Introduction
Non-alcoholic fatty liver disease (NAFLD) is
becoming one of the most prevalent chronic liver diseases in modern
countries, increasing rapidly as a result of recent upward trends
in obesity and life-style changes (1). A subset of NAFLD patients go on to
develop non-alcoholic steatohepatitis (NASH) by progression of
steatosis and necro-inflammatory changes in the liver, leading to
an increase in the incidence of hepatocellular carcinoma (2). Mortality in NAFLD patients has been
reported to be independently associated with the stage of liver
fibrosis (3), and it is important
to prevent the progression of liver fibrosis in NAFLD patients.
Recently, several drugs have been developed and have entered phase
2 or 3 clinical trials, but no effective drugs against NAFLD are
yet available. Therefore, it is important to clarify the mechanism
of liver fibrosis in NAFLD in order to identify therapeutic
targets.
To identify clinical factors associated with the
progression of liver fibrosis in NAFLD patients, several genome
wide association studies (GWAS) have recently been performed
worldwide. A single nucleotide polymorphism (SNP) at
rs738409 in patatin-like phospholipase domain containing
3 (PNPLA3) was identified as having strong associations
with prevalence and disease progression in NAFLD and NASH (4–7). A SNP
in transmembrane 6 superfamily 2 (TM6SF2) was also
identified as a potential contributor to NAFLD pathogenesis
(8,9). The SNP rs58542926 in
TM6SF2 is significantly associated with incidence of NAFLD
and with fibrosis stage (10–13).
TM6SF2 protein is highly expressed in the small intestine and liver
and plays a role in lipid synthesis and secretion of
triglyceride-rich lipoproteins in the liver (14–19).
TM6SF2 rs58542926 (C>T), a coding SNP that causes an
amino acid substitution at codon 167 (E167K), is considered to lead
to a loss of function and to accelerate hepatic steatosis (20). However, although lipids are
metabolized in hepatocytes and may accumulate in these cells, liver
fibrosis is strongly associated with hepatic stellate cells (HSCs)
(21). The influence of the coding
SNP in TM6SF2 on the function of HSCs has not been
clarified.
HSCs are normally activated in response to
stimulation by inflammatory cytokines, such as transforming growth
factor beta 1 (TGFβ1), and by pathogen-associated molecular
patterns, such as lipopolysaccharides (21). Activated HSCs transform into
myofibroblasts, and alpha-smooth muscle actin (αSMA) expression is
upregulated in the transformed myofibroblasts (21,22).
Activation of HSCs leads to secretion of extra-cellular matrix
proteins such as collagen type 1 into the sinusoids, resulting in
collagen accumulation and progression of liver fibrosis (21–23).
Although the impacts of genetic factors on clinical features of
NAFLD and hepatocyte functions have been analyzed, the impacts of
genetic factors on HSCs have not been examined. In the present
study, we explored the role of TM6SF2 SNP rs58542926
in liver fibrosis using an in vitro activated HSC model.
Materials and methods
Construction of TM6SF2 expression
plasmids
Human TM6SF2 mRNA was amplified from LX-2
cells and cloned into p3×FLAG-CMV-10 vector (Sigma-Aldrich). The
cloned plasmid containing the wild-type CC genotype at rs58542926
in TM6SF2 gene was designated as p3FLAG/TM6SF2-WT.
Subsequently, a modified plasmid, designated as p3FLAG/TM6SF2-MT,
was generated by introducing a C-to-T point mutation at rs58542926
in TM6SF2 to create an amino acid substitution [glutamic
acid (E) to lysine (K)] in the TM6SF2 gene using the QuikChange
Site-Directed Mutagenesis kit (Agilent Technologies).
Cell culture
LX-2 cells from a human hepatic stellate cell line,
which were provided by Dr Mutsumi Miyauchi (Hiroshima University,
Hiroshima, Japan), were grown in Dulbecco's modified Eagle's medium
(DMEM) supplemented with 10% (v/v) fetal bovine serum at 37°C and
under 5% CO2. Mycoplasma testing was done before and
after the experiment.
Each TM6SF2 expression plasmid was transiently
transfected into LX-2 cells by FuGENE HD Transfection reagent
(Promega) in accordance with the instructions supplied by the
manufacturer. Twenty-four hours after transfection, transfected
cells were stimulated with 10 ng/ml of TGFβ1 for 48 h, and then the
cells were harvested and stored at −80°C until use.
Quantification of mRNA expression
level
Total RNA was extracted from collected LX-2 cells
using RNeasy Mini kit (Qiagen) and reverse-transcribed using
ReverTra Ace (Toyobo Co., Ltd.) and random primer in accordance
with the instructions supplied by the manufacturer. αSMA or
TM6SF2 mRNA levels were quantified from the resulting cDNA
by quantitative PCR using the 7300 Real-Time PCR System (Applied
Biosystems), with the expression of GAPDH serving as a control.
