Introduction
Coronary artery ischemic disease is prevalent
worldwide (1), it affects >15
million adults in the United States (2). A lack of coronary blood supply caused
by thrombosis or the acute alteration of coronary atherosclerotic
plaques contributes to myocardial ischemia (MI) (3). The early restoration of blood flow is
a common treatment strategy (4);
however, it can also cause further severe cardiac damage, which is
referred to as myocardial ischemia-reperfusion injury (MIRI)
(5,6). Therefore, it is of great importance
to further investigate safe and effective novel therapeutic
treatments to prevent MIRI and improve the clinical outcomes of
acute MI.
It is well established that mitochondria are the
powerhouses of the cell due to their crucial role in generating ATP
(7), but they are also important
regulators of programmed cell death pathways (8). Properties of mitochondrial
dysfunction include a reduction in energy charge (EC), the opening
of the mitochondrial permeability transition pore (mPTP), the
release of cytochrome c and Ca2+ overload
(9). These properties subsequently
lead to mitochondrial membrane depolarization, the homeostatic
imbalance between apoptotic proteins, and ultimately cardiomyocyte
death (10). Reactive oxygen
species (ROS), of which mitochondria are the predominant source,
induce MIRI through mitochondrial DNA (mtDNA) damage, reducing
energy production and inhibiting protein synthesis through a
vicious circle of mitochondrial damage (11,12).
In addition, mitochondria are dynamic organelles that continually
alter their morphology by undergoing routine fission and fusion
events (13,14), which requires specifically
controlled proteins; for example, dynamin-related protein 1 (Drp
1), which is a cytosolic GTPase that serves a fundamental role in
mitochondrial fission by translocating to the outer mitochondrial
membrane to generate the force necessary for mitochondrial fission
(15,16), and mitofusin 1 (Mfn1) and mitofusin
2 (Mfn2), which are required for the membrane remodeling processes
necessary for mitochondrial fusion (17,18).
Autophagy is an evolutionarily conserved,
lysosome-dependent degradation process that is activated during
MIRI (19). It has been reported
that autophagy serves a dual role in MIRI:A slight induction of
autophagy promotes cell survival during ischemia, whilst a
significant increase induces cell death during reperfusion
(20,21). Although a small amount of autophagy
can maintain cell function, excessive autophagy was found to
promote myocardial injury due to the consumption of cellular
constituents (22). Mammalian
target of rapamycin (mTOR) kinase is an important regulator of the
classical autophagy pathway (23,24),
and autophagy related 5 (Atg5) and microtubule-associated protein
1A/1B-light chain 3 (LC3) conjugation systems are required for the
formation of autophagic vesicles (25). Eukaryotic initiation factor
4E-binding protein 1 (4E-BP1) binds to eukaryotic initiation factor
4E (eIF4E) (26). The
phosphorylation of 4E-BP1 disrupts the assembly of the eIF4E/4E-BP1
complex, which initiates eIF4E-dependent translation, and thereby
the activation of cap-dependent mRNA translation (27). In addition to regulating cell cycle
checkpoints and apoptosis (28),
p53 has also been demonstrated to mediate the transactivation of
autophagy inducers (29,30).
Hyperbaric oxygen (HBO) therapy is the clinical
application of pure oxygen at a higher pressure (usually 2–3 times
atmospheric pressure) in a chamber to treat ischemia- or
hypoxia-associated diseases, such as coronary heart disease,
cerebral infarction and carbon monoxide poisoning (31–34).
HBO therapy has been widely agreed by major hyperbaric craft
groups, such as the Undersea and Hyperbaric Medical Society or the
U.S. Food and Drug Administration (35). In addition, data from our previous
studies demonstrated that HBO exerted neuroprotective effects in
certain animal models; for example, HBO combined with Madopar
protected against 6-hydroxydopamine-induced Parkinson's disease in
rats (36) and HBO treatment also
alleviated the withdrawal symptoms induced by morphine dependence
(37). Moreover, HBO prevented the
cognitive impairments induced by D-galactose (38,39).
The protective effects of HBO are mainly associated with the
increased induction of antioxidant enzymes and ischemic tolerance,
as well as the inhibition of cell apoptosis and the modulation of
neurotransmitters (36–39). Previously, our group discovered
that HBO preconditioning protected against MI and improved cardiac
function (40), of which the
underlying mechanisms were associated with the reduction of oxygen
stress, the correction of energy metabolism and the inhibition of
apoptosis. However, the effect of HBO treatment on mitochondria,
and the interaction between mitochondrial dysfunction and
autophagy, remain unclear. The present study aimed to investigate
the effects of HBO treatment in a rat model of MIRI, established by
the ligation of the left anterior descending (LAD) artery, through
analyzing mitochondrial function and the mTOR-mediated autophagy
pathway.
Materials and methods
Animal studies
All animal experimental procedures were approved by
the Animal Ethical Committee of Guangxi Medical University. A total
of 60 healthy Sprague-Dawley rats of both sexes (ratio, 1:1;
weight, 180–220 g; age, 6 weeks) were obtained from the
Experimental Animal Centre of Guangxi Medical University (Guangxi,
China). Animals were housed under controlled conditions at a
temperature of 25±2°C and a relative humidity of 60±10%, with a
12-h light/dark cycle. Food and water were available ad
libitum.
Experimental design and groupings
According to previous guidelines (41), following 1 week of acclimatization
to the laboratory conditions, the 60 rats were divided into the
following three groups (20 rats/group) using the random number
table method: i) Sham group; ii) ischemia-reperfusion (IR) group
(MIRI model); and iii) HBO group.
MIRI was inflicted by the occlusion of the LAD
coronary artery followed by reperfusion, according to a previous
study (40). Briefly, rats were
anaesthetized with 30 mg/kg sodium pentobarbital (i.p.) and
mechanically ventilated using an animal respirator (respiration
rate, 70 breaths/min; tidal volume, 6–8 ml/kg; Shanghai Alcott
Biotech Co., Ltd.). The chest was then opened and the heart was
exposed. The LAD artery was ligated using a 5–0 silk suture for 30
min and released to allow reperfusion for 1 h.
Rats in the IR and HBO groups were subjected to a 30
min LAD ligation followed by reperfusion for 1 h, whilst the sham
group received encircling of the LAD artery with a suture, but no
ligation. Before the surgical procedure, rats in the HBO group were
pretreated with HBO for 1 h (0.25 MPa) for 14 days (once daily) in
a hyperbaric chamber (Yantai Hongyuan Co., Ltd.), as previously
described (38).
At the end of the reperfusion, all rats were
immediately anesthetized using 30 mg/kg sodium pentobarbital (i.p.)
and were subsequently euthanized through exsanguination by
collecting 8 ml blood from the abdominal aorta. Death was confirmed
following the detection of a still heartbeat, and when breathing
had stopped. Blood samples were centrifuged at 302 × g for 10 min
at 4°C, and the supernatant was collected and stored at −80°C for
use in the biochemical assays. The heart was promptly removed from
the rats and the infarct was isolated on an ice box for further
measurements.
Determination of ATP, ADP, AMP and
cytochrome c levels in MI tissue
Myocardial samples (n=5) were homogenized in
ice-cold physiological saline (10%, w/v) and centrifuged at 3,354 ×
g for 10 min at 4°C to collect the supernatant. ATP and ADP levels
were determined using ATP and ADP assay kits, respectively
(colorimetric method; cat. nos. ab83355 and ab83359; Abcam),
according to the manufacturer's protocols. AMP and cytochrome
c levels were determined using an AMP ELISA kit (cat. no.
tw045885; Shanghai Tongwei Biological Technology, Co., Ltd.) and a
cytochrome c ELISA kit (cat. no. ab210575; Abcam),
respectively, and a SpectraMax Plus 384 microplate reader
(Molecular Devices, LLC). The EC was calculated using the following
formula: (ATP+0.5 ADP)/(ATP+ADP+AMP).
Measurement of intracellular ROS
levels and opening of the mPTP
Intracellular ROS and mPTP openings were detected
using dihydroethidium (DHE; 10 µM) and calcein-AM fluorescent
probes as permeabilization reagents, respectively (n=5). PBS
solution (37°C, 1 min) was used as blocking reagent. Briefly, fresh
frozen myocardial specimens were cut into 10-µm sections using a
Leica CM1950 frozen section machine (Leica Microsystems GmbH).
According to the manufacturer's protocol, sections were incubated
with 10 µM DHE (cat. no. GMS10111.2; Genmed Scientific, Inc.) and
10 µM Calcein-AM (cat. no. GMS12705, Genmed Scientific, Inc.) for
~30 min at 37°C and subsequently washed with 0.1 mol/l PBS
solution. Stained sections were visualized using a fluorescence
microscope at a magnification of ×200 (Olympus Corporation). The
average fluorescence intensity was calculated using Image-ProPlus
version 6.0 software (Media Cybernetics, Inc.).
