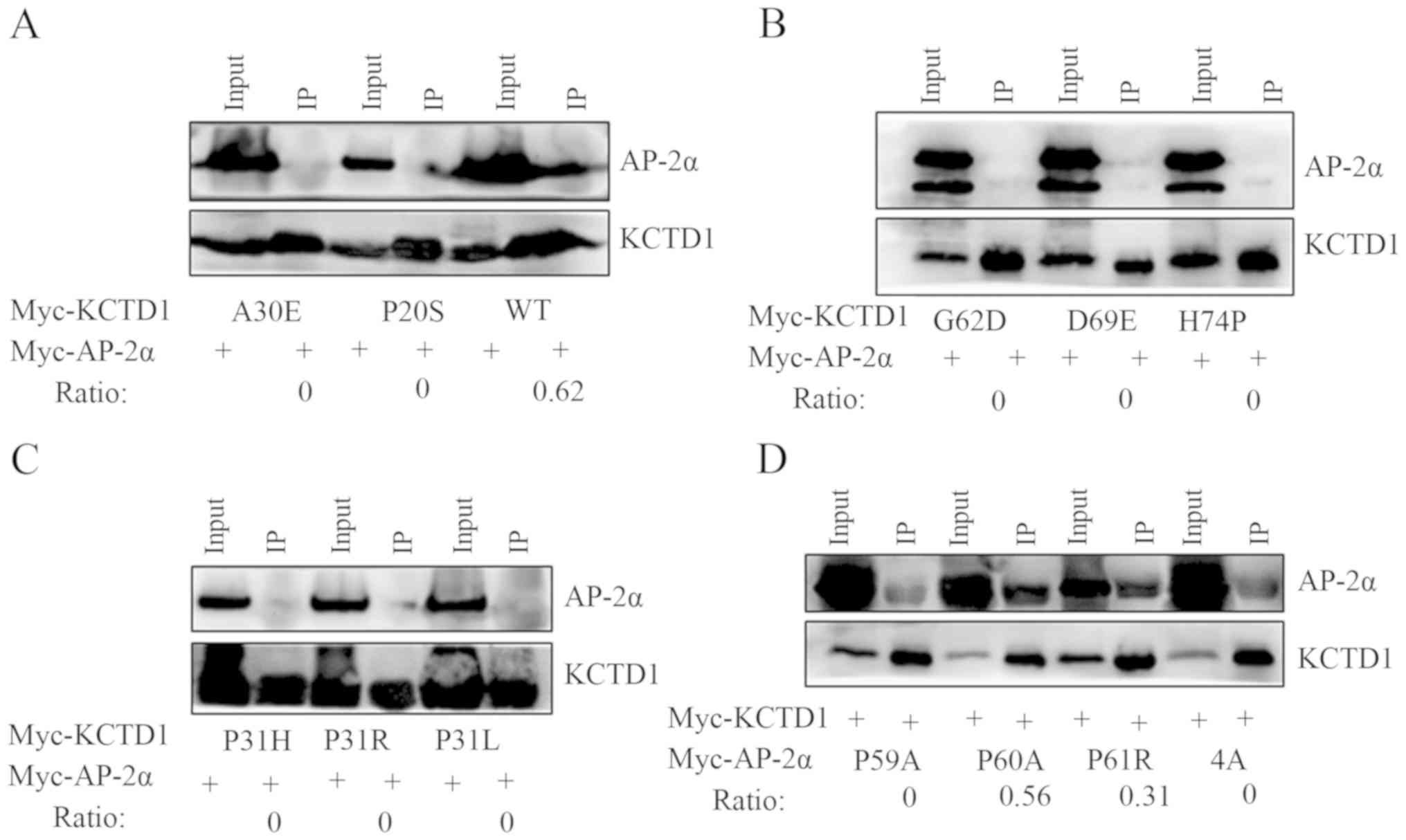
Protein interactions are altered
between KCTD1 mutants/wild-type AP-2α and wild-type KCTD1/AP-2α
P59A mutant
Coimmunoprecipitation analysis was performed to
investigate in vitro interactions between AP-2α and KCTD1
mutants. Anti-KCTD1 antibodies immunoprecipitated the wild-type
KCTD1/AP-2α complex but did not precipitate the KCTD1 mutant/AP-2α
protein complex (Fig. 4A-C).
