Introduction
Ferroptosis has emerged as a novel type of regulated
cell death (RCD) in various diseases, particularly in hepatic or
renal ischemia-reperfusion injury (IRI). The occurrence of
ferroptosis is based on iron overload, which generates reactive
oxygen species (ROS) and lipid peroxides, primarily
phosphatidylethanolamine-OOH (PE-OOH) in vivo (1–3).
Ferroptosis was identified in 2012 and was originally reported to
be associated with mutant RAS cancer cells 1 (4,5).
Although the mechanism of ferroptosis has a relatively specific
description, primarily including iron-dependent accumulation of
lipid ROS and the consumption of plasma membrane polyunsaturated
fatty acids (PUFAs) (5), the role
of ferroptosis in cancer, heart, liver or kidney injury, and
neurotoxicity remains unclear (6).
Severe hepatic IRI may lead to serious impairment of liver function
or even acute liver failure (7,8).
Therefore, it is critical to prevent hepatic IRI, especially in
liver transplantation, due to the high risk of urgent
re-transplantation (9). Distinct
from other types of RCD (apoptosis, necroptosis and autophagy),
ferroptosis is characterized by resulting oxidative damage in the
mitochondria, which exerts harmful effects on hepatic
ischemia-reperfusion (6,8). Furthermore, these injuries can be
prevented by the ferroptosis-specific inhibitor ferrostatin-1
(Fer-1) and by iron chelators (6,8).
Therefore, ferroptosis is a potential target for preventing and
treating hepatic IRI. Thus, exploration of the exact mechanisms of
ferroptosis in liver cell death is required.
The present review firstly introduces the types of
hepatic IRI and subsequently describes the molecular mechanisms of
liver IRI. Thirdly, the general mechanisms of ferroptosis and the
role of ferroptosis in hepatic IRI are discussed; primarily
including the inflammatory response and oxidative stress. In the
final part of this review, several therapeutic strategies
associated with ferroptosis are described in detail.
Hepatic IRI
Types of liver IRI
Hepatic IRI is still a long-standing problem in
clinical conditions that occurs in hepatic resection surgery, liver
transplantation and during states of shock. Two main types of
hepatic IRI exist, including warm and cold IRI. Warm IRI, initiated
by hepatocellular injury, occurs ischemia at routine temperature
and is generally present in liver transplantation surgery or
different forms of trauma or shock, and might lead to liver
failure, or even bring the outcome of multiorgan failure (10). Cold IRI starts at the injury of
endothelial cells in hepatic sinusoidal and microcirculation
disorders with the temperature of liver decreasing rapidly and
uniformly, which develops during in vitro preservation and
is usually accompanied by warm IRI in the process of liver
transplantation surgery (10,11).
Although the two IRI types might possess distinct initial cellular
targets, they do share similar pathophysiological processes,
including local inflammatory innate immune activation (10,12,13),
and expression of fibronectin (FN) in endothelial cells is a
prominent feature of the liver injury response (14). At present, there is no evidence that
hot or cold IRI causes different types of cell death.
In addition, IRI can be divided into two phases,
ischemia and reperfusion, which are primarily the result of
oxidative stress accompanied by nutritional deficiency, loss of
blood flow, inflammation and other conditions (15). Such trauma primarily causes
autophagy in liver cells, including apoptosis and necrosis
(16). There is evidence that
various markers of autophagy are elevated throughout the entire IRI
process (17). Among them,
iron-mediated death is primarily believed to be associated with
oxidative stress from ROS, especially during blood reperfusion
(18).
Ferroptosis is a type of iron-dependent oxidative
cell death characterized by accumulation of intracellular ROS,
which will be discussed in more detail. Furthermore, iron-mediated
cell death is an important form of autophagy (19,20).
Molecular mechanisms of liver IRI
ROS (such as OH- and HOO-), chemically reactive
species containing oxygen, are an important cause of initial liver
injury (21) and are originally
produced in Kupffer cells, which kill hepatocytes through lipid
peroxidation, DNA oxidation and enzymatic degeneration (22). The pathway regulated by ROS that
promotes apoptosis contains different molecules and transporters.
Initially, ROS activates apoptosis signal-regulating kinase 1
(ASRK1) through TNF-receptor-associated factor 2 (TRAF-2) that
leads to c-Jun-N-terminal kinase (JNK), which directly regulates
the activities of pro- and anti-apoptotic mitochondrial proteins
through different phosphorylation events or via upregulating
pro-apoptotic genes through the trans-activation of specific
transcription factors (23). In
addition, tumour necrosis factor-α (TNF-α), subsequently released
by ROS, can increase the damage after IRI by promoting extra
release of inflammatory cytokines and creating positive feedback
circuits, which leads to organ damage. Furthermore, TNF-α regulates
the production of gangliochemical genes and adhesion molecules that
are absorbed into the liver, and these neutrophils are eventually
responsible for the subsequent stage of injury. In addition to
TFN-α, other proinflammatory cytokines, such as IL-1β (24), IL-12 (25), IL-18 and IL-6 (10), are critical for the hepatic
inflammatory response. Furthermore, IL-12 is indispensable for
fully producing TNF-α in the liver and the ensuing inflammatory
response, which was confirmed using neutralizing antibodies or
IL-12 knockout mice to eliminate IL-12 (10). These injuries eventually lead to
biliary microcirculatory disorders and apoptosis of biliary
epithelial cells.
Relevant proinflammatory signalling pathways include
nuclear factor κB (NF-κB), which is activated by proinflammatory
cytokines, such as IL-1 and TNF-α.
Of note, ROS can also stimulate NF-κB to promote
hepatic IRI (26,27). Hence, antioxidants decrease the
expression of pro-inflammatory genes by inhibiting the activation
of NF-κB (28–31). On the other hand, superoxide
formation in endothelial cells (32) and hepatocytes (33) was recently shown to originate from a
phagocyte-type nicotinamide adenine dinucleotide phosphate (NADPH)
oxidase. Therefore, inhibiting Rac1, a member of the Rho family of
small GTPases that can regulate this oxidase, attenuates
intracellular oxidant stress and protects against hepatocyte injury
during the early reperfusion phase (33).
