Introduction
Cardiac fibrosis is a common pathophysiological
condition and a key step in the occurrence and maintenance of the
majority of cardiovascular diseases, including atrial fibrillation
(AF), hypertension and heart failure (1,2).
Fibrosis promotes systolic and diastolic dysfunction, as well as
heart rhythm disturbances. Therefore, suppression of cardiac
fibrosis may impede cardiac remodeling progression and may be a
therapeutic target for AF and other types of cardiovascular disease
(3).
Cardiac fibrosis is a complicated process associated
with myocardial infarction, pressure and/or volume overload,
mechanical stress, metabolic dysfunction and aging (4–6). One
of the most important cellular and molecular mechanisms of cardiac
fibrosis is differentiation of fibroblasts into myofibroblasts,
which is characterized by the expression of secretory and
contractile proteins, such as α-smooth muscle actin (α-SMA) and
matrix metalloproteinases (MMPs), and production of large amounts
of extracellular matrix (ECM) components, such as collagen I and
III (1,7). Activated myofibroblasts regulate
matrix and cardiomyocyte remodeling. It has been reported that
activation of the renin-angiotensin system (RAS) is involved in the
pathogenesis of cardiac fibrosis remodeling (2,8,9).
Furthermore, studies have shown that angiotensin II (AngII)
increases cardiac fibroblast proliferation and migration, as well
as collagen expression levels (10,11).
Previous studies have suggested that a variety of cell signaling
pathways, such as TGF-β and mitogen-activated protein kinases
(MAPKs) pathways, are involved in the differentiation,
proliferation and migration of cardiac fibroblasts (1,12–14).
TGF-β-activated kinase and downstream p38 MAPK, ERK1/2 and Janus
kinase (JNK) pathways are associated with AngII- or TGF-β-mediated
activation of proliferation and migration of cardiac fibroblasts
(15). Therefore, the inhibition of
AngII-mediated activation of myofibroblasts may be an important
strategy for treating cardiac fibrosis.
P21-activated kinases (PAKs) are a group of
serine/threonine protein kinases that are activated by cell
division cycle 42 and Rac family small GTPase 1. The PAK family
consists of six members, namely PAK1-6 (16–18).
PAK1, 2 and 3 are abundantly expressed in the human heart and PAK1
is involved in actin-based cytoskeletal remodeling (19). Previous studies have shown that PAK1
contributes to TGF-β-induced cancer cell proliferation, migration
and invasion (20–23); these effects have also been reported
in cardiomyocytes. Therefore, it has been demonstrated that PAK1
serves a key role in protection against cardiac hypertrophy
(24). However, the role of PAK1 in
cardiac fibroblasts has not been fully investigated. Therefore, the
present study aimed to investigate the role of PAK1 in cardiac
fibroblasts and the underlying mechanisms.
Materials and methods
Chemicals and reagents
IPA-3, a direct non-ATP-competitive PAK1 inhibitor,
was purchased from MedChemExpress (cat. no. HY-15663). AngII was
obtained from Sigma-Aldrich (cat. no. 4474-91-3; Merck KGaA).
RNA interference
The short hairpin (sh)RNA lentiviral particle
package for PAK1 interference (PAK1-shRNA) was purchased from
Shanghai Jikai Gene Chemical Technology Co., Ltd. Human cardiac
fibroblasts (HCFs) were cultured to 60–70% confluence and then
transduced with shRNA package (1×106 cells transduced
with 5 µl lentivirus) according to the manufacturer's instructions.
Following transduction for 48–72 h, the cells were used for
subsequent experiments. The knocked-down mRNA and protein levels of
PAK1 were determined using reverse transcription-quantitative PCR
(RT-qPCR) and western blot analysis, respectively. The target
sequence of PAK1-shRNA was 5′-CCGCTTGCTTCAAACATCAAA-3′. The
sequence of negative control (NC)-shRNA (scrambled control) was
5′-TTCTCCGAACGTGTCACGT-3′.
Cell culture
Adult HCFs and fibroblast medium-2 were purchased
from ScienCell Research Laboratories, Inc. (cat. no. 6320;
sciencellonline.com/human-cardiac-fibroblasts-juvenile-atrial.html).
Cell passage did not exceed 7–10 generations.
Cell migration assay
Fibroblast migration ability was measured using
wound healing and Transwell migration assays. Briefly, fibroblasts
were cultured to 60–70% confluence in 3.5-cm diameter dishes. Cells
were starved of serum for 12 h, then divided into the following
groups: i) Control; ii) AngII; iii) IPA-3; and iv) AngII + IPA-3 or
i) NC; ii) AngII; iii) PAK1-shRNA; and iv) AngII + PAK1-shRNA for
wound healing and Transwell migration assay. The multiplicity of
infection of PAK1-shRNA package by lentiviral particles in the
present study was difficult to determine because human adult
cardiac fibroblasts can proliferate. In order to ensure the
efficiency of virus interference, confluence of the cells was
decreased for PAK1-shRNA silencing. In order to determine the wound
area, a thin scratch (wound) was made in the central area of the
dish using 1-ml pipette tips. Following washing with fibroblast
medium-2 to remove detached cells, fibroblasts were cultured with
fresh medium with 5% fetal bovine serum. Images of the scratch
wounds were captured with a light microscope (magnification, ×4) at
0 and 24 h, and the wound width was calculated using the following
formula: (T0 - T24/T0) × 100%, where T0 and T24 indicated the width
of the wound area at the start time (0 h) and end point (24 h),
respectively. For the Transwell migration assay, fibroblasts
(density, 3×104) were rehydrated in 100 µl fibroblast
medium-2 supplemented with 0.1% bovine serum albumin. Following
digestion and resuspension in serum-free fibroblast medium-2,
fibroblasts were placed into a Costar Transwell chamber with 8.0-µm
pore size membrane (Corning, Inc.). Fibroblasts were seeded into
6-well plates containing 200 µl fibroblast medium-2 supplemented
with 5% fetal bovine serum at 37°C with 5% CO2 for 12 h.