Expression levels were compared using the Wilcoxon signed-rank
test. Amplification was performed in a 25 µl reaction mixture
containing 12.5 µl SYBR-Green PCR Master Mix (Applied Biosystems),
5 pmol of forward primer, 5 pmol of reverse primer, and 1 µl of
cDNA solution. After incubation for 2 min at 50°C, the sample was
denatured for 10 min at 95°C, followed by a PCR cycling program
consisting of 40 cycles of 15 sec at 95°C, 30 sec at 55°C and 60
sec at 60°C. The following primer sequences were used: αSMA;
5′-CTCATTTTCAAAGTCCAGAGCTACA-3′ and 5′-AGCGTGGCTATTCCTTCGT-3′,
TM6SF2; 5′-TGAAGCCCACCACATAGCTG-3′ and 5′-CGGTCTACAGCTTGTCCCAT-3′,
GAPDH; 5′-GAAGGTGAAGGTCGGAGTC-3′ and
5′-GAAGATGGTGATGGGATTTC-3′.
Automated capillary western
blotting
LX-2 cells, transfected with TM6SF2 expression
plasmids and treated with TGFβ1, were cooled on ice and dissolved
with RIPA-like buffer [50 mM Tris-HCl (pH 8.0), 0.1% SDS, 1% NP-40,
150 mM sodium chloride, and 0.5% sodium deoxycholate] containing
protease inhibitor cocktail (Sigma-Aldrich). Cell lysates were
transferred onto capillary western immunoassay using Wes system
(ProteinSimple). The proteins were detected with anti-TM6SF2 rabbit
polyclonal antibody (Thermo Fisher Scientific, Inc.), anti-αSMA
rabbit monoclonal antibody (Cell Signaling Technology Japan), or
anti-GAPDH rabbit polyclonal antibody (Santa Cruz Biotechnology),
followed by anti-rabbit immunoglobulin (GE Healthcare). Signal
intensities were quantified using Compass software (ProteinSimple)
and were corrected by GAPDH and analyzed by Mann-Whitney U
test.
Knockdown of TM6SF2 by siRNA
treatment
TM6SF2 siRNA was designed by siDirect (http://sidirect2.rnai.jp) using the TM6SF2 mRNA
sequence (NM_001001524) as a reference. The designed siRNA sequence
was as follows: 5′-AAAAUUCCGGUAUCUCUUCCU-3′,
5′-GAAGAGAUACCGGAAUUUUGG-3′. Prepared siRNAs were transfected into
LX-2 cells by electroporation using the Neon transfection system
(Thermo Fisher Scientific, Inc.) at 1,100 mV for 30 msec followed
by 24-h incubation with serum-free medium.
Immunocytochemistry
LX-2 cells that had been transfected with TM6SF2
expression plasmid or treated with siRNA were incubated for 48 h
were fixed with 4% (v/v) paraformaldehyde and stained with
