Introduction
Acute lung injury (ALI) is a disease with high
incidence and mortality rates and can cause acute respiratory
distress syndrome (ARDS), which seriously threatens human health
(1–3). A prospective epidemiological study in
1999–2000 estimated an annual incidence of ALI and ARDS of 190,000
adult patients in the United States of America (1). As a critical disease, ALI has
attracted increasing attention in clinical practice. The primary
manifestations of ALI are alveolar capillary system damage,
increased pulmonary vascular permeability, overactivation of
macrophages and neutrophils, excessive inflammatory response and
increased reactive oxygen species (ROS) production, which
eventually lead to respiratory function damage (4). Previous evidence has revealed that
inflammation and oxidative stress are associated with ALI (5).
The mechanisms of inflammation and oxidative stress
work in different ways. MAPKs serve as typical
inflammation-associated signals and comprise ERK-1/2, p38 and JNK
(6). JNK is primarily involved in
inflammation and oxidative stress (6). JNK mediates activation of NF-κB
signaling, the main upstream regulator of proinflammatory cytokines
in ALI (7). Activated NF-κB/p65
translocates to the nucleus to regulate a number of inflammatory
cytokines (8,9) and promote oxidative stress (10). Previous studies have demonstrated
that, following paraquat poisoning, JNK is activated in the lung,
and the JNK inhibitor SP600125 can alleviate ALI caused by paraquat
(11,12). Another JNK inhibitor, JNK-IN-8
(13), has an inhibitory effect on
the regulation of NF-κB signaling activation by JNK, and can
inhibit inflammation and improve neurological function in ischemic
brain injury (14). These studies
(6–14) suggest that JNK inhibitors serve a
major role in the molecular pathology of numerous types of disease.
Nevertheless, to the best of our knowledge, the exact role of
JNK-IN-8 in inflammation and oxidative stress associated with ALI
is not clear.
JNK-IN-8, as an effective and specific JNK
inhibitor, has a notable inhibitory effect on the JNK/NF-κB
signaling pathway (14,15) which is associated with inflammation
and oxidative stress in ALI. Therefore, its impacts may extend to
ALI. However, to the best of our knowledge, the in vitro and
in vivo effects of JNK-IN-8 on lipopolysaccharide
(LPS)-induced ALI have not been reported. Therefore, the aim of the
present study was to investigate the protective effects of JNK-IN-8
in LPS-induced ALI, and the association between its molecular
mechanism and inflammation and oxidative stress.
Materials and methods
Reagents and antibodies
JNK-IN-8 was purchased from MedChemExpress. Mouse
TNF-α (cat. no. CSB-E04741m), IL-6 (cat. no. CSB-E04639m) and IL-1β
(cat. no. CSB-E08054m) ELISA kits were purchased from Cusabio
Technology LLC. Alexa Fluor® 488 anti-mouse Ly-6G (cat.
no. 127625) and 594 anti-mouse F4/80 antibody (cat. no. 123140)
were purchased from BioLegend, Inc. Specific primary antibodies
against NF-κB p65 (cat. no. 8242), phosphorylated (p)-p65 (Ser536;
cat. no. 3303), JNK (cat. no. 9252), p-JNK (Thr183/Tyr185; cat. no.
4668) and β-actin (cat. no. 3700) were purchased from Cell
Signaling Technology, Inc.
Animals
A total of 75 animals (age, 8 weeks) received humane
care in 2019, according to the Guide for the Care and Use of
Laboratory Animals (16). Male
specific pathogen-free grade C57BL/6 mice (weight, 18–22 g) were
provided by Shanghai SLAC Laboratory Animal Co., Ltd., and were
housed (5 mice/individually vented cage) using a 12-h light/dark
cycle under optimal temperature (20–26°C) and humidity (40–70%).
Mice were provided with free access to food and water. Prior to the
experiment, the mice were habituated to the environment for ≥1
weeks. The specific criteria of humane endpoints were selected
according to the Guide for the Care and Use of Laboratory Animals:
Eighth Edition (17). The humane
endpoints included dying (indicated by weak response to leg
clamping, very faint heartbeat, irregular breathing, depression and
hypothermia), loss of appetite (>24 h) and weakness (unable to
feed, drink or stand). Euthanasia was performed according to the
American Veterinary Medical Association Guidelines for the
Euthanasia of Animals: 2013 Edition (18). The displacement rate used for
CO2 inhalation euthanasia was 20% to avoid distress.
Standard evidence of death included dilated pupils and absence of
heartbeat, as well as failure to respond to a toe pinch or touch of
the eye. Cervical dislocation was performed following euthanasia to
ensure death. All animal experiments were approved by the Ethics
Committee of Zhejiang University (approval no. ZJU20170913;
Hangzhou, China).
Establishment of the ALI mouse
model
The ALI mouse model was established by intratracheal
administration of LPS (20 µg/50 µl PBS). A total of 24 mice were
divided into four groups at random (n=6/group): Sham operation
(equivalent PBS via intraperitoneal injection), JNK-IN-8, LPS and
LPS + JNK-IN-8 group. Mice in the JNK-IN-8 and LPS + JNK-IN-8
groups received JNK-IN-8 (10 mg/kg) via intraperitoneal injection.
After 1 h, mice in the LPS + JNK-IN-8 group also received LPS
treatment. LPS (0.4 mg/kg; Sigma-Aldrich; Merck KGaA) was injected
into the mice in the LPS and LPS + JNK-IN-8 groups via the trachea
following anesthetization with sevoflurane (induction, 5%;
maintenance, 2.5%), as previously described (18). The physical condition of animals was
monitored every 2 h and sacrificed at 6 h after LPS injection by
CO2 inhalation. Subsequently, bronchoalveolar lavage
fluid (BALF) and lung tissues were collected and stored at −80°C
until use. In the ALI model, no mice died during the experiment. In
the survival experiment, 48 mice were divided into four groups at
random (n=12/group): Sham operation (equivalent PBS via
intraperitoneal injection), JNK-IN-8, LPS and LPS + JNK-IN-8 group.
Mice in the JNK-IN-8 and LPS + JNK-IN-8 groups received JNK-IN-8
(10 mg/kg) via intraperitoneal injection. After 1 h, mice in the
LPS and LPS + JNK-IN-8 groups were given a lethal dose of LPS (20
mg/kg). Animals were monitored every 6 h and the survival rate was
recorded every 12 h for 72 h, as previously described (19). In the survival experiment, 15 mice
died due to LPS-induced ALI and two were euthanized upon reaching a
humane endpoint of dying. All surviving mice were euthanized by
CO2 inhalation followed by cervical dislocation after
the experiment.
Inflammatory cell count in BALF
C57BL/6 mice used for the collection of BALF were
euthanized by CO2 inhalation, after which an
intermediate incision was made to open the chest to expose the
trachea, as previously described (20). The alveoli were washed twice with
0.5 ml PBS during endotracheal intubation to collect 0.8 ml BALF.
The numbers of inflammatory cells (specifically, total living
cells, ly6g+ neutrophils and F4/80+
macrophages) were counted using flow cytometry (BD Biosciences).
Cells in BALF (500 µl) were stained with ly6g for neutrophils and
F4/80 for macrophages, as previously described (21,22).
Subsequently, flow cytometry was used to count the cells. The
ly6g+ and F4/80− cells were considered to be
‘neutrophils’. The ly6g− and F4/80+ cells
were considered to be ‘macrophages’. The ly6g− and
F4/80− cells were considered to be other cells. The sum
of the three types of cells was the number of ‘total living cells’.
