Introduction
Ischemic stroke refers to a type of cerebrovascular
disease caused by the occlusion of a cerebral blood vessel,
disrupting the supply of nutrients and oxygen to the brain and
ultimately leading to brain tissue necrosis and neurological damage
(1). As the second leading cause
of death in industrialized countries, ischemic injury accounts for
87% of all strokes (2). It has
been reported that ~20% of stroke survivors need institutional
care, and 15–30% of stroke survivors remain permanently disabled
(3). Intravenous recombinant
tissue plasminogen activator is an effective treatment of ischemia;
however, only 5% patients are eligible for treatment, and when
administered for >4.5 h, the risk of intracranial hemorrhage may
exceed its benefit (4). Previous
studies have demonstrated that cerebral ischemia may lead to
apoptosis and neuronal damage in the ischemic area (5), although the subtle underlying
mechanisms are not fully understood. Therefore, clarifying the
molecular mechanism of cerebral ischemia may provide new insights
into ischemic stroke. MicroRNAs (miRNAs/miRs) are small non-coding
single-stranded RNA molecules ~22 nucleotides long that regulate
gene expression after transcription by inhibiting translation or
inducing the degradation of the target gene mRNA (6). In recent years, increasing studies
have suggested that miRNAs serve vital roles in ischemic stroke
(7). In addition, experimental
therapies based on miRNAs have been developed to assist post-stroke
neurological recovery and mitigate ischemic brain injury (8,9).
Chang et al (10) have
reported that miR-195 ameliorates ischemic stroke through
inhibiting neuronal apoptosis in a middle cerebral artery occlusion
(MCAO) model of brain ischemia. miR-183, which belongs to the
evolutionarily conserved miRNA cluster, is located on human
chromosome 7 and serves a number of functions in key cellular
processes, including the development of neurons (11). Lin et al (12) have demonstrated that ischemic
post-conditioning increases miR-183-5p levels in the murine liver
compared with those in ischemia-reperfusion (I/R) mice.
Furthermore, miR-183-5p upregulation alleviates liver injury after
I/R by targeting apoptotic protease-activating factor 1. Gong et
al (13) have reported that
knockdown of long non-coding RNA maternally expressed 3 upregulates
the levels of miR-183-5p, thus protecting H9c2 rat cardiomyocytes
from hypoxic injury. However, to the best of our knowledge, whether
miR-183-5p mitigates cerebral ischemia injury has not been
reported.
The phosphatase and tensin homolog (PTEN) gene,
located on human chromosome 19, encodes a dual protein phosphatase
enzyme that dephosphorylates protein and lipid substrates (14). A previous study has revealed
increased expression levels of PTEN in MCAO or oxygen-glucose
deprivation (OGD)-induced ischemic stroke (15). In addition, knockdown of miR-183-5p
upregulated PTEN expression in multiple types of tumor (including
synovial sarcoma, rhabdomyosarcoma and colon cancer) and T
cell-dependent autoimmune diseases (16,17).
Based on these findings, the current study hypothesized that
miR-183-5p may reduce cerebral ischemic injury by regulating PTEN.
This hypothesis was verified by inducing transient focal cerebral
ischemia in mice exposed to MCAO and exposing Neuro-2A
neuroblastoma (N2A) cells to OGD.
Materials and methods
Animal model of focal cerebral
ischemia
In the present study, a total of 112 male 8-week-old
C57BL/6 mice were purchased from Changsheng Bio-technology Co.,
Ltd. All mice were maintained in a controlled environment with a
temperature of 21–23°C and a humidity of 45–55% under a 12-h
light/dark cycle with free access to food and water. After adaptive
feeding for 1 week, mice were randomly divided into the sham group
(n=32) and the experimental group (n=80). Animals in the
experimental group were subjected to cerebral ischemia using the
MCAO method as previously described (18). Mice were anesthetized with 50 mg/kg
pentobarbital sodium. The common carotid artery and the vagus nerve
were exposed by a midline neck incision. Then, the internal carotid
artery (ICA) and external carotid artery (ECA) were bluntly
isolated. After a small opening was made between the ligation (at
the distal end of ECA) and the clipping (at the proximal end of ECA
and CCA), a plug (diameter, 0.22–0.23 mm) was inserted to block the
blood flow of the middle cerebral artery, followed by suturing of
the skin. The plug was pulled out 1 h after ischemia, and
subsequent experiments were performed 24 h after reperfusion. For
the sham surgery, all procedures were identical, with the exception
that the plug was not inserted. All procedures using laboratory
animals were in accordance with the Institutional Animal Care and
Use Committee of Binzhou People's Hospital (approval no.
LYP200).
The mice were randomly divided into four groups: i)
Sham (n=32); ii) MCAO (n=32); iii) MCAO + agomiR-183-5p (n=24); and
iv) MCAO + agomiR-negative control (NC) (n=24). In each group, mice
were used to evaluate mRNA and protein expression (n=8), determine
the cerebral infarct area (n=8), evaluate cerebral edema (n=8) and
determine neurological scores by histopathological analysis of the
ischemic penumbra (n=8).
AgomiR-183-5p transfection into the
mouse brain
The miR-183-5p agomir (agomiR-183-5p;
5′-UAUGGCACUGGUAGAAUUCACU-3′) and its negative control (agomiR-NC;
5′-UUCUCCGAACGUGUCACGUTT-3′) were obtained from Shanghai GenePharma
Co., Ltd. In vivo transfection was performed as previously
described (19): The stereotaxic
coordinates were 0.5 mm posterior and 1.0 mm lateral to the bregma,
and 2.5–3.0 mm ventral to the surface of the skull. To assess the
neurological dysfunction and neuronal damage in mice following
MACO, agomiR-183-5p or agomiR-NC was mixed with the
Entranster™-in vivo transfection reagent (Engreen Biosystem
Co., Ltd.) and subsequently injected intracerebroventricularly into
the experimental group mice (the MCAO + agomiR-183-5p and MCAO +
agomiR-NC groups) at a rate of 0.2 µl/min using a mini-pump prior
to cerebral ischemia. For the MCAO group, an equal amount of
Entranster™-in vivo was injected. After 24 h, cerebral
ischemia was induced in the experimental group via MCAO.
Neurological scoring
Neurological deficits were evaluated immediately
after reperfusion to verify the MCAO model. Deficits were scored
using an 18-point scoring system as previously described (20). The scoring system was six parts: i)
Spontaneous activity in the cage for 5 min; ii) limb activity
symmetry; iii) forelimb symmetry; iv) climbing in metal wire cages;
v) torso touching; and vi) vibrissa responses. The scores of each
test ranged between 0 and 3, with a score of 0 indicating a normal
neurological response.
Brain edema assay
Mice were sacrificed under deep anesthesia (150
mg/kg pentobarbital sodium; death was confirmed by lack of
breathing and heartbeat) at 24 h post-reperfusion, and the brain
tissue was harvested. Brain edema was analyzed using the wet/dry
method as previously described (21). Brain water content was calculated
using the following formula: [(wet weight-dry weight)/wet weight]
×100%.