Moreover, consistent with luciferase assay results, KCTD1 formed a
complex with AP-2α P60A and P61R, but not with AP-2α P59A or 4A
(Fig. 4D). These results
demonstrated that the KCTD1 mutants did not interact with AP-2α,
although some nucleic co-localization was observed for the AP-2α
and KCTD1 mutants, which indicated that KCTD1 mutations in the BTB
domain reduce its suppressive effects on AP-2α. However, KCTD1
continued to inhibit the transcriptional activation of AP-2α P60A
and P61R and formed a complex through protein-protein
interaction.
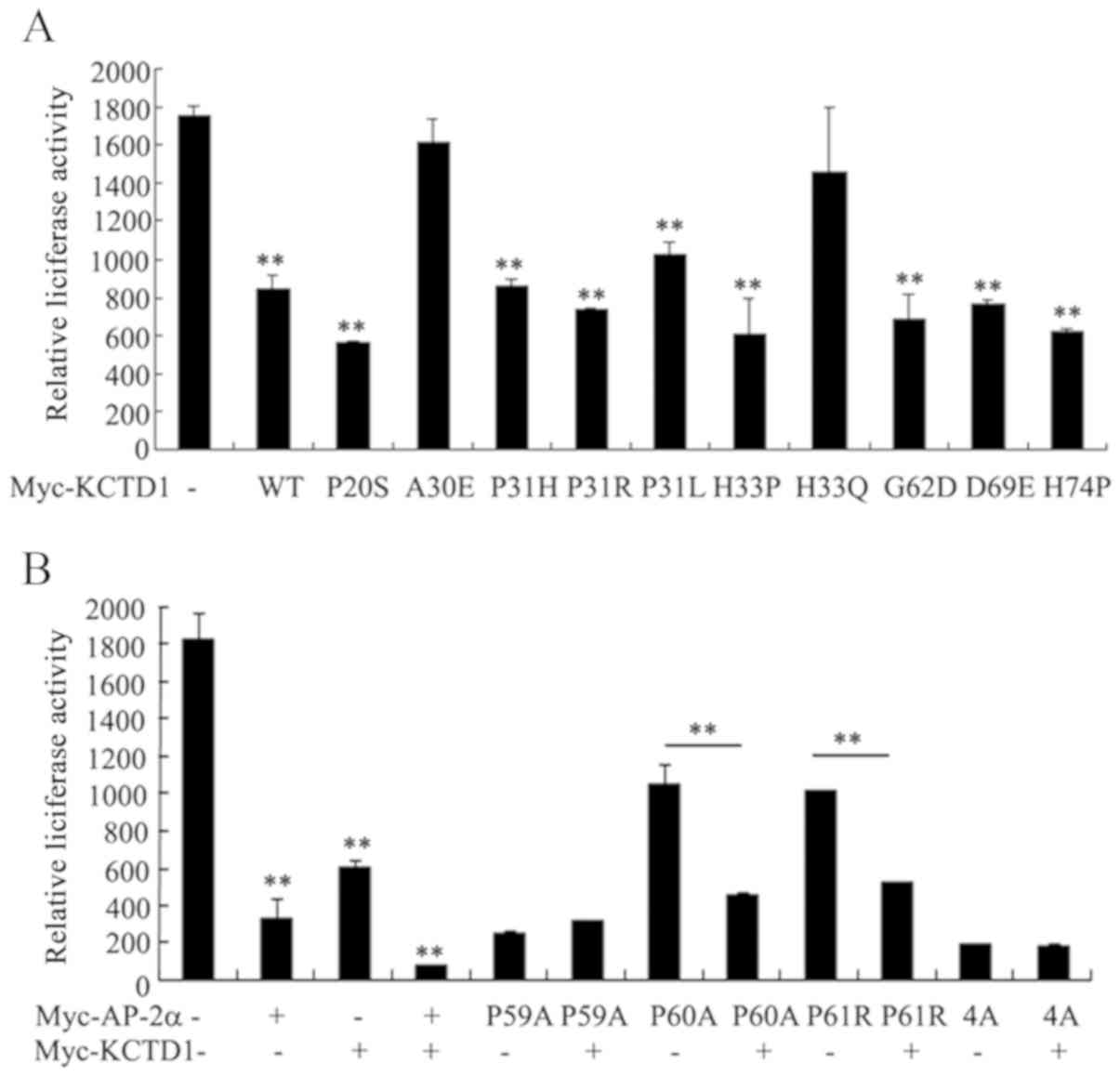
Differential regulation of KCTD1 and
AP-2α mutants on the Wnt/β-catenin signaling pathway
Wnt/β-catenin signaling serves a key role in
embryonic development and various diseases including bone disease
including osteoporosis-pseudoglioma syndrome, cardiometabolic
disease and Parkinson's disease (22–25).