Transmission electron microscopy
Myocardial infarct tissue (~1 mm3; n=5)
was fixed in 2.5% glutaraldehyde overnight at 4°C, rinsed with PBS
(pH 7.2) and subsequently fixed in 1% osmium tetroxide for 3 h at
4°C. Samples were dehydrated in an increasing concentration ethanol
series, then embedded in epoxy resin overnight at 60°C. Embedded
sections were cut into 70-nm thick slices and stained with 3%
uranium acetate and lead citrate for 15 min at 37°C. The
ultrastructures of myocardial cells, including the mitochondria,
intercalated discs, myofilaments and autophagosomes, were observed
using a Hitachi H-7650 transmission electron microscope at
magnifications of ×15,000 and ×30,000 (Hitachi High-Technologies
Corporation) and analyzed using RADIUS 2.0 (EMSIS GmbH).
Immunohistochemistry of 4E-BP1,
Atg5and mTOR expression levels
At the end of reperfusion, myocardial tissue (n=5)
was fixed in 4% buffered paraformaldehyde solution overnight at
4°C. After processing with routine histological procedures
(dehydration, transparent, dipped wax and embedding),
paraffin-embedded tissues were cut into 4 µm-thick sections. The
tissue sections were subsequently deparaffinized with xylene at
37°C and rehydrated in a descending series of alcohol. Sections
were incubated in 0.01 M sodium citrate buffer solution (pH 6.0)
for ~15 min at 100°C for antigen retrieval and then blocked in 10%
goat serum (cat. no. 701323A; Beijing Zhongsan Jinqiao
Biotechnology Co., Ltd.) for 15 min at room temperature. Tissue
sections were incubated with primary antibodies against 4E-BP1
(1:100; cat. no. ab131453; Abcam), Atg5 (1:100; cat. no. ab227084;
Abcam) and mTOR (1:100; cat. no. ab32028; Abcam) overnight at 4°C.
Following the primary incubation, sections were incubated with a
biotinylated goat anti-rabbit immunoglobulin G secondary antibody
(100 µl per section) for 25 min at 37°C and then a
streptavidin-biotin complex (100 µl per section) for 15 min at 37°C
(both from the same kit; cat. no. SAP-9100; Beijing Zhongsan
Jinqiao Biotechnology Co., Ltd.) for 20 min at room temperature.
The slides were subsequently stained with 3,3′-diaminobenzidine for
15 min and 4E-BP1-, Atg5- and mTOR-positive cells were observed
under a light microscope (magnification, ×200; Olympus
Corporation). In total, five fields of each section were captured
using a Leica DM6000 digital camera (Leica Microsystems GmbH).
Measurement of mtDNA copy number
DNA was extracted from ischemic myocardial tissue
using a mitochondrial DNA isolation kit (Abnova) (7). PCR amplification (TaKaRa Ex Taq
HSasDNA polymerase; Takara Bio, Inc.) and analysis were performed
using a 7500 Real-Time PCR system equipped with SDS software v2.0
(Applied Biosystems; Thermo Fisher Scientific, Inc.). The thermal
cycling conditions were: Initial denaturation at 95°C for 30 sec,
followed by 40 cycles of 95°C for 5 sec, 55°C for 30 sec and 72°C
for 30 sec. The following primer pairs were used for the PCR: mtDNA
(238 bp) forward, 5′-CCCCTGCTATAACCCAATACA-3′ and reverse,
5′-CCAAACCCTGGAAGAATTAAGA-3′; GAPDH (98 bp) forward,
5′-TGCTGTAGCCATATTCATTGT-3′ and reverse,
5′-CCATTCTTCCACCTTTGATGCT-3′. mtDNA copy number was normalized to
GAPDH (n=5).
Reverse transcription-quantitative PCR
(RT-qPCR)
RT-qPCR was performed according to previously
described method (38). Briefly,
total RNA was extracted from the ischemic myocardial tissue using
TRIzol® reagent (Invitrogen; Thermo Fisher Scientific,
Inc.), according to the manufacturer's instructions. RNA was
quantified using a spectrophotometer (Thermo Fisher Scientific,
Inc.) at an optical density of 260/280 nm. A total of 1 µg RNA was
reverse-transcribed into cDNA using the PrimeScriptTM RT Reagent
kit with gDNA Eraser (Takara Bio, Inc.), according to the
manufacturer's protocol. The following RT temperature protocol was
used: 37°C for 15 min, followed by 85°C for 5 sec and 4°C for 5
min. The qPCR reaction was subsequently performed in a reaction
mixture containing 2 µl cDNA (500 ng/µl), 0.8 µl forward primer (10
µM), 0.8 µl reverse primer (10 µM), 10 µl 2X SYBR Premix Ex Taq™ II
(Takara Bio, Inc.), 0.4 µl =50X ROX Reference Dye or Dye II (Takara
Bio, Inc.) and 6 µl RNase-Free dH2O (final volume: 20
µl) using a 7500 Real Time PCR system (Applied Biosystems; Thermo
Fisher Scientific, Inc.). The primer pairs used for the qPCR were
synthesized by Sangon Biotech Co., Ltd. and are presented in
Table I. The following
thermocycling conditions were used for the qPCR: 95°C for 30 sec;
40 cycles of 95°C for 5 sec and 60°C for 30 sec. Expression levels
were quantified using the 2−ΔΔCq method (42) and GAPDH and β-actin were used as
the internal reference controls (n=5).
 | Table I.Primer sequences for reverse
transcription-quantitative PCR. |
Table I.
Primer sequences for reverse
transcription-quantitative PCR.
| Gene | Primer sequence
(5′→3′) |
|---|
| NADH dehydrogenase
subunit 1 | F:
CGGCTCCTTCTCCCTACAA |
|
| R:
ATGGTCCTGCGGCGTATT |
| Cytochrome c | F:
CCCCTGCTATAACCCAATACA |
|
| R:
CCAAACCCTGGAAGAATTAAGA |
| GAPDH | F:
TGTTGCTGTAGCCATATTCATTGT |
|
| R:
CCATTCTTCCACCTTTGATGCT |
| Dynamin-related
protein 1 | F:
CGTAGTGGGAACTCAGAGCA |
|
| R:
TGGACCAGCTGCAGAATAAG |
| Mitofusin 1 | F:
GCTGCATACAGACAGACAGCCT |
|
| R:
GGTAATGACCTGTCTCAGGGCT |
| Mitofusin 2 | F:
GAACTTGTGTCTTGCATTTGGC |
|
| R:
TGCAGGCCTAACTCCTCCCAC |
| β-actin | F:
CCTCTATGCCAACACAGTGC |
|
| R:
ATACTCCTGCTTGCTGATCC |
| mTOR | F:
GTGTGGCAAGAGCGGCAGAC |
|
| R:
TGTTGGCAGAGGATGGTCAAGTTG |
| p53 | F:
GTCACCTCCACACCTCCACCTG |
|
| R:
TGCCTGTCGTCCAGATACTCAGC |
| GAPDH | F:
GGAGAAGGAGCAGGAGAATC |
|
| R:
GAGACAGACAGGAGGTGATG |
Statistical analysis
Statistical analysis was performed using the
Statistics Package for Social Science in SPSS version 13.0 software
(SPSS, Inc.). Data are expressed as the mean ± SD. Statistical
differences between groups was determined using a one-way ANOVA
followed by a LSD post hoc test for multiple comparisons. P<0.05
was considered to indicate a statistically significant
difference.