2,3,5-Triphenyltetrazoliumchloride
(TTC) staining
The area of cerebral infarction was measured as
previously described (22). Brain
tissues from mice were frozen at −20°C for 1–2 h. The olfactory
bulb and the cerebellum were removed, and the tissues were sliced
into five 1-mm thick coronal sections and stored on ice. The
sections were immersed in 1% TTC (Shanghai Aladdin Bio-chem
Technology Co., Ltd.) and stained for 10–15 min at 37°C in the
dark, during which the slices were continuously turned to ensure
even coverage. The cerebral infarct area was photographed using a
digital camera and quantified by Image-pro plus 6.0 software (Media
Cybernetics, Inc.).
Nissl staining
Samples of the ischemic penumbra were obtained from
mice 24 h after reperfusion. Neuronal damage in the ischemic
penumbra was assessed using Nissl staining as previously described
(23). Samples were fixed with 4%
paraformaldehyde for 15 min at room temperature, embedded in
paraffin, dewaxed in xylene and rehydrated with graded ethanol (95,
85 and 75%). The samples were cut coronally with a thickness of 4
µm. Following rinsing with distilled water, the sections were
stained with 0.5% crystal violet (Sinopharm Chemical Reagent Co.,
Ltd.) for 10 min at room temperature. Images of the ischemic
penumbra were observed under an Olympus BX53 light microscope
(Olympus Corporation) at ×200 magnification. The number of neurons
was quantified by a professional researcher, who was blind to the
grouping.
Fluoro-Jade B (FJB) staining
FJB staining was performed using an FJB staining kit
(EMD Millipore) according to the manufacturer's instructions.
Coronary sections (4 µm) from the ischemic penumbra were visualized
using an Olympus BX53 fluorescence microscope at magnification,
×200 (Olympus Corporation) to evaluate degenerating neurons.
FJB-positive neurons were counted by a professional researcher, who
was blind to the grouping.
Cell culture
Mouse N2A cells were purchased from Procell Life
Science & Technology Co., Ltd. and cultured in Dulbecco's
Modified Eagle's Medium (DMEM; Thermo Fisher Scientific, Inc.)
containing high glucose (HG) and 10% fetal bovine serum (Thermo
Fisher Scientific, Inc.) at 37°C in a humidified atmosphere of 5%
CO2.
OGD and reoxygenation
To simulate ischemic-like conditions in
vitro, N2A cells were seeded on 6-well plates (4×105
cells/well) and incubated at 37°C with 5% CO2 for 24 h.
Subsequently, HG medium was replaced with glucose-free DMEM and
incubated at 37°C in an anaerobic chamber containing 95%
N2 and 5% CO2 for 3 h. Following OGD
exposure, cells were returned to HG medium under normoxic
conditions for 24-h reoxygenation. Control cells not exposed to OGD
were maintained in DMEM containing HG at 37°C with 5%
CO2 and 95% O2.
Cell transfection
N2A cells were plated into a 6-well plate
(4×105 cells/well), cultured overnight, and were then
transfected with 100 pmol agomiR-183-5p, antagomiR-183-5p or their
negative controls at room temperature for 48 h using
Lipofectamine® 2000 (Invitrogen; Thermo Fisher
Scientific, Inc.) according to the manufacturer's instructions.
Additionally, a PTEN-overexpression pcDNA3.1 plasmid (1 µg;
Invitrogen; Thermo Fisher Scientific, Inc.) was transfected alone
or co-transfected with 50 pmol agomiR-183-5p into N2A cells using
Lipofectamine® 2000 (Invitrogen; Thermo Fisher
Scientific, Inc.) according to the manufacturer's instructions. An
empty pcDNA3.1 vector was used as a negative control. OGD was
performed 48 h after transfection.
Luciferase activity assay
TargetScan (http://www.targetscan.org) was used to predict the
potential targets of miR-183-5p, and an interaction between
miR-183-5P and PTEN was identified. N2A cells were incubated in
12-well plates at 90% confluence for 24 h. Cells were then
co-transfected with the pMIR-reporter luciferase vector, which
included the wild-type or mutant PTEN-3′-untranslated region (UTR)
miR-183-5p binding site 1 or 2 and agomiR-NC or agomiR-183-5p using
Lipofectamine® 2000 (Invitrogen; Thermo Fisher
Scientific, Inc.) according to the manufacturer's instructions.
After 48-h transfection, luciferase activity was detected by the
Luciferase Assay kit (Promega Corporation) according to the
manufacturer's instructions. Luciferase activity was normalized to
the Renilla luciferase activity.
Reverse transcription-quantitative
(RT-q)PCR
Total RNA was extracted from the ischemic penumbra
or N2A cells using the RNApure kit (BioTeke Corporation) according
to the manufacturer's instructions. To determine miR-183-5p or PTEN
mRNA expression, reverse transcription was performed using M-MLV
Reverse Transcriptase (2641A, Takara Biotechnology Co., Ltd.)
according to the manufacturer's directions. Real-time PCR was
subsequently conducted using Taq™ HS Perfect Mix (Takara
Biotechnology Co., Ltd.) and SYBR® Green (BioTeke
Corporation). The forward and reverse primers of miR-183-5p, U19,
PTEN and β-actin used for real-time PCR were as follows:
miR-183-5p, 5′-GCGGCTATGGCACTGGTAGAA-3′ and
5′-GTGCAGGGTCCGAGGTATTC-3′; U19, 5′-TGTGGAGTTGGTCCTGGTCT-3′ and
5′-GTGCAGGGTCCGAGGTATTC-3′; PTEN, 5′-GACCATAACCCACCACAGC-3′ and
5′-CATTACACCAGTCCGTCCCT-3′; β-actin, 5′-AATCGTGCGTGACATCAA-3′ and
5′-AGAAGGAAGGCTGGAAAA-3′. Relative miR-183-5p was calculated using
the 2−ΔΔCq method (24)
and normalized to U19. Relative PTEN expression was calculated
using the same method and normalized to β-actin.
Cell Counting Kit-8 (CCK-8) assay
Cell viability was assessed using a CCK-8 kit
(Sigma-Aldrich; Merck KGaA) 24 h after OGD according to the
manufacturer's instructions. Briefly, N2A cells from different
groups, including i) Control; ii) OGD; iii) OGD + agomiR-NC; iv)
OGD + agomiR-183-5p; v) OGD + agomiR-183-5p + vector; and vi) OGD +
agomiR-183-5p + PTEN-overexpression plasmid, were plated in 96-well
plates (3×103 cells/well). Following the addition
of·CCK-8 solution to each well, the cells were incubated for 2 h at
37°C with 95% O2 and 5% CO2. Optical density
values were measured at 450 nm using an ELX-800 microplate reader
(BioTeke Corporation).
Flow cytometry assay
OGD-induced apoptosis was detected using an
Annexin-V/propidium iodide (PI) apoptosis detection kit (Beyotime
Institute of Biotechnology) according to the manufacturer's
instructions. Briefly, N2A cells from each group were harvested
after centrifugation at 90 × g for 5 min at room temperature and
washed twice with PBS. Next, the cells were stained with Annexin
V-FITC and PI for 15 min in the dark. Finally, apoptosis was
analyzed using a flow cytometer (NovoCyte, Aceabio Biosciences,
Inc.) and NovoExpress software version 1.3.1 (Aceabio Biosciences,
Inc.).