Subsequently, fibroblasts on the inner side of the membrane were
removed with a cotton bud; migrated cells on the outer side of the
membrane were stained with 0.5% crystal violet for 10 min at room
temperature, then observed with a light microscope (magnification,
×4) and counted manually.
Cell proliferation assay
CCK-8 kit was used to analyze cell proliferation in
the present study (25). HCFs
(density, 1×104 cells/well) were cultured in 96-well
plates and grown to 50% confluence at 37°C with 5% CO2.
Subsequently, the cells were cultured in serum-free fibroblast
medium-2 for 12 h followed by treatment with 1 µM AngII in the
presence or absence of 5 µM IPA-3 for 24 h at 37°C with 5%
CO2. Following incubation for 24 h, the cells were
treated with 10 µl CCK-8 solution (Nanjing Jiangcheng
Bioengineering Institute) at 37°C with 5% CO2 for 4 h
according to the manufacturer's instructions. Finally, the optical
density of each well at a wavelength of 450 nm was measured using a
Spectramax M5 microplate reader (Molecular Devices, LLC).
RT-qPCR
Total RNA from cells was isolated with TRIzol
(Qiagen China Co., Ltd.) and reverse transcribed into cDNA using
the ReverTra Ace qPCR RT Master Mix (Toyobo Life Science) as
follows: 37°C for 15 min; 50°C for 5 min; and 98°C for 5 min.
Subsequently, the expression levels of PAK1 in PAK1-shRNA-treated
cells were quantified by qPCR using SYBR Green (Qiagen China Co.,
Ltd.) as follows: 95°C for 10 sec; 52°C for 10 sec; and 72°C for 10
sec for 40 cycles. The following primer pairs were used: PAK1:
Forward, 5′-TCCGCCAGATGCTTTGACCC-3′ and reverse,
5′-AGCCTCCAGCCAAGTATTCC-3′; and GAPDH: Forward,
5′-GTGGACCTGACCTGCCGTCT-3′ and reverse, 5′-GGAGGAGTGGGTGTCGCTGT-3′.
The expression levels of PAK1 were normalized to those of GAPDH and
calculated using the 2−ΔΔCq method (26).
Western blot analysis
In order to determine the protein expression levels,
cells were lysed with ice-cold RIPA buffer (Beyotime Institute of
Biotechnology) supplemented with protease and phosphatase inhibitor
cocktail (Thermo Fisher Scientific, Inc.), and the protein
concentration was measured via the bicinchoninic acid method. Cell
lysate (30 µg) was resolved by 5% SDS-PAGE and transferred onto
PVDF membranes. Following blocking for 2 h with 5% non-fat dry milk
in TBS-T [20.00 mM Tris (pH 8.0), 150.00 mM NaCl, 0.05% Tween-20]
at room temperature, membranes were incubated with primary
antibodies against α-SMA, collagen І (both 1:2,000; cat. nos.
GB11044 and GB11022, respectively; both Servicebio, Inc.); JNK,
phosphorylated (p)-JNK; ERK, p-ERK; p38, p-p38 (all 1:1,000; cat.
nos. 9252, 9255, 4696, 4370, 8690 and 4511, respectively; all Cell
Signaling Technology, Inc.) and GAPDH (1:5,000; cat. no. G9295;
Sigma-Aldrich; Merck KGaA) at 4°C overnight. The PVDF membrane was
then incubated with horseradish peroxidase (HRP)-conjugated
anti-mouse and anti-rabbit IgG and secondary antibodies (both
1:2,000; cat. nos. 7076 and 7074, respectively; both Cell Signaling
Technology, Inc.) at room temperature for 1 h and treated with
Chemiluminescent HRP Substrate (cat. no. WBKLS0500; EMD Millipore).
Images were captured and immunoreactive bands were quantified using
Quantity One software v4.6.6 (Bio-Rad Laboratories, Inc.).
Statistical analysis
Data are expressed as the mean ± SEM. Results were
analyzed by one-way ANOVA followed by Tukey's posthoc comparison.
P<0.05 was considered to indicate a statistically significant
difference.