Gates were artificially set according to the cell community
location. Finally, the data were analyzed using FlowJo software
(version 10; Tree Star, Inc.).
Wet/dry (W/D) weight ratio of lung
tissue in the ALI model
Following euthanasia, the right lung tissue was
collected and the wet weight was recorded. Subsequently, the lung
tissue was covered with foil and placed in an incubator at 80°C
until the weight remained stable to obtain the dry weight. Finally,
the W/D ratio of the lung tissues was calculated to evaluate the
degree of pulmonary edema.
Histology evaluation
At 6 h after LPS injection, the thoraces of the mice
in the ALI model were opened under deep anesthesia, and the mice
were perfused with 50 ml 0.01 M PBS (pH, 7.4) followed by 4%
paraformaldehyde in 0.1 M PBS (pH, 7.4) through the ascending
aorta. Subsequently, the left upper lung lobe was placed in 4%
paraformaldehyde, dehydrated with ethanol and embedded in paraffin.
Lung tissues were cut into slices (thickness, 4–5 µm) and then
subjected to hematoxylin and eosin and periodic acid Schiff
staining. Pathological changes were observed under an optical light
microscope (magnification, ×200) and two experienced pathologists
blindly scored the changes, as previously described (23): 0, normal tissue; 1, minimal
inflammatory change; 2, no obvious damage to the lung architecture;
3, thickening of the alveolar septae; 4, formation of nodules or
areas of pneumonitis that distorted the normal architecture; and 5,
total obliteration of the field.
ELISA
Expression levels of cytokines, including IL-1β,
IL-6 and TNF-α, in BALF, lung tissue and cell supernatant were
detected using ELISA kits according to the manufacturer's
protocols.
Cell culture
Mouse macrophage (RAW264.7) cells were purchased
from the American Type Culture Collection. RAW264.7 cells were
cultured in DMEM (Gibco; Thermo Fisher Scientific, Inc.)
supplemented with 10% FBS (Gibco; Thermo Fisher Scientific, Inc.)
with 5% CO2 and 37°C. Injecting thioglycolate into the
abdominal cavity of mice creates a sterile inflammatory environment
in which macrophages accumulate and can be obtained by lavage of
the abdominal cavity (8). A total
of three 8-week-old male C57BL/6 mice were injected
intraperitoneally with 3 ml 3% thioglycolate (Sigma-Aldrich; Merck
KGaA), as previously described (24). After 3 days, the mice were
sacrificed by CO2 inhalation followed by cervical
dislocation. The abdomen was soaked with 75% alcohol for 5 min at
room temperature, then a small incision along the midline was made
with sterile scissors. Subsequently, 10 ml cold Dulbecco's PBS was
injected into the peritoneal cavity. The lavages were collected by
centrifugation at 1,500 × g for 10 min at room temperature and
resuspended in RPMI-1640 (Gibco; Thermo Fisher Scientific, Inc.)
supplemented with 10% FBS (Gibco; Thermo Fisher Scientific, Inc.).
Cells were then incubated at 37°C for 6 h and washed with PBS to
remove the non-adherent cells. The remaining adherent cells were
used as the peritoneal macrophages in subsequent experiments. In
order to confirm these isolated cells to be macrophages,
LPS-induced M1 polarization experiments were performed. For
macrophages, including TNF-α, IL-6 and IL-1β, these M1 polarization
indexes increase following LPS stimulation (3). In order to investigate the effect of
JNK-IN-8 on the inflammatory response of macrophages, cells were
pretreated with JNK-IN-8 at 5% CO2 and 37°C for 1 h,
followed by treatment with LPS (100 ng/ml) at 5% CO2 and
37°C for 6 h.
Cell viability
Following treatment, cell viability was assessed
using a Cell Counting Kit-8 (CCK-8) cell proliferation/cytotoxicity
assay kit (Beyotime Institute of Biotechnology). Primary
macrophages and RAW 264.7 cells were seeded into 96-well plates at
a density of 2×103 cells/well at 5% CO2 and
37°C for 24 h. Cells were then treated with increasing
concentrations of JNK-IN-8 (0.00, 0.39, 0.78, 1.56, 3.13, 6.25,
12.50, 25.00, 50.00 and 100.00 µM) at 5% CO2 and 37°C
for 24 h. At the end of the experimental period, cells were
incubated with CCK-8 reagent and absorbance at a wavelength of 450
nm was measured using an ELX800 absorbance microplate reader. The
experiments were performed in triplicate.
Calculation of biochemical
parameters
Lung samples were homogenized and dissolved in
extraction buffer from commercially available assay kits (Beijing
Solarbio Science & Technology Co., Ltd.) to measure lactate
dehydrogenase (LDH; cat. no. BC0685), malondialdehyde (MDA; cat.
no. BC0025), glutathione (GSH; cat. no. BC1175), superoxide
dismutase (SOD; cat. no. BC0170) and myeloperoxidase (MPO; cat. no.
BC0073) levels, according to the manufacturer's protocols.
Reverse transcription-quantitative
(RT-q)PCR assay
Total RNA from cells/tissue was extracted using the
RNeasy Mini kit (Qiagen). After evaluating the quality of the RNA
based on the A260/A280 ratio, cDNA was synthesized using 1 mg RNA
from each sample, 2 ml 5X PrimeScript RT Master Mix (Takara Bio,
Inc.), and 4 ml RNase-free distilled water in a total volume of 10
ml. RT-qPCR was performed using an ABI Prism 7500 system (Applied
Biosystems; Thermo Fisher Scientific, Inc.) with SYBR Green QPCR
Master Mix (Takara Bio, Inc.). The total volume (20 µl) of each PCR
reaction consisted of 10 µl SYBR-Green QPCR Master Mix, 6 µl ddH2O,
2 µl cDNA and 10 µM each of forward and reverse primers. RT-qPCR
was performed using the following thermocycling conditions: 95°C
for 10 min, followed by 40 cycles of 95°C for 10 sec, 60°C for 20
sec and 72°C for 20 sec, and a final extension at 72°C for 1 min.
GAPDH served as the internal control. The murine primer sequences
were as follows: GAPDH forward, 5′-ACCCAGAAGACTGTGGATGG-3′ and
reverse, 5′-CACATTGGGGGTAGGAACAC-3′; TNF-α forward,
5′-CGGGCAGGTCTACTTTGGAG-3′ and reverse, 5′-ACCCTGAGCCATAATCCCCT-3′;
IL-6 forward, 5′-AACGATGATGCACTTGCAGA-3′ and reverse,
5′-TGTGACTCCAGCTTATCTCTTGG-3′; and IL-1β forward,
5′-TGCCACCTTTTGACAGTGATG-3′ and reverse,
5′-CAAAGGTTTGGAAGCAGCCC-3′. The experiments were performed in
triplicate.
Western blot analysis
Nucleoprotein and total protein were extracted from
lung tissue or cells using RIPA reagent (Beijing Solarbio Science
& Technology Co., Ltd.). A BCA protein assay kit (Thermo Fisher
Scientific, Inc.) was used to determine the protein concentration
of samples from lysed lung tissues or cells. Each well of a 10%
SDS-PAGE gel was loaded with an equal volume of protein, and the
proteins were separated by SDS-PAGE, after which they were
transferred to a PVDF membrane. After incubation with 5% milk at
room temperature for 1 h, the membrane was incubated with primary
antibodies (all 1:1,000; all Cell Signaling Technology, Inc.),
including anti-NF-κB p65 (cat. no. 8242), anti-p-NF-κB p65 at
Ser536 (cat. no. 3033), anti-β-actin (cat. no. 3700) and anti-Lamin
B (cat. no. 12255), at 4°C overnight. Then, the membrane was
incubated with corresponding secondary antibodies (1:2,000; cat.
no. 6990; Cell Signaling Technology, Inc.) at room temperature for
1 h. The protein bands were visualized using an ECL western blot
kit (Bio-Rad Laboratories, Inc.) and visualized on a ChemiDoc XRS
System (Bio-Rad Laboratories, Inc.). Protein expression was
calculated by measuring the optical density using Image Lab
software (version 3.0, Bio-Rad Laboratories, Inc.). Target protein
expression levels were normalized to those of β-actin.