Western blotting
Total protein was extracted from the ischemic
penumbra or N2A cells and quantified using a BCA kit (Beyotime
Institute of Biotechnology) according to the manufacturer's
instructions. Equal amounts of protein (30 µg per lane) were
separated on 10–15% sodium dodecyl sulfate-polyacrylamide gels and
transferred to polyvinylidene fluoride membranes (Thermo Fisher
Scientific, Inc.). The membranes were blocked with 5% (m/v) bovine
serum albumin (Biosharp Life Sciences) for 1 h at room temperature
and incubated at 4°C overnight with the following primary
antibodies: Anti-B-cell lymphoma-extra-large (Bcl-xl; 1:1,000; cat.
no. 2762; Cell Signaling Technology, Inc.), anti-B-cell lymphoma-2
(Bcl-2; 1:500; cat. no. 12789-1-AP; ProteinTech Group, Inc.),
anti-Bcl-2-associated X protein (Bax; 1:500; cat. no. 50599-2-Ig;
ProteinTech Group, Inc.), anti-cleaved caspase-3 (1:1,000; cat. no.
9654; Cell Signaling Technology, Inc.), anti-pro caspase-3
(1:1,000; cat. no. 9662; Cell Signaling Technology, Inc.),
anti-PTEN (1:1,000; cat. no. A11193; ABclonal Biotech Co., Ltd.) or
anti-β-actin (1:2,000; cat. no. 60008-1-Ig; ProteinTech Group,
Inc.). After washing with TBS + 0.1% Tween-20, the membranes were
incubated for 40 min at 37°C with horseradish peroxidase-labeled
goat anti-rabbit IgG (1:10,000; cat. no. SA00001-2; ProteinTech
Group, Inc.) or goat anti-mouse IgG (1:10,000; cat. no. SA00001-1;
ProteinTech Group, Inc.) secondary antibodies. The signal bands of
the membranes were visualized using an ECL kit (Shanghai 7sea
Biotech Co., Ltd.) according to the manufacturer's protocol and
quantified by Gel-Pro-Analyzer 4.0 software (Media Cybernetics,
Inc.).
Statistical analysis
Data are presented as the mean ± SD, and each
experiment was repeated ≥3 times. Statistical analyses were
performed by GraphPad Prism 8.0 (GraphPad Software, Inc.) using
one-way ANOVA followed by Tukey's test for multiple comparisons or
two-tailed unpaired Student's t-tests for comparisons between two
groups. P<0.05 was considered to indicate a statistically
significant difference.
Results
Expression of miR-183-5p in the MCAO
mouse model
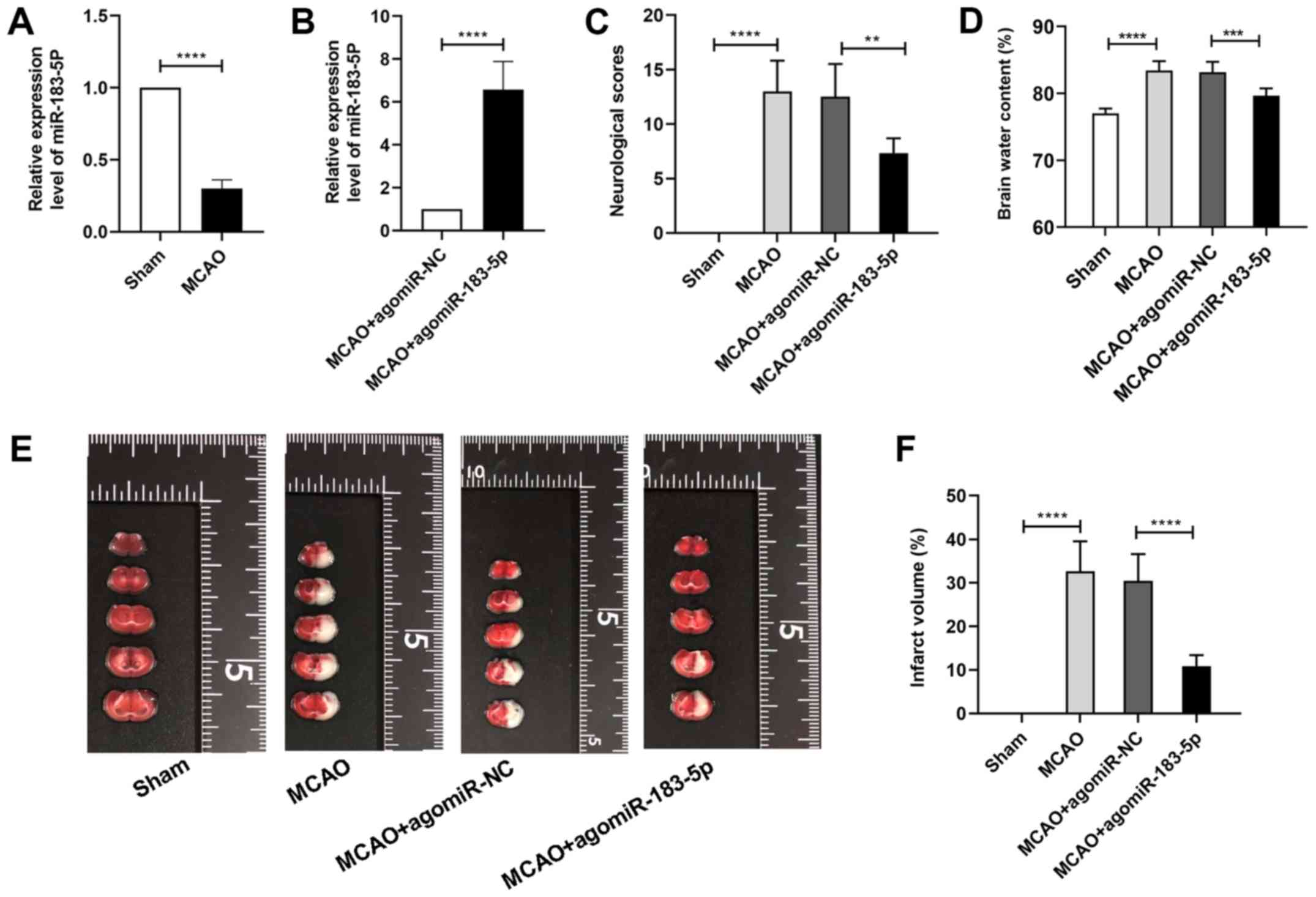
To identify the expression of miR-183-5p in
vivo before and after ischemia induction, mice were subjected
to MCAO (~25% mortality). The results of the RT-qPCR assay
demonstrated that the expression of miR-183-5p was significantly
decreased after MCAO (P<0.05; Fig.
1A) compared with that in the sham group, indicating that
downregulation of miR-183-5p expression in vivo was
associated with cerebral ischemia.
Effects of miR-183-5p on the
neurological score, cerebral edema and infarct area
To explore the role of miR-183-5p on cerebral
ischemia, agomiR-NC or agomiR-183-5p was injected into the murine
cerebra prior to MCAO. RT-qPCR analysis revealed that compared with
agomiR-NC, transfection with agomiR-183-5p significantly
upregulated the expression of miR-183-5p in the MCAO group
(P<0.05; Fig. 1B). Neurological
score and brain water content analyses were performed to assess the
neurological function and brain edema, respectively, and TTC
staining was performed to determine the area of cerebral
infarction. As demonstrated in Fig.