anti-TM6SF2 antibody. The bound antibodies were detected with an
Alexa 594-conjugated antibody against rabbit IgG (1:2,000)
(Molecular Probes). Nuclei were counterstained with bisbenzimide H
33258 (Hoechst 33258; Abcam). The stained cells were examined using
a Fluoview FV10i microscope (Olympus Co.). Fluorescence intensities
of TM6SF2 were compared using the Mann-Whitney U test.
Statistical analysis
All experiments were performed in triplicate wells.
All data are expressed as the mean ± standard deviation (SD) and
are presented relative to control. Pairwise differences between
groups were examined for statistical significance using the
Mann-Whitney U test. Univariate or multivariable differences among
three or more groups were estimated using one-way or two-way ANOVA
with Tukey's post-hoc multiple comparison test. P<0.05 was
considered to indicate a statistically significant difference.
Statistical analysis was performed using IBM SPSS Statistics for
Windows, version 22.0 (IBM Corp.).
Results
TM6SF2 regulates αSMA expression in
LX-2 cells
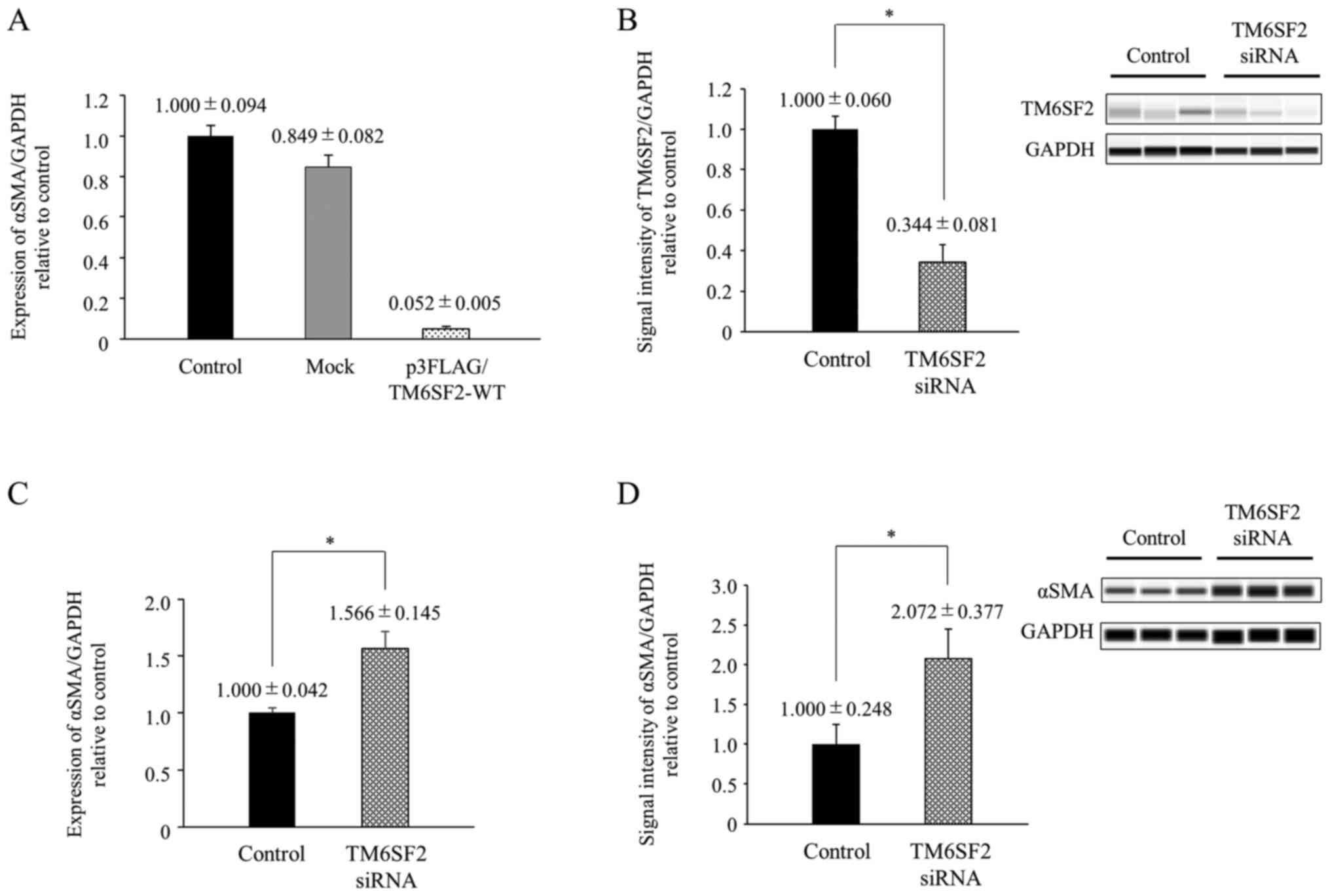
To analyze the impact of TM6SF2 on HSC
activation, p3FLAG/TM6SF2-WT plasmid was transiently transfected
into LX-2 cells, and the induction of alpha-smooth muscle actin
(αSMA) level was compared. Although αSMA level was not changed by
transfection with empty vector (mock), αSMA mRNA expression
was significantly suppressed in the presence of TM6SF2 expression
plasmids (one-way ANOVA, P<0.05, respectively) (Fig. 1A).
To verify this result, we also analyzed the
association between TM6SF2 and αSMA by knocking down
TM6SF2. The siRNA targeted to TM6SF2 was transfected into
LX-2 cells by electroporation, and intracellular αSMA level was
compared 24 h after siRNA treatment. TM6SF2 protein expression in
LX-2 cells was suppressed to 34.4% by treatment with siRNA
(Fig. 1B), and immunostaining of
TM6SF2 exhibited the same results (Figs. S1 and 2). αSMA expression in TM6SF2-knocked down
cells was 1.5~2.0-fold elevated compared to control cells in both
mRNA and protein levels (Fig. 1C and
D). A similar tendency was also observed in intracellular
collagen type 1 alpha 1 (COL1A1) expression measured by
real-time PCR (Fig. S3). These
results suggest that TM6SF2 downregulates αSMA expression in
HSCs.