Brief overview of ferroptosis
Ferroptosis is a form of RCD that is dependent on
iron and ROS, and is initiated by the failure of glutathione
biosynthesis or the inactivation of glutathione peroxidase 4
(GPX4), an antioxidant enzyme that depends on glutathione, thus
resulting in lipid peroxidation, the consumption of PUFAs, and
eventual cell death (4).
Ferroptosis has distinct features at the morphological,
biochemical, and genetic level compared with other forms of RCD,
including necroptosis, apoptosis and autophagy. Small molecules,
such as erastin, ras-selective lethal small molecule (RSL3), high
concentrations of glutamate, and sulfasalazine are known to reduce
ferroptosis, while α-tocopherol, ferrostatin-1, liproxstatin-1,
glutathione, zileuton and iron chelators (such as deferasirox,
deferiprone, chelation with deferoxamine and 1,10-phenanthroline)
are inhibitors involved in relevant mechanisms of ferroptosis that
contribute to hepatic IRI (6,34,35).
However, how ferroptosis plays an essential role in cell injury is
not well understood. Moreover, the detailed signalling pathway that
lie between IRI induction and ferroptosis activation remains
unknown. Validation is still required to understand all the
molecules on this pathway, and the known details are summarized,
which is still the inevitable limitation of this review.
The ferroptotic signalling pathway
The activation of mitogen-activated protein kinase
(MAPK), iron metabolism and lipid peroxidation signalling pathways
are currently known to contribute to ferroptotic cell death
(36). However, it has been
reported that the MAPK pathway was associated to to cancer cell
death, which inhibits ferroptosis induced by erastin by blocking
the Ras/Raf/MEK/ERK pathway in Ras-mutated cancer cells (8). Hence, iron and ROS signalling pathways
are primarily described in the present review.
Iron and ferroptosis
The principal role of iron is to transport oxygen in
the haematological system. Iron possesses two forms in cells:
Fe2+ and Fe3+. The Fe2+ absorbed
into the blood is oxidized to Fe3+ by ceruloplasmin, and
Fe3+ is transported to the tissues after binding with
transferrin or is absorbed into the cell through the membrane
transferrin receptor (TfR), then localised to the internal body,
and Fe3+ is reduced to Fe2+ by the
ferrireductase activity of six-transmembrane epithelial antigen of
prostate 3 (STEAP3). Finally, release of Fe2+ from the
endosome to a labile iron pool is mediated by divalent metal
transporter 1 (DMT1) in the cytoplasm. Excess iron is kept in the
monocyte-macrophage system of the liver, spleen, bone marrow and
other organs in the form of ferritin and hemosiderin. Membrane
protein ferroportin, an iron efflux pump that oxidizes
Fe2+ into Fe3+, transmits signals to mediate
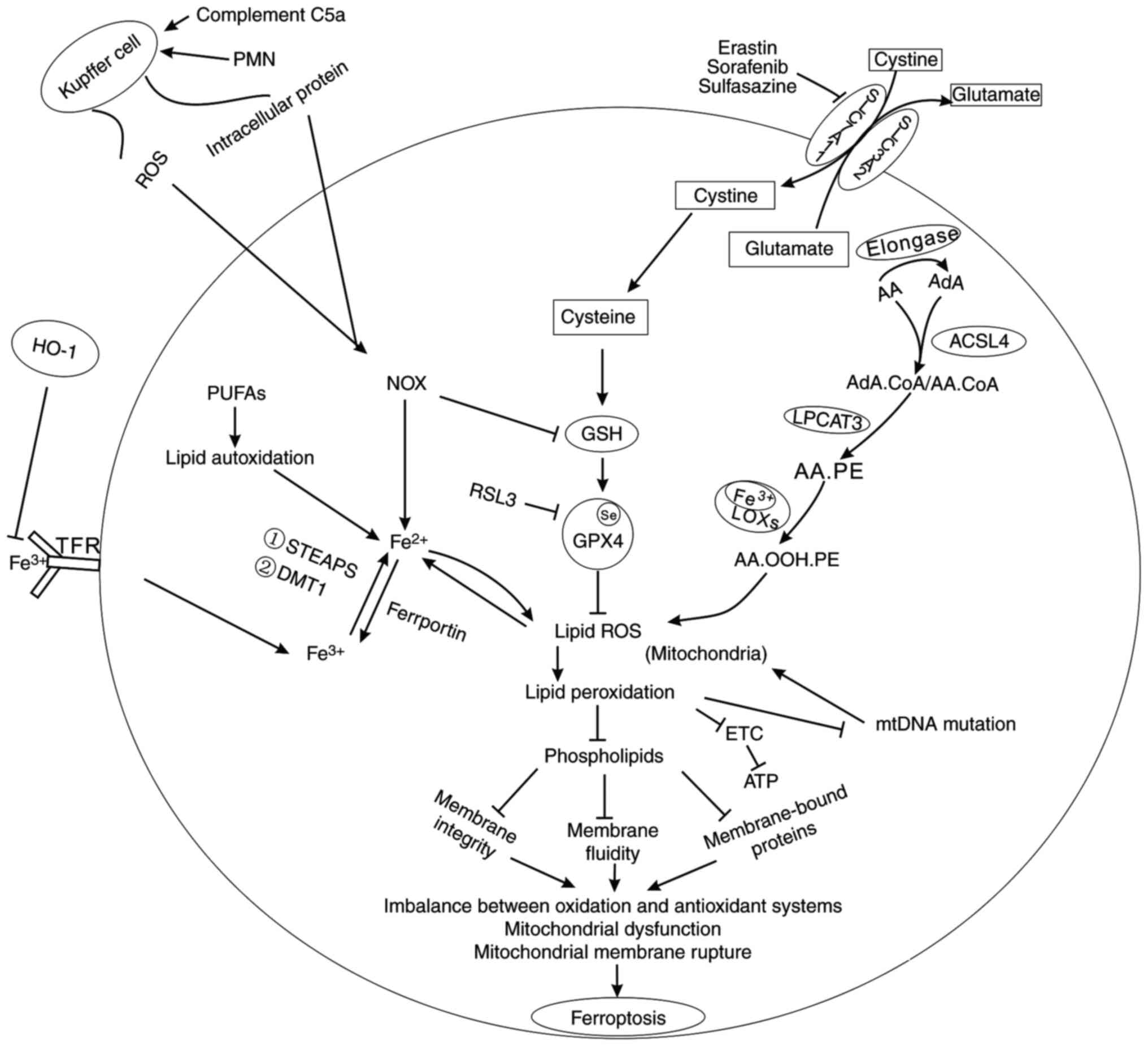
iron output (Fig. 1).