Statistical analysis
Data are presented as the mean ± SD of three
independent repeats. SPSS 16.0 software (SPSS, Inc.) was used for
statistical analysis. Figures were created using Prism 5.0 software
(GraphPad Software, Inc.). Survival data were analyzed with
Kaplan-Meier plots and log-rank tests. Differences among the
experimental groups were compared by one-way ANOVA and then
analyzed by Tukey's multiple comparison test. The inflammation
score was analyzed using a Kruskal-Wallis test followed by Dunn's
post hoc test. P<0.05 was considered to indicate a statistically
significant difference.
Results
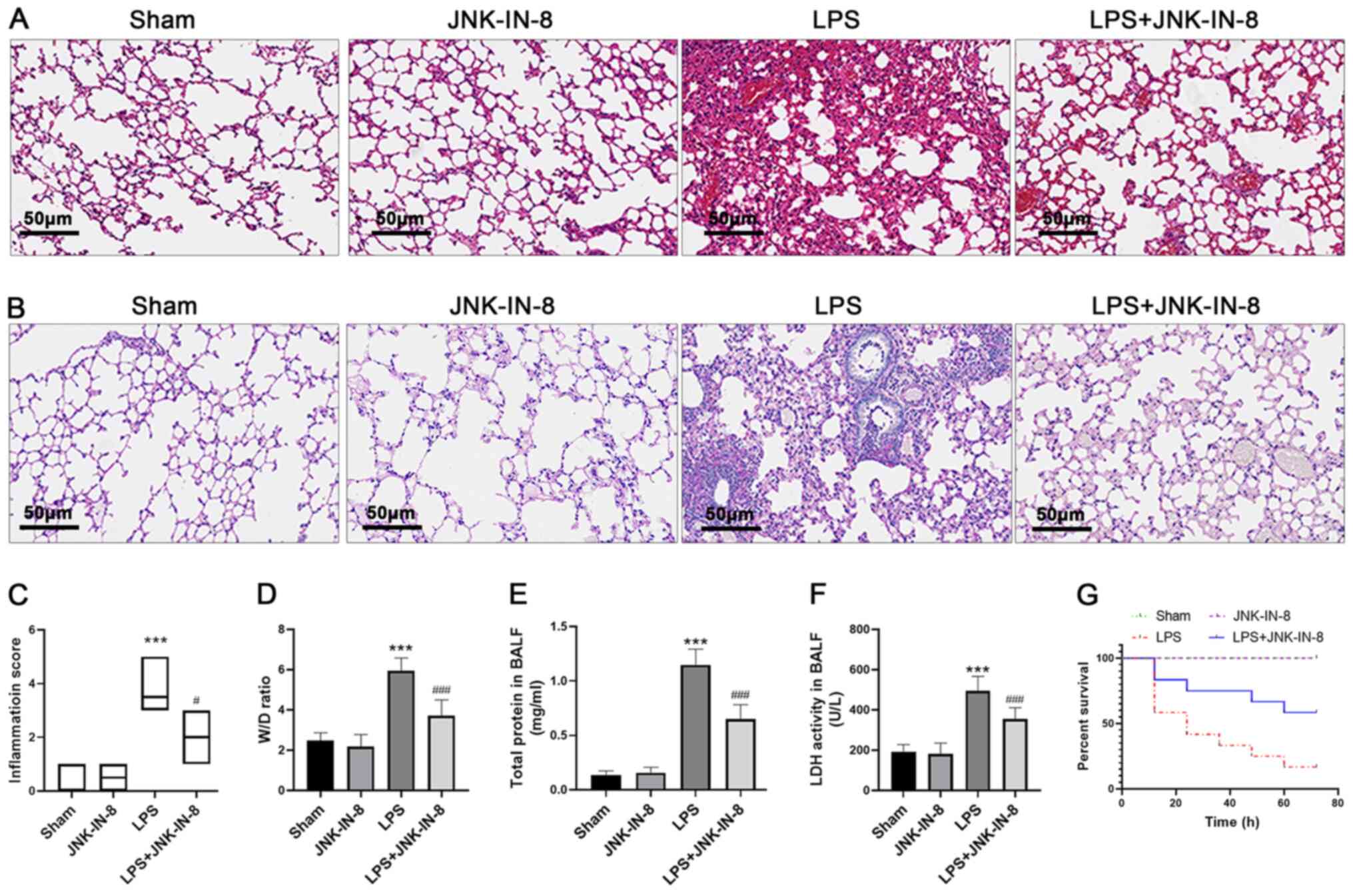
JNK-IN-8 decreases lung injury induced
by LPS in mice
Compared with the Sham and JNK-IN-8 groups, LPS
caused marked pathological changes by increasing the accumulation
of inflammatory cell damage in alveolar tissue (Fig. 1A and B). JNK-IN-8 treatment
alleviated severe histopathological changes caused by LPS,
including inflammatory cell infiltration, tissue destruction and
glycogen deposition. In accordance with this finding, the lung
injury score of the LPS + JNK-IN-8 treatment group was
significantly lower than that of the LPS group (Fig. 1C). The W/D ratio of lung tissue and
total protein levels in BALF (which represent the severity of
pulmonary edema) were significantly higher in the LPS group than in
the Sham and JNK-IN-8 groups, whereas pretreatment with JNK-IN-8
significantly decreased the W/D ratio and total protein levels
(Fig. 1D and E). Furthermore,
JNK-IN-8 treatment decreased LDH activity in the BALF of
LPS-challenged mice (Fig. 1F).
Pretreatment with JNK-IN-8 improved the survival rate within 72 h
of lethal injection of LPS (Fig.
1G). These findings suggest that JNK-IN-8 alleviated
pathological damage of the lungs induced by LPS.
JNK-IN-8 decreases the inflammatory
response and oxidative stress in the lungs of mice treated with
LPS
Subsequently, the role of JNK-IN-8 in the
inflammatory response and oxidative stress in the lungs of mice
challenged with LPS was determined. The mRNA expression levels of
TNF-α, IL-6 and IL-1β in lung tissue were significantly decreased
by JNK-IN-8 pretreatment (Fig. 2A),
and this also decreased the serum levels of TNF-α, IL-6 and IL-1β
(Fig. 2B). In addition, JNK-IN-8
treatment effectively inhibited the increase in MPO activity and
MDA generation caused by LPS (Fig.
2C). It also significantly decreased LPS-induced SOD depletion
(Fig. 2C), suggesting that JNK-IN-8
treatment decreased the inflammatory response and oxidative stress
in mice with LPS-induced ALI.