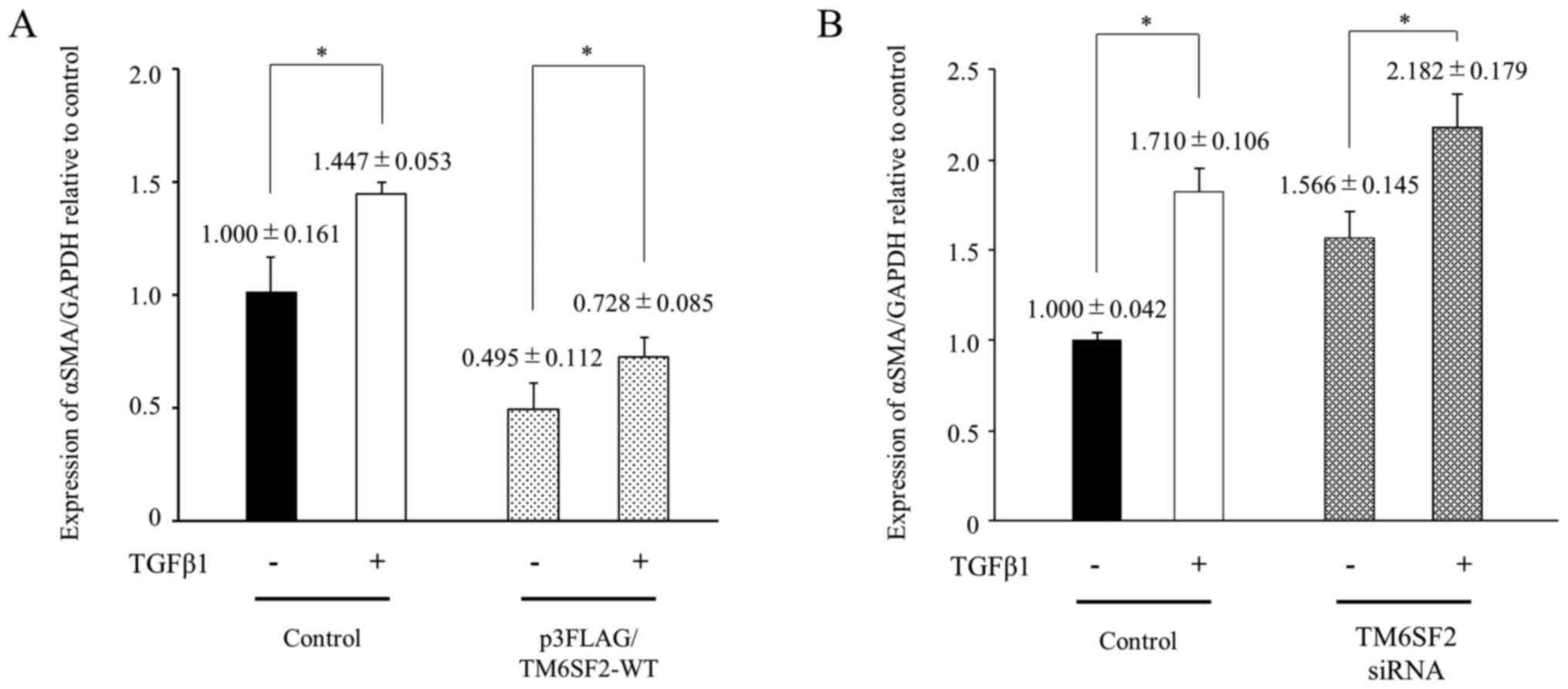
TM6SF2 suppresses αSMA induction by
TGFβ1 in LX-2 cells
To analyze the influence of TM6SF2 on αSMA
expression under TGFβ1 stimulation, changes in αSMA expression in
LX-2 cells were compared after TGFβ1 treatment. LX-2 cells were
transfected with p3FLAG/TM6SF2-WT plasmid. Twenty-four hours after
transfection, the cells were treated with 10 ng/ml of TGFβ1 for 48
h, and intracellular αSMA induction was analyzed by quantitative
PCR. αSMA mRNA levels in TM6SF2-overexpressed LX-2 cells
were significantly suppressed and failed to increase to control
levels following TGFβ1 stimulation (Fig. 2A). A similar tendency was also
observed in the siRNA experiment. The αSMA expression level in
TGFβ1-stimulated LX-2 cells were similar in TM6SF2-knock down LX-2
cells, and the expression of αSMA was enhanced under TGFβ1
stimulation (Fig. 2B). These
results suggested that HSCs could be additionally activated via
TGFβ1 stimulation under lower expression of TM6SF2.
The impact of TM6SF2 phenotype on αSMA
induction
To analyze the impact of the substitution at TM6SF2
amino acid 167, αSMA expression was compared between LX-2 cells
transfected with p3FLAG/TM6SF2-WT plasmid and with p3FLAG/TM6SF2-MT
plasmid. αSMA mRNA expression in cells transfected with
p3FLAG/TM6SF2-WT plasmid was lower than that with p3FLAG/TM6SF2-MT
plasmid (Fig. 3A; P<0.05).
Similarly, αSMA protein expression was >50% suppressed following
transfection with p3FLAG/TM6SF2-WT plasmid compared to transfection
with p3FLAG/TM6SF2-MT plasmid (Fig.
3B; P<0.05).