Fe2+ has the feature of a catalyst, which can transfer
electrons and participate in various oxidation-reduction reactions,
while Fe3+ primarily exists in the process of
transportation and storage.
However, iron overload is recognized as poisonous to
cells, since the transferred electrons are given to O2
and H2O2 to produce superoxide anions and
hydroxyl radicals, which exert harmful influences on biological
macromolecules, such as nucleic acids, proteins and lipids
(37). Moreover, Fe2+
can oxidize organics combined with H2O2 to
generate ROS by the Fenton reaction (3,37). As
Wang et al (38) stated,
hepatocytes and macrophages are sensitive to extracellular iron
levels, and a high-iron diet in mice could trigger ferroptotic cell
death. Additionally, shock protein family B member 1 (HSPB1)
inhibits ferroptosis through decreasing intracellular iron levels
and upholding glutathione (GSH) in its reduced form. Furthermore,
TfR1-mediated iron uptake is inhibited by HSPB1, which blocks the
endocytosis and recycling of transferrin to decrease intracellular
iron levels (39–41). These studies suggest that iron plays
a critical role during ferroptosis, although the role of iron in
the signalling pathway of ferroptosis remains poorly understood. To
date, in addition to the Fenton reaction by Fe2+, there
is an additional source involved in the iron-dependent accumulation
of lipid ROS in ferroptosis: Lipid peroxidation controlled by
iron-containing lipoxygenases (LOXs) (Fig. 1) (42).
LOXs are a family of non-haem, iron-containing
enzymes, and most of them catalyse the deoxygenation of PUFAs, such
as arachidonic acid (AA) and linolenic acid, in lipids containing a
cis, cis−1,4-pentadiene into cell signalling agents
(43–45). As one type of PUFA, AA is converted
to adrenoyl (AdA) under the action of elongase (2), and then on the endoplasmic reticulum
or mitochondrial outer membrane, AA and AdA are catalysed by
acyl-CoA synthetase long-chain family 4 (ACSL4) to form
AdA-CoA/AA-CoA, which is next esterified to AA-PE under the action
of lysophosphatidylcholine acyltransferase 3 (LPCAT3), finally
forming AA-OOH-PE, a cell death signal of ferroptosis, under the
oxidation of iron-containing LOXs (46,47).
However, the role of iron in regulating LOXs relies on
phosphorylase kinase G2 (PHKG2), which activates glycogen
phosphorylase (GP) to release glucose-1-phosphate from glycogen,
promoting the phosphorylation of LOXs to synthesise lipid
peroxides. Furthermore, glycogen primarily exists in liver and
muscle tissues; therefore, glycogen breakdown might be an important
factor in ferroptosis in liver or muscle injury, although no
current studies have confirmed this. In conclusion, iron-containing
LOXs are required for ferroptosis in the reaction of lipid
peroxidation, and inhibition of ACSL4 and LPCAT3 may decrease
oxidation of some sensitive fatty acids in the membrane. However,
more studies are required to further explain the detailed role of
iron in mediating LOX activity.
ROS and ferroptosis
ROS are primarily located in the mitochondria during
electron transport (48), and
iron-dependent lipid ROS produced by the two aforementioned sources
mediate lipid peroxidation that further promotes the accumulation
of lipid peroxides (Fig. 1)
(6). Furthermore, peroxides lead to
fundamental changes in lipids, especially phospholipids, which are
essential for maintaining the integrity of the mitochondrial
membrane architecture. In addition, peroxides can affect the
fluidity of lipids, thus blocking receptor clustering and
propagating inflammatory signalling (49). The peroxidation of phospholipids
also inactivates membrane-bound proteins, ultimately causing
destruction of the membrane (50).
In addition, the occurrence of ROS accompanies single electron
leakage of oxidative phosphorylation in the mitochondria, thus
decreasing the yield of ATP and inhibiting cell survival (49). Thus, increased mitochondrial ROS can
destroy the integrity of the electron transport chain, causing
respiratory chain dysfunction (51). Moreover, lipid peroxidation products
lead to mtDNA damage, further contributing to mitochondrial
mutations, whereas mutations in the mitochondria further increase
levels of ROS with toxic effects (49,51),
resulting in a vicious circle. On the one hand, stable aldehyde
peroxidation products from PUFAs, such as 4-hydroxynonenal and
malondialdehydes, are involved in mtDNA mutations or deletions
(52). Although aldehydes have
significant toxic effects, some enzymes in vivo, such as
cytochrome P450 (CYP), aldehyde dehydrogenase and aldo-keto
reductases, can metabolize them to less toxic compounds (53). Evidence shows that members of CYP3A
and CYP4A oxidize 4-hydroxynonenal (38). However, when mitochondrial
dysfunction and increased ROS occur, oxidation and antioxidant
systems lose balance in vivo, and the mitochondrial crest
decreases or disappears. The outer wall of the mitochondrial
membrane ruptures, which is a typical feature of ferroptosis that
differentiates it from apoptosis, necroptosis and autophagy
(6). In conclusion, it is
postulated that ROS-induced ferroptosis depends on changes in the
mitochondria.
Ferroptosis in hepatic IRI
Recent evidence has shown that ferroptosis is
associated with the pathogenesis of various diseases, such as
neoplastic diseases and ischemic injury to the brain, heart, liver,
kidney and intestine (6–8,54–56).
Furthermore, several studies have indicated that inhibitors of
ferroptosis, such as ferrostatins-1 and liproxstatins-1, protect
against cell death in the liver, kidney, brain and heart ischemic
injury in mouse models (55–57).