Results
Effect of HBO pretreatment on
mitochondrial function
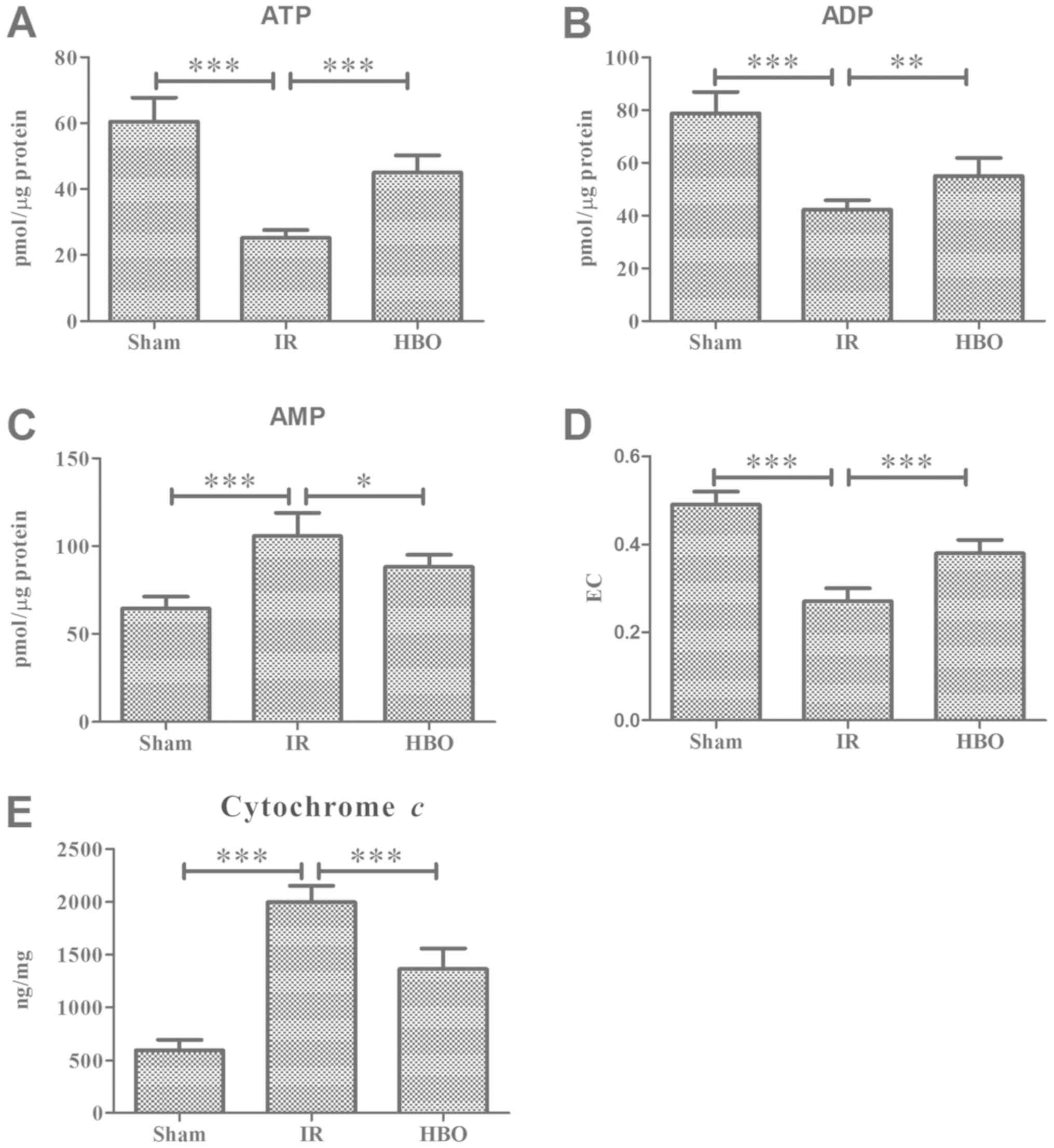
The levels of AMP in the MI tissue were
significantly increased by 164% in the IR group compared with the
sham group (105.8±13.16 vs. 64.4±6.88; F=24.280; P<0.001;
Fig. 1C), whereas the ATP and ADP
levels were significantly decreased by 58.28% (25.2±2.39 vs.
60.4±7.37; F=53.378; P<0.001; Fig.
1A) and 46.19% (42.4±3.36 vs. 78.8±8.17; F=40.84; P<0.001;
Fig. 1B), respectively. In the HBO
group compared with the IR group, the AMP levels were reduced by
16.64% (88.2±6.80; P<0.05), and the ATP and ADP levels were
increased by 78.57% (45.0±5.24; P<0.001) and 29.72% (55.0±6.89;
P<0.01), respectively. Moreover, the level of EC was
significantly decreased following MIRI surgery (IR group) compared
with the sham group (0.27±0.03 vs. 0.49±0.03; F=63.275; P<0.001;
Fig. 1D), whereas the pretreatment
with HBO in the HBO group significantly increased the EC level
compared with the IR group (0.38±0.03; P<0.001).
To determine whether HBO pretreatment influenced the
inner mitochondrial membrane, the activity of cytochrome c
was analyzed at the end of reperfusion. The levels of cytochrome
c in the MI tissue were 2.36-fold higher in the IR group
compared with the sham group (1,994.8±155.9 vs. 594.0±98.9;
F=102.836; P<0.001; Fig. 1E);
however, these levels were partly recovered in the HBO group
(1,363.0±194.11; P<0.001; Fig.
1E).
Effect of HBO pretreatment on
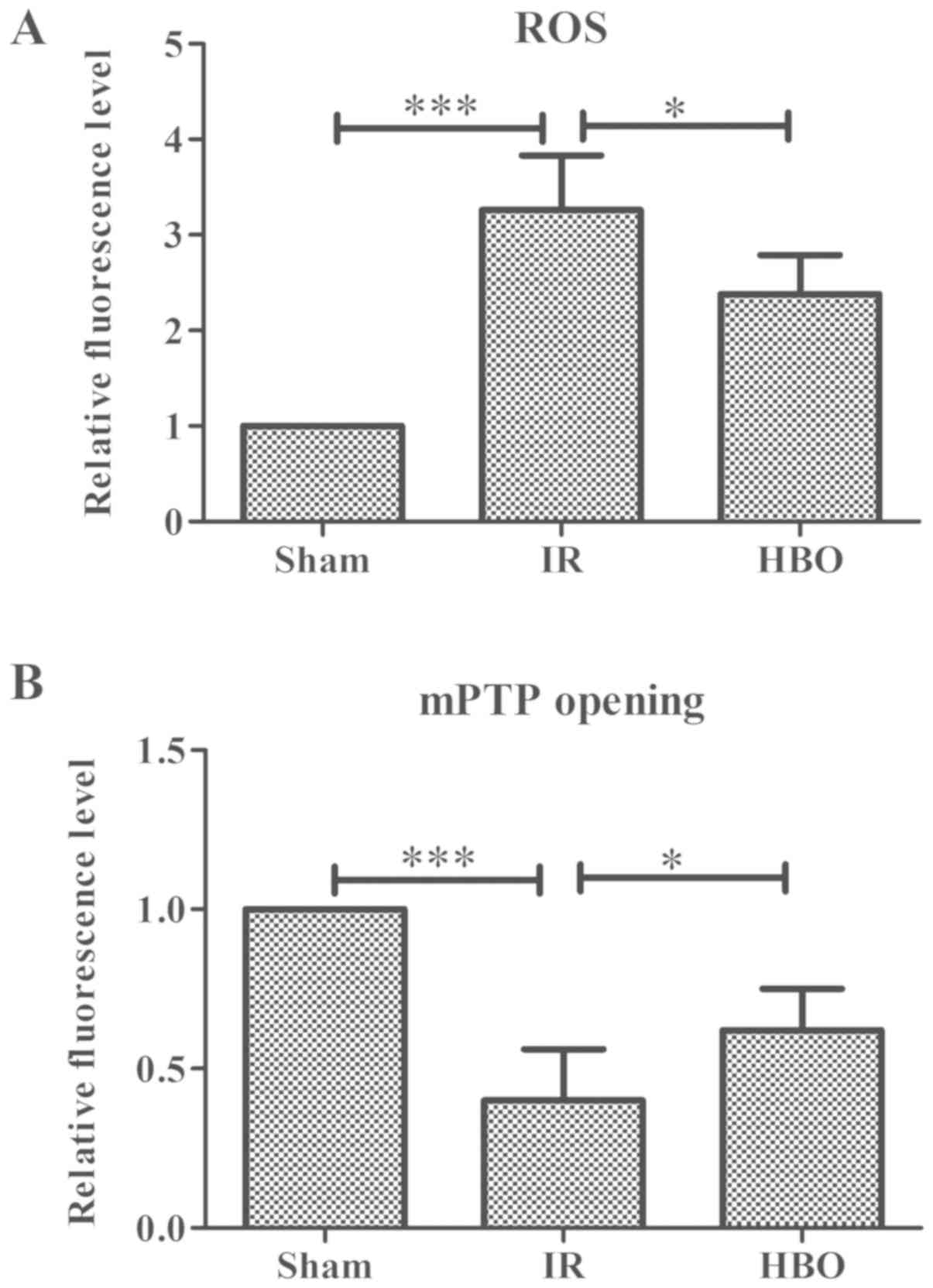
intracellular ROS levels and mPTP opening
The levels of ROS and mPTP opening in myocardial
tissue was investigated in response to MIRI using frozen sections
and fluorescent probes. The relative fluorescent level of ROS in
the IR group was significantly increased compared with the sham
group (F=39.325; P<0.001; Fig.
2A), whereas the levels were significantly decreased following
HBO pretreatment (P<0.05). Compared with the sham group, the
relative fluorescence level of calcein-AM in IR rats was
significantly reduced (F=32.905; P<0.001; Fig. 2B), whereas it was significantly
increased in the HBO group (P<0.05).