Results
IPA-3-mediated inhibition of PAK1
attenuates AngII-induced migration and transdifferentiation of
HCFs
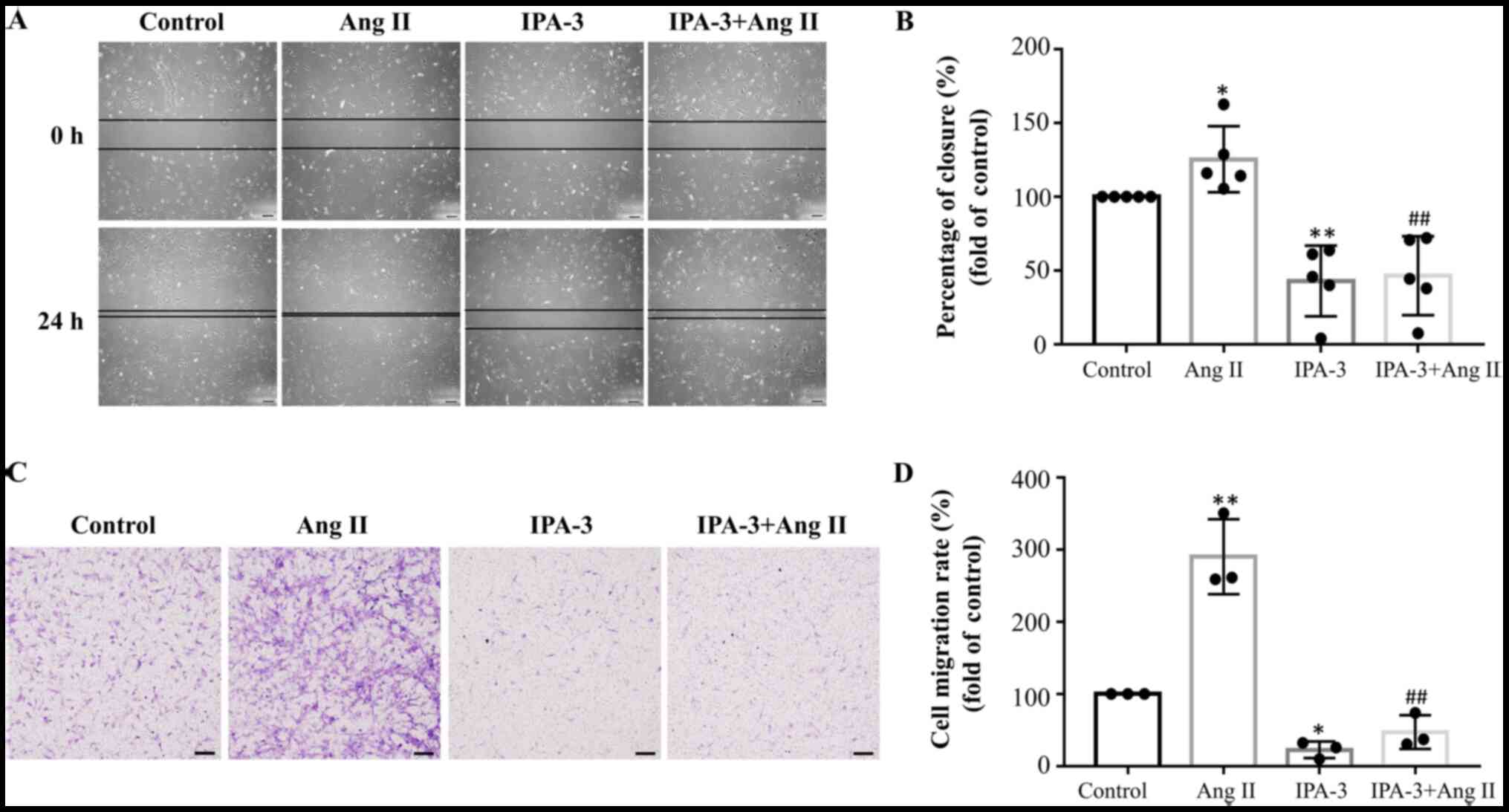
The effect of PAK1 on AngII-induced migration of
HCFs was first investigated. Growth and migration of HCFs were
decreased using a PAK1 inhibitor, IPA-3 (concentration, 5 µM) or
PAK1-shRNA, respectively (Figs. 1
and 2). Exposure of HCFs to 1 µM
AngII for 24 h increased their migration ability, as indicated by
Transwell and wound healing migration assays (Fig. 1A and C). Compared with the control
group, treatment of HCFs with AngII significantly increased wound
closure (P<0.05; n=5) and cell migration rate (P<0.01; n=3;
Fig. 1B and D). Furthermore,
co-treatment of AngII-treated HCFs with 5 µM IPA-3 decreased wound
closure (P<0.01, n=5) and relative cell migration rate
(P<0.01; n=3) compared with the AngII-treated group (Fig. 1B and D).
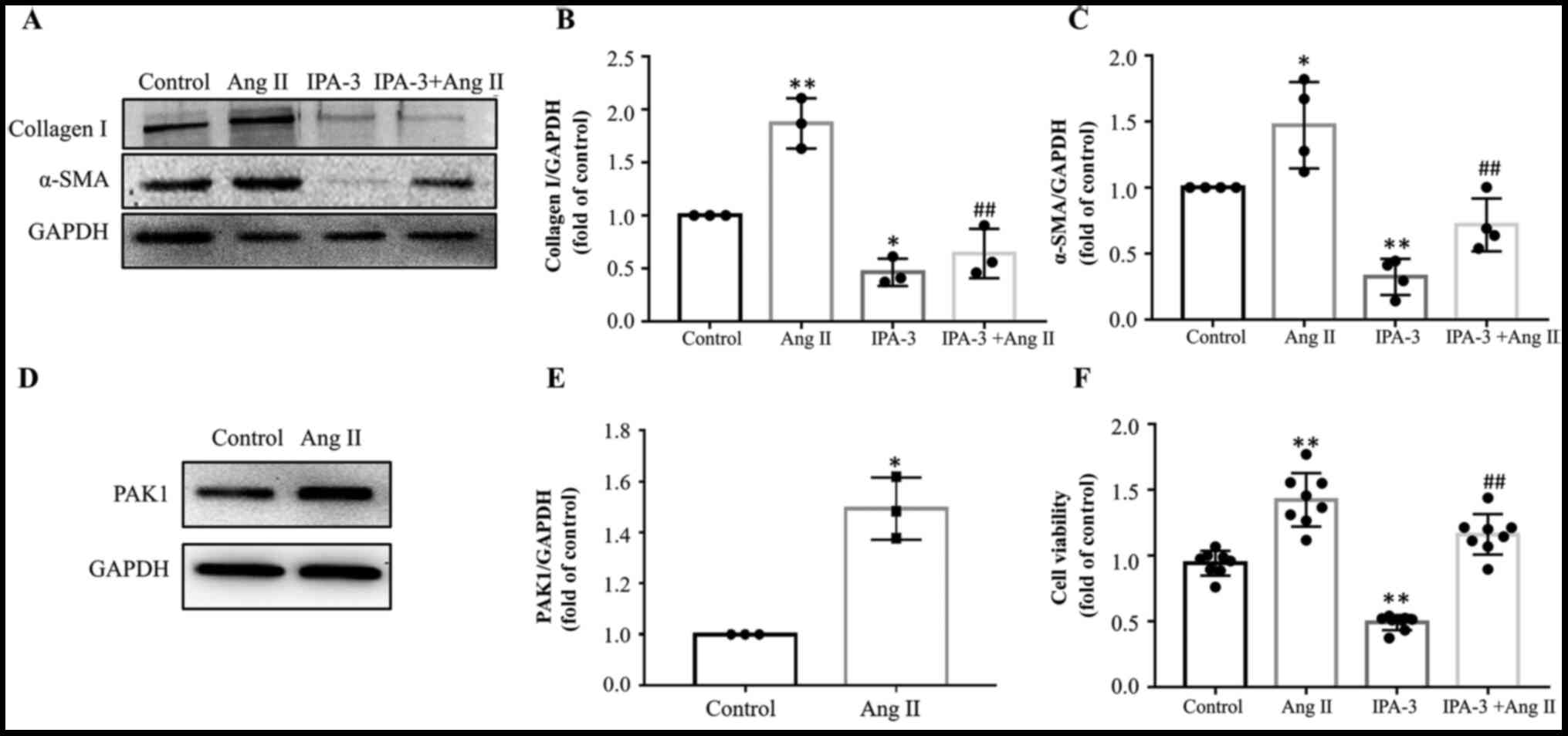
Collagen I and α-SMA were used as markers of
transdifferentiation of fibroblasts into myofibroblasts. Treatment