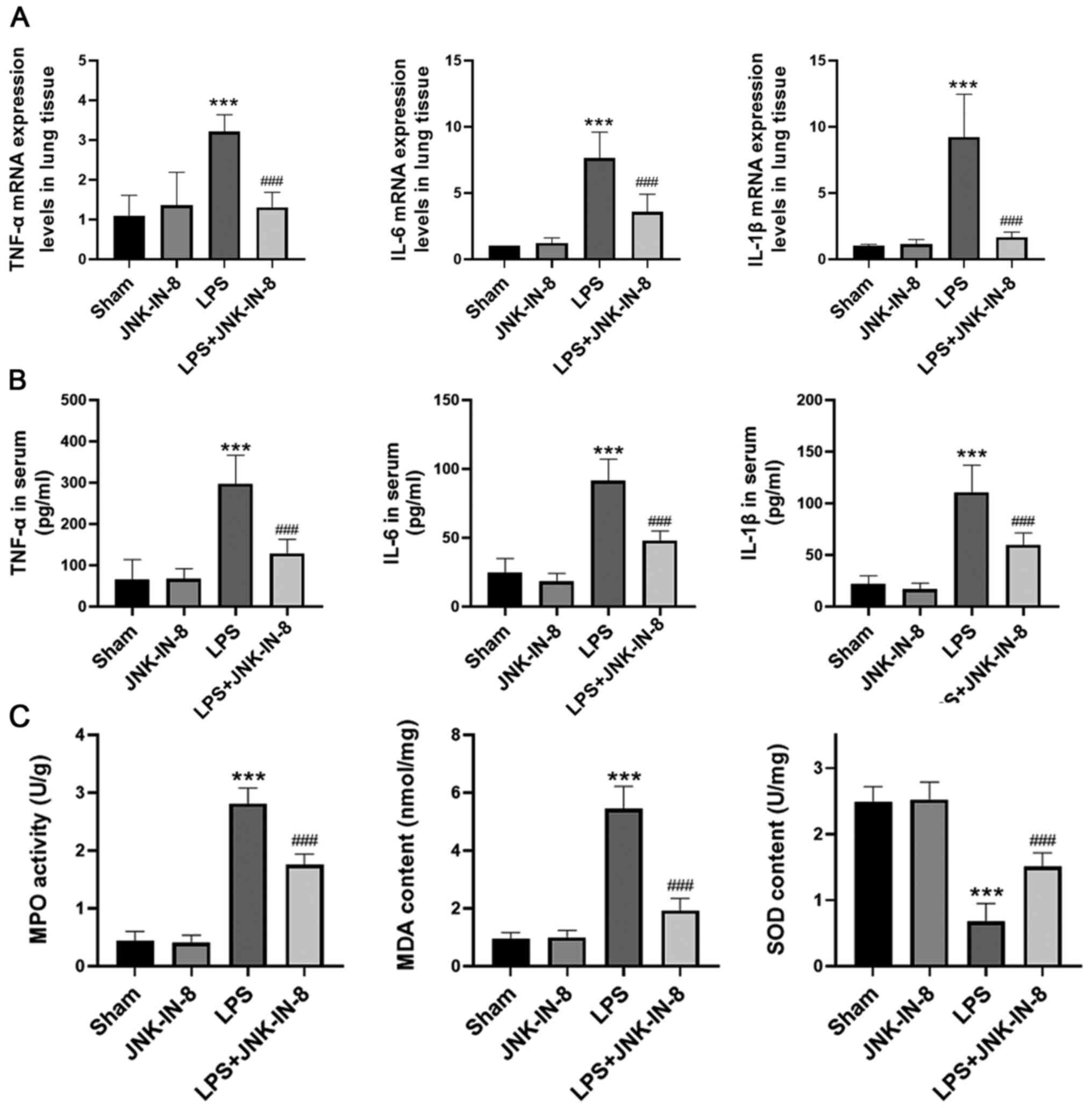 | Figure 2.JNK-IN-8 decreases inflammatory
responses and oxidative stress in ALI mice. Mice with LPS-induced
ALI were pretreated with JNK-IN-8. TNF-α, IL-6 and IL-1β (A) mRNA
levels in lung tissue samples, and (B) protein levels in serum were
determined by reverse transcription-quantitative PCR and ELISA,
respectively. (C) MPO activity, SOD and MDA content, in lung tissue
samples were examined. Data are presented as the mean ± SD (n=6).
***P<0.001 vs. Sham; ###P<0.001 vs. LPS. ALI,
acute lung injury; LPS, lipopolysaccharide; MPO, myeloperoxidase;
SOD, superoxide dismutase; MDA, malondialdehyde. |
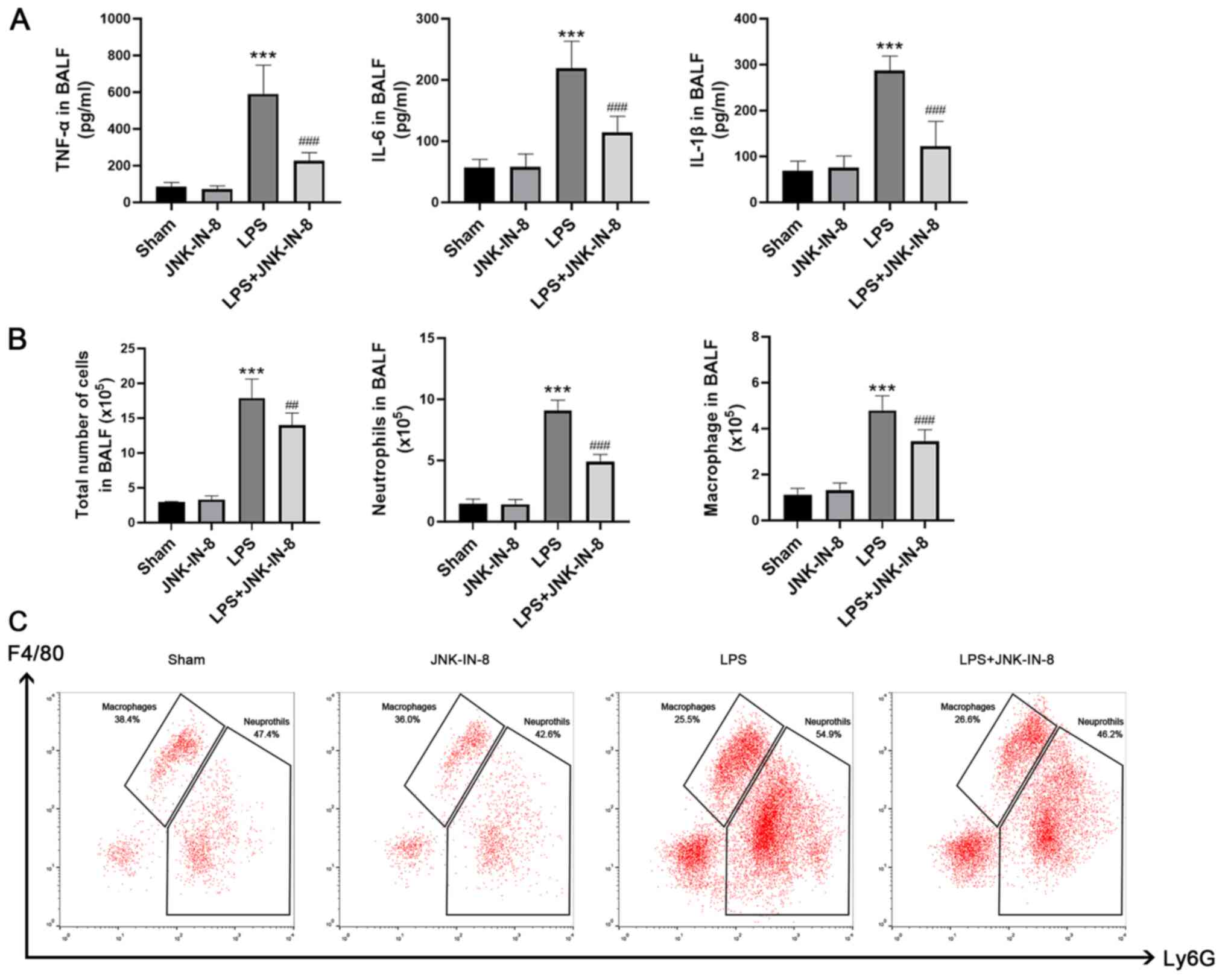
Pretreatment with JNK-IN-8 significantly decreased
the levels of TNF-α, IL-6 and IL-1β in BALF compared with those in
the LPS group (Fig. 3A). Compared
with the Sham group, the numbers of total cells, neutrophils and
macrophages in BALF were increased following LPS stimulation, and
this change was inhibited by pretreatment with JNK-IN-8 (Fig. 3B and C).