The present study investigated the effects of KCTD1 mutants on the
canonical Wnt/β-catenin signaling pathway. The TOPFLASH reporter
plasmid was transfected into 293 cells in the presence or absence
of pCMV-Myc-KCTD1 wild-type or KCTD1 mutants. The results
demonstrated that wild-type KCTD1 overexpression markedly
suppressed the Wnt pathway-responsive TOPFLASH reporter gene
activity (Fig. 5A). In addition,
the majority of KCTD1 mutants strongly inhibited the
transcriptional activity of the TOPFLASH reporter. KCTD1 A30E or
H33Q mutants had no effect on the transcriptional activity of the
TOPFLASH reporter, but eight other KCTD1 mutants significantly
decreased the transcription activity of the TOPFLASH reporter gene
to a similar extent as the wild-type KCTD1. In the H33Q KCTD1
mutant, the change from polar and charged His33 to a polar residue,
Gln (Q), did not inhibit TOPFLASH reporter activity. By contrast,
the change from His to Pro in the H33P mutant inhibited the
TOPFLASH reporter activity. Collectively, these results suggested
that Ala 30 and His 33 were important for KCTD1-mediated regulation
of Wnt/β-catenin signaling (Fig.
5A). These data indicate that the pathogenic mechanisms
underlying certain KCTD1 mutants are primarily due to suppressed
Wnt/β-catenin signaling, resulting in specific abnormal phenotypes
in SEN syndrome.
AP-2α has been reported to inhibit TOPFLASH reporter
activity in a manner similar to KCTD1 (17,18).
AP-2α P59A and AP-2α 4A suppressed TOPFLASH activity to a similar
degree as wild-type AP-2α (Fig.
5B); AP-2α P60A and P61R also partially inhibited TOPFLASH
reporter activity. Notably, inhibition of TOPFLASH reporter
activity by AP-2α P60A and P61R was enhanced in cells
co-transfected with wild-type KCTD1, but KCTD1 had no effect on the
TOPFLASH reporter activity of AP-2α P59A or 4A (Fig. 5B). These results supported the
hypothesis that the AP-2α P59A mutant downregulates the
Wnt/β-catenin signaling pathway, which is unaffected by KCTD1.
Finally, western blotting was performed to measure
the influence of AP-2α and KCTD1 proteins on the Wnt/β-catenin
pathway. KCTD1 and AP-2α overexpression (individually and in
combination) decreased β-catenin expression levels (Fig. 6A); downstream CCND1 expression
levels were also downregulated. Moreover, KCTD1 and AP-2α decreased
the survival of the SW480 cells, and 20 µM chemotherapeutic drug
cisplatin (DDP) exhibited a stronger inhibitory effect on the
viability of SW480 cells transfected with KCTD1 and/or AP-2α,
compared with the normal saline (NS) group (Fig. 6B). These results demonstrated that
KCTD1 and AP-2α downregulated β-catenin expression levels in the
canonical Wnt pathway and inhibited SW480 cell viability,
suggesting KCTD1 and AP-2α might regulate these syndromes through
the Wnt/β-catenin pathway.