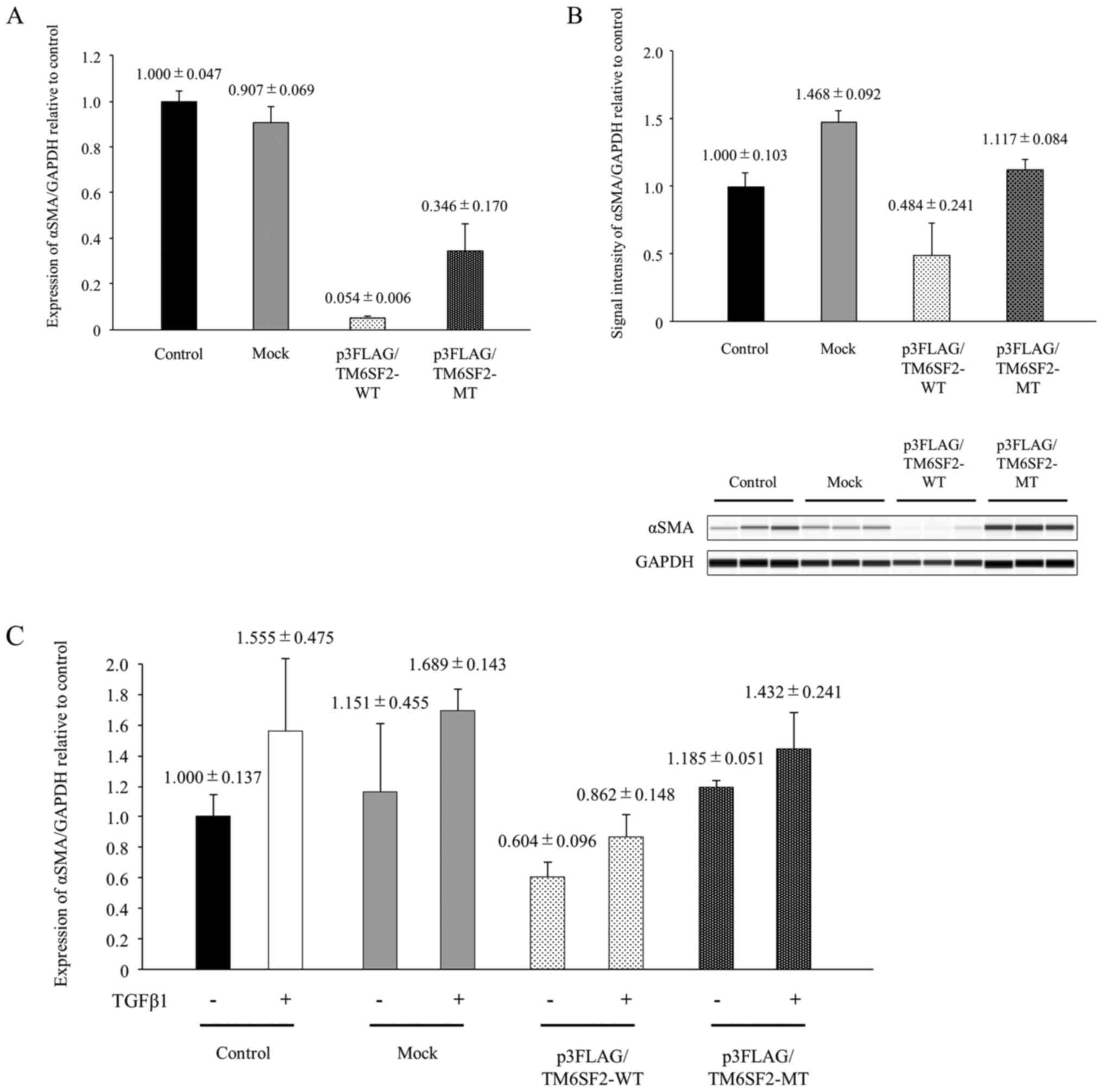 | Figure 3.The impact of TM6SF2 phenotype on
αSMA induction in LX-2 cells. (A) The cloned TM6SF2 expression
plasmid consisting of p3FLAG/TM6SF2-WT and p3FLAG/TM6SF2-MT and
empty vector (Mock) were transiently transfected into LX-2 cells,
followed by 24 h of incubation. Intracellular αSMA expression was
measured using quantitative PCR, with the expression of GAPDH
serving as a control. Experiments were performed in triplicate
wells. (B) Cloned TM6SF2 expression plasmids consisting of
p3FLAG/TM6SF2-WT and p3FLAG/TM6SF2-MT and empty vector (Mock) were
transiently transfected into LX-2 cells, followed by 24 h of
incubation. LX-2 lysates were transferred onto a automated
capillary western blotting system. Anti-TM6SF2 antibody or
anti-GAPDH antibody were applied, followed by anti-rabbit
immunoglobulin. Signal intensity was corrected by GAPDH, as shown
in the bar graph. Experiments were performed in triplicate wells.
(C) Cloned TM6SF2 expression plasmids consisting of
p3FLAG/TM6SF2-WT and p3FLAG/TM6SF2-MT and empty vector (Mock) were
transiently transfected into LX-2 cells, followed by 24 h of
incubation. LX-2 cells were stimulated with or without 10 ng/ml of
TGFβ1 for 48 h. Intracellular αSMA expression was measured using
quantitative PCR, with GAPDH as a control. Experiments were
performed in triplicate wells. TM6SF2, transmembrane 6 superfamily
2; TGFβ1, transforming growth factor β1; αSMA, α-smooth muscle
actin; MT, mutant type; WT, wild-type. |
LX-2 cells that had been transfected with or without
TM6SF2 expression plasmid were stimulated by TGFβ1, and
intracellular αSMA induction was analyzed by quantitative PCR.
Although αSMA mRNA levels were suppressed by TM6SF2 expression,
αSMA expression level in TM6SF2 E167K isoform
(p3FLAG/TM6SF2-MT)-overexpressed LX-2 cells recovered and reached
levels similar to those of control or mock cells after TGFβ1
stimulation (Fig. 3C). Cell
transfection and TGFβ1 stimulation independently affected αSMA
expression in LX-2 cells (two-way ANOVA; P<0.05), and, in
particular, overexpression of p3FLAG/TM6SF2-WT plasmid
significantly affected αSMA expression (Tukey's post-hoc multiple
comparison test; P<0.05). A similar tendency was also observed
in intracellular COL1A1 expression estimated by real-time
PCR (Fig. S4). These results
suggest that basal αSMA expression in HSCs with TM6SF2 wild-type
might be low but might be upregulated by TGFβ1 to a much higher
level than HSCs with the TM6SF2 E167K isoform.