JNK-IN-8 decreases LPS-induced
inflammatory cytokine production and oxidative stress in primary
murine peritoneal macrophages and RAW264.7 cells in vitro
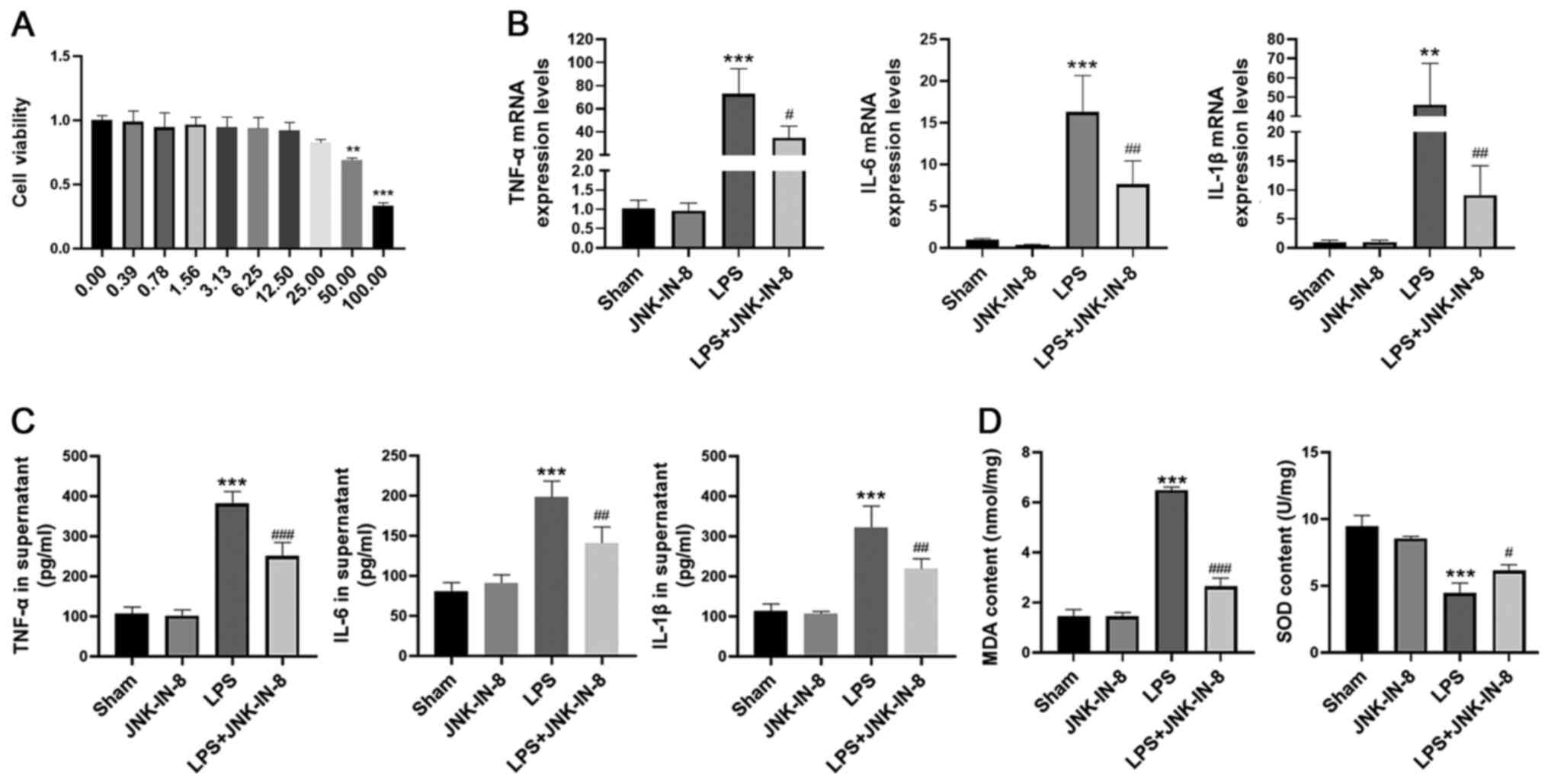
Before assessing the effects of JNK-IN-8 on primary
murine peritoneal macrophages, the cytotoxic effect of JNK-IN-8 on
cells was assessed using a CCK-8 assay (Fig. 4A). No significant cytotoxic effects
of JNK-IN-8 were observed at concentrations of ≤12.50 µM in primary
macrophages (Fig. 4A). Based on
these data, a maximal concentration of 10 µM was selected to
analyze the effects of JNK-IN-8 in primary macrophages.
In order to investigate the anti-inflammatory effect
of JNK-IN-8, primary macrophages were pretreated with JNK-IN-8 for
1 h and then stimulated with LPS (100 ng/ml) for 6 h. mRNA
expression levels and secretion of TNF-α, IL-6 and IL-1β in the LPS
group were significantly increased compared with those in the Sham
group, but these were decreased in the LPS + JNK-IN-8 group
compared with the LPS group (Fig. 4B
and C). Subsequently, the role of JNK-IN-8 in oxidative stress
was investigated. JNK-IN-8 pretreatment significantly decreased MDA
content and inhibited the LPS-induced decrease in SOD activity
(Fig. 4D) in primary
macrophages.