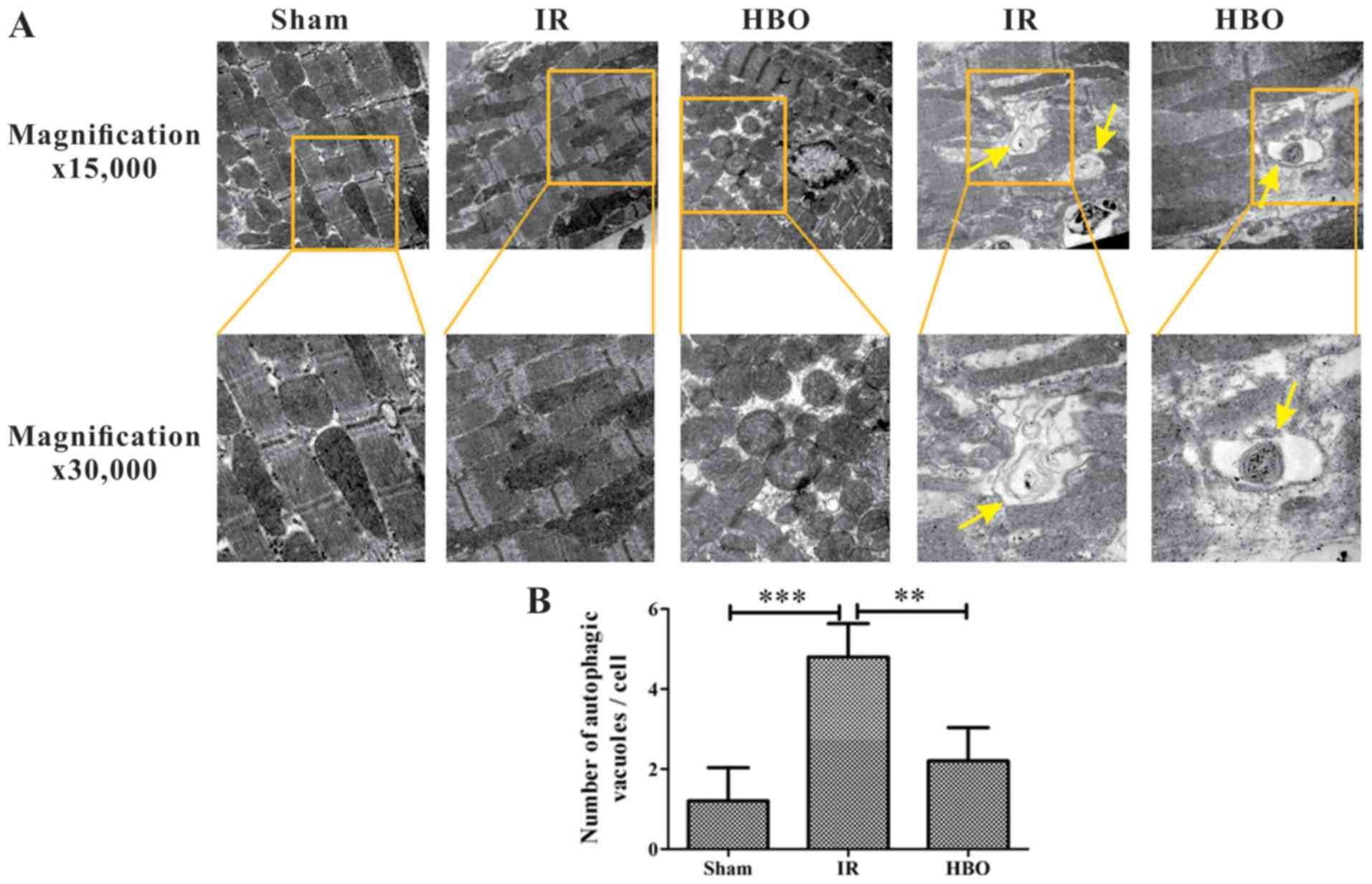
Morphological changes of myocardial
cells
Autophagic vesicles were rarely observed in the sham
group. In order to highlight the contrast of the groups, only
Fig. 3A is shown as a
representative of the sham group. Rats in the sham group
demonstrated a normal myofilament distribution, with clear light
and shade, complete intercalated disks and an intact, normal
morphology of mitochondria (Fig.
3A); however, in the IR group, myocardial cells exhibited signs
of myofilament disorders, sarcoplasmic reticulum dilatation and
mitochondrial damage (Fig. 3A), in
addition to significantly increased numbers of autophagic vesicles
compared with the sham group (Fig.
3B). Notably, these ultrastructural changes observed in the IR
group were improved in the HBO group, which exhibited only a small
number of autophagic vacuoles and slight damage to the mitochondria
(Fig. 3) compared with the IR
group.
Effect of HBO pretreatment on 4E-BP1,
Atg5 and mTOR expression levels in myocardial tissue
As indicated by the staining with brown granules,
4E-BP1 and Atg5 protein expression was mainly observed to be
located in the cytoplasm, whereas mTOR protein was located in the
nucleus (Fig. 4A). The staining
intensity of 4E-BP1 was significantly decreased in the IR group
compared with the sham group (F4E-BP1=44.685;
P<0.001; Fig. 4A and B),
whereas the Atg5 staining intensity was significantly increased
(FAtg5=338.150; P<0.001; Fig. 4A and C). Notably, HBO pretreatment
significantly alleviated these alterations observed in the IR group
(P<0.01 or P<0.001; Fig.
4A-C). In addition, the number of mTOR-positive cells was
significantly decreased in the IR group compared with the sham
group (F=106.408; P<0.001; Fig. 4A
and D), whilst the number of mTOR-positive cells were
significantly increased in the HBO group compared with the IR group
(P<0.001).
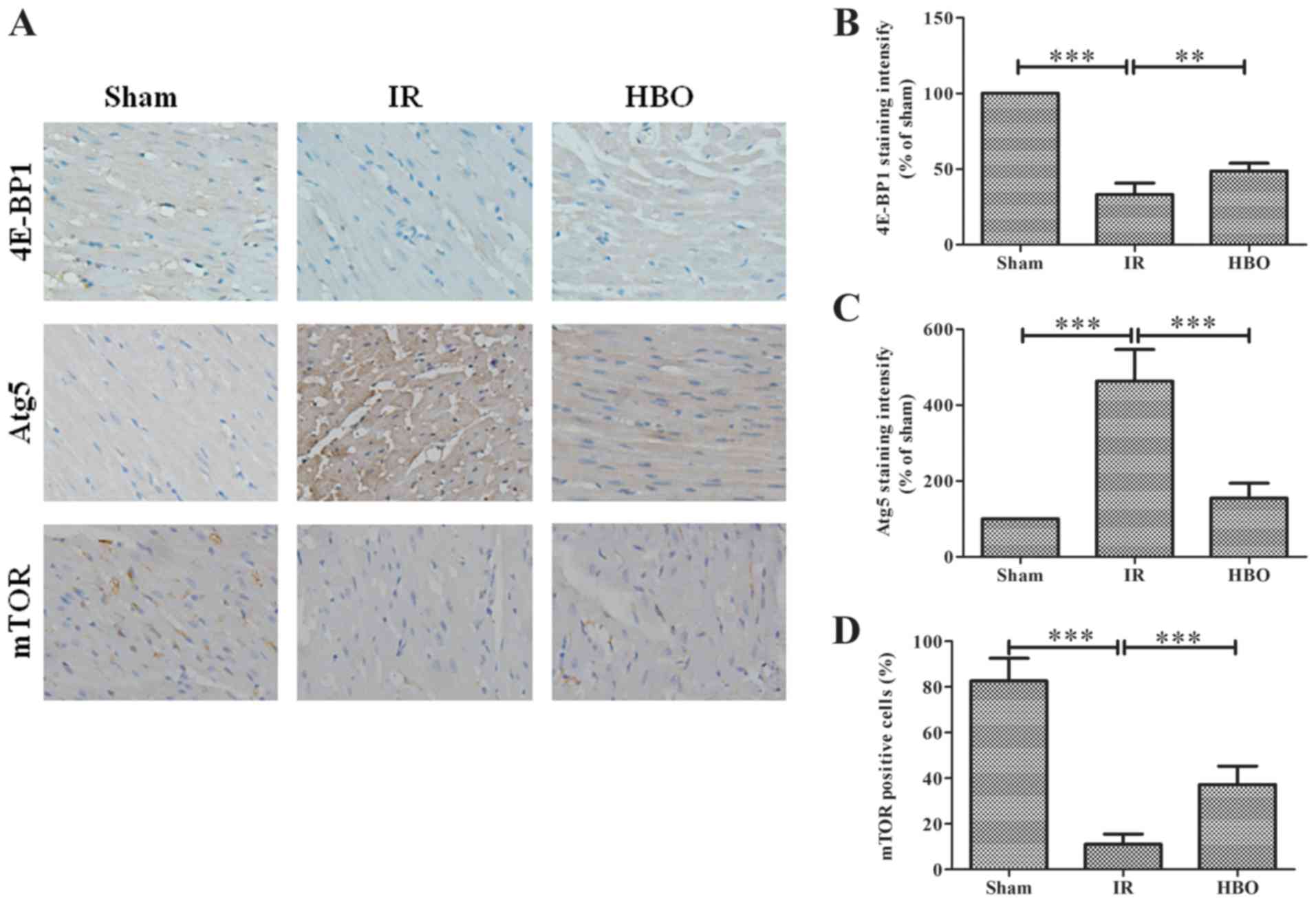 | Figure 4.Effect of HBO pretreatment on 4E-BP1,
Atg5 and mTOR expression levels in myocardial tissue. (A)
Immunohistochemical staining of myocardial sections to detect the
expression levels of 4E-BP1, Atg5 and mTOR in the sham, IR and HBO
groups. Nuclei were stained blue and the target proteins were
stained brown. Magnification, ×400. Semi-quantification of (B)
4E-BP1, (C) Atg5 and (D) mTOR expression levels in the sham, IR and
HBO groups. Data are normalized to the sham group and presented as
percentages. Data are presented as the mean ± SD (n=5).