1C, the neurological scores of the mice in the MCAO group were
markedly increased compared with those of mice in the sham group.
Additionally, agomiR-183-5p significantly decreased the
neurological scores compared with those in the MCAO + agomiR-NC
group (P<0.05). As demonstrated in Fig. 1D-F, brain water content and infarct
area (demonstrated as white coloration) were significantly
increased in the MCAO group compared with that in the sham group,
and these effects were relieved by agomiR-183-5p compared with
those in the MCAO + agomiR-NC group (P<0.05).
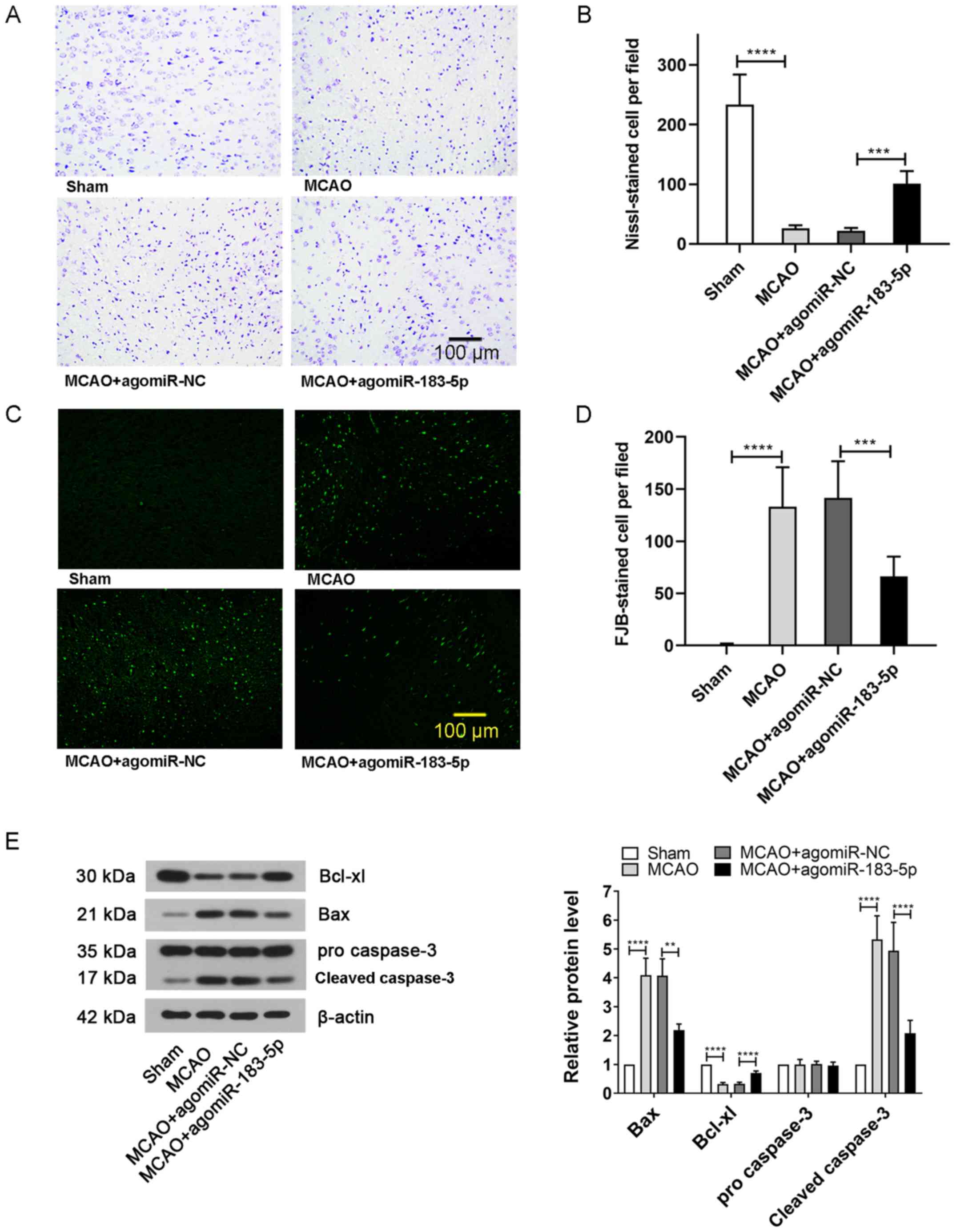
Effects of miR-183-5p on neuronal
damage and apoptosis
To further study the extent of neuronal damage,
Nissl and FJB staining of brain tissues was performed. The results
of Nissl staining demonstrated that the number of normal neurons
was markedly decreased following MCAO treatment compared with that
in the sham-treated mice; however, the numbers were increased after
injecting mice with agomiR-183-5p compared with that in the MCAO +
agomiR-NC group (P<0.05; Fig. 2A
and B). Consistent with this result, the number of FJB-positive
cells observed in the MCAO group was significantly higher compared
with that in the sham group, but significantly reduced after
injection with agomiR-183-5p (P<0.05; Fig. 2C and D). In addition, the neuronal
expression of apoptosis marker Bcl-xl was markedly inhibited and
the levels of Bax and cleaved caspase-3 were increased in the MCAO
group compared with that in the sham group; however, agomiR-183-5p
reversed the MACO-induced expression of apoptosis-related proteins
(P<0.05; Fig. 2E). These
results suggested that miR-183-5p effectively alleviated the brain
damage caused by cerebral ischemia.