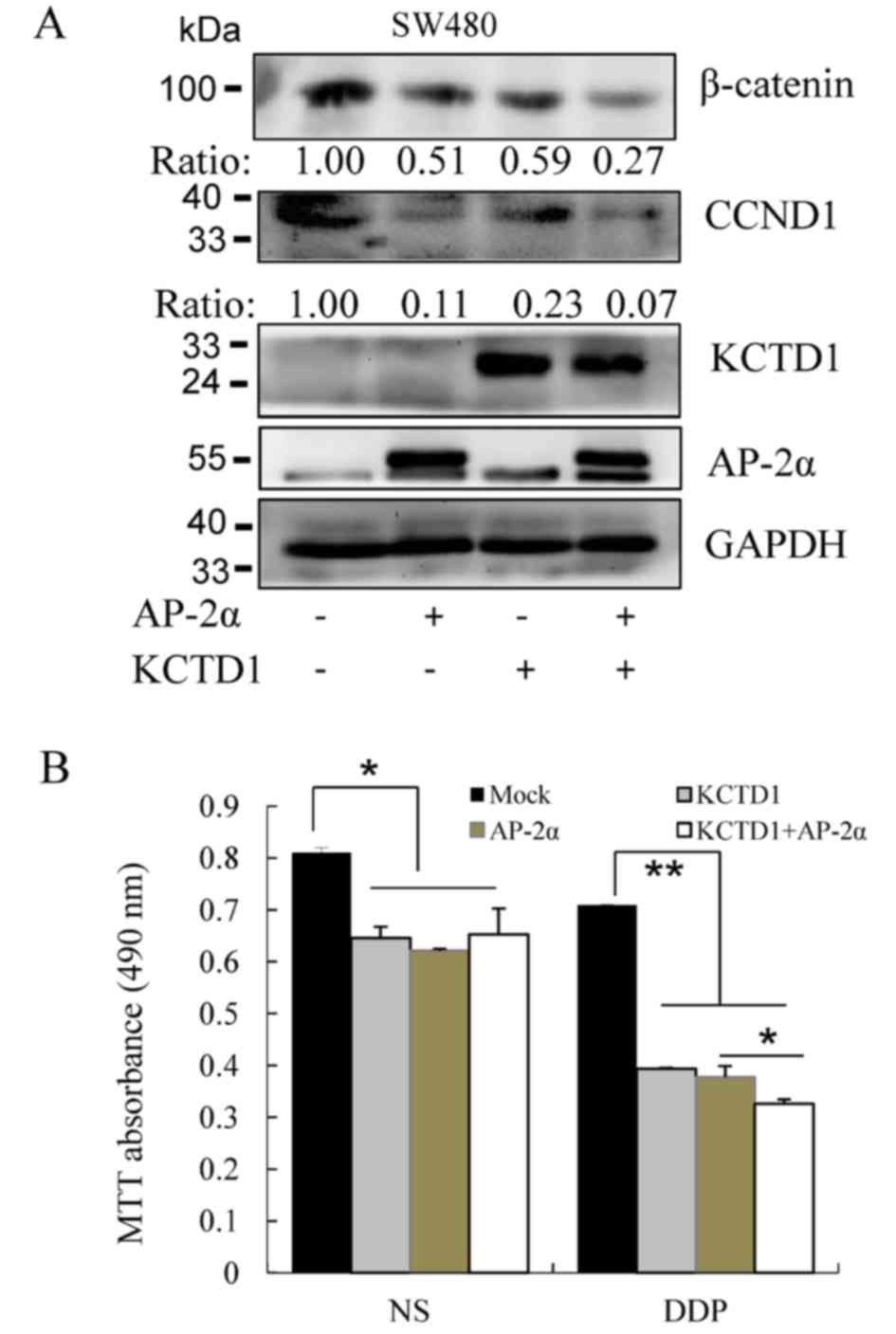 | Figure 6.KCTD1 and AP-2α decrease SW480 cell
viability by downregulating β-catenin expression levels. (A) SW480
cells were transiently transfected with pCMV-Myc (Mock),
pCMV-Myc-KCTD1 and/or pCMV-Myc-AP-2α plasmids. At 24 h after
transfection, total cell lysates were harvested and detected by
western blotting using mouse monoclonal antibodies against
β-catenin, Myc-tag, CCND1 and GAPDH. GAPDH served as a loading
control for protein normalization. (B) Effects of overexpressed
KCTD1, AP-2α or DDP treatment on SW480 cells. SW480 cells were
transiently transfected with pCMV-Myc-KCTD1 and/or pCMV-Myc-AP-2α
plasmids and treated with or without 20 µM DDP for 24 h. Cell
viability was determined by MTT assays and absorbance was measured
at 490 nm. *P<0.05, **P<0.01 compared with controls (the Mock
group or the AP-2α group). KCTD1, potassium-channel
tetramerization-domain-containing 1; CCND1, cyclin D1; DDP,
cisplatin; NS, normal saline. |
Discussion
The BTB domain is crucial for transcriptional
regulation; proteins with a BTB domain primarily function as
transcriptional repressors (26–28).