**P<0.01; ***P<0.001. IR, ischemia/reperfusion; HBO,
hyperbaric oxygen; Atg5, autophagy-related 5; 4E-BP1, eukaryotic
initiation factor 4E-binding protein 1; mTOR, mammalian target of
rapamycin. |
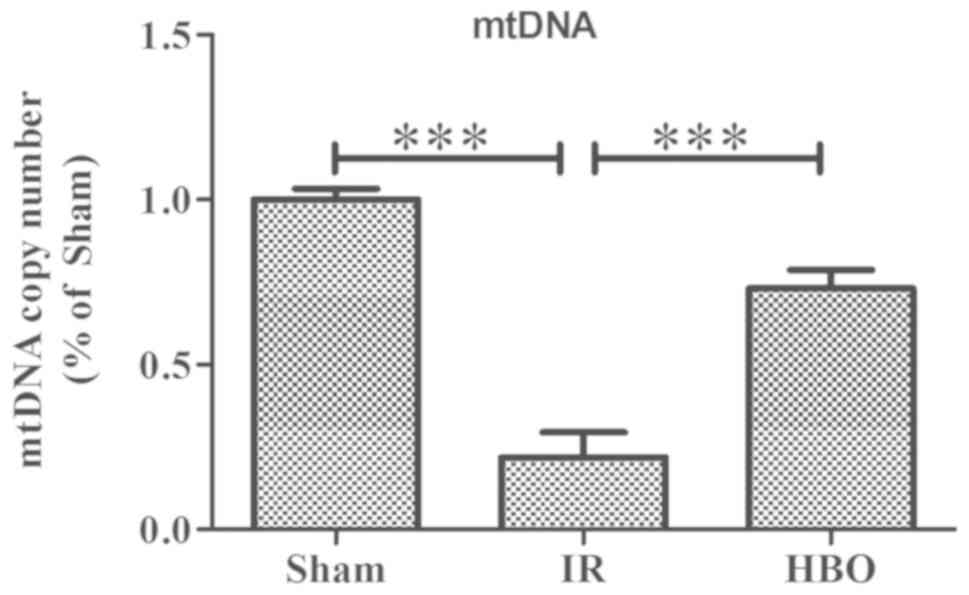
Effect of HBO pretreatment on mtDNA
damage
A significantly decreased mtDNA copy number was
observed in the myocardial tissue of the IR group compared with the
sham group (F=260.906; P<0.001; Fig. 5). In contrast, the HBO group had
significantly increased mtDNA copy numbers compared with the IR
group (P<0.001).
Effect of HBO pretreatment on the mRNA
expression levels of mitochondrial dynamic- and autophagy-related
genes
The mRNA expression levels of NADH dehydrogenase
subunit 1 (ND1), Mfn1 and Mnf2 were significantly decreased by
~68.42, 71.69 and 66.58%, respectively, in the IR group compared
with the sham group (ND1, 0.24±0.05 vs. 0.76±0.09; F=46.316;
P<0.001; Mfn1, 0.24±0.06 vs. 0.85±0.05; F=75.189; P<0.001;
Mfn2, 0.25±0.06 vs. 0.74±0.05; F=61.543; P<0.001; Fig. 6A, D and E). The mRNA expression
levels of cytochrome c and Drp1+ were 117 and 212% higher,
respectively, in the IR group compared with the sham group,
respectively (cytochrome c, 0.72±0.06 vs. 0.33±0.09;
F=27.699; P<0.001; Drp1, 0.76±0.11 vs. 0.24±0.06; F=45.180;
P<0.001; Fig. 6B and C).
Notably, the mRNA expression levels of ND1, Mfn1, cytochrome
c, Drp1 and Mfn2 were significantly reversed in rats in the
HBO group compared with rats in the IR group (ND1, 0.62±0.11,
P<0.001; Mfn1, 0.54±0.11, P<0.001; cytochrome c,
0.58±0.10, P<0.05; Drp1, 0.54±0.08, P<0.01; Mfn2, 0.50±0.09;
P<0.01).
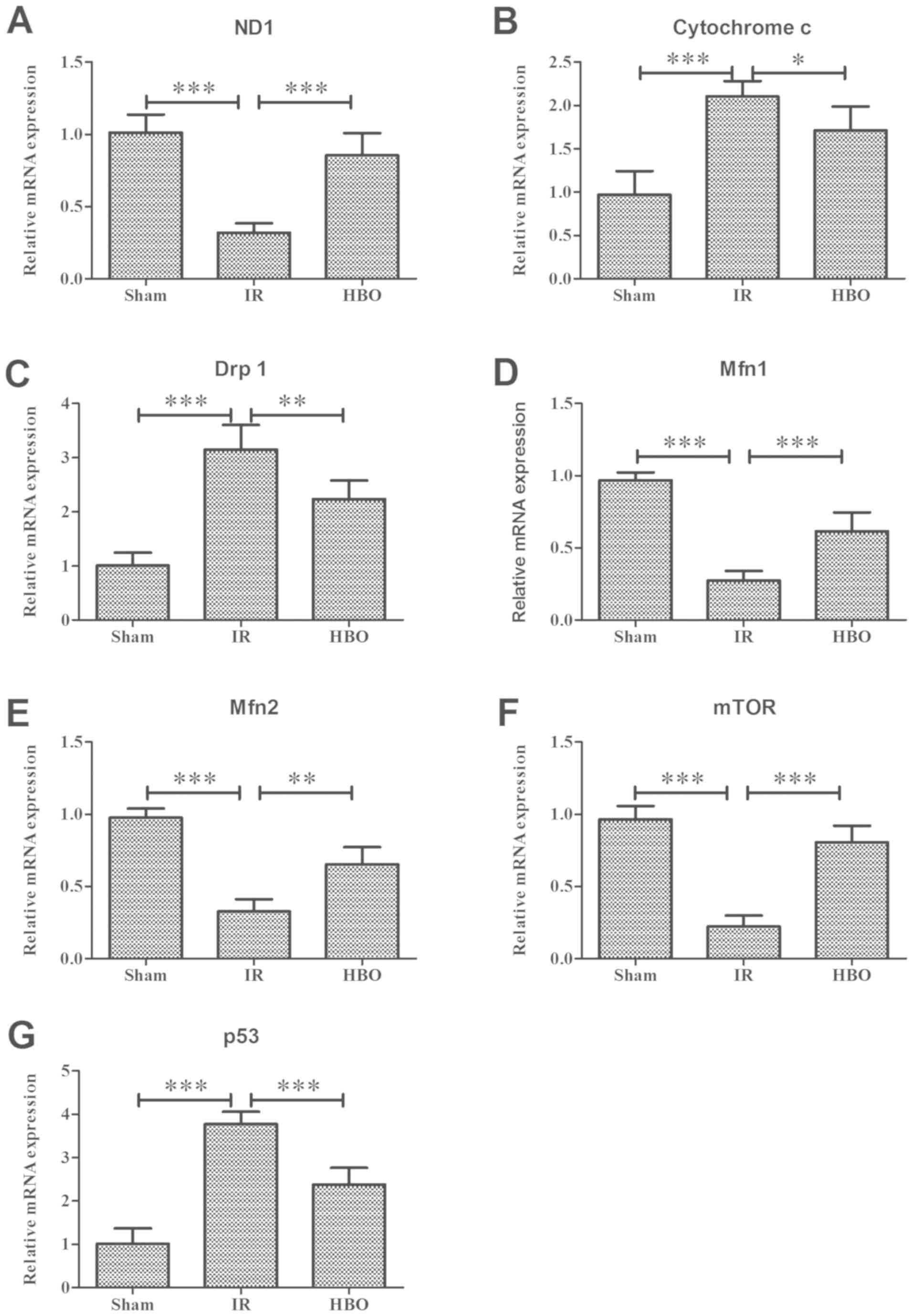 | Figure 6.Statistical analysis of reverse
transcription-quantitative PCR results. (A-E) Effect of HBO
pretreatment on the mRNA expression levels of mitochondrial
dynamics-related genes: (A) ND1, (B) cytochrome c, (C) Drp1,
(D) Mfn1 and (E) Mfn2 in the sham, IR and HBO groups. Effect of HBO
pretreatment on the expression levels of autophagy-related genes:
(F) mTOR and (G) p53 in the sham, IR and HBO groups. Data are
presented as the mean ± SD (n=5). *P<0.05; **P<0.01;
***P<0.001. HBO, hyperbaric oxygen; IR, ischemia/reperfusion;
ND1, NADH dehydrogenase subunit 1; Drp1, dynamin-related protein 1;
Mfn1, mitofusin 1; Mfn2, mitofusin 2. |
The expression levels of autophagy-related genes
were subsequently investigated; significantly decreased expression
levels of mTOR mRNA (0.17±0.06 vs. 0.73±0.07; F=84.943; P<0.001;
Fig. 6F) and significantly
increased expression levels of p53 mRNA (0.83±0.06 vs. 0.22±0.08;
F=80.131; P<0.001; Fig. 6G)
were observed in the IR group compared with the sham group.