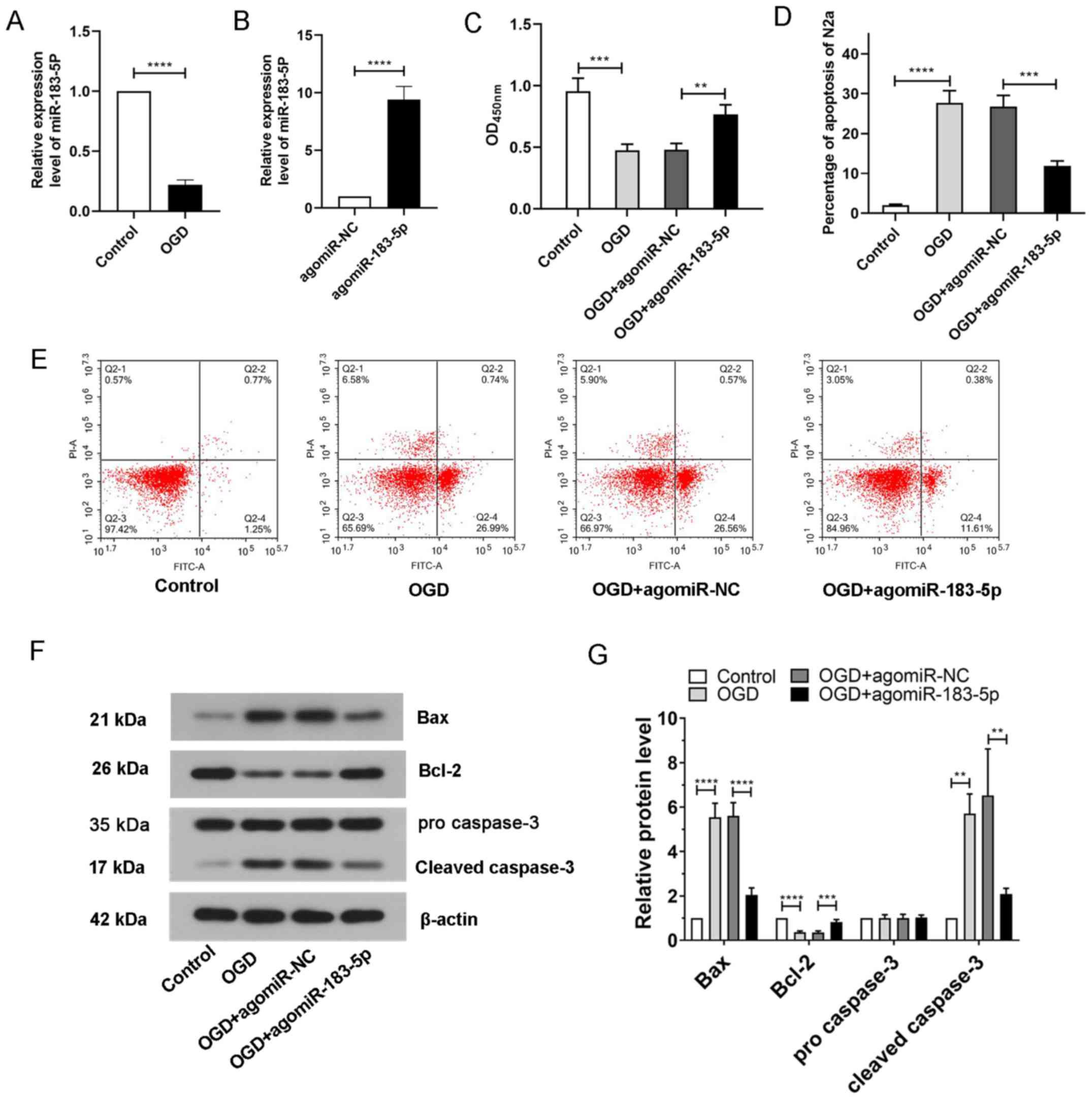
Expression of miR-183-5p in N2A cells
exposed to OGD
To simulate ischemia-like conditions in
vitro, N2A cells were exposed to OGD followed by reoxygenation.
The in vitro expression of miR-183-5p before and after
ischemia was detected by RT-qPCR. The results demonstrated that the
expression of miR-183-5p after OGD treatment was significantly
decreased compared with that in the control group (P<0.05;
Fig. 3A), suggesting that in
vitro miR-183-5p expression was associated with ischemia.
Effects of miR-183-5p on the viability
of N2A cells exposed to OGD
To explore the role of miR-183-5p in N2A cells,
agomiR-NC or agomiR-183-5p was transfected into N2A cells 48 h
prior to OGD treatment. The expression levels of miR-183-5p in N2A
cells was upregulated after transfection with agomiR-183-5p
compared with that in the agomiR-NC group (P<0.05; Fig. 3B). The viability of N2A cells
exposed to OGD was subsequently measured using a CCK-8 assay. As
presented in Fig. 3C, N2A cell
viability was significantly decreased after exposure to OGD
compared with that of the control cells, but transfection with
agomiR-183-5p significantly increased N2A cell viability,
indicating that miR-183-5p may promote cell survival
(P<0.05).
Effects of miR-183-5p on apoptosis in
N2A cells exposed to OGD
To analyze the apoptosis of N2A cells, flow
cytometry and western blotting were carried out. The results of the
flow cytometry assay demonstrated that the apoptotic rates in
untreated N2A cells were low, whereas apoptosis was significantly
increased following exposure to OGD. However, the increase in
apoptosis was suppressed in N2A cells transfected with
agomiR-183-5p compared with that in cells transfected with
agomiR-NC (P<0.05; Fig. 3D and
E). The results of western blotting analysis demonstrated that
the expression levels of the apoptosis-related markers Bax and
cleaved caspase-3 were significantly upregulated in N2A cells
exposed to OGD, and the expression levels of Bcl-2 upon OGD
exposure were downregulated compared with those in the control
group; transfection with agomiR-183-5p reversed these changes in
apoptosis-related markers (Fig. 3F and
G; P<0.05). These results suggested that miR-183-5p
inhibited apoptosis in N2A cells.
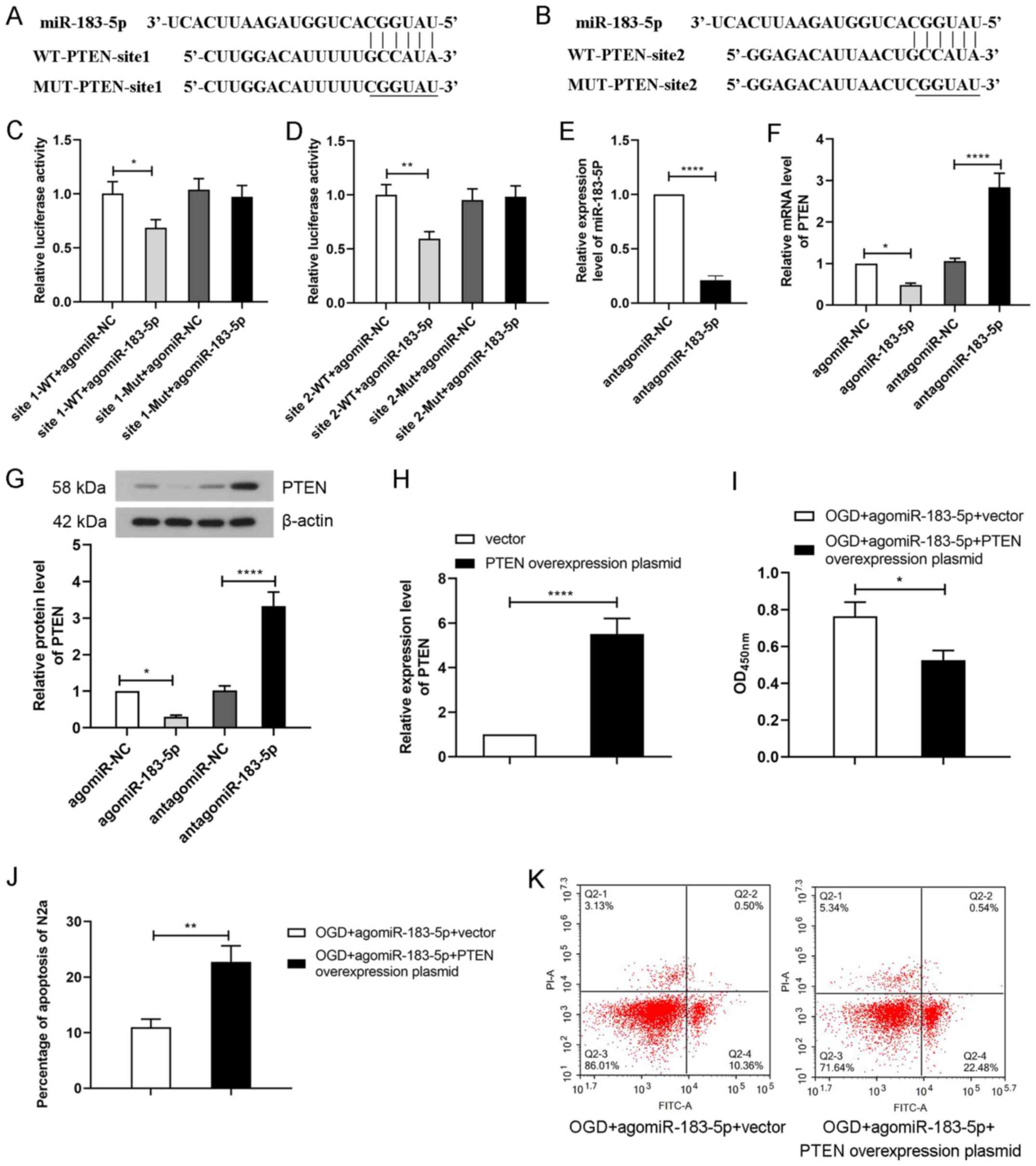
PTEN is the target gene of
miR-183-5p
To further elucidate the mechanism of miR-183-5p in
cerebral ischemia-induced apoptosis, bioinformatics analysis was
used to predict its target genes (http://www.targetscan.org). The results revealed that
PTEN was predicted to be a target gene of miR-183-5p. Binding sites
between PTEN-3′-UTR and miR-183-5p were elucidated, and their
sequences are presented in Fig. 4A and
B. To verify the interaction between miR-183-5p and PTEN, N2A
cells were co-transfected with agomiR-NC or agomiR-183-5p and
assessed by performing a luciferase reporter assay. As demonstrated
in Fig. 4C and D, transfection
with agomiR-183-5p significantly decreased the luciferase activity
of the wild-type reporter gene containing PTEN-3′-UTR site 1 or 2
(P<0.05). No significant differences were observed in the
luciferase activity of the mutated reporter gene containing
PTEN-3′-UTR site 1 or 2 following agmiR-183-5p treatment. These
results suggested that miR-183-5p could directly bind to the target
gene PTEN.