of HCFs with AngII significantly upregulated α-SMA (P<0.05, n=4)
and collagen I (P<0.01, n=3) expression levels, whereas
co-treatment with IPA-3 downregulated AngII-mediated upregulation
of both proteins (P<0.01; Fig.
3A-C). AngII also significantly increased the expression levels
of PAK1 in fibroblasts (Fig. 3D and
E). Finally, CCK-8 assay revealed that IPA-3 significantly
inhibited AngII-induced cell proliferation (P<0.05, n=8;
Fig. 3F).
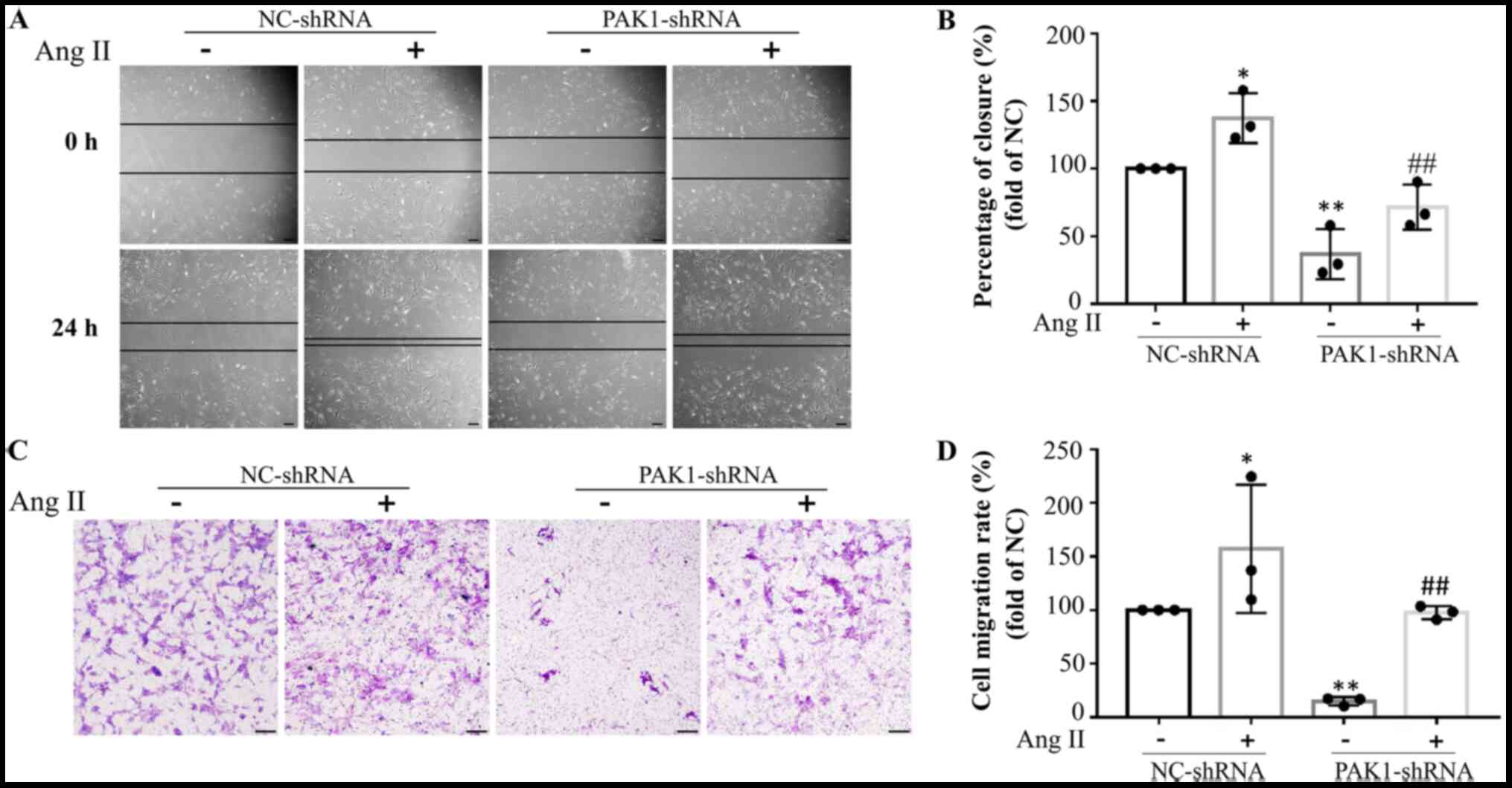
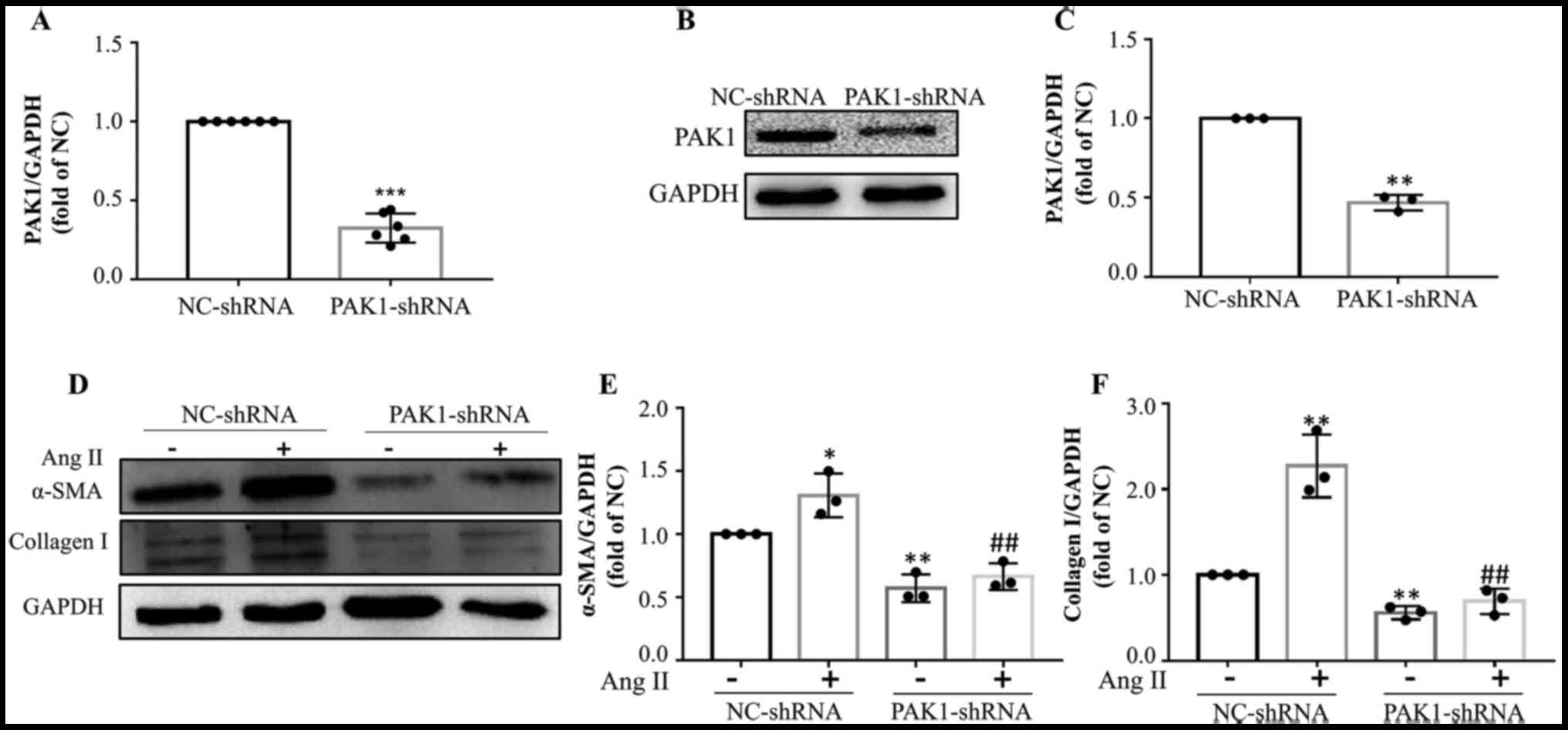
PAK1 knockdown attenuates
AngII-induced migration and transdifferentiation of HCFs
Subsequently, the effect of PAK1-knockdown
(PAK1-shRNA) on AngII-induced migration and transdifferentiation of
HCFs was investigated. Interference rate of PAK1-shRNA is presented
in Fig. 4A-C. PAK1-shRNA
significantly decreased the mRNA (Fig.
4A) and protein levels (Fig. 4B and
C) of PAK1 in HCFs. Similar results were observed between HCFs
exposed to IPA-3 and HCFs transduced with PAK1-shRNA. Transwell and
wound healing migration assays and western blot analysis
demonstrated that PAK1 knockdown significantly inhibited
AngII-induced wound closure (Fig. 2A
and B) and cell migration (Fig. 2C
and D) and PAK1 knockdown significantly (Fig. 4A-C) decreased the expression of
collagen I and α-SMA (Fig. 4D-F),
respectively.
JNK/c-Jun pathway is involved in the
effects of PAK1 on HCFs
Furthermore, the underlying signaling pathway
associated with the effect of PAK1 on fibroblasts was investigated.
The results showed that the JNK/c-Jun pathway is involved in the
inhibitory effect of PAK1 on AngII-induced migration and
transdifferentiation of HCFs (Fig.
5). Treatment of HCFs with AngII increased phosphorylation of
JNK and its downstream molecule c-Jun, whereas PAK1 inhibition with
IPA-3 (Fig. 5D-F) or PAK1-shRNA
(Fig. 5A-C) significantly
attenuated the effect of AngII. Knockdown of PAK1 with PAK1-shRNA
significantly decreased AngII-induced phosphorylation of JNK and
c-Jun (Fig. 5A-C). Similarly, IPA-3
significantly decreased AngII-induced phosphorylation of JNK and
c-Jun (Fig. 5D-F).