Notably, rats pretreated with HBO exhibited significantly increased
mTOR expression levels (0.61±0.09; P<0.001) and significantly
decreased expression levels of p53 (0.52±0.08; P<0.001) compared
with the IR group.
Discussion
The present study investigated the effects of HBO
pretreatment on rats undergoing MIRI. In our previous study, it was
observed that HBO preconditioning attenuated MIRI in rats, mainly
through reducing oxygen stress damage, correcting energy metabolism
and inhibiting cell apoptosis (40). In the present study, it was
identified that HBO pretreatment protected the integrity of the
mitochondria and decreased cardiomyocyte autophagy, as observed
using a transmission electron microscope. Mechanistically, these
findings suggested that HBO may participate in regulating
mitochondrial function and inhibiting autophagy through the mTOR
signaling pathway. In addition, the results indicated that HBO may
exert an important protective function against MIRI, with our study
providing experimental data for the clinical application of
HBO.
Mitochondrial function serves important roles in the
MIRI process (43); excessive
oxidative stress results in a collapse in the mitochondrial
membrane structure and mitochondrial membrane potential
(Δψm) (44). Following
a period of ischemia, reperfusion of the oxygen supply is found to
produce an excessive amount of ROS, which triggers oxidative stress
(45). Our previous study
demonstrated that oxidative stress parameters, such as superoxide
dismutase, malondialdehyde and glutathione peroxidase, were
activated in MIRI model rats (40). Consistent with our previous study,
in the present study MIRI promoted higher relative fluorescence
levels of ROS in the myocardial tissue. In addition, in IR rats,
ATP and ADP levels were decreased, whereas AMP levels were
increased, and the EC was also decreased in IR rats. Notably, HBO
pretreatment was observed to significantly reverse all of these
changes.
Due to energy homeostasis dysfunction, the loss of
ATP leads to the disruption of ionic pumps systems, such as
Na+-K+-ATPase and
Ca2+-Mg2+-ATPase. The latter results in
increased intracellular and mitochondrial Ca2+, which
was found to further accelerate ATP depletion (46). Our previous study found that the
function of Na+-K+-ATPase and
Ca2+-Mg2+-ATPase tended to be disrupted in
MIRI rats, as well as the regulation effect of HBO on the two
ATPase (40). Factors, including
the decreased levels of ATP, influx of extracellular
Ca2+ and oxidative stress (47,48),
are suggested to accelerate the opening of the mPTP. Additionally,
abnormal activities during mitochondrial fission and fusion may
reduce the Δψm, which has been found to lead to the
elimination of these mitochondria through autophagy (49). In the present study, the opening of
the mPTP in intact cells was investigated by monitoring the
fluorescence of mitochondrial-entrapped calcein. Using the MIRI
model rats, a rapid decrease in calcein-AM fluorescence was
observed in the IR group, which suggested the increased opening of
the mPTP. In contrast, HBO pretreatment inhibited mPTP opening, as
evidenced by increased fluorescence intensity. As a subunit of
oxidative phosphorylation, cytochrome c detaches from the
mitochondrial inner membrane and leaks into the cytoplasm following
the opening of the mPTP (50,51).
In accordance with these findings, our data demonstrated that the
levels of cytochrome c were significantly increased in the
IR group compared with the sham group, whereas HBO pretreatment was
able to dampen these changes.
In the mitochondrial genome, mtDNA comprises a naked
circular strand of DNA of 16.5 kb (52), which encodes 13 polypeptides
involved in oxidative phosphorylation (53). The results from the present study
indicated that mtDNA damage occurred, as its corresponding
transcription level was decreased during MIRI, which was consistent
with the excessive cellular ROS production observed in the
cardiomyocytes. Notably, mtDNA copy number and ND1 expression
levels were increased following HBO pretreatment in rats subjected
to MIRI. ND1 is encoded by the light chain of mtDNA, thus it
reflects the transcription level of mtDNA (54). In addition, previous studies have
reported that mitochondrial fission and fusion are important events
in MIRI (55,56). In fact, the inactivation of Drp1
and the activation of Mfn2 helped improve the mitochondrial dynamic
parameters and delayed the development of MIRI (57). In the present study, the results
implied that the balance between mitochondrial fission and fusion
was disrupted by the mRNA increasing expression level of Drp1,
while decreasing expression levels of Mnf1 and Mnf2 in MIRI.
However, Drp1 expression levels were subsequently decreased with
HBO pretreatment, while Mnf1 and Mnf2 expression levels were
increased following HBO pretreatment. These findings suggested that
HBO pretreatment may provide cardiac protection against MIRI by
improving mitochondrial function, including inhibiting the
production of ROS and cytochrome c, increasing the opening
of the mPTP, increasing the EC and regulating fission- and
fusion-associated gene expression levels.
In addition to mitochondrial dysfunction, autophagy
has been recently reported as a novel regulatory target during MIRI
(58,59); for example, one previous study
observed that excessive autophagy during reperfusion triggered
myocardial cell death (22). In
the present study, it was found that MIRI dramatically induced
autophagosome formation, as evidenced by transmission electron
microscopy, which was consistent with a previous study (60). As the core kinase protein of the
mTOR complex 1 (mTORC1), the activation of mTOR promotes the
phosphorylation of its downstream effectors, including p70S6K and
4E-BP1, thus inducing cell growth and proliferation (61,62).
Moreover, mTOR represses unc-51-like autophagy activating kinase 1
(ULK1) through its phosphorylation on Ser757, which leads to the
inhibition of autophagy (63). In
addition, AMP-activated protein kinase (AMPK) is a crucial sensor
for maintaining energy homeostasis (64), and it can also promote autophagy by
directly activating ULK1 through phosphorylation on Ser317/777
under the conditions of glucose starvation (65). The current study suggested that HBO
pretreatment may inhibit MIRI-induced autophagy through regulating
the mTOR-mediated autophagy pathway, by increasing the expression
levels of mTOR and 4E-BP1and decreasing the expression levels of
Atg5 and p53. Notably, numerous studies have demonstrated that
crosstalk between autophagy and apoptosis exists (66,67).
Our previous study has demonstrated that the Bax family members,
caspase cascade, and cardiomyocyte apoptosis were inhibited
following HBO pretreatment (40).
Thus, these results suggested that the cardioprotective mechanism
of HBO may be involved in inhibiting mTOR-mediated autophagy.
Nonetheless, the present study has numerous
limitations. It was observed that mitochondrial dysfunction and
excessive autophagy occurred in the MIRI model following 30 min of
ischemia and 1 h of reperfusion, as evidenced by transmission
electron microscopy, immunohistochemistry and the analysis of mRNA
expression levels. These findings have also been reported in
numerous previous studies (20,21,68,69).
In the present study, HBO pretreatment was found to restore
mitochondrial function and inhibit cardiomyocyte autophagy;
therefore, it was hypothesized that the protective effect of HBO
pretreatment may be related to the modulation of mitochondrial
function and cell autophagy. However, the mechanism by which HBO
pretreatment affects the cardiomyocyte mitochondria and autophagy
remains to be further investigated. Secondly, several experimental
methods were used to analyze the mitochondrial function, including
EC, cytochrome c levels, intracellular ROS production, the
opening of the mPTP, mitochondrial ultrastructure, mtDNA copy
number and the mRNA expression levels of Drp 1, Mfn1 and Mfn2. In
future experiments, experimental methods investigating the
bioenergetics and the status of oxidative phosphorylation will be
used to further determine the mitochondrial function.