BTB proteins, such as Bcl-6 and promyelocytic leukemia zinc-finger
(PLZF), are involved in a number of developmental processes, such
as limb formation, gastrulation, ovary morphogenesis, eye
development and hematopoietic stem cell fate determination
(29–33). The Bcl6 mutant does not interact
with co-repressors through its BTB domain, which results in
defective B cell proliferation and survival, disrupted formation of
germinal centers and suppressed affinity maturation of
immunoglobulin (34).
Specifically, residues 33–35 form the floor of the pocket and
residues 63, 64 and 66–68 form each wall in the secondary structure
of PLZF BTB domain (35). All
known KCTD mutants in SEN syndrome are caused by missense
mutations, and the BTB pocket is formed by a number of residues
(3). KCTD1 mutations associated
with SEN syndrome abrogate KCTD1 transcriptional repression
activity on transcription factor AP-2α (13). These missense mutations result in
the loss of KCTD1 function in a dominant-negative manner. This is
consistent with the present study results, which demonstrated that
alterations in the KCTD1 BTB domain had no effect on the
transcriptional repression of AP-2α owing to a lack of interaction,
despite a degree of co-localization. Notably, the present study
revealed that KCTD1 H33P was localized in a punctate pattern in the
cells. These findings were consistent with a previous study in
which amyloid precursor protein (APP) exhibited a punctate pattern
of immunofluorescence indicative of internalization from the cell
surface to an endosomal compartment (36), indicating that KCTD1 H33P may serve
important roles in internalization by endocytosis. To investigate
the function of KCTD1 mutants in maintaining a normal tissue
phenotype, as well as the regulatory association between KCTD1
mutants and Wnt/β-catenin signaling, normal cell lines, such as 293
cells, were selected as previously described (18). The majority of KCTD1 BTB domain
mutants suppressed the Wnt/β-catenin pathway, indicating that
substitutions of amino acids in cases of SEN syndrome do not wholly
disrupt transcriptional repression caused by BTB domain mutations
in KCTD1, although only KCTD1 A30E and H33Q displayed unchanged
TOPFLASH reporter activity. It was hypothesized that KCTD1
mutations that cause SEN syndrome may result in abnormal regulation
of AP-2α and developmental defects.
Char syndrome is characterized by patent ductus
arteriosis, facial dysmorphism and incurving fifth fingers
(37). The PY motif in AP-2
protein family is associated with Char syndrome as autosomal
dominant diseases (7). The PY
motif is conserved in the transcription activation domain of AP-2
proteins and KCTD1 binds to this transactivation domain. KCTD1
inhibited the transcriptional activation of AP-2α by three-fold,
whereas KCTD1 only suppressed the transcriptional activity of AP-2α
P60A and P61R by one-half, and did not decrease the transcriptional
activity of AP-2α P59A (Fig. 1D),
indicating that regulatory the AP-2α mutation could been influenced
by differential regulation of KCTD1.
The Wnt/β-catenin signaling pathway serves a key
role in embryonic developmental processes (22,38,39)
and carcinogenesis (40–42). Truncations in APC are frequently
found in familial adenomatous polyposis, a dominantly inherited
disease characterized by polyps in the colon and rectum (43,44).
Mutations in β-catenin and APC cause sporadic colon cancer and
other types of tumor, such as liver cancer and intraductal
papillary neoplasms of the bile duct (45–47).
KCTD1 directly binds to β-catenin, enhances its degradation and
inhibits the canonical Wnt/β-catenin signaling pathway (18). In the present study, AP-2α and
KCTD1 downregulated β-catenin expression levels in individual
transfection experiments and decreased SW480 cell viability,
although both of these proteins exhibited stronger inhibitory
effects on β-catenin protein levels and cell viability in
co-transfection experiments, potentially due to the negative
regulatory association between AP-2α and KCTD1. The chemotherapy
drug cisplatin enhanced the influence of both genes on SW480 cell
viability.
In summary, the present data demonstrated that KCTD1
mutants causing SEN syndrome did not bind AP-2α and exhibited no
inhibitory effects on AP-2α, and that KCTD1 was not associated with
AP-2α P59A-induced Char syndrome. The interactions between KCTD1
mutants/wild-type AP-2α or wild-type KCTD1/AP-2α mutants were
abrogated as a potential effect of altered localization, including
KCTD1 mutants P20S, P31H, H33P and AP-2α mutants P59A and 4A.