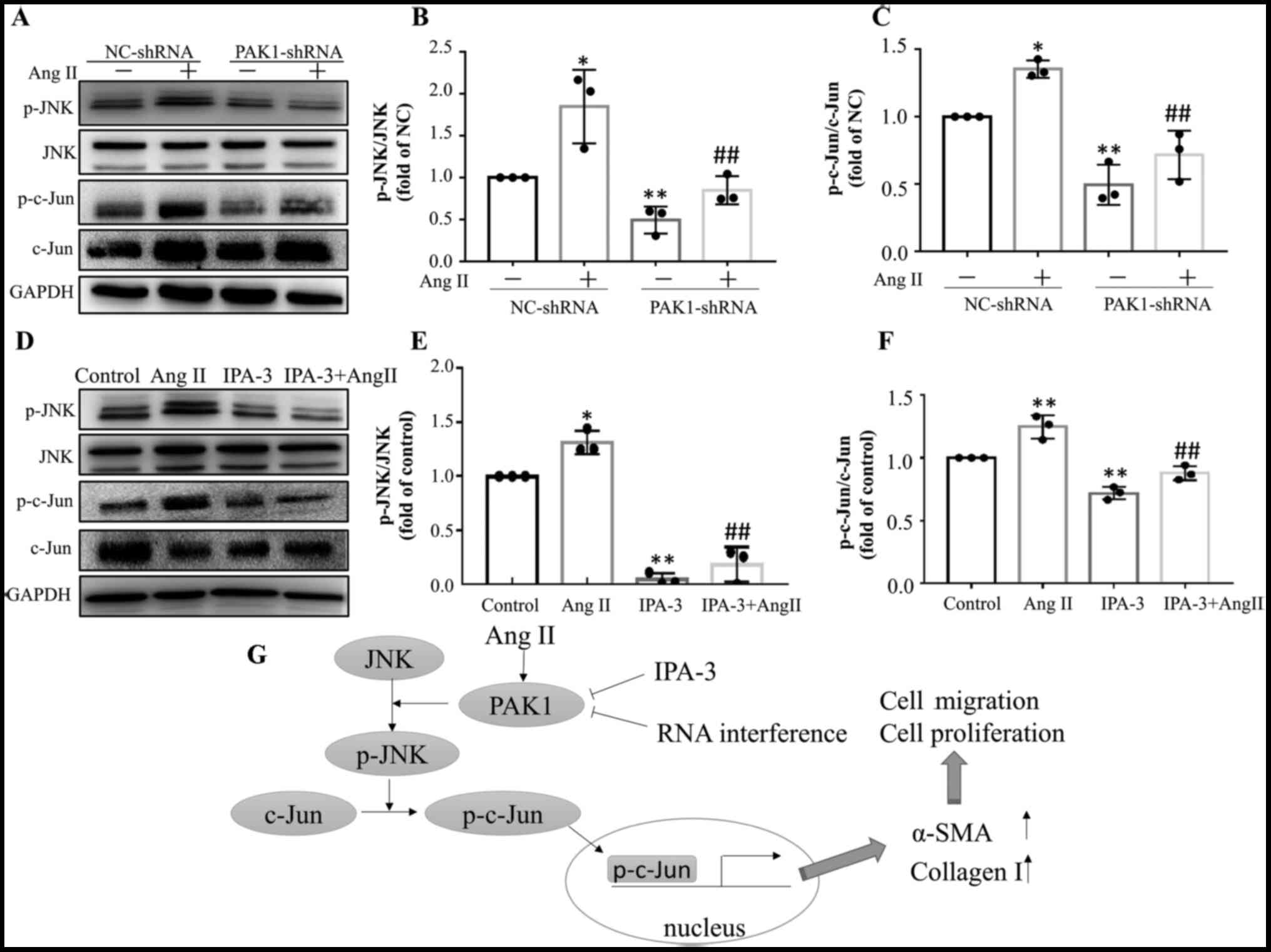 | Figure 5.JNK/c-Jun signaling pathway mediates
the effects of PAK1 in HCFs. (A) Representative images of western
blot assays and histograms showing that transduction of HCFs with
PAK1-shRNA attenuated AngII-induced phosphorylation of (B) JNK and
(C) c-Jun. *P<0.05, **P<0.01 vs. NC. ##P<0.01
vs. AngII. (D) Representative images of western blot assays and
histograms showing that PAK1 inhibition with IPA-3 (5 µM)
attenuated AngII-induced phosphorylation of (E) JNK and (F) c-Jun.
*P<0.05, **P<0.01 vs. control. ##P<0.01 vs.
AngII. (G) Mechanism underlying the effect of PAK1 on the migration
and transdifferentiation of HCFs. Data are presented as the mean
fold change ± SEM. JNK, Janus kinase; PAK1, p21-activated kinase 1;
HCF, human cardiac fibroblast; sh, short hairpin; AngII,
angiotensin II; NC, negative control; p, phosphorylated; α-SMA, α
smooth muscle actin. |
Discussion
Activation of myofibroblasts is a key step in
cardiac fibrosis (1,6,27). The
present study showed that p21-activated kinase 1 (PAK1) contributed
to angiotensin II (AngII)-mediated activation of myofibroblasts,
whereas its inhibition significantly attenuated the effects of
AngII on human cardiac fibroblasts (HCFs) (Fig. 5G).
Activation of the renin-angiotensin system (RAS),
particularly AngII, is involved in numerous types of cardiovascular
disease by promoting cardiac fibroblast differentiation into
myofibroblasts (28). Therefore, in
the present study AngII-activated myofibroblasts were used to
investigate the underlying mechanism of PAK1 in HCFs.