In conclusion, the present study demonstrated that
HBO pretreatment effectively protected rat hearts from MIRI. This
effect may be related to the restoration of mitochondrial function
and the inhibition of cardiomyocyte autophagy. Thus, these findings
suggested that HBO treatment may be a useful agent for the
mitigation of MIRI.
Acknowledgements
The authors would like to thank Dr Jianquan Li, from
the Guangxi Medical University, China, for his technical support in
immunohistochemistry.
Funding
This present study was supported by the National
Natural Science Foundation of China (grant nos. 81701089 and
81960246), The Guangxi Natural Science Foundation (grant no.
2017GXNSFBA198010) and the Guangxi Sanitation Research Project
(grant nos. Z2016582 and Z20201096).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request
Authors' contributions
WC and LL wrote the manuscript. WC, LL, XC, XP, and
CC performed the experiments. ZN collected the data and analyzed
the data. CC designed the study, revised the manuscript and funded
the research. All authors read and approved the final
manuscript.
Ethics approval and consent to
participate
All animal experimental procedures were approved by
the Animal Ethical Committee of Guangxi Medical University.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Nabel EG and Braunwald E: A tale of
coronary artery disease and myocardial infarction. N Engl J Med.
366:54–63. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Writing Group Members, Mozaffarian D,
Benjamin EJ, Go AS, Arnett DK, Blaha MJ, Cushman M, Das SR,
Ferranti SD, Després JP, et al: Executive summary: Heart disease
and stroke statistics-2016 update: A report from the American Heart
Association. Circulation. 133:447–454. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Buja LM: Myocardial ischemia and
reperfusion injury. Cardiovasc Pathol. 14:170–175. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Levisman J and Price MJ: Update on the
guidelines for the management of ST-elevation myocardial
infarction. Am J Cardiol. 115 (Suppl 5):A3–A9. 2015. View Article : Google Scholar
|
|
5
|
Yellon DM and Hausenloy DJ: Myocardial
reperfusion injury. N Engl J Med. 357:1121–1135. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Ibáñez B, Heusch G, Ovize M and Van de
Werf F: Evolving therapies for myocardial ischemia/reperfusion
injury. J Am Coll Cardiol. 65:1454–1471. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Lee MS: Role of mitochondrial function in
cell death and body metabolism. Front Biosci (Landmark Ed).
21:1233–1244. 2016. View
Article : Google Scholar : PubMed/NCBI
|
|
8
|
Shen YQ, Guerra-Librero A, Fernandez-Gil
BI, Florido J, García-López S, Martinez-Ruiz L, Mendivil-Perez M,
Soto-Mercado V, Acuña-Castroviejo Dario, Ortega-Arellano H, et al:
Combination of melatonin and rapamycin for head and neck cancer
therapy: Suppression of AKT/mTOR pathway activation, and activation
of mitophagy and apoptosis via mitochondrial function regulation. J
Pineal Res. 64:e124612017. View Article : Google Scholar
|
|
9
|
Zhou H, Hu SY, Jin QH, Shi C, Zhang Y, Zhu
PJ, Ma Q, Tian F and Chen YD: Mff-dependent mitochondrial fission
contributes to the pathogenesis of cardiac microvasculature
ischemia/reperfusion injury via induction of mROS-Mmediated
cardiolipin oxidation and HK2/VDAC1 disassociation-involved mPTP
opening. J Am Heart Assoc. 6:e0053282017. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Shires SE and Gustafsson AB: Mitophagy and
heart failure. J Mol Med (Berl). 93:253–262. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Saito T and Sadoshima J: Molecular
mechanisms of mitochondrial autophagy/mitophagy in the heart. Circ
Res. 116:1477–1490. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Scarffe LA, Stevens DA, Dawson VL and
Dawson TM: Parkin and PINK1: Much more than mitophagy. Trends
Neurosci. 37:315–324. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Hausenloy DJ and Scorrano L: Targeting
cell death. Clin Pharmacol Ther. 82:370–373. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Liesa M, Palacin M and Zorzano A:
Mitochondrial dynamics in mammalian health and disease. Physiol
Rev. 89:799–845. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Ingerman E, Perkins EM, Marino M, Mears
JA, McCaffery JM, Hinshaw JE and Nunnari J: Dnm1 forms spirals that
are structurally tailored to fit mitochondria. J Cell Biol.
170:1021–1027. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Smirnova E, Griparic L, Shurland DL and
van der Bliek AM: Dynamin-related protein Drp1 is required for
mitochondrial division in mammalian cells. Mol Biol Cell.
12:2245–2256. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Rapaport D, Brunner M, Neupert W and
Westermann B: Fzo1p is a mitochondrial outer membrane protein
essential for the biogenesis of functional mitochondria in
Saccharomyces cerevisiae. J Biol Chem. 273:20150–20155. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Santel A and Fuller MT: Control of
mitochondrial morphology by a human mitofusin. J Cell Sci.
114:867–874. 2001.PubMed/NCBI
|
|
19
|
Hao M, Zhu S, Hu L, Zhu H, Wu X and Li Q:
Myocardial ischemic postconditioning promotes autophagy against
ischemia reperfusion injury via the activation of the
nNOS/AMPK/mTOR pathway. Int J Mol Sci. 18:6142017. View Article : Google Scholar
|
|
20
|
Jian J, Xuan F, Qin F and Huang R:
Bauhinia championii flavone inhibits apoptosis and autophagy via
the PI3K/Akt pathway in myocardial ischemia/reperfusion injury in
rats. Drug Des Devel Ther. 9:5933–5945. 2015.PubMed/NCBI
|
|
21
|
Xuan F and Jian J: Epigallocatechin
gallate exerts protective effects against myocardial
ischemia/reperfusion injury through the PI3K/Akt pathway-mediated
inhibition of apoptosis and the restoration of the autophagic flux.
Int J Mol Med. 38:328–336. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Hariharan N, Zhai P and Sadoshima J:
Oxidative stress stimulates autophagic flux during
ischemia/reperfusion. Antioxid Redox Signal. 14:2179–2190. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Kang R, Zeh HJ, Lotze MT and Tang D: The
Beclin 1 network regulates autophagy and apoptosis. Cell Death
Differ. 18:571–580. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Yu L, McPhee CK, Zheng LX, Mardones GA,
Rong YG, Peng JY, Mi N, Zhao Y, Liu ZH and Wan FY: Termination of
autophagy and reformation of lysosomes regulated by mTOR. Nature.
465:942–946. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Han YF, Zhao YB, Li J, Li L, Li YG, Li SP
and Li ZD: Stat3-Atg5 signal axis inducing autophagy to alleviate
hepatic ischemia-reperfusion injury. J Cell Biochem. 119:3440–3450.
2018. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Pópulo H, Lopes JM and Soares P: The mTOR
signalling pathway in human cancer. Int J Mol Sci. 13:1886–1918.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Bramham CR, Jensen KB and Proud CG: Tuning
specific translation in cancer metastasis and synaptic memory:
Control at the MNK-eIF4E axis. Trends Biochem Sci. 41:847–858.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Foster SS, De S, Johnson LK, Petrini JH
and Stracker TH: Cell cycle- and DNA repair pathway-specific
effects of apoptosis on tumor suppression. Proc Natl Acad Sci USA.
109:9953–9958. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Tasdemir E, Maiuri MC, Galluzzi L, Vitale
I, Djavaheri-Mergny M, D'Amelio M, Criollo A, Morselli E, Zhu C,
Harper F, et al: Regulation of autophagy by cytoplasmic p53. Nat
Cell Biol. 10:676–687. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Crighton D, Wilkinson S, O'Prey J, Syed N,
Smith P, Harrison PR, Gasco M, Garrone O, Crook T and Ryan KM:
DRAM, a p53-induced modulator of autophagy, is critical for
apoptosis. Cell. 126:121–134. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Dekleva M, Neskovic A, Vlahovic A,
Putnikovic B, Beleslin B and Ostojic M: Adjunctive effect of
hyperbaric oxygen treatment after thrombolysis on left ventricular
function in patients with acute myocardial infarction. Am Heart J.
148:0312004. View Article : Google Scholar
|
|
32
|
Bennett MH, Lehm JP and Jepson N:
Hyperbaric oxygen therapy for acute coronary syndrome. Cochrane
Database Syst Rev. 7:CD0048182015.
|
|
33
|
Rusyniak DE, Kirk MA, May JD, Kao LW,
Brizendine EJ, Welch JL, Cordell WH and Alonso RJ: Hyperbaric
oxygen therapy in acute ischemic stroke: Results of the hyperbaric
oxygen in acute ischemic stroke trial pilot study. Stroke.