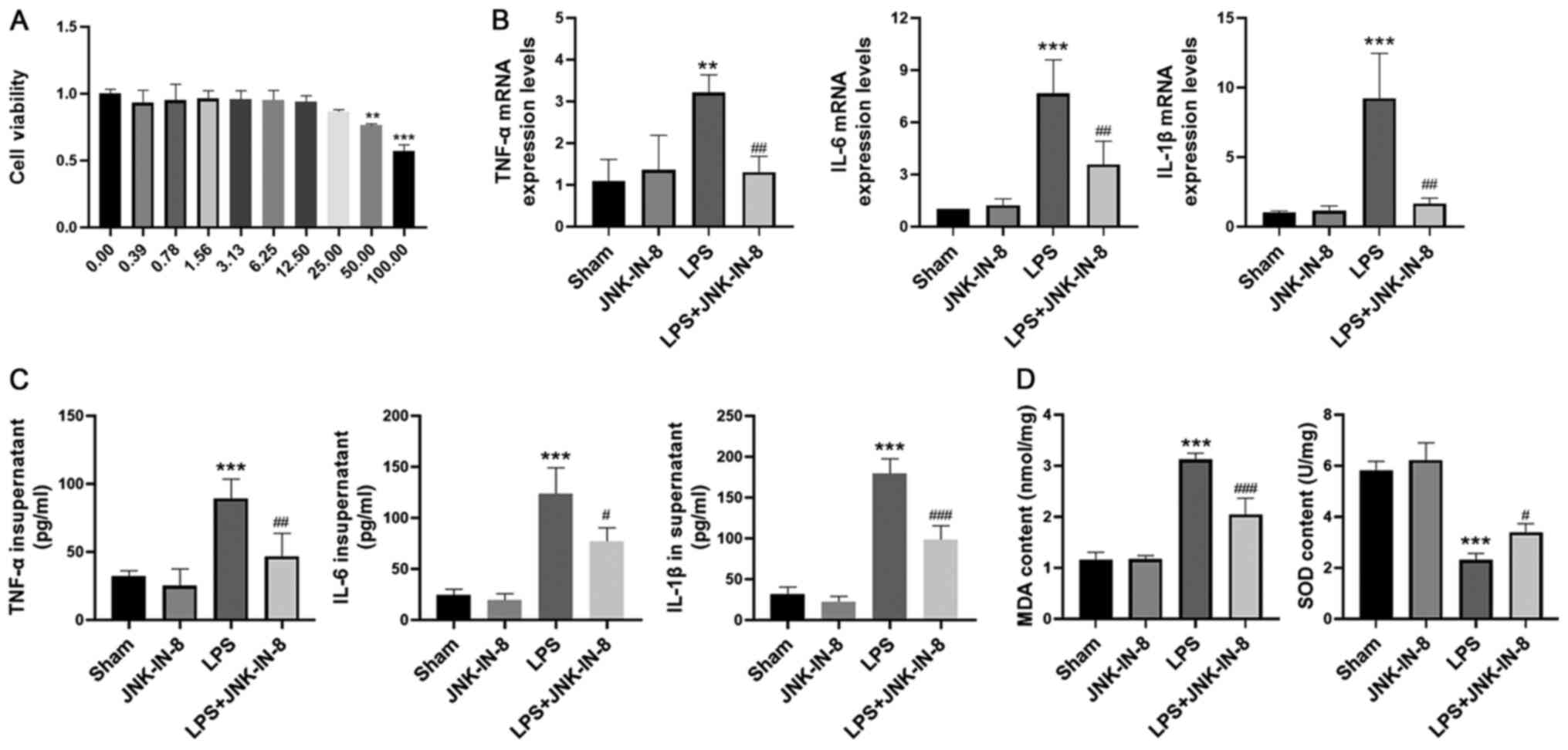
The effects of JNK-IN-8 on the macrophage cell line
RAW264.7 were assessed. In the CCK-8 assay, no cytotoxic effects of
JNK-IN-8 were observed at concentrations ≤12.50 µM in RAW264.7
cells (Fig. 5A). RAW264.7 cells
were cultured and treated with JNK-IN-8 in vitro. The trend
was the same as that of primary macrophages. RAW264.7 cells were
pretreated with JNK-IN-8 for 1 h and then stimulated with LPS (100
ng/ml) for 6 h. The gene expression levels and secretion of TNF-α,
IL-6 and IL-1β were decreased by JNK-IN-8 pretreatment compared
with those in the LPS group (Fig. 5B
and C). JNK-IN-8 administration significantly decreased the MDA
content and inhibited the LPS-induced decrease in SOD activity
(Fig. 5D).
JNK-IN-8 regulates JNK and NF-κB
activation to affect inflammation and oxidative stress in
LPS-treated cells
In order to investigate the mechanism by which
JNK-IN-8 inhibits inflammation, its effects on the JNK/NF-κB
signaling pathway were analyzed. A significant increase was
observed in the protein levels of p-JNK, as well as p-NF-κB p65, in
LPS-stimulated lung tissues, and JNK-IN-8 treatment significantly
inhibited this increase (Fig. 6A and
B). JNK-IN-8 also decreased the phosphorylation of JNK and
NF-κB p65, which was stimulated by LPS in a dose- and
time-dependent manner in primary macrophages in vivo
(Fig. 6C-F). Further experiments
revealed that JNK-IN-8 treatment exhibited a significant inhibitory
effect on the nuclear translocation of p65 induced by LPS (Fig. 6G and H). In conclusion, these data
provided evidence that JNK-IN-8 decreased LPS-induced injury via
the JNK/NF-κB signaling pathway.
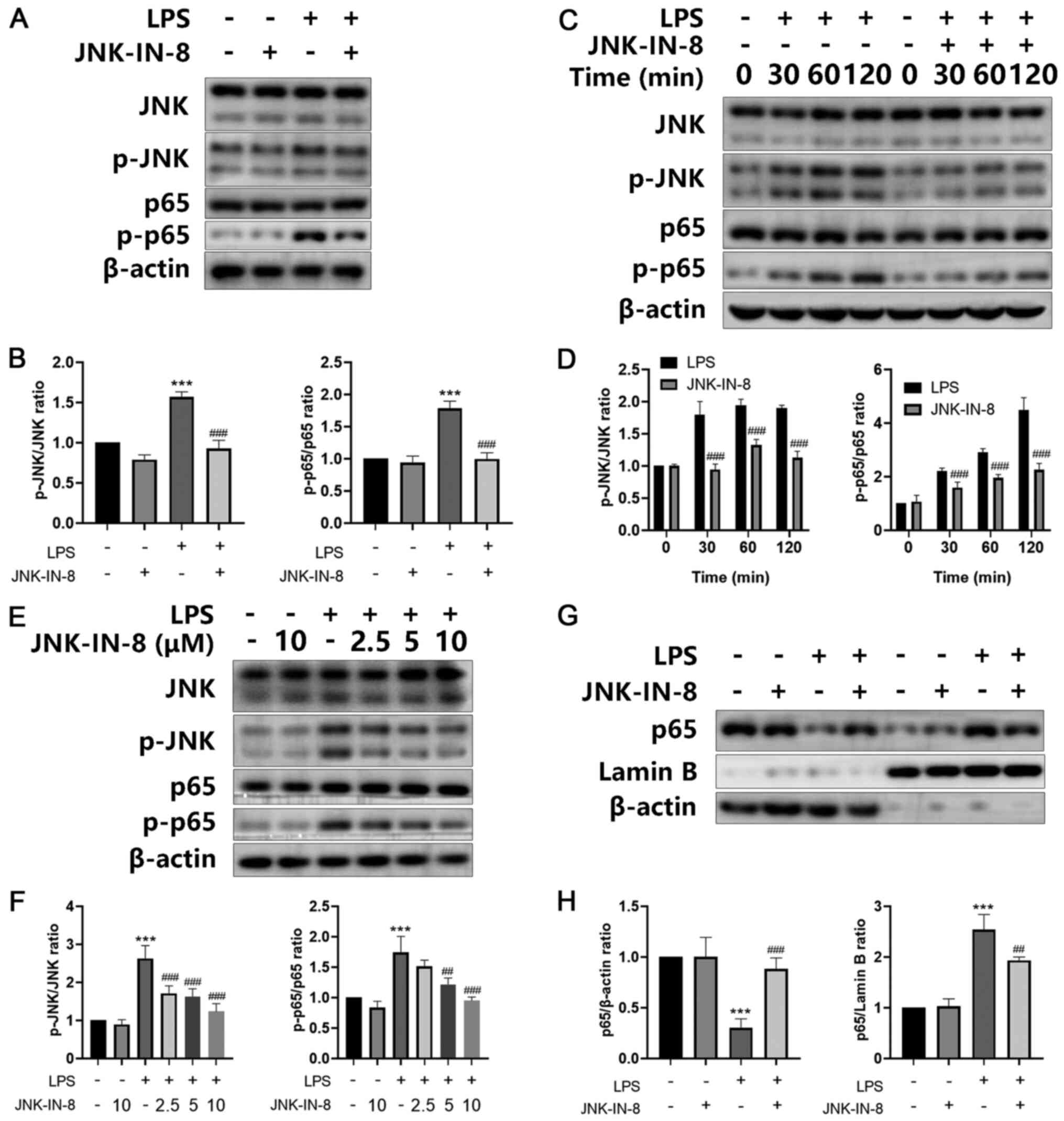 | Figure 6.JNK-IN-8 suppresses the JNK/NF-κB
signaling pathway in LPS-induced acute lung injury in vivo
and in vitro. (A) Primary macrophages stimulated by LPS and
JNK-IN-8 (10 mM) for 2 h were extracted and subjected to western
blot analysis. The protein expression levels of JNK, p-JNK, p65 and
p-p65 were assessed and (B) analyzed by densitometry. (C) Primary
macrophages, stimulated with LPS in the presence or absence of
JNK-IN-8 (10 mM) for 0, 30, 60 or 120 min, were extracted and
subjected to western blot analysis. The protein expression levels
of JNK, p-JNK, p65 and p-p65 were assessed and (D) analyzed by
densitometry. (E) Protein expression levels of JNK, p-JNK, p65 and
p-p65 in lung tissue were assessed and (F) analyzed by
densitometry. β-actin was used as the internal loading control. (G)
Primary macrophages were stimulated with LPS in the presence or
absence of JNK-IN-8 (10 mM) for 2 h. Cytoplasmic and nuclear NF-κB
p65 protein expression levels were determined and (H) analyzed by
densitometry. β-actin and Lamin B were used as internal loading
controls. Data are presented as the mean ± SD (n=3). ***P<0.001
vs. Sham; ##P<0.01 and ###P<0.001 vs.