Discussion
NAFLD and NASH are progressive liver diseases
characterized by accumulation of fat in human hepatocytes and an
increased risk of cirrhosis or hepatocellular carcinoma. The number
of patients is increasing worldwide, accompanied by recent upward
trends in obesity, westernized high-fat oral intake, gut dysbiosis,
inadequate exercise, and comorbid metabolic disorders like diabetes
mellitus (2,24). To identify factors associated with
NAFLD, clinical studies have concluded that the prevalence,
prognosis, and progression or severity of disease is significantly
associated with SNPs in PNPLA3 (rs738409) and
TM6SF2 (rs58542926) (4,5,11,25,26).
Several studies have shown that the rs738409SNP in
PNPLA3 causes a loss-of-function amino acid substitution
(I148M) in PNPLA3 that affects regulation of lipid droplets
in human hepatocytes and retinol metabolism in human HSCs,
resulting in positive modulation of HSC activation (27,28).
However, the functional impact of the coding SNP in TM6SF2
has not been sufficiently clarified. Although it has been reported
that TM6SF2 is highly expressed in the liver, kidney, brain, and
small intestine and that the E167K amino acid substitution TM6SF2
(rs58542926) interferes with localization to the endoplasmic
reticulum due to protein misfolding (14,20),
the association between the existence of the coding SNP in
TM6SF2 and activation of HSCs has not been fully
elucidated.
We first analyzed the association between TM6SF2 and
the activation of human HSCs. Intracellular αSMA mRNA expression in
LX-2 cells was suppressed by TM6SF2 overexpression, and its
expression was increased by knocking down TM6SF2 (Fig. 1A and D). Since similar results were
observed in the other experiments (Fig.
2A and B), these data suggest that TM6SF2 negatively regulates
HSC activation.
In the progression of liver fibrosis, it is well
known that TGFβ1, secreted directly by HSCs or by activated Kupffer
cells, could activate HSCs, triggering transformation of HSCs to
myofibroblasts (22). Thus, we
analyzed the impact of TM6SF2 on HSC activation via TGFβ1
signaling. Although intracellular αSMA expression in both control
cells and TM6SF2 over-expressed cells was significantly upregulated
by TGFβ1 treatment, αSMA expression in TM6SF2 overexpressed LX-2
cells was significantly lower than that in control LX-2 cells after
TGFβ1 treatment (Fig. 2A). Similar
results were observed in TM6SF2 knock down cells (Fig. 2B). A similar tendency was also
observed in intracellular COL1A1 expression measured using
real time PCR (Figs. S3 and
4). These results indicate that
lower TM6SF2 expression could activate HSCs and that TGFβ1 could
enhance this HSC activation.
Subsequently, we analyzed the functional impact of
the coding SNP in TM6SF2 in vitro. Normal HSCs have lipid
droplets containing retinol. However, once HSCs are activated,
lipid droplets are diminished, and retinyl ester is degraded in the
endoplasmic reticulum (ER) in HSCs (29). Since the amino acid substitution
(E167K) in TM6SF2 (rs58542926 SNP) causes TM6SF2 to fail to
localize to the ER (18,19), we propose that amino acid
substitution E167K in TM6SF2 could induce HSC activation by
disrupting homeostasis in the ER. When TM6SF2 wild-type or mutant
type (E167K) were overexpressed in LX-2 cells, intracellular αSMA
in LX-2 cells that overexpressed wild-type TM6SF2 decreased more
than those that overexpressed mutant TM6SF2 (Fig. 3). Furthermore, αSMA expression in
TM6SF2-mutant-overexpressed LX-2 cells increased to similar levels
as control LX-2 cells after treatment with TGFβ1. Although the
precise regulation of TM6SF2 in HSCs could not be determined in
this study, our results suggest that the TM6SF2 E167K isoform
affects HSC sensitivity by enhancing the response to TGFβ1.
In the present study, we demonstrated the impact of
an amino acid substitution in TM6SF2 on liver fibrosis using LX-2
cells. Although the impact of this TM6SF2 coding SNP on
liver fibrosis might not be strong considering the hazard ratio
calculated for this SNP by GWAS studies, we consider that these
results could help to clarify the role of TM6SF2 and the impact of
the TM6SF2 SNP on the progression of liver fibrosis in NAFLD
and NASH patients.
TM6SF2 negatively affects αSMA expression in HSCs,
and the TM6SF2 E167K isoform associated with the rs58542926
SNP might affect HSC activation sensitivity. These results suggest
that TM6SF2 might play a role in the process of HSC activation and
liver fibrosis in NASH.
Supplementary Material
Supporting Data
Acknowledgements
Not applicable.