34:571–574. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Liu WC, Yang SN, Wu CW, Chen LW and Chan
JY: Hyperbaric oxygen therapy alleviates carbon monoxide
poisoning-induced delayed memory impairment by preserving
brain-derived neurotrophic factor-dependent hippocampal
neurogenesis. Crit Care Med. 44:e25–39. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Fife CE, Eckert KA and Workman WT: Ethical
issues, standards, and quality control in the practice of
hyperbaric medicine. Springer; Switzerland: Textbook of Hyperbaric
Medicine. pp. 597–608. 2016
|
|
36
|
Pan X, Chen C, Huang J, Wei H and Fan Q:
Neuroprotective effect of combined therapy with hyperbaric oxygen
and madopar on 6-hydroxydopamine-induced Parkinson's disease in
rats. Neurosci Lett. 600:220–225. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Chen CX, Fan QP, Nong ZH, Chen W, Li YX,
Huang LY, Feng DR, Pan XR and Lan SY: Hyperbaric oxygen attenuates
withdrawal symptoms by regulating monoaminergic neurotransmitters
and NO signaling pathway at nucleus accumbens in morphine-dependent
rats. Neurochem Res. 43:531–539. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Chen CX, Huang LY, Nong ZH, Li YX, Chen W,
Huang JP, Pan XR, Wu GW and Lin YZ: Hyperbaric oxygen prevents
cognitive impairments in mice induced by D-galactose by improving
cholinergic and anti-apoptotic functions. Neurochem Res.
42:1240–1253. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Chen X, Li Y, Chen W, Nong Z, Huang J and
Chen C: Protective effect of hyperbaric oxygen on cognitive
impairment induced by D-galactose in mice. Neurochem Res.
41:3032–3041. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Chen C, Chen W, Nong Z, Ma Y, Qiu S and Wu
G: Cardioprotective effects of combined therapy with hyperbaric
oxygen and diltiazem pretreatment on myocardial
ischemia-reperfusion injury in rats. Cell Physiol Biochem.
38:2015–2029. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Bøtker HE, Hausenloy D, Andreadou I,
Antonucci S, Boengler K, Davidson SM, Deshwal S, Devaux Y, Lisa FD,
Sante MD, et al: Practical guidelines for rigor and reproducibility
in preclinical and clinical studies on cardioprotection. Basic Res
Cardiol. 113:018–0696. 2018. View Article : Google Scholar
|
|
42
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Qiao X, Jia S, Ye J, Fang X, Zhang C, Cao
Y, Xu C, Zhao L, Zhu Y, Wang L and Zheng M: PTPIP51 regulates mouse
cardiac ischemia/reperfusion through mediating the mitochondria-SR
junction. Sci Rep. 7:453792017. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Cook SA, Sugden PH and Clerk A: Regulation
of bcl-2 family proteins during development and in response to
oxidative stress in cardiac myocytes: Association with changes in
mitochondrial membrane potential. Circ Res. 85:940–949. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Farber JL: Mechanisms of cell injury by
activated oxygen species. Environ Health Perspect. 10 (Suppl
10):S17–S24. 1994. View Article : Google Scholar
|
|
46
|
Kalogeris T, Baines CP, Krenz M and
Korthuis RJ: Cell biology of ischemia/reperfusion injury. Int Rev
Cell Mol Biol. 298:229–317. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Zorov DB, Juhaszova M and Sollott SJ:
Mitochondrial reactive oxygen species (ROS) and ROS-induced ROS
release. Physiol Rev. 94:909–950. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Zhang T, Zhang Y, Cui MY, Jin L, Wang YM,
Lv FX, Liu YL, Zheng W, Shang HB, Zhang J, et al: CaMKII is a RIP3
substrate mediating ischemia- and oxidative stress-induced
myocardial necroptosis. Nat Med. 22:175–182. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Twig G, Elorza A, Molina AJA, Mohamed H,
Wikstrom JD, Walzer G, Stiles L, Haigh SE, Katz S, Las G, et al:
Fission and selective fusion govern mitochondrial segregation and
elimination by autophagy. EMBO J. 27:433–446. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Brooks C, Wei Q, Cho SG and Dong Z:
Regulation of mitochondrial dynamics in acute kidney injury in cell
culture and rodent models. J Clin Invest. 119:1275–1285. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Estaquier J and Arnoult D: Inhibiting
Drp1-mediated mitochondrial fission selectively prevents the
release of cytochrome c during apoptosis. Cell Death Differ.
14:1086–1094. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Lander ES and Lodish H: Mitochondrial
diseases: Gene mapping and gene therapy. Cell. 61:925–926. 1990.
View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Borst P and Grivell LA: The mitochondrial
genome of yeast. Cell. 15:705–723. 1978. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Haendeler J, Dröse S, Büchner N, Jakob S,
Altschmid J, Goy C, Spyridopoulos L, Zeiher AM, Brandt U and
Dimmeler S: Mitochondrial telomerase reverse transcriptase binds to
and protects mitochondrial DNA and function from damage.
Arterioscler Thromb Vasc Biol. 29:929–935. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Ong SB, Subrayan S, Lim SY, Yellon DM,
Davidson SM and Hausenloy DJ: Inhibiting mitochondrial fission
protects the heart against ischemia/reperfusion injury.
Circulation. 121:2012–2022. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Ong SB, Hall AR and Hausenloy DJ:
Mitochondrial dynamics in cardiovascular health and disease.
Antioxid Redox Signal. 19:400–414. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Zhang Y, Zhang L, Zhang Y, Fan X, Yang WW,
Yu BY, Kou JP and Li F: YiQiFuMai powder injection attenuates
coronary artery ligation-induced heart failure through improving
mitochondrial function via regulating ROS generation and CaMKII
signaling pathways. Front Pharmacol. 10:3812019. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Ma H, Guo R, Yu L, Zhang Y and Ren J:
Aldehyde dehydrogenase 2 (ALDH2) rescues myocardial
ischaemia/reperfusion injury: Role of autophagy paradox and toxic
aldehyde. Eur Heart J. 32:1025–1038. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Ma X, Liu H, Foyil SR, Godar RJ,
Weinheimer CJ and Diwan A: Autophagy is impaired in cardiac
ischemia-reperfusion injury. Autophagy. 8:1394–1396. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Dong W, Yang R, Yang J, Ding J, Wu H and
Zhang J: Resveratrol pretreatment protects rat hearts from
ischemia/reperfusion injury partly via a NALP3 inflammasome
pathway. Int J Clin Exp Pathol. 8:8731–8741. 2015.PubMed/NCBI
|
|
61
|
Rabanal-Ruiz Y, Otten EG and Korolchuk VI:
mTORC1 as the main gateway to autophagy. Essays Biochem.
61:565–584. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Yang Z and Klionsky DJ: Mammalian
autophagy: Core molecular machinery and signaling regulation. Curr
Opin Cell Biol. 22:124–131. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Dan E, Joungmok K, Shaw RJ and Kun-Liang
G: The autophagy initiating kinase ULK1 is regulated via opposing
phosphorylation by AMPK and mTOR. Autophagy. 7:643–644. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Hardie DG: AMP-activated/SNF1 protein
kinases: Conserved guardians of cellular energy. Nat Rev Mol Cell
Biol. 8:774–785. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Kim J, Kundu M, Viollet B and Guan KL:
AMPK and mTOR regulate autophagy through direct phosphorylation of
Ulk1. Nat Cell Biol. 13:132–141. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Luo S and Rubinsztein DC: Apoptosis blocks
Beclin 1-dependent autophagosome synthesis: An effect rescued by
Bcl-xL. Cell Death Differ. 17:268–277. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Yang B and Zhao S: Polydatin regulates
proliferation, apoptosis and autophagy in multiple myeloma cells
through mTOR/p70s6k pathway. Onco Targets Ther. 10:935–944. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Liu CY, Zhang YH, Li RB, Zhou LY, An Tao,
Zhang RC, Zhai M, Huang Y, Yan KW, Dong YH, et al: lncRNA CAIF
inhibits autophagy and attenuates myocardial infarction by blocking
p53-mediated myocardin transcription. Nat Commun. 9:292018.
View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Guo X, Jiang H, Yang J, Chen J, Yang J,
Ding JW, Li S, Wu H and Ding HS: Radioprotective 105 kDa protein
attenuates ischemia/reperfusion-induced myocardial apoptosis and
autophagy by inhibiting the activation of the TLR4/NF-κB signaling
pathway in rats. Int J Mol Med. 38:885–893. 2016. View Article : Google Scholar : PubMed/NCBI
|