However, the majority of KCTD1 and AP-2α mutants inhibited TOPFLASH
reporter activity to different extents, and wild-type KCTD1 and
AP-2α downregulated β-catenin expression levels and decreased SW480
cell viability. Further investigation using an animal model is
required to fully elucidate the regulatory mechanisms of these
proteins in SEN and Char syndrome.
Supplementary Material
Supporting Data
Acknowledgements
The authors would like to thank Dr Limin Li (Hunan
Normal University, Changsha, China) for critical editing of the
manuscript.
Funding
The present study was supported by The National
Natural Science Foundation of China (grant no. 81872256, 81770389
and 81272190), and The Cooperative Innovation Center of Engineering
and New Products for Developmental Biology of Hunan Province (grant
no. 20134486).
Availability of data and materials
The datasets used and analyzed during this study are
available from the corresponding author on reasonable request.
Authors' contributions
XD designed the present study and wrote the
manuscript. LH, LC, WH, ZY, LY, XL, QL and JQ performed the
experiments and analyzed the data. All authors read and approved
the final version of the manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Finlay AY and Marks R: An hereditary
syndrome of lumpy scalp, odd ears and rudimentary nipples. Br J
Dermatol. 99:423–430. 1978. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
van Steensel MA, Celli J, van Bokhoven JH
and Brunner HG: Probing the gene expression database for candidate
genes. Eur J Hum Genet. 7:910–919. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Marneros AG, Beck AE, Turner EH, McMillin
MJ, Edwards MJ, Field M, de Macena Sobreira NL, Perez ABA, Fortes
JAR, Lampe AK, et al: Mutations in KCTD1 cause scalp-ear-nipple
syndrome. Am J Hum Genet. 92:621–626. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Zhang J, Hagopian-Donaldson S, Serbedzija
G, Elsemore J, Plehn-Dujowich D, McMahon AP, Flavell RA and
Williams T: Neural tube, skeletal and body wall defects in mice
lacking transcription factor AP-2. Nature. 381:238–241. 1996.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Ahituv N, Erven A, Fuchs H, Guy K,
Ashery-Padan R, Williams T, de Angelis MH, Avraham KB and Steel KP:
An ENU-induced mutation in AP-2alpha leads to middle ear and ocular
defects in doarad mice. Mamm Genome. 15:424–432. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Sletten LJ and Pierpont ME: Familial
occurrence of patent ductus arteriosus. Am J Med Genet. 57:27–30.
1995. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Zhao F, Weismann CG, Satoda M, Pierpont
ME, Sweeney E, Thompson EM and Gelb BD: Novel TFAP2B mutations that
cause char syndrome provide a genotype-phenotype correlation. Am J
Hum Genet. 69:695–703. 2001. View
Article : Google Scholar : PubMed/NCBI
|
|
8
|
Li H, Sheridan R and Williams T: Analysis
of TFAP2A mutations in branchio-oculo-facial syndrome indicates
functional complexity within the AP-2α DNA-binding domain. Hum Mol
Genet. 22:3195–3206. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Milunsky JM, Maher TA, Zhao G, Roberts AE,
Stalker HJ, Zori RT, Burch MN, Clemens M, Mulliken JB, Smith R and
Lin AE: TFAP2A mutations result in branchio-oculo-facial syndrome.
Am J Hum Genet. 82:1171–1177. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Milunsky JM, Maher TM, Zhao G, Wang Z,
Mulliken JB, Chitayat D, Clemens M, Stalker HJ, Bauer M, Burch M,
et al: Genotype-phenotype analysis of the branchio-oculo-facial
syndrome. Am J Med Genet A 155A. 22–32. 2011. View Article : Google Scholar
|
|
11
|
Dumitrescu AV, Milunsky JM, Longmuir SQ
and Drack AV: A family with branchio-oculo-facial syndrome with
primarily ocular involvement associated with mutation of the TFAP2A
gene. Ophthalmic Genet. 33:100–106. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Thomeer HG, Crins TT, Kamsteeg EJ,
Buijsman W, Cruysberg JR, Knoers NV and Cremers CWRJ: Clinical
presentation and the presence of hearing impairment in
branchio-oculo-facial syndrome: A new mutation in the TFAP2A gene.