In addition, to further determine the effects of
miR-183-5p on PTEN, agomiR-NC, agomiR-183-5p, antagomiR-NC or
antagomiR-183-5p was transfected into N2A cells. Transfection with
antagomiR-183-5p decreased the levels of miR-183-5p in N2A cells
compared with those in the antagomir-NC group (P<0.05; Fig. 4E). The results of RT-qPCR and
western blotting revealed that transfection with agomiR-183-5p
significantly decreased PTEN levels, whereas antagomiR-183-5p
significantly increased PTEN levels compared with those in the
respective NC groups (P<0.05; Fig.
4F and G). These results suggested that miR-183-5p inhibited
PTEN protein expression by binding to its 3′-UTR.
PTEN overexpression partially reverses
the effect of miR-183-5p
To determine the effect of miR-183-5p/PTEN signaling
on OGD-treated N2A cells, a PTEN overexpression plasmid was
transfected alone or co-transfected with agomiR-183-5p into N2A
cells prior to OGD. The result of RT-qPCR demonstrated that
transfection with the PTEN overexpression plasmid markedly
increased the PTEN levels compared with those in cells transfected
with the empty vector (P<0.05; Fig.
4H). The survival of N2A cells exposed to OGD was subsequently
analyzed by CCK-8 and flow cytometry assays. As presented in
Fig. 4I, N2A cell viability was
significantly decreased after transfection with PTEN-overexpression
plasmid compared with the control vector plasmids (P<0.05). In
addition, the apoptotic rate of OGD-treated N2A cells was
significantly increased following co-transfection with the PTEN
overexpression plasmid and agomiR-183-5p compared with that of
cells transfected with agomiR-183-5p and the control vector
(P<0.05; Fig. 4J and K). These
results suggested that miR-183-5p inhibited apoptosis and increased
cell viability by negatively regulating PTEN in N2A cells.
Discussion
A mouse model of the cerebral I/R is a commonly used
experimental method for investigating the pathophysiology of
ischemia-induced brain injury (25). C57BL/6 mice are an extensively used
strain in cerebral I/R experiments since they can be easily
genetically manipulated (26). The
current study evaluated the role and regulatory mechanism of
miR-183-5p in cerebral ischemia. The expression of miR-183-5p was
decreased and brain damage was increased in mice following ischemia
model establishment compared with those in the sham group. Similar
results were observed in an ischemia model in N2A cells, where
miR-183-5p expression was reduced and apoptosis was increased
compared with those in untreated cells. In further experiments,
following transfection with agomiR-183-5p, cerebral ischemia injury
and apoptosis levels were reduced compared with those in the MCAO +
agomiR-NC group. In addition, PTEN was identified as a direct
target of miR-183-5p, and the expression of PTEN was negatively
regulated by miR-183-5p. Following PTEN overexpression, the
neuroprotective effects of miR-183-5p were reduced.
A previous study has demonstrated that the majority
of neurons in the area of ischemic injury undergo necrosis or
apoptosis (20). In contrast to
necrosis, apoptosis is a method of programmed cell death that is
regulated by a variety of molecular mechanisms (27). Therefore, apoptotic cells can be
salvaged by treatment, with neuronal damage also being alleviated.
Yin et al (28) have
demonstrated that inhibition of miR-497 attenuated apoptosis and
neuronal damage by enhancing the expression of certain
antiapoptotic proteins, including Bcl-2 and Bcl-2-family protein
(Bcl-w). Zhang et al (29)
have revealed that following transfection with miR-378 agomir,
upregulation of miR-378 attenuated ischemic injury by negatively
regulating the apoptosis-associated protein caspase-3. Although the
regulation of miR-183 expression is considered to be effective for
treating acute myocardial infarction or liver damage caused by
ischemia (12,13), reports detailing miR-183 treatment
in cerebral ischemia are unknown. As a member of the miR-183 gene
cluster, the normal expression of miR-183-5p is important for
nervous system function (30). A
previous study has revealed that 17 miRNAs, including miR-183-5p,
were downregulated in infarcted brain areas following focal
cerebral ischemia in rats according to microRNA sequencing compared
with healthy tissues (31). The
results of the present study demonstrated decreased levels of
miR-183-5p in MCAO models of cerebral ischemia injury and
OGD-treated N2A cells compared with those in the respective control
groups.
Brain injury caused by cerebral ischemia can lead to
neurological deficit symptoms and local infarction (29). A previous study has demonstrated
that neurological scores and cerebral edema can reflect the degree
of brain damage (32).
Additionally, neuronal damage and infarct size may visually
indicate the extent of brain damage (33). MCAO causes motor and cognitive
impairments reflecting the dysfunction in the frontal and parietal
lobes (34). To assess the
neurological dysfunction and neuronal damage in mice following
MACO, agomiR-183-5p was injected intracerebroventricularly into the
experimental group mice; injection site was located in the cerebral
cortex around the frontal and parietal lobes. A previous report has
detailed the intracerebral injection of miR-183-5p mimics and its
efficient amelioration of neuropathic pain by targeting the
tandem-pore-domain potassium channel TREK-1 (35). In congruence with these results,
the neurological scores and brain edema assay results of the
current study, along with TTC, Nissl and FJB staining, revealed
that the transfection with agomiR-183-5p alleviated brain injury by
reducing neuronal injury, brain water content and brain infarction
size. Similarly, in vitro experiment results demonstrated
that N2A cells underwent apoptosis following exposure to OGD.