In a study by Yamada et al (8), the role of ferroptosis in hepatic IRI
was explored, which established a murine model of hepatic
ischemia-reperfusion injury and found that upregulation of the
ferroptosis marker Ptgs2, lipid peroxidation and liver damage were
induced by hepatic ischemia reperfusion. Moreover, all of these
liver cell injuries can be prevented when super-inducing the
ferroptosis-specific inhibitor Fer-1 and by iron chelation
(8,54). Thus, it seems that iron overload is
a critical factor for hepatic IRI, and the pathogenesis of hepatic
IRI is partly attributed to ferroptosis (8).
Role of iron in inflammation in
hepatic IRI
At present, it is widely accepted that hepatic IRI
is characterized by an excessive inflammatory response, release of
inflammatory cytokines and chemokines, as well as neutrophil and
macrophages infiltration. In addition, IL-1β is the decisive factor
that drives many sterile inflammatory diseases (58), especially in hepatic IRI (Fig. 2). However, there are two stages in
the release process of IL-1β: The synthesis of pro-IL-1β and the
maturation of IL-1β. Regarding the priming process, Toll-like
receptor 4 (TLR4) binds to the ligands (such as heat shock
proteins, fibronectin, fibrinogen, high mobility group box 1,
hyaluronan and heparin sulphate) to produce signal transduction
(59) and then through NF-κB
activation, induces pro-IL-1β synthesis. Concerning the maturation
of IL-1β, it is reported that receptor family pyrin domain
containing 3 (NLRP3) inflammasomes play an important role (60–62).
Firstly, NLRP3 inflammasomes, containing adaptor molecule
apoptosis-associated speck-like protein, containing a caspase
recruitment domain, cysteine protease caspase-1 and NLRP3,
contribute to the activation of caspase-1. Secondly, pro-IL-1β is
processed into its mature form by caspase-1 as an IL-1β converting
enzyme, and caspase-1 induces the release of IL-1β. Finally, IL-1β
induces expression of IL-6, TNF-α, Ccl2, Cxcl1 and Cxcl2, leading
to tissue injury in the ischemia-reperfusion liver (63).
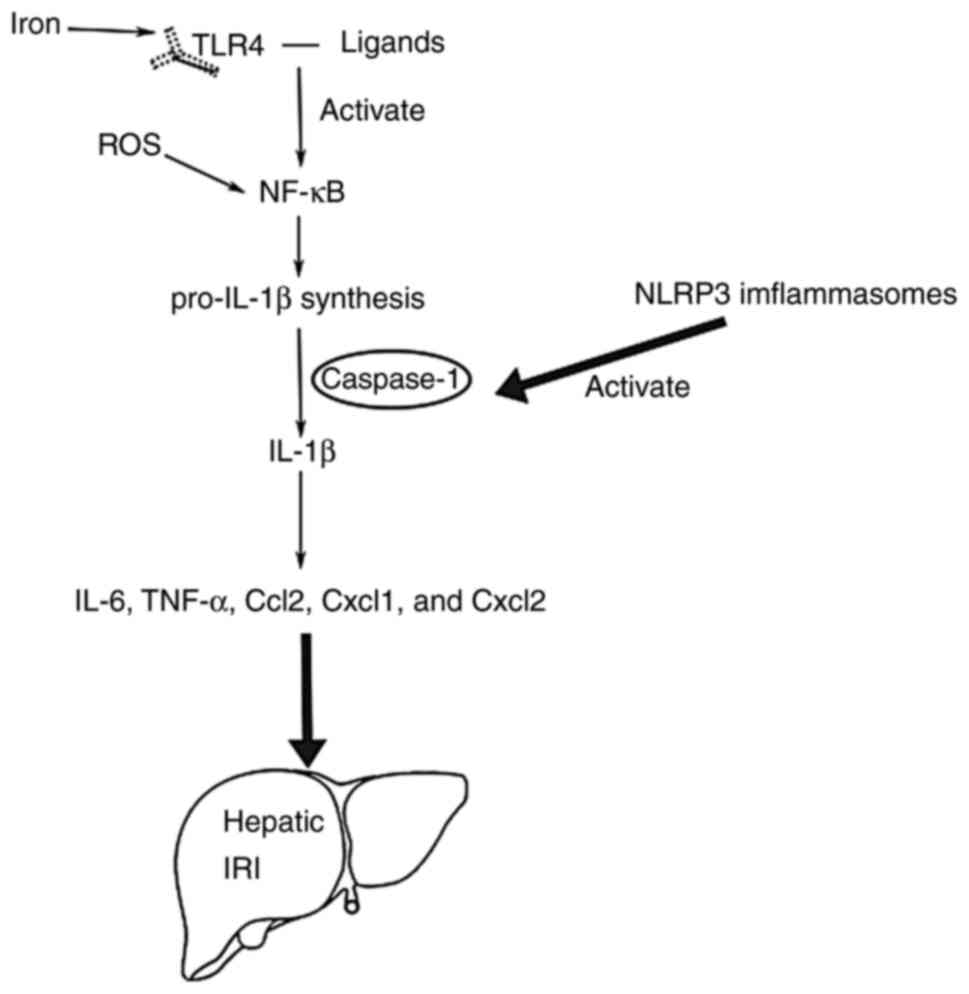 | Figure 2.The role of IL-1β in hepatic IRI. Two
stages in the release process of IL-1β: i) The synthesis of
pro-IL-1β; and ii) the maturation of IL-1β. Finally, IL-1β induces
expression of IL-6, TNF-α, Ccl2, Cxcl1, and Cxcl2, leading to
tissue injury in the ischemia-reperfusion liver. IRI,
ischemia-reperfusion injury; IL, interleukin; TNF-α, tumor necrosis
factor-α; Ccl2, chemokine (C-C motif) ligand 2; Cxcl1, chemokine
(C-X-C motif) ligand 1; Cxcl2, chemokine (C-X-C motif) ligand 2;
TLR4, Toll-like receptor 4; NF-κB, transcription factors nuclear
factor κB; NLRP3, pyrin domain containing 3. |
By performing real-time RT-PCR analysis, a previous
investigation evaluated the expression of inflammatory cytokines
and cell markers to confirm the association between iron overload
and inflammatory response in the liver (8). The results showed that inflammatory
cytokines and cell markers were significantly inhibited by Fer-1.