Ann Otol Rhinol Laryngol. 119:806–814. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Ding X, Luo C, Zhou J, Zhong Y, Hu X, Zhou
F, Ren K, Gan L, He A, Zhu J, et al: The interaction of KCTD1 with
transcription factor AP-2alpha inhibits its transactivation. J Cell
Biochem. 106:285–295. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Zarelli VE and Dawid IB: Inhibition of
neural crest formation by Kctd15 involves regulation of
transcription factor AP-2. Proc Natl Acad Sci USA. 110:2870–2875.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Heffer A, Marquart GD, Aquilina-Beck A,
Saleem N, Burgess HA and Dawid IB: Generation and characterization
of Kctd15 mutations in zebrafish. PLoS One. 12:e01891622017.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Dutta S and Dawid IB: Kctd15 inhibits
neural crest formation by attenuating Wnt/beta-catenin signaling
output. Development. 137:3013–3018. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Li Q and Dashwood RH: Activator protein
2alpha associates with adenomatous polyposis coli/beta-catenin and
inhibits beta-catenin/T-cell factor transcriptional activity in
colorectal cancer cells. J Biol Chem. 279:45669–45675. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Li X, Chen C, Wang F, Huang W, Liang Z,
Xiao Y, Wei K, Wan Z, Hu X, Xiang S, et al: KCTD1 suppresses
canonical Wnt signaling pathway by enhancing β-catenin degradation.
PLoS One. 9:e943432014. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Ding X, Fan C, Zhou J, Zhong Y, Liu R, Ren
K, Hu X, Luo C, Xiao S, Wang Y, et al: GAS41 interacts with
transcription factor AP-2beta and stimulates AP-2beta-mediated
transactivation. Nucleic Acids Res. 34:2570–2578. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Ding X, Yang Z, Zhou F, Wang F, Li X, Chen
C, Li X, Hu X, Xiang S and Zhang J: Transcription factor AP-2α
regulates acute myeloid leukemia cell proliferation by influencing
hoxa gene expression. Int J Biochem Cell Biol. 45:1647–1656. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Ding XF, Luo C, Ren KQ and Zhang J, Zhou
JL, Hu X, Liu RS, Wang Y, Gao X and Zhang J: Characterization and
expression of a human KCTD1 gene containing the BTB domain, which
mediates transcriptional repression and homomeric interactions. DNA
Cell Biol. 27:257–265. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Clevers H: Wnt/beta-catenin signaling in
development and disease. Cell. 127:469–480. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Baron R and Kneissel M: WNT signaling in
bone homeostasis and disease: From human mutations to treatments.
Nat Med. 19:179–192. 2013. View
Article : Google Scholar : PubMed/NCBI
|
|
24
|
Gay A and Towler DA: Wnt signaling in
cardiovascular disease: Opportunities and challenges. Curr Opin
Lipidol. 28:387–396. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Ng L, Kaur P, Bunnag N, Suresh J, Sung
ICH, Tan QH, Gruber J and Tolwinski NS: WNT signaling in disease.
Cells. 8:8262019. View Article : Google Scholar
|
|
26
|
Li X, Peng H, Schultz DC, Lopez-Guisa JM,
Rauscher FJ III and Marmorstein R: Structure-function studies of
the BTB/POZ transcriptional repression domain from the
promyelocytic leukemia zinc finger oncoprotein. Cancer Res.
59:5275–5282. 1999.PubMed/NCBI
|
|
27
|
Fedele M, Benvenuto G, Pero R, Majello B,
Battista S, Lembo F, Vollono E, Day PM, Santoro M, Lania L, et al:
A novel member of the BTB/POZ family, PATZ, associates with the
RNF4 RING finger protein and acts as a transcriptional repressor. J
Biol Chem. 275:7894–7901. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Hoatlin ME, Zhi Y, Ball H, Silvey K,
Melnick A, Stone S, Arai S, Hawe N, Owen G, Zelent A and Licht JD:
A novel BTB/POZ transcriptional repressor protein interacts with
the fanconi anemia group C protein and PLZF. Blood. 94:3737–3747.
1999. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Kelly KF and Daniel JM: POZ for
effect--POZ-ZF transcription factors in cancer and development.
Trends Cell Biol. 16:578–587. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Kojima S, Hatano M, Okada S, Fukuda T,
Toyama Y, Yuasa S, Ito H and Tokuhisa T: Testicular germ cell
apoptosis in Bcl6-deficient mice. Development. 128:57–65.