AgomiR-183-5p inhibited OGD-induced N2A cell apoptosis and
increased cell viability. Extrinsic and intrinsic pathways are
recognized to be involved in apoptosis, which relies on alterations
in the expression levels of Bcl-2 and caspase family proteins,
including Bcl-2, Bcl-xl, Bax and caspase-3 (36,37).
The results of the present study demonstrated that following
transfection with agomiR-183-5p, apoptosis was decreased, and the
cell survival rate was increased compared with those in the
agomiR-NC-transfected group. In addition, the expression levels of
Bax and cleaved caspase-3 were decreased, whereas those of Bcl-2
and Bcl-xl were increased in the agomiR-183-5p group compared with
those in the agomiR-NC group. As genes that inhibit apoptosis,
Bcl-2 and Bcl-xl attenuate the role of the pro-apoptotic gene Bax;
therefore, in most cells, apoptosis is controlled by Bcl-2 family
proteins (38). In the current
study, transfection with agomiR-183-5p promoted Bcl-2 expression,
which in turn led to a decrease in Bax expression, thereby
maintaining a high cell survival rate. Similarly, as a
pro-apoptotic protein (39),
caspase-3 expression was decreased following transfection with
agomiR-183-5p in the present study. This result was in agreement
with a previous report, in which treatment of glabridin decreased
caspase-3 transcriptional activity and inhibited
staurosporine-induced rat cortical neuronal apoptosis (40). Therefore, the results of the
present study suggested that miR-183-5p may regulate apoptosis, as
it is associated with the expression of Bcl-2, Bax and cleaved
caspase-3 expression.
PTEN is a tumor suppressor gene with phosphatase
activity that modulates cell survival, apoptosis and metabolism by
negatively regulating the PI3K/Akt signaling pathway (41). Increasing studies have confirmed
that the inhibition of PTEN exerts neuroprotective effects in
cerebral ischemic injury (42,43).
In MCAO and OGD models, it has been determined that PTEN is highly
expressed and contributes to the development of ischemic stroke
through the PI3K/Akt pathway (42,43).
The bioinformatics analysis in the present study revealed that PTEN
was a target of miR-183-5p. A previous study has also revealed that
PTEN is a target of miR-130a and can reverse the protective effects
of miR-130a on cerebral I/R damage in vivo and vitro
(44), indicating that PTEN serves
an important role in the response to ischemia. Similarly, the
results of the present study demonstrated that miR-183-5p and PTEN
formed a negative feedback loop, and that PTEN overexpression
inhibited the protective effects exerted by miR-183-5p on cell
viability, which in turn increased apoptosis. The current results
only suggested that miR-183-5p decreased cerebral ischemic injury
by targeting PTEN; however, PTEN expression and the effects of
agomiR-183-5p on PTEN activation after MCAO or OGD are not clear
and require further study.
In conclusion, the present study primarily clarified
the function and mechanism of miR-183-5p in cerebral ischemia.
Transfection with agomiR-183-5p inhibited apoptosis and increased
cell viability by negatively regulating the expression of PTEN,
thus mitigating cerebral ischemic injury. These findings may
contribute to the further understanding of the mechanism of
cerebral ischemia and may provide novel insights into ischemic
injury treatment.
Acknowledgements
Not applicable.
Funding
No funding was received.
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
JL conceived and designed the study. LZ, SL, SZ, JY
and XF prepared the materials, performed the experiments, collected
and analyzed the data. LZ drafted the manuscript. XZ performed the
supplementary experiments and revised the manuscript. All authors
read and approved the final manuscript.
Ethics approval and consent to
participate
All the procedures in the animal experiments
followed the guide for the care and use of laboratory animals, and
this study was approved by Institutional Animal Care and Use
Committee of Binzhou People's Hospital (approval no. LYP200).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Singh V, Roth S, Veltkamp R and Liesz A:
HMGB1 as a key mediator of immune mechanisms in ischemic stroke.
Antioxid Redox Signal. 24:635–651. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Zerna C, Hegedus J and Hill MD: Evolving
treatments for acute ischemic stroke. Circ Res. 118:1425–1442.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Lakhan SE, Kirchgessner A and Hofer M:
Inflammatory mechanisms in ischemic stroke: Therapeutic approaches.
J Transl Med. 7:972009. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Fonarow GC, Smith EE, Saver JL, Reeves MJ,
Bhatt DL, Grau-Sepulveda MV, Olson DM, Hernandez AF, Peterson ED
and Schwamm LH: Timeliness of tissue-type plasminogen activator
therapy in acute ischemic stroke: Patient characteristics, hospital
factors, and outcomes associated with door-to-needle times within
60 minutes. Circulation. 123:750–758. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Rami A and Kogel D: Apoptosis meets
autophagy-like cell death in the ischemic penumbra: Two sides of
the same coin? Autophagy. 4:422–426. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Hammond S: An overview of microRNAs. Adv
Drug Deliv Rev. 87:3–14. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Li G, Morris-Blanco KC, Lopez MS, Yang T,
Zhao H, Vemuganti R and Luo Y: Impact of microRNAs on ischemic
stroke: From pre- to post-disease. Prog Neurobiol. 163-164:59–78.
2018. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Birch D, Britt BC, Dukes SC, Kessler JA
and Dizon ML: MicroRNAs participate in the murine oligodendroglial
response to perinatal hypoxia-ischemia. Pediatr Res. 76:334–340.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Majdi A, Mahmoudi J, Sadigh-Eteghad S,
Farhoudi M and Shotorbani SS: The interplay of microRNAs and
post-ischemic glutamate excitotoxicity: An emergent research field
in stroke medicine. Neurol Sci. 37:1765–1771. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Chang L, Zhang W, Shi S, Peng Y, Wang D,
Zhang L and Zhang J: microRNA-195 attenuates neuronal apoptosis in
rats with ischemic stroke through inhibiting KLF5-mediated
activation of the JNK signaling pathway. Mol Med. 26:312020.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Pierce ML, Weston MD, Fritzsch B, Gabel
HW, Ruvkun G and Soukup GA: Microrna-183 family conservation and
ciliated neurosensory organ expression. Evol Dev. 10:106–113. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Lin HC, Liu SY, Yen EY, Li TK and Lai IR:
microRNA-183 mediates protective postconditioning of the liver by
repressing Apaf-1. Antioxid Redox Signal. 26:583–597. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Gong L, Xu H, Chang H, Tong Y, Zhang T and
Guo G: Knockdown of long non-coding RNA MEG3 protects H9c2 cells
from hypoxia-induced injury by targeting microRNA-183. J Cell
Biochem. 119:1429–1440. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Garcia-Junco-Clemente P and Golshani P:
PTEN: A master regulator of neuronal structure, function, and
plasticity. Commun Integr Biol. 7:e283582014. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Miao SY, Miao SM, Cui RT, Yu AL and Miao
ZJ: SETD5-AS1 stimulates neuron death in stroke via promoting PTEN
expression. Eur Rev Med Pharmacol Sci. 22:6035–6041.
2018.PubMed/NCBI
|
|
16
|
Sarver AL, Li L and Subramanian S:
MicroRNA miR-183 functions as an oncogene by targeting the
transcription factor EGR1 and promoting tumor cell migration.