Furthermore, the infiltration of neutrophils and macrophages was
also apparently inhibited (8). This
implies that ferroptosis in liver cell death might be closely
associated with the inflammatory reaction in hepatic IRI. Further
evidence demonstrated that iron is central to many aspects of the
innate immune response, including ROS generation and host
inflammatory regulation (1). Iron
overload causes metabolic disturbance, leading to an increased
susceptibility to infection and triggering the inflammatory
response as the oversaturation of host transferrin leads to
defective nutritional immunity (64). In a healthy individual, in
vivo iron is a stable condition, and excess iron accumulation
can lead to the production of ROS. Regarding the role of iron in
inflammation in hepatic IRI, several studies have demonstrated that
the TLR4-activated inflammatory response is modulated by iron, as
well as increasing oxidative stress through the generation of
reactive oxygen and nitrogen species (65,66).
As previously stated, induction of pro-IL-1β synthesis is required
for NF-κB activation, while ROS can activate the transcription
factor NF-κB (67). Furthermore,
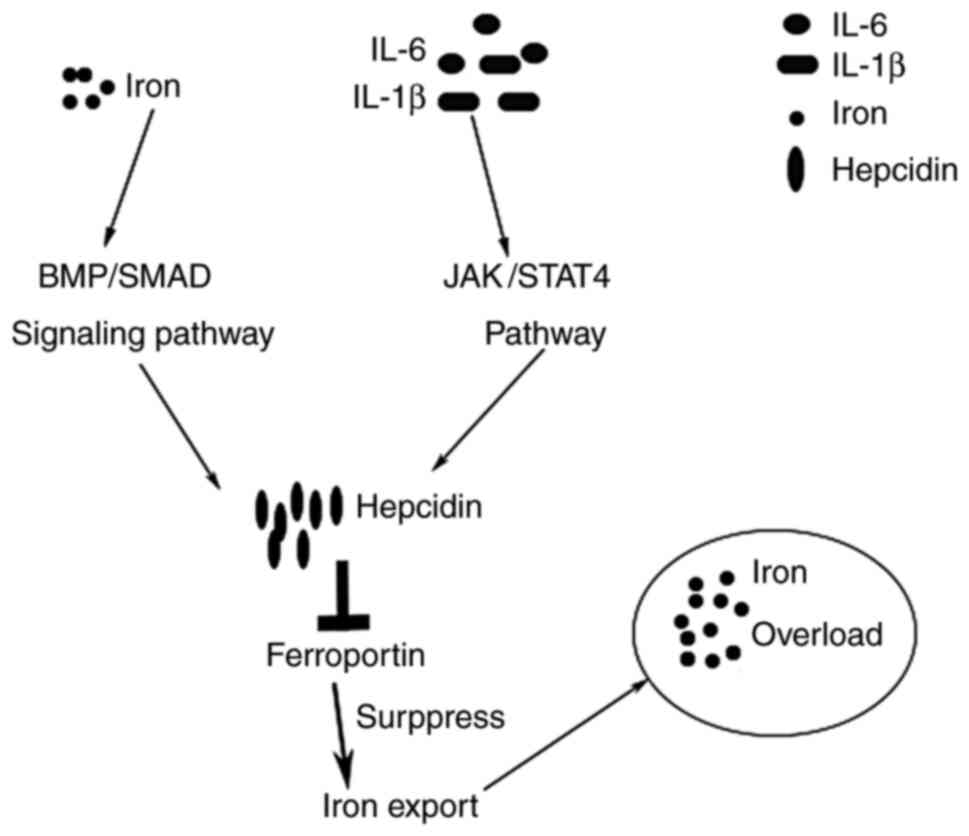
systemic iron homeostasis is regulated in the liver, and the
hepatic hormone hepcidin is the central regulator (68). There are two pathways to increase
the expression of hepcidin at the transcriptional level, including
inflammatory cytokines, such as IL-1β and IL-6 via the JAK/STAT3
pathway, and iron via the BMP/Smad signalling pathway (Fig. 3) (69). Increased hepcidin downregulates the
level of ferroportin, the sole known iron exporter on the cell
surface of hepatocytes, so that intracellular iron levels increase
due to suppression of iron export (69). Hence, it is conjectured that the
inflammatory response triggered in hepatic IRI induces iron
overload in hepatocytes. In other words, ferroptosis greatly
contributes to the pathogenesis of hepatic IRI.
Role of iron in oxidative damage in
hepatic IRI
In order to continue exploring the role of iron in
hepatic IRI, oxidative damage in liver cell injury is described in
detail in the present review. In the mechanism of ferroptosis
occurrence, iron-dependent accumulation of lipid ROS can be
produced from GSH depletion and NADPH-dependent lipid peroxidation
(5,70). Low levels of ROS, including hydrogen
peroxide (H2O2), superoxide anions
(O2−) and hydroxyl radicals (-OH) (71–73),
play an indispensable role in various molecular biological
processes, such as intracellular messaging and molecular pathways
in cellular progression (cell growth, differentiation and death) or
immunity (74), the arrest of
growth, and defence against microorganisms and apoptosis (75,76).