LPS. LPS, lipopolysaccharide; p-, phosphorylated. |
Discussion
Excessive inflammation and oxidative stress are key
factors that regulate the development of ALI. The expression levels
of proinflammatory regulators can be enhanced by oxidative stress
(5). Furthermore, inflammatory
cells induce the overproduction of ROS in a similar manner, thus
forming a cycle to promote the occurrence and development of ALI
(25–27). JNK-IN-8 was used in the present
study to demonstrate that inhibition of JNK may be a potential
therapy for the treatment of ALI. The present in vivo
experiments revealed that inhibition of JNK by JNK-IN-8 attenuated
ALI in an LPS-induced mouse model. Furthermore, in vitro
experiments demonstrated that JNK-IN-8 inhibited secretion of
inflammatory factors and oxidative stress in LPS-stimulated mouse
peritoneal macrophages, as well as RAW264.7 cells, by inhibiting
JNK/NF-κB signaling. Therefore, it was concluded that JNK-IN-8
inhibited the activation of JNK, as well as NK-κB, both in
vivo and in vitro. This indicated that JNK contributed
to the regulation of inflammation and oxidative stress.
Furthermore, this could offer a novel strategy for treating
ALI.
LPS is a well-known agent for inducing ALI (28). LPS stimulates macrophage activation
and inflammatory cell infiltration via the JNK and NF-κB signaling
pathways, causing uncontrolled release of inflammatory cytokines
and oxidative stress (29–30). JNK signaling has been reported to be
associated with inflammation and oxidative stress. As a result, JNK
is a promising candidate for novel therapeutic targets of
inflammation and oxidative stress (6). In inflammatory diseases, a number of
JNK-associated synthetic inhibitors, such as the micromolecules
SP600125 (31) and CC-930 (32), have been reported. Shen et al
(12) reported that SP600125
treatment suppresses the JNK/AP-1 signaling pathway to decrease
oxidative stress and the inflammatory response, thus decreasing the
apoptosis of A549 cells. Notably, different JNK inhibitors exhibit
numerous physiological characteristics due to the different types
of JNKs (11–14). Therefore, further identification of
selective JNK inhibitors is a key goal.
JNK-IN-8, the first irreversible JNK inhibitor to be
described, suppresses IL-1β-stimulated c-Jun phosphorylation in
IL-1R cells (13). A recent study
revealed that JNK-IN-8 inhibits neuroinflammation in ischemic brain
injury by inhibiting JNK/NF-κB (14). In the present study, JNK-IN-8 also
effectively inhibited JNK/NF-κB signaling to suppress IL-1β, TNF-α
and IL-6 expression levels and release in primary peritoneal
macrophages following LPS treatment. These data demonstrated that
the JNK/NF-κB signaling pathway serves an important role in
inflammatory disease. In addition, the inhibition of JNK-mediated
NF-κB activation by JNK-IN-8 may be a potential target for
treatment of inflammatory diseases.
Excessive oxidative stress is involved in the
pathogenesis of ALI (29). When
lung tissue is damaged in ALI, levels of MDA, a marker of lipid
peroxidation, increase, whereas those of SOD, an antioxidant
enzyme, decrease (26). The present
results demonstrated that LPS increased the MDA content and
decreased the activity of SOD in lung tissues and primary
peritoneal macrophages. JNK-IN-8 inhibited the increase in MDA
content and decreased the LPS-induced SOD activity in vivo
and in vitro, indicating that JNK-IN-8 alleviated the
oxidative stress induced by LPS and protected against lung
damage.
In the present study, JNK-IN-8 contributed to the
regulation of inflammation and oxidative stress following ALI.
JNK-IN-8 treatment decreased LPS-induced inflammation and oxidative
stress activation both in vivo and in vitro. TNF-α,
IL-1β and IL-6 were upregulated following LPS stimulation, whereas
treatment with JNK-IN-8 decreased LPS-induced upregulation of
TNF-α, IL-1β and IL-6, suggesting that JNK-IN-8 treatment
contributed to the decrease in inflammation. Compared with the
control group, JNK-IN-8 inhibited JNK phosphorylation. This finding
indicated that LPS-induced JNK activation was prevented by
JNK-IN-8. In addition, JNK-IN-8 decreased NF-κB signaling
activation. In summary, the present study demonstrated that
targeting JNK-IN-8 is a potent prospective therapy for the
treatment of ALI.
Acknowledgements
Not applicable.
Funding
No funding was received.
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
JD designed and performed the experiments, analyzed
the data and wrote the manuscript. GW designed the experiments. GW,
HL, NL and JX interpreted the data. JX conceptualized and
supervised the study. All authors read and approved the final
manuscript.
Ethics approval and consent to
participate
All animal experiments were approved by the Ethics
Committee of Zhejiang University (approval no. ZJU20170913).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Matthay MA, Ware LB and Zimmerman GA: The
acute respiratory distress syndrome. J Clin Invest. 122:2731–2740.
2012. View
Article : Google Scholar : PubMed/NCBI
|
|
2
|
Li WW, Wang TY, Cao B, Liu B, Rong YM,
Wang JJ, Wei F, Wei LQ, Chen H and Liu YX: Synergistic protection
of matrine and lycopene against lipopolysaccharide induced acute
lung injury in mice. Mol Med Rep. 20:455–462. 2019.PubMed/NCBI
|
|
3
|
Yao H, Sun Y, Song S, Qi Y, Tao X, Xu L,
Yin L, Han X, Xu Y, Li H, et al: Protective effects of dioscin
against lipopolysaccharide-induced acute lung injury through
inhibition of oxidative stress and inflammation. Front Pharmacol.
8:1202017. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Jiang K, Guo S, Yang C, Yang J, Chen Y,
Shaukat A, Zhao G, Wu H and Deng G: Barbaloin protects against
lipopolysaccharide (LPS)-induced acute lung injury by inhibiting
the ROS-mediated PI3K/AKT/NF-kB pathway. Int Immunopharmacol.