2001.PubMed/NCBI
|
|
31
|
Shaffer AL, Yu X, He Y, Boldrick J, Chan
EP and Staudt LM: BCL-6 represses genes that function in lymphocyte
differentiation, inflammation, and cell cycle control. Immunity.
13:199–212. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Barna M, Hawe N, Niswander L and Pandolfi
PP: Plzf regulates limb and axial skeletal patterning. Nat Genet.
25:166–172. 2000. View
Article : Google Scholar : PubMed/NCBI
|
|
33
|
Costoya JA, Hobbs RM, Barna M, Cattoretti
G, Manova K, Sukhwani M, Orwig KE, Wolgemuth DJ and Pandolfi PP:
Essential role of Plzf in maintenance of spermatogonial stem cells.
Nat Genet. 36:653–659. 2004. View
Article : Google Scholar : PubMed/NCBI
|
|
34
|
Huang C, Hatzi K and Melnick A:
Lineage-specific functions of Bcl-6 in immunity and inflammation
are mediated by distinct biochemical mechanisms. Nat Immunol.
14:380–388. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Ahmad KF, Engel CK and Prive GG: Crystal
structure of the BTB domain from PLZF. Proc Natl Acad Sci USA.
95:12123–12128. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Carey RM, Balcz BA, Lopez-Coviella I and
Slack BE: Inhibition of dynamin-dependent endocytosis increases
shedding of the amyloid precursor protein ectodomain and reduces
generation of amyloid beta protein. BMC Cell Biol. 6:302005.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Char F: Peculiar facies with short
philtrum, duck-bill lips, ptosis and low-set ears-a new syndrome?
Birth Defects Orig Artic Ser. 14:303–305. 1978.PubMed/NCBI
|
|
38
|
Marston DJ, Roh M, Mikels AJ, Nusse R and
Goldstein B: Wnt signaling during caenorhabditis elegans embryonic
development. Methods Mol Biol. 469:103–111. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Munoz-Descalzo S, Hadjantonakis AK and
Arias AM: Wnt/ß-catenin signalling and the dynamics of fate
decisions in early mouse embryos and embryonic stem (ES) cells.
Semin Cell Dev Biol. 47-48:101–109. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Wang B, Tian T, Kalland KH, Ke X and Qu Y:
Targeting Wnt/β-catenin signaling for cancer immunotherapy. Trends
Pharmacol Sci. 39:648–658. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Wang W, Smits R, Hao H and He C:
Wnt/β-catenin signaling in liver cancers. Cancers (Basel).
11:9262019. View Article : Google Scholar
|
|
42
|
Jung YS and Park JI: Wnt signaling in
cancer: Therapeutic targeting of Wnt signaling beyond β-catenin and
the destruction complex. Exp Mol Med. 52:183–191. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Nishisho I, Nakamura Y, Miyoshi Y, Miki Y,
Ando H, Horii A, Koyama K, Utsunomiya J, Baba S and Hedge P:
Mutations of chromosome 5q21 genes in FAP and colorectal cancer
patients. Science. 253:665–669. 1991. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Nakamura Y, Nishisho I, Kinzler KW,
Vogelstein B, Miyoshi Y, Miki Y, Ando H, Horii A and Nagase H:
Mutations of the adenomatous polyposis coli gene in familial
polyposis coli patients and sporadic colorectal tumors. Princess
Takamatsu Symp. 22:285–292. 1991.PubMed/NCBI
|
|
45
|
Suraweera N, Robinson J, Volikos E,
Guenther T, Talbot I, Tomlinson I and Silver A: Mutations within
Wnt pathway genes in sporadic colorectal cancers and cell lines.
Int J Cancer. 119:1837–1842. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Colnot S: Focusing on beta-catenin
activating mutations to refine liver tumor profiling. Hepatology.
64:1850–1852. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Fujikura K, Akita M, Ajiki T, Fukumoto T,
Itoh T and Zen Y: Recurrent mutations in APC and CTNNB1 and
activated Wnt/β-catenin signaling in intraductal papillary
neoplasms of the bile duct: A whole exome sequencing study. Am J
Surg Pathol. 42:1674–1685. 2018. View Article : Google Scholar : PubMed/NCBI
|