Funding
The present study was supported by grants-in-aid for
scientific research and development from the Ministry of Health,
Labor and Welfare and Ministry of Education Culture Sports Science
and Technology (JSPS KAKENHI grant no. 18K15814) and Japan Agency
for Medical Research and Development (grant no. JP20fk0210040). The
present study was also supported by the Gilead Sciences Research
Scholars Program in Liver Disease by Gilead Sciences, Inc. The
funders had no role in study design, data collection and analysis,
decision to publish, or preparation of the manuscript.
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
SL, EM and KC conceived the study. GNM, AO, DM and
HAC made contributions to the design of the experiment. SL and EM
analyzed and interpreted the experimental data. TN, KO, YT, TU, KM,
HF, MY, TK and AH were involved in analyzing the data and revising
the manuscript. MT performed western blotting experiments and
edited the manuscript. DM and AO performed part of the
immunostaining experiments and checked gene expression analysis
data. MI and HA designed the study and confirmed the quality of
experimental data. CNH contributed to statistical analysis and
proofreading. MT and CNH were major contributors in editing the
manuscript. KC revised the manuscript and gave the final approval
of the version to be submitted. All authors read and approved the
final manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Glossary
Abbreviations
Abbreviations:
|
HCS
|
hepatic stellate cell
|
|
TM6SF2
|
transmembrane 6 superfamily 2
|
|
TGFβ1
|
transforming growth factor β1
|
|
αSMA
|
α-smooth muscle actin
|
|
SNP
|
single nucleotide polymorphism
|
References
|
1
|
Eslam M, Valenti L and Romeo S: Genetics
and epigenetics of NAFLD and NASH: Clinical impact. J Hepatol.
68:268–279. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Kleiner DE, Brunt EM, Van Natta M, Behling
C, Contos MJ, Cummings OW, Ferrell LD, Liu YC, Torbenson MS,
Unalp-Arida A, et al: Design and validation of a histological
scoring system for nonalcoholic fatty liver disease. Hepatology.
41:1313–1321. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Angulo P, Kleiner DE, Dam-Larsen S, Adams
LA, Bjornsson ES, Charatcharoenwitthaya P, Mills PR, Keach JC,
Lafferty HD, Stahler A, et al: Liver fibrosis, but no other
histologic features, is associated with long-term outcomes of
patients with nonalcoholic fatty liver disease. Gastroenterology.
149:389–397. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Romeo S, Kozlitina J, Xing C, Pertsemlidis
A, Cox D, Pennacchio LA, Boerwinkle E, Cohen JC and Hobbs HH:
Genetic variation in PNPLA3 confers susceptibility to nonalcoholic
fatty liver disease. Nat Genet. 40:1461–1465. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Speliotes EK, Yerges-Armstrong LM, Wu J,
Hernaez R, Kim LJ, Palmer CD, Gudnason V, Eiriksdottir G, Garcia
ME, Launer LJ, et al: Genome-wide association analysis identifies
variants associated with nonalcoholic fatty liver disease that have
distinct effects on metabolic traits. PLoS Genet. 7:e10013242011.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Singal AG, Manjunath H, Yopp AC, Beg MS,
Marrero JA, Gopal P and Waljee AK: The effect of PNPLA3 on fibrosis
progression and development of hepatocellular carcinoma: A
meta-analysis. Am J Gastroenterol. 109:325–334. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Hotta K, Yoneda M, Hyogo H, Ochi H,
Mizusawa S, Ueno T, Chayama K, Nakajima A, Nakao K and Sekine A:
Association of the rs738409 polymorphism in PNPLA3 with liver
damage and the development of nonalcoholic fatty liver disease. BMC
Med Genet. 11:1722010. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Sookoian S, Castaño GO, Scian R, Mallardi
P, Fernández Gianotti T, Burgueño AL, San Martino J and Pirola CJ:
Genetic variation in transmembrane 6 superfamily member 2 and the
risk of nonalcoholic fatty liver disease and histological disease
severity. Hepatology. 61:515–525. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Pirola CJ and Sookoian SL: The dual and
opposite role of the TM6SF2-rs58542926 variant in protecting
against cardiovascular disease and conferring risk for nonalcoholic
fatty liver: A meta-analysis. Hepatology. 62:1742–1756. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Macaluso FS, Maida M and Petta S: Genetic
background in nonalcoholic fatty liver disease: A comprehensive
review. World J Gastroenterol. 21:11088–11111. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Goffredo M, Caprio S, Feldstein AE,
D'Adamo E, Shaw MM, Pierpont B, Savoye M, Zhao H, Bale AE and
Santoro N: Role of TM6SF2 rs58542926 in the pathogenesis of
nonalcoholic pediatric fatty liver disease: A multiethnic study.
Hepatology. 63:117–125. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Liu YL, Reeves HL, Burt AD, Tiniakos D,
McPherson S, Leathart JB, Allison ME, Alexander GJ, Piguet AC, Anty
R, et al: TM6SF2 rs58542926 influences hepatic fibrosis progression
in patients with Non-alcoholic fatty liver disease. Nat Commun.