In contrast, high or and/or inadequate removal of lipid ROS from
Kupffer cells is the cardinal factor in vascular and parenchymal
cell oxidative damage during reperfusion, which occurs by inducing
oxidant stress (77). According to
current studies, there are distinct factors participating oxidant
stress. First, complement and polymorphonuclear neutrophils (PMNs)
participate in the process of oxidant stress induced by Kupffer
cells (Fig. 1) (78). Kupffer cells release intracellular
proteins and ROS during hepatic ischemia, inducing the activation
of complement and leading to slight initial injury. Complement, on
the one hand, activates and further stimulates Kupffer cells to
produce ROS, and on the other hand, directly or indirectly causes
activation and generation of PMNs in the liver through aggravating
the initial injury induced by Kupffer cells. Moreover, the
mechanism of complement-induced activation of neutrophils has been
described in other studies, which demonstrated that complement
factors, such as C5a, can recruit neutrophils into sinusoids by
upregulating the Mac-1 receptor on circulating neutrophils
(79,80). However, the generation of PMNs in
the liver also causes hepatocyte injury, and this damage further
promotes the activation of complement and PMNs, stimulating the
Kupffer cells to produce reactive oxygen that contributes to the
oxidative damage in hepatic IRI (81). An investigation using cobra venom
factor (CVF) that effectively inhibits complement activation
through the classical and alternate pathway (82,83),
induced the depletion of complement and a novel soluble complement
receptor type 1, significantly attenuating the increase of plasma
alanine aminotransferase (ALT) activities (81). This experiment confirms that
complement exerts an indispensable role in Kupffer cell-induced
oxidative injury. Secondly, a current view suggested that Kupffer
cells could be potentially activated during the ischemic period,
resulting in the Kupffer cells generating superoxides, such as the
superoxide anion radical (O2−), when subjected to
reoxygenation (84). This process
is likely to be activated by nitric oxide (NOX), while the oxidase
also stimulates the activation of ferroptosis by inhibiting
glutathione biosynthesis or GPX4 (4). Moreover, NOX induces lipid
peroxidation, producing many complex products, such as epoxides,
hydroperoxides, and carbonyl compounds (85). Lipid peroxidation primarily targets
cellular membranes, then peroxide PUFAs of membrane phospholipids,
and finally causes structural and functional tissue damage due to
the disintegration of the cellular membrane (86). In hepatic IRI, this mechanism
exacerbates erythrocyte functions, impairing membrane integrity
(87) and significantly altering
erythrocyte deformability (86).
Diminished erythrocyte deformability not only attenuates oxygen
transport capacity of the erythrocytes but also affects the
survival of circulating erythrocytes (87,88).
Furthermore, another product of lipid peroxidation is
malondialdehyde, which can react with DNA and as a result is toxic
and mutagenic (88). Ultimately,
malondialdehyde is substantially generated in the liver and results
in the death of hepatic parenchyma cells. An antioxidant defence
system containing glutathione peroxidase (GPX), ascorbic acid
(vitamin C), superoxide dismutase (SOD), a-tocopherol (vitamin E),
catalase (CAT), and GSH also exists in the body to fight against
the generation of free radicals by eliminating superoxide anions
and hydrogen peroxides (89–92).
When the balance between oxidation and antioxidant systems is
disrupted, increased lipid peroxidation can induce oxidative stress
(89). As previously reported, iron
promotes ferroptosis by lipid peroxidation in hepatic IRI (6,8,36). To
affirm this mechanism, a mouse ischemic model was given iron
chelation with deferoxamine treatment, which decreased the liver
iron content and serum ferritin levels (8).
Other types of regulated cell death
In addition to ferroptosis, there are other types of
regulated cell death, such as apoptosis, necrosis and autophagy.
Apoptosis, a form of programmed cell death, leads to characteristic
cell changes including blebbing, cell shrinkage, nuclear
fragmentation, chromatin condensation, chromosomal DNA
fragmentation, and global mRNA decay, which finally leads to the
formation of apoptotic bodies and phagocytosis of the apoptotic
bodies by adjacent parenchymal cells, neoplastic cells or
macrophages (93). The pathways
that initiate apoptosis are categorized as intrinsic or extrinsic,
which are initiated by different types of stimuli, and finally
through pro-apoptotic proteins to activate caspase-9 and caspase-8,
respectively (94–96). Bcl-2 family members and cell death
receptor/ligand (FasL/FasR and TNF-α/TNFR1) are the main molecules
of the main apoptosis signal pathway (95,97,98).
In contrast to apoptosis, necrosis, a passive type of RCD is
initiated by external physical or chemical factors, and mainly
characterized by swelling of cytoplasm and mitochondria, loss of
plasma membrane integrity, resulting in the release of
pro-inflammatory factors and the inflammation in the surrounding
tissue (99). Similar to the
extrinsic signaling pathway of apoptosis, necrosis also is
initiated by cell death receptor/ligand (FasL/FasR and
TNF-α/TNFR1), which forms a death-inducing signaling complex (DISC)
with procaspase-8 by recruiting Fas-associated death-domain and
receptor-interacting serine/threonine-protein kinase 1 (RIPK1)
(94). Differentially, apoptosis
originates from the activation of caspase-8 by the complex, while
necrosis is caused by deubiquitinated RIPK1 recruiting RIPK3
through the RIP homotypic interaction motif interaction and
phosphorylation of mixed-lineage kinase domain-like (MLKL) protein
when caspase-8 activity is inhibited. The oligomerization of
phosphorylated MLKL seems to bind to high-order inositol phosphate
(IP), which is then transferred to the plasma membrane to induce
cytolysis, resulting in the release of pro-inflammatory
damage-associated molecular proteins. Besides, it also activates
NLRP3, then leads to the secretion of interleukin (IL)-1β and IL-18
(100). It is common that the
inflammatory responses activated by ferroptosis and necrosis both
involve the participation of molecules such as IL-1β and TNF-α.
However, ferroptosis accounts more for lipid peroxidation caused by
iron overload, and the accumulation of ROS leads to the hepatocyte
mitochondrial membrane permeability.
As aforementioned, hepatic IRI is divided into two
processes: Ischemia and reperfusion. In the process of liver
ischemia, it mainly causes hypoxia and energy depletion, and the
reperfusion process causes oxidative stress and inflammatory
reaction. Both of these processes will lead to apoptosis and
necrosis, and finally result in autophagy. Autophagy is another
type of RCD regarding to a process that cytoplasmic substances are
transported to lysosomes, autophagy-related protein forms
autophagosomes, and finally the components contained are degraded.
More importantly, autophagy plays a critical role in regulating
liver metabolism, energy production and quality control checkpoints
as organelles such as mitochondria (101). However, a study shows that
autophagy is also associated with hepatocyte death (102). Autophagy can be divided into three
types: i) autophagy-associated cell death; ii) autophagy-mediated
cell death, and iii) autophagy-dependent cell death (103). For the first two types, autophagy
plays a minor role in the mechanism of cell death, thus should
depend on other types of cell death, such as apoptosis, necrosis
and ferroptosis. The third type of autophagy can independently
mediate the mechanism of cell death, so in the process of
hepatocyte death, apoptosis, necrosis, autophagy and ferroptosis
can be either interdependent or independently mediated.