The activation of myofibroblasts is associated with
overexpression of α-smooth muscle actin (α-SMA) and collagen I and
II (29). Consistent with previous
studies, the present study demonstrated that treatment of HCFs with
AngII significantly upregulated α-SMA and collagen I expression
levels (8,30). Furthermore,
inhibition/downregulation of PAK1 activity using IPA-3 inhibitor or
shRNA interference, respectively, attenuated AngII-mediated
overexpression of α-SMA and collagen I. These findings indicate
that PAK1 may be involved in AngII-mediated differentiation of
HCFs. The present study also found that treatment with IPA-3 or
PAK1-shRNA in the absence of AngII inhibited the migration and
proliferation of fibroblasts. These data indicated that PAK1 was
involved in the basic properties of fibroblasts, such as
proliferation and migration.
The activation of myofibroblasts is characterized by
increased cell migration and proliferation ability. Activation of
RAS or upregulation of AngII increase cardiac fibroblast migration
and proliferation (31). Herein,
Transwell and wound healing migration assays also confirmed the
effect of AngII on the migration ability of HCFs. Additionally,
inhibition or downregulation of PAK1 significantly decreased
AngII-induced migration and proliferation of HCFs.
It has been documented that numerous signaling
pathways are involved in AngII-mediated activation of
myofibroblasts, such as the TGFβ1/Smad and MAPK pathways (14,32).
However, the mechanism underlying the effect of PAK1 in
AngII-activated HCFs remains unknown. c-Jun is an important
downstream molecule of the JNK pathway and promotes the
proliferation of fibroblasts (33).
The results of the present study demonstrated that the JNK/c-Jun
pathway mediated the effects of PAK1 on AngII-induced
myofibroblasts.
To the best of our knowledge, the present study is
the first to demonstrate that PAK1 contributes to AngII-mediated
activation of myofibroblasts via the JNK/c-Jun pathway (Fig. 5G). The effect of PAK1 on fibroblasts
was different from that observed in cardiomyocytes in previous
study (20,34). Therefore, it was hypothesized that
PAK1 downregulation may decrease the AngII-induced differentiation,
proliferation and migration of myofibroblasts, thus providing a
novel approach in preventing cardiac fibrosis. In the present
study, the role of PAK1 was investigated only in vitro using
an HCF cell line; therefore, in vivo studies using a
PAK1-knockout mouse model should be performed to further
investigate the role of PAK1 in myofibroblasts.
Acknowledgements
Not applicable.
Funding
The present study was supported by the National
Natural Science Foundation of China (grant nos. 31300948 and
81670310) and the Outstanding Youth Foundation of Sichuan province
of China (grant no. 2020JDJQ0047).
Availability of data and materials
The datasets generated and/or analyzed during the
current study are available from the corresponding author on
reasonable request.
Authors' contributions
XT, YZ and ZF designed the experiment. YZ, YX, TL,
and PZ performed the experiments. XT, YZ, YX and TC performed the
data analysis. XT, YZ and ZF wrote the manuscript. All authors read
and approved the final manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Frangogiannis NG: Cardiac fibrosis: Cell
biological mechanisms, molecular pathways and therapeutic
opportunities. Mol Aspects Med. 65:70–99. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Prabhu SD and Frangogiannis NG: The
Biological Basis for Cardiac Repair After Myocardial Infarction:
From Inflammation to Fibrosis. Circ Res. 119:91–112. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Ma ZG, Yuan YP, Wu HM, Zhang X and Tang
QZ: Cardiac fibrosis: New insights into the pathogenesis. Int J
Biol Sci. 14:1645–1657. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Humeres C and Frangogiannis NG:
Fibroblasts in the Infarcted, Remodeling, and Failing Heart. JACC
Basic Transl Sci. 4:449–467. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Ji Y, Qiu M, Shen Y, Gao L, Wang Y, Sun W,
Li X, Lu Y and Kong X: MicroRNA-327 regulates cardiac hypertrophy
and fibrosis induced by pressure overload. Int J Mol Med.
41:1909–1916. 2018.PubMed/NCBI
|
|
6
|
Kong P, Christia P and Frangogiannis NG:
The pathogenesis of cardiac fibrosis. Cell Mol Life Sci.
71:549–574. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Weber KT: Fibrosis in hypertensive heart
disease: Focus on cardiac fibroblasts. J Hypertens. 22:47–50. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Frangogiannis NG: Cardiac fibrosis: Cell
biological mechanisms, molecular pathways and therapeutic
opportunities. Mol Aspects Med. 65:70–99. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Ma H, Kong J, Wang YL, Li JL, Hei NH, Cao
XR, Yang JJ, Yan WJ, Liang WJ, Dai HY and Dong B:
Angiotensin-converting enzyme 2 overexpression protects against
doxorubicin-induced cardiomyopathy by multiple mechanisms in rats.
Oncotarget. 8:24548–24563. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Chen T, Li M, Fan X, Cheng J and Wang L:
Sodium Tanshinone IIA Sulfonate Prevents Angiotensin II–Induced
Differentiation of Human Atrial Fibroblasts into Myofibroblasts.