64:140–150. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Chen X, Zhang Y, Wang W, Liu Z, Meng J and
Han Z: Mesenchymal stem cells modified with heme oxygenase-1 have
enhanced paracrine function and attenuate
lipopolysaccharide-induced inflammatory and oxidative damage in
pulmonary microvascular endothelial cells. Cell Physiol Biochem.
49:101–122. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Pearson G, Robinson F, Beers Gibson T, Xu
BE, Karandikar M, Berman K and Cobb MH: Mitogen-activated protein
(MAP) kinase pathways: Regulation and physiological functions.
Endocr Rev. 22:153–183. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Romashko J III, Horowitz S, Franek WR,
Palaia T, Miller EJ, Lin A, Birrer MJ, Scott W and Mantell LL: MAPK
pathways mediate hyperoxia-induced oncotic cell death in lung
epithelial cells. Free Radic Biol Med. 35:978–993. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Dong L, Zhou Y, Zhu ZQ, Liu T, Duan JX,
Zhang J, Li P, Hammcok BD and Guan CX: Soluble epoxide hydrolase
inhibitor suppresses the expression of triggering receptor
expressed on myeloid cells-1 by inhibiting NF-κB activation in
murine macrophage. Inflammation. 40:13–20. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Chen XY, Dou YX, Luo DD, Zhang ZB, Li CL,
Zeng HF, Su ZR, Xie JH, Lai XP and Li YC: β-Patchoulene from
patchouli oil protects against LPS-induced acute lung injury via
suppressing NF-κB and activating Nrf2 pathways. Int
Immunopharmacol. 50:270–278. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Kuo MY, Liao MF, Chen FL, Li YC, Yang ML,
Lin RH and Kuan YH: Luteolin attenuates the pulmonary inflammatory
response involves abilities of antioxidation and inhibition of MAPK
and NFkB pathways in mice with endotoxin-induced acute lung injury.
Food Chem Toxicol. 49:2660–2666. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Liu Z, Wang Y, Zhao H, Zheng Q, Xiao L and
Zhao M: CB2 receptor activation ameliorates the proinflammatory
activity in acute lung injury induced by paraquat. Biomed Res Int.
2014:9717502014.PubMed/NCBI
|
|
12
|
Shen H, Wu N, Wang Y, Han X, Zheng Q, Cai
X, Zhang H and Zhao M: JNK inhibitor SP600125 attenuates
paraquat-induced acute lung injury: An in vivo and in vitro study.
Inflammation. 40:1319–1330. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Zhang T, Inesta-Vaquera F, Niepel M, Zhang
J, Ficarro SB, Machleidt T, Xie T, Marto JA, Kim ND, Sim T, et al:
Discovery of potent and selective covalent inhibitors of JNK. Chem
Biol. 19:140–154. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Zheng J, Dai Q, Han K, Hong W, Jia D, Mo
Y, Lv Y, Tang H, Fu H and Geng W: JNK-IN-8, a c-Jun N-terminal
kinase inhibitor, improves functional recovery through suppressing
neuroinflammation in ischemic stroke. J Cell Physiol.
235:2792–2799. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Chen X, Li X, Zhang W, He J, Xu B, Lei B,
Wang Z, Cates C, Rousselle T and Li J: Activation of AMPK inhibits
inflammatory response during hypoxia and reoxygenation through
modulating JNK-mediated NF-κB pathway. Metabolism. 83:256–270.
2018. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
National Research Council: Guide for the
Care and Use of Laboratory Animals - French version. The National
Academies Press; Washington, DC: pp. p1341996
|
|
17
|
National Research Council: Guide for the
Care and Use of Laboratory Animals. 8th edition. The National
Academies Press; Washington, DC: pp. p2462010
|
|
18
|
Leary S: AVMA Guidelines for the
Euthanasia of Animals: 2013 Edition. American Veterinary Medical
Association; Schaumburg, IL: pp. p2012013
|
|
19
|
Wang F, Fu X, Wu X, Zhang J, Zhu J, Zou Y
and Li J: Bone marrow derived M2 macrophages protected against
lipopolysaccharide-induced acute lung injury through inhibiting
oxidative stress and inflammation by modulating neutrophils and T
lymphocytes responses. Int Immunopharmacol. 61:162–168. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Grailer JJ, Haggadone MD, Sarma JV,
Zetoune FS and Ward PA: Induction of M2 regulatory macrophages
through the β2-adrenergic receptor with protection during
endotoxemia and acute lung injury. J Innate Immun. 6:607–618. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Zou Y, Bao S, Wang F, Guo L, Zhu J, Wang
J, Deng X and Li J: FN14 blockade on pulmonary microvascular
endothelial cells improves the outcome of sepsis-induced acute lung
injury. Shock. 49:213–220. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Gan T, Yang Y, Hu F, Chen X, Zhou J, Li Y,
Xu Y, Wang H, Chen Y and Zhang M: TLR3 regulated poly I:C-Induced
neutrophil extracellular traps and acute lung injury partly through
p38 MAP kinase. Front Microbiol. 9:31742018. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Tan W, Zhang C, Liu J and Miao Q:
Regulatory T-cells promote pulmonary repair by modulating T helper
cell immune responses in lipopolysaccharide-induced acute
respiratory distress syndrome. Immunology. 157:151–162. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Zhu X, Zou Y, Wang B, Zhu J, Chen Y, Wang
L, Li J and Deng X: Blockade of CXC chemokine receptor 3 on
endothelial cells protects against sepsis-induced acute lung
injury. J Surg Res. 204:288–296. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Huang XT, Liu W, Zhou Y, Sun M, Yang HH,
Zhang CY and Tang SY: Galectin-1 ameliorates
lipopolysaccharide-induced acute lung injury via AMPK-Nrf2 pathway
in mice. Free Radic Biol Med. 146:222–233. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Jing W, Chunhua M and Shumin W: Effects of
acteoside on lipopolysaccharide-induced inflammation in acute lung
injury via regulation of NF-kB pathway in vivo and in vitro.
Toxicol Appl Pharmacol. 285:128–135. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Jiang W, Luo F, Lu Q, Liu J, Li P, Wang X,
Fu Y, Hao K, Yan T and Ding X: The protective effect of Trillin
LPS-induced acute lung injury by the regulations of inflammation
and oxidative state. Chem Biol Interact. 243:127–134. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Lin WC, Chen CW, Huang YW, Chao L, Chao J,
Lin YS and Lin CF: Kallistatin protects against sepsis-related
acute lung injury via inhibiting inflammation and apoptosis. Sci
Rep. 5:124632015. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Chen H, Bai C and Wang X: The value of the
lipopolysaccharide-induced acute lung injury model in respiratory
medicine. Expert Rev Respir Med. 4:773–783. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Sarma JV and Ward PA: Oxidants and redox
signaling in acute lung injury. Compr Physiol. 1:1365–1381.
2011.PubMed/NCBI
|
|
31
|
Chen Y, Liu K, Zhang J, Hai Y, Wang P,
Wang H, Liu Q, Wong CCL, Yao J, Gao Y, et al: JNK phosphorylates
the Neh6 domain Of Nrf2 and downregulates cytoprotective genes in
acetaminophen-induced liver injury. Hepatology. 2020:
|
|
32
|
Reich N, Tomcik M, Zerr P, Lang V, Dees C,
Avouac J, Palumbo K, Horn A, Akhmetshina A, Beyer C, et al: Jun
N-terminal kinase as a potential molecular target for prevention
and treatment of dermal fibrosis. Ann Rheum Dis. 71:737–745. 2012.
View Article : Google Scholar : PubMed/NCBI
|