Interestingly, studies have shown that there is a link between the
activation of autophagy and the development of ferroptosis, a
process known as ‘ferritinophagy’, which is characterized by
autophagy degradation of ferritin. In this process, the nuclear
receptor coactivator 4, a selective cargo receptor for the turnover
of ferritin, enables the maintenance of iron homeostasis, then
results in iron overload and promotes the development of
ferroptosis through the degradation of ferritin (104,105).
Current therapeutic strategies in hepatic
IRI
Currently, some potential therapeutic strategies
have been reported for ferroptosis regulation in liver
ischemia-reperfusion, primarily including antioxidants and
iron-removing molecules (such as desferoxamine, ferrostatin-1,
liproxstatin-1, α-tocopherol, ascorbic acid, GSH, alpha lipoic
acid, gadolinium chloride, zileuton and gadolinium chloride).
Desferoxamine
As aforementioned, iron overload promotes lipid
peroxidation and is involved in the inflammatory response of
hepatic IRI, leading to ferroptosis in liver cells. Desferoxamine
is an iron chelator that can decrease the levels of intracellular
iron. Experimental studies on hepatic ischemia models using
desferoxamine pretreatment have shown beneficial effects, such as
decreasing the liver iron content, decreasing serum ferritin
levels, and restoring total GSH levels, in response to warm or cold
hepatic ischemia (8,106,107).
Ferrostatin-1
Ferrostatin-1 is a first-generation ferrostatin that
inhibits ferroptosis by interfering with ROS accumulation from
lipid peroxidation (7,8). Mechanistically, to fight against
ferroptosis, a previous study (108) demonstrated that anti-ferroptotic
activity of fer-1 primarily depends on the scavenging of initiating
alkoxyl radicals produced by ferrous iron from lipid
hydroperoxides, and moreover, when fer-1 attenuates lipid
peroxidation, its levels are not significantly consumed. The
mechanism underlying this effect is not currently understood, and
more molecular studies are needed to explain it. In addition to
ferrostatin-1, there exits second- and third-generation
ferrostatins that are more stable, exhibiting increased metabolic
stability in the plasma. All of the third-generation ferrostatins
are significantly protective against tissue injury, including acute
kidney injury and IRI, in vivo (6).
Liproxstatin-1
Liproxstatin-1 is a potent ferroptosis inhibitor in
Gpx4−/− cells that acts by preventing ROS accumulation.
Liproxstatin-1 also inhibits ferroptosis in a mouse model of liver
tissue injury induced by ischemia-reperfusion (57). A previous study reported that
liproxstatin-1 decreases voltage-dependent anion channel 1 levels
and restores GPX4 levels to protect against ischemia-reperfusion
(109). Furthermore,
post-treatment with liproxstatin-1 protects mitochondrial
structural integrity (109).
Although liproxstatin-1 decreases ROS levels, it does not affect
Ca2+-induced mitochondrial permeability transition pore
opening. Moreover, compared to fer-1, liproxstatin-1 has relatively
stronger potency. Liproxstatin-1 also suppresses
ferroptosis-inducing agents (FINs), comprising RSL3, erastin, and
BODIPY 581/591 C11 oxidation (57).
Hence, liproxstatin-1 may represent an extremely promising
therapeutic drug in hepatic IRI.
α-Tocopherol
α-Tocopherol is a type of membrane and extracellular
antioxidant, also called vitamin E. It helps to prevent free
radicals from damaging hepatic cells and serves as an inhibitor of
protein kinase C and lipid peroxidation that increases GSH levels
(110,111). The efficacy of protecting liver
cells during ischemia-reperfusion was shown in an animal
experiment, which indicated that the group treated with
α-tocopherol exhibited a significantly higher survival rate
(110). Another study showed that
pretreatment with high doses of α-tocopherol (30 and 300 mg/kg of
body weight administered intramuscularly) enhanced ATP levels,
attenuated lipid peroxidation, and prevented the loss of hepatic
glutathione (111–113). Furthermore, α-tocopherol has shown
beneficial effects in both cold and warm IRI, decreases
mitochondrial damage induced by oxidative stress (113,114). Low doses of α-tocopherol can also
protect against liver cell death if combined with gadolinium
chloride (GdCl3) or ischemic preconditioning (IPC)
(110,115).
Ascorbic acid
Ascorbic acid, also known as vitamin C, is a vital
antioxidant with strong inhibition of lipid peroxidation and ROS
scavenging ability (116).
Ascorbic acid conveys an electron(s) to ROS, providing
site-specific protection against oxidative stress (117). The clotting factors can be used to
access acute liver cell damage, and after treatment with ascorbic
acid, the activity of clotting factors I, II, V, VII, and X showed
significant improvement (116).
Furthermore, ascorbic acid avoids the oxidative degradation of
vitamin E (a type of antioxidant) by reacting directly with
intermediates of tocopherol oxidation, as well as free radicals
(118). Another study treated rats
with ascorbic acid (100 mg/kg, i.v.) 5 min before sustained
ischemia, and IPC and ascorbic acid synergistically attenuated
mitochondrial damage during reperfusion due to decreased oxidant
stress (119). During the process
of ferroptosis in hepatic IRI, iron reacts with hydrogen peroxide
to form hydroxyl-like radicals, hydroxyl and ferric ions, and these
products are reduced by ascorbic acid, which inhibits
iron-dependent Fenton reactions (120). In accordance with another study,
it was clearly demonstrated that serum aminotransferase levels,
lipid peroxidation, the loss of bile flow and cholate output were
inhibited by ascorbic acid doses of 30 and 100 mg/kg but were
promoted by a dose of 1,000 mg/kg (120). Therefore, low doses of ascorbic
acid (30 and 100 mg/kg) have antioxidant effects, while high doses
(1,000 mg/kg) have pro-oxidant effects; thus, the dose should be
adjusted when ascorbic acid is applied. Moreover, the therapeutic
window might be appropriate over a short time prior to or just at
the beginning of reperfusion (121).