Cancer Res. 70:9570–9580. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Thiel J, Alter C, Luppus S, Eckstein A,
Tan S, Führer D, Pastille E, Westendorf AM, Buer J and Hansen W:
MicroRNA-183 and microRNA-96 are associated with autoimmune
responses by regulating T cell activation. J Autoimmun. 96:94–103.
2019. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Guo D, Ma J, Li T and Yan L: Up-regulation
of miR-122 protects against neuronal cell death in ischemic stroke
through the heat shock protein 70-dependent NF-kappaB pathway by
targeting FOXO3. Exp Cell Res. 369:34–42. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Ni J, Wang X, Chen S, Liu H, Wang Y, Xu X,
Cheng J, Jia J and Zhen X: MicroRNA let-7c-5p protects against
cerebral ischemia injury via mechanisms involving the inhibition of
microglia activation. Brain Behav Immun. 49:75–85. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Chen J, Sanberg PR, Li Y, Wang L, Lu M,
Willing AE, Sanchez-Ramos J and Chopp M: Intravenous administration
of human umbilical cord blood reduces behavioral deficits after
stroke in rats. Stroke. 32:2682–2688. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Yoon S, Woo SU, Kang JH, Kim K, Kwon MH,
Park S, Shin HJ, Gwak HS and Chwae YJ: STAT3 transcriptional factor
activated by reactive oxygen species induces IL6 in
starvation-induced autophagy of cancer cells. Autophagy.
6:1125–1138. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Wang X, Wang S, Wang J, Guo H, Dong Z,
Chai L, Hu L, Zhang Y, Wang H and Chen L: Neuroprotective effect of
xueshuantong for injection (Lyophilized) in transient and permanent
rat cerebral ischemia model. Evid Based Complement Alternat Med.
2015:1346852015. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Gong SJ, Chen LY, Zhang M, Gong JX, Ma YX,
Zhang JM, Wang YJ, Hu YY, Sun XC, Li WB and Zhang Y: Intermittent
hypobaric hypoxia preconditioning induced brain ischemic tolerance
by up-regulating glial glutamate transporter-1 in rats. Neurochem
Res. 37:527–537. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Woodruff TM, Thundyil J, Tang SC, Sobey
CG, Taylor SM and Arumugam TV: Pathophysiology, treatment, and
animal and cellular models of human ischemic stroke. Mol
Neurodegener. 6:112011. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Yonekura I, Kawahara N, Nakatomi H, Furuya
K and Kirino T: A model of global cerebral ischemia in C57 BL/6
mice. J Cereb Blood Flow Metab. 24:151–158. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Doyle KP, Simon RP and Stenzelpoore MP:
Mechanisms of ischemic brain damage. Neuropharmacology. 55:310–318.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Yin KJ, Deng Z, Huang H, Hamblin M, Xie C,
Zhang J and Chen YE: miR-497 regulates neuronal death in mouse
brain after transient focal cerebral ischemia. Neurobiol Dis.
38:17–26. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Zhang N, Zhong J, Han S, Li Y, Yin Y and
Li J: MicroRNA-378 alleviates cerebral ischemic injury by
negatively regulating apoptosis executioner caspase-3. Int J Mol
Sci. 17:14272016. View Article : Google Scholar
|
|
30
|
Banks SA, Pierce ML and Soukup GA:
Sensational MicroRNAs: Neurosensory roles of the MicroRNA-183
family. Mol Neurobiol. 57:358–371. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Duan X, Gan J, Peng DY, Bao Q, Xiao L, Wei
L and Wu J: Identification and functional analysis of microRNAs in
rats following focal cerebral ischemia injury. Mol Med Rep.
19:4175–4184. 2019.PubMed/NCBI
|
|
32
|
Ye Y, Jin T, Zhang X, Zeng Z, Ye B, Wang
J, Zhong Y, Xiong X and Gu L: Meisoindigo protects against focal
cerebral ischemia-reperfusion injury by inhibiting NLRP3
inflammasome activation and regulating microglia/macrophage
polarization via TLR4/NF-κB signaling pathway. Front Cell Neurosci.
13:5532019. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Park SJ, Nam KW, Lee HJ, Cho EY, Koo U and
Mar W: Neuroprotective effects of an alkaloid-free ethyl acetate
extract from the root of Sophora flavescens Ait. against focal
cerebral ischemia in rats. Phytomedicine. 16:1042–1051. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Truong DT, Venna VR, McCullough LD and
Fitch RH: Deficits in auditory, cognitive, and motor processing
following reversible middle cerebral artery occlusion in mice. Exp
Neurol. 238:114–121. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Shi DN, Yuan YT, Ye D, Kang LM, Wen J and
Chen HP: MiR-183-5p alleviates chronic constriction injury-induced
neuropathic pain through inhibition of TREK-1. Neurochem Res.
43:1143–1149. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Pilchova I, Klacanova K, Chomova M,
Tatarkova Z, Dobrota D and Racay P: Possible contribution of
proteins of Bcl-2 family in neuronal death following transient
global brain ischemia. Cell Mol Neurobiol. 35:23–31. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Datta A, Sarmah D, Mounica L, Kaur H,
Kesharwani R, Verma G, Veeresh P, Kotian V, Kalia K, Borah A, et
al: Cell death pathways in ischemic stroke and targeted
pharmacotherapy. Transl Stroke Res. 26–Mar;2020.(Epub ahead of
print). View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Cory S, Huang DC and Adams JM: The Bcl-2
family: Roles in cell survival and oncogenesis. Oncogene.
22:8590–8607. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Massieu L, Moran J and Christen Y: Effect
of Ginkgo biloba (EGb 761) on staurosporine-induced neuronal death
and caspase activity in cortical cultured neurons. Brain Res.
1002:76–85. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Yu XQ, Xue CC, Zhou ZW, Li CG, Du YM,
Liang J and Zhou SF: In vitro and in vivo neuroprotective effect
and mechanisms of glabridin, a major active isoflavan from
Glycyrrhiza glabra (licorice). Life Sci. 82:68–78. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Oudit GY, Sun H, Kerfant BG, Crackower MA,
Penninger JM and Backx PH: The role of phosphoinositide-3 kinase
and PTEN in cardiovascular physiology and disease. J Mol Cell
Cardiol. 37:449–471. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Xing Y, Wang MM, Feng YS, Dong F and Zhang
F: Possible involvement of PTEN signaling pathway in the
anti-apoptotic effect of electroacupuncture following ischemic
stroke in rats. Cell Mol Neurobiol. 38:1453–1463. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Zhao D, Chen J, Zhang Y, Liao HB, Zhang
ZF, Zhuang Y, Pan MX, Tang JC, Liu R, Lei Y, et al: Glycine confers
neuroprotection through PTEN/AKT signal pathway in experimental
intracerebral hemorrhage. Biochem Biophys Res Commun. 501:85–91.
2018. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Zheng T, Shi Y, Zhang J, Peng J, Zhang X,
Chen K, Chen Y and Liu L: MiR-130a exerts neuroprotective effects
against ischemic stroke through PTEN/PI3K/AKT pathway. Biomed
Pharmacother. 117:1091172019. View Article : Google Scholar : PubMed/NCBI
|