5:43092014. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Dongiovanni P, Petta S, Maglio C,
Fracanzani AL, Pipitone R, Mozzi E, Motta BM, Kaminska D, Rametta
R, Grimaudo S, et al: Transmembrane 6 superfamily member 2 gene
variant disentangles nonalcoholic steatohepatitis from
cardiovascular disease. Hepatology. 61:506–514. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Li TT, Li TH, Peng J, He B, Liu LS, Wei
DH, Jiang ZS, Zheng XL and Tang ZH: TM6SF2: A novel target for
plasma lipid regulation. Atherosclerosis. 268:170–176. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
O'Hare EA, Yang R, Yerges-Armstrong LM,
Sreenivasan U, McFarland R, Leitch CC, Wilson MH, Narina S, Gorden
A, Ryan KA, et al: TM6SF2 rs58542926 impacts lipid processing in
liver and small intestine. Hepatology. 65:1526–1542. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Luukkonen PK, Zhou Y, Nidhina Haridas PA,
Dwivedi OP, Hyötyläinen T, Ali A, Juuti A, Leivonen M, Tukiainen T,
Ahonen L, et al: Impaired hepatic lipid synthesis from
polyunsaturated fatty acids in TM6SF2 E167K variant carriers with
NAFLD. J Hepatol. 67:128–136. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Prill S, Caddeo A, Baselli G, Jamialahmadi
O, Dongiovanni P, Rametta R, Kanebratt KP, Pujia A, Pingitore P,
Mancina RM, et al: The TM6SF2 E167K genetic variant induces lipid
biosynthesis and reduces apolipoprotein B secretion in human
hepatic 3D spheroids. Sci Rep. 9:115852019. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Mahdessian H, Taxiarchis A, Popov S,
Silveira A, Franco-Cereceda A, Hamsten A, Eriksson P and van't
Hooft F: TM6SF2 is a regulator of liver fat metabolism influencing
triglyceride secretion and hepatic lipid droplet content. Proc Natl
Acad Sci USA. 111:8913–8918. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Smagris E, Gilyard S, BusuRay S, Cohen JC
and Hobbs HH: Inactivation of TM6SF2, a gene defective in fatty
liver disease, impairs lipidation but not secretion of very low
density lipoproteins. J Biol Chem. 291:10659–10676. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Kozlitina J, Smagris E, Stender S,
Nordestgaard BG, Zhou HH, Tybjærg-Hansen A, Vogt TF, Hobbs HH and
Cohen JC: Exome-wide association study identifies a TM6SF2 variant
that confers susceptibility to nonalcoholic fatty liver disease.
Nat Genet. 46:352–356. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Li JT, Liao ZX, Ping J, Xu D and Wang H:
Molecular mechanism of hepatic stellate cell activation and
antifibrotic therapeutic strategies. J Gastroenterol. 43:419–428.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Dewidar B, Mayer C, Dooley S and
Meindl-Beinker AN: TGF-β in hepatic stellate cell activation and
liver fibrogenesis-updated 2019. Cells. 8:14192019. View Article : Google Scholar
|
|
23
|
Carpino G, Morini S, Corradini G,
Corradini S, Franchitto A, Merli M, Siciliano M, Gentili F, Onetti
Muda A, Berloco P, et al: Alpha-SMA expression in hepatic stellate
cells and quantitative analysis of hepatic fibrosis in cirrhosis
and in recurrent chronic hepatitis after liver transplantation. Dig
Liver Dis. 37:349–356. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Henao-Mejia J, Elinav E, Jin C, Hao L,
Mehal WZ, Strowig T, Thaiss CA, Kau AL, Eisenbarth SC, Jurczak MJ,
et al: Inflammasome-mediated dysbiosis regulates progression of
NAFLD and obesity. Nature. 482:179–185. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Eslam M, Mangia A, Berg T, Chan HL, Irving
WL, Dore GJ, Abate ML, Bugianesi E, Adams LA, Najim MA, et al:
Diverse impacts of the rs58542926 E167K variant in TM6SF2 on viral
and metabolic liver disease phenotypes. Hepatology. 64:34–46. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Sookoian S and Pirola CJ: Meta-analysis of
the influence of TM6SF2 E167K variant on plasma concentration of
aminotransferases across different populations and diverse liver
phenotypes. Sci Rep. 6:277182016. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Pingitore P, Dongiovanni P, Motta BM,
Meroni M, Lepore SM, Mancina RM, Pelusi S, Russo C, Caddeo A, Rossi
G, et al: PNPLA3 overexpression results in reduction of proteins
predisposing to fibrosis. Hum Mol Genet. 25:5212–5122.
2016.PubMed/NCBI
|
|
28
|
Bruschi FV, Claudel T, Tardelli M,
Caligiuri A, Stulnig TM, Marra F and Trauner M: The PNPLA3 I148M
variant modulates the fibrogenic phenotype of human hepatic
stellate cells. Hepatology. 65:1875–1890. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Testerink N, Ajat M, Houweling M, Brouwers
JF, Pully VV, van Manen HJ, Otto C, Helms JB and Vaandrager AB:
Replacement of retinyl esters by polyunsaturated triacylglycerol
species in lipid droplets of hepatic stellate cells during
activation. PLoS One. 7:e34945K2012. View Article : Google Scholar
|