Oxid Med Cell Longev. 2018:67125852018. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Dong X, Yu S, Wang Y, Yang M, Xiong J, Hei
N, Dong B, Su Q and Chen J: (Pro)renin receptor-mediated myocardial
injury, apoptosis, and inflammatory response in rats with diabetic
cardiomyopathy. J Biol Chem. 294:8218–8226. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Li R, Xiao J, Qing X, Xing J, Xia Y, Qi J,
Liu X, Zhang S, Sheng X, Zhang X, et al: Sp1 Mediates a Therapeutic
Role of MiR-7a/b in Angiotensin II–Induced Cardiac Fibrosis via
Mechanism Involving the TGF-β and MAPKs Pathways in Cardiac
Fibroblasts. PLoS One. 10:e01255132015. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Działo E, Tkacz K and Błyszczuk P:
Crosstalk between the TGF-β and WNT signalling pathways during
cardiac fibrogenesis. Acta Biochim Pol. 65:341–349. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Gu J, Liu X, Wang QX, Tan HW, Guo M, Jiang
WF and Zhou L: Angiotensin II increases CTGF expression via
MAPKs/TGF-β1/TRAF6 pathway in atrial fibroblasts. Exp Cell Res.
318:2105–2115. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Trial J and Cieslik KA: Changes in cardiac
resident fibroblast physiology and phenotype in aging. Am J Physiol
Heart Circ Physiol. 315:H745–H755. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Rane CK and Minden A: P21 activated kinase
signaling in cancer. Semin Cancer Biol. 54:40–49. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Sun X, Su VL and Calderwood DA: The
subcellular localization of type I p21-activated kinases is
controlled by the disordered variable region and polybasic
sequences. J Biol Chem. 294:14319–14332. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Rudolph J, Crawford JJ, Hoeflich KP and
Wang W: Inhibitors of p21-activated kinases (PAKs). J Med Chem.
58:111–29. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Wang Y, Wang S, Lei M, Boyett M, Tsui H,
Liu W and Wang X: The p21-activated kinase 1 (Pak1) signalling
pathway in cardiac disease: From mechanistic study to therapeutic
exploration. Br J Pharmacol. 175:1362–1374. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Al-Azayzih A, Gao F and Somanath PR: P21
activated kinase-1 mediates transforming growth factor β1-induced
prostate cancer cell epithelial to mesenchymal transition. Biochim
Biophys Acta. 1853:1229–1239. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Chen QY, Xu LQ, Jiao DM, Yao QH, Wang YY,
Hu HZ, Wu YQ, Song J, Yan J and Wu LJ: Silencing of Rac1 modifies
lung cancer cell migration, invasion and actin cytoskeleton
rearrangements and enhances chemosensitivity to antitumor drugs.
Int J Mol Med. 28:769–776. 2011.PubMed/NCBI
|
|
22
|
Yang Y, Du J, Hu Z, Liu J, Tian Y, Zhu Y,
Wang L and Gu L: Activation of Rac1-PI3K/Akt is required for
epidermal growth factor-induced PAK1 activation and cell migration
in MDA-MB-231 breast cancer cells. J Biomed Res. 25:237–245. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Zhang C, Lee HJ, Shrivastava A, Wang R,
McQuiston TJ, Challberg SS, Pollok BA and Wang T: Long-Term In
Vitro Expansion of Epithelial Stem Cells Enabled by Pharmacological
Inhibition of PAK1-ROCK-Myosin II and TGF-β Signaling. Cell Rep.
25:598–610.e5. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Liu W, Zi M, Naumann R, Ulm S, Jin J,
Taglieri DM, Prehar S, Gui J, Tsui H, Xiao RP, et al: Pak1 as a
novel therapeutic target for antihypertrophic treatment in the
heart. Circulation. 124:2702–2715. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Buttke TM, McCubrey JA and Owen TC: Use of
an aqueous soluble tetrazolium/formazan assay to measure viability
and proliferation of lymphokine-dependent cell lines. J Immunol
Methods. 157:233–240. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Mazzei M, Vascellari M, Zanardello C,
Melchiotti E, Vannini S, Forzan M, Marchetti V, Albanese F and
Abramo F: Quantitative real time polymerase chain reaction
(qRT-PCR) and RNAscope in situ hybridization (RNA-ISH) as effective
tools to diagnose feline herpesvirus-1-associated dermatitis. Vet
Dermatol. 30:491–e147. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Talman V and Ruskoaho H: Cardiac fibrosis
in myocardial infarction-from repair and remodeling to
regeneration. Cell Tissue Res. 365:563–581. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Holtz J: Pathophysiology of heart failure
and the renin-angiotensin-system. Basic Res Cardiol. 88 (Suppl
1):183–201. 1993.PubMed/NCBI
|
|
29
|
Petrov VV, Fagard RH and Lijnen PJ:
Stimulation of collagen production by transforming growth
factor-beta1 during differentiation of cardiac fibroblasts to
myofibroblasts. Hypertension. 39:258–263. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Harada M, Luo X, Qi XY, Tadevosyan A,
Maguy A, Ordog B, Ledoux J, Kato T, Naud P, Voigt N, et al:
Transient receptor potential canonical-3 channel-dependent
fibroblast regulation in atrial fibrillation. Circulation.
126:2051–2064. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Alex L and Frangogiannis NG: The Cellular
Origin of Activated Fibroblasts in the Infarcted and Remodeling
Myocardium. Circ Res. 122:540–542. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Hirsch AT, Pinto YM, Schunkert H and Dzau
VJ: Potential role of the tissue renin-angiotensin system in the
pathophysiology of congestive heart failure. Am J Cardiol.
66:D22–D332. 1990. View Article : Google Scholar
|
|
33
|
Sabapathy K, Hochedlinger K, Nam SY, Bauer
A, Karin M and Wagner EF: Distinct roles for JNK1 and JNK2 in
regulating JNK activity and c-Jun-dependent cell proliferation. Mol
Cell. 15:713–725. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Harms FL, Kloth K, Bley A, Denecke J,
Santer R, Lessel D, Hempel M and Kutsche K: Activating Mutations in
PAK1, Encoding p21-Activated Kinase 1, Cause a Neurodevelopmental
Disorder. Am J Hum Genet. 103:579–591. 2018. View Article : Google Scholar : PubMed/NCBI
|