GSH
GSH is a thiol-containing compound, oxidizing
sulfhydryl group of cysteine that exerts antioxidant effects
(122). It is a substrate of GPX4,
and GSH depletion results in inactivation of GPX4, contributing to
ferroptosis by accumulation of ROS from lipid peroxidation
(70). Therefore, pretreatment with
GSH directly scavenges ROS (123).
The GSH/GSSG redox system majorly regulates intracellular redox
status (112) and giving GSH in
advance may promote intracellular reduction response to prevent
oxidative damage. However, there exit some administrative
limitations. For instance, whether intracellular GSH is simply
available and its ability to decrease GSSG and to what extent are
not completely understood (124).
A previous analysis demonstrated significant protection for
hepatocytes in both warm and cold liver ischemia by intravenous
glutathione administration (doses over 100 mol/h/kg) (125). It is assumed that GSH will become
an additional therapeutic approach for ferroptosis during hepatic
IRI.
α lipoic acid (ALA)
ALA is a natural compound that occurs in
vivo. ALA is an antioxidant that provides protection against
damage to the body's cells. Because of its antioxidant and
oxidant-scavenging properties, ALA may protect the liver against
oxidative injury (126), and this
function has been corroborated in rat liver that underwent 90 min
of warm ischemia (127). ALA was
shown to significantly decrease levels of AST, increase ATP
content, and lower apoptotic hepatocyte injury by improving
expression of anti-apoptotic proteins to decrease hepatic injury
(128). Moreover, ALA also
protects against IRI caused by cirrhosis or steatosis due to
improving cholinesterase activity in the serum (128). Furthermore, another study reported
additional findings for ALA in the treatment of hepatic IRI,
including decreasing levels of TNF-α and IL-1β, reversing
myeloperoxidase activity (indicating increased neutrophil
infiltration to the tissue), and maintaining regular morphology of
the central vein and hepatocytes (126). As a powerful direct chain-breaking
antioxidant, ALA strengthens the antioxidant potency of both
ascorbate and vitamin E (129).
Currently, ALA is a potential strategy to protect against hepatic
IRI.
CVF
CVF has been affirmed to be beneficial for the
protection of hepatocytes during ischemia-reperfusion in clinical
settings (81,130). CVF is a stable complement
inhibitor from cobra venom that binds to factor B as a structural
and functional analogue of the complement component C3b, forming
the bimolecular complex CVF/Bb through the cleavage of factor D
(130). CVF/Bb is a C3/C5
convertase that simultaneously cleaves complement C3 and C5
(131–133). Thus, continuous activation of C3
and C5 leads to the depletion of complement components and inhibits
their activation. In a final analysis, through subsequently
suppressing the release of inflammatory mediators, such as TNF-α
and IL-1β, oxidant stress induced by Kupffer cells and hepatic cell
apoptosis was decreased to attenuate hepatic injury (78,130).
However, the window of time available for therapeutic intervention
to block complement-mediated inflammatory responses and oxidant
stress in hepatic cells should be reviewed due to the recovery of
complement activity and regeneration in hepatocytes (78,130).
As an anticomplement protein, CVF may represent a novel therapy to
improve multiple organ injury induced by ischemia-reperfusion.
Zileuton
Zileuton inhibits the biosynthesis of leukotrienes
(LTB4, LTC4, LTD4 and LTE4) because it is an active inhibitor of
5-lipoxygenase. Zileuton decreases glutamate-induced ROS
accumulation, significantly inhibiting glutamate- and
erastin-induced ferroptosis (134).
Gadolinium chloride
GdCl3, a rare earth metal, is a
protective intervention in a rat hepatic reperfusion injury model
that inhibits Kupffer cell activation (115,135). It has been shown that pretreatment
with GdCl3 for hepatic IRI enhances the survival rate
(115), decreases neutrophil
infiltration and myeloperoxidase activity (136), and decreases platelet aggregation
in cold-perfusion liver (137).
Additional experiments have concluded that GdCl3
promotes recovery of hepatic function (135,138), prevents sinusoid endothelial cell
apoptosis (137), inhibits the
formation of free radicals, and attenuates lipid peroxidation
(139,140). However, the use of
GdCl3 may induce side effects (115), including significant loss of bile
flow, altered hepatocellular integrity (increased serum enzyme
activities), and inhibition of phagocytic activity in Kupffer
cells. In addition, inhibition of Kupffer cells damages host
defences (141,142) because the ability to clear
bacterial lipopolysaccharides from the blood is deranged. Hence,
the dose of GdCl3 should be kept as low as possible due
to potential adverse effects, and it is necessary to monitor
hepatic function when using GdCl3 for treatment
(115).
Conclusions and future perspectives
Hepatic IRI is a complex process that involves
various pathways and is complicated by a range of factors.
Therefore, it is important to understand the pathophysiological
pathways involved in liver damage during ischemia-reperfusion. This
review discussed the detailed mechanism of ferroptosis, a novel and
determinant type of regulated cell death, and concluded that it
involves differential activation of various signal transduction
pathways. Iron primarily participates in the relevant inflammatory
response, stimulating the release of inflammatory and cytokines,
and induces iron-dependent lipid peroxidation that generates
oxidative damage to hepatic cells. Currently, ferroptosis still
comprises some challenges, including the lack of a specific marker
for animal studies and clinical settings (7). Additional studies about ferroptosis in
hepatic IRI are required to better understand the presence of
ferroptosis in the liver. This novel type of cell death in hepatic
IRI will provide more precise therapeutic targets and will be
advantageous for developing new clinical therapeutic methods.
Acknowledgements
Not applicable.
Funding
The present work was supported by the National
Nature Science Foundation of China (grant no. 82060450), and the
Nature Science Foundation of Jiangxi province of China (grant nos.
20181BAB205050, 20192BAB205072 and 20171BCB23086).
Availability of data and materials
Not applicable.
Authors' contributions
LL, GM and DH conceived and designed the study; LL
prepared the original draft of the manuscript; LL and GM reviewed
and edited the manuscript; and DH supervised the study and acquired
the funding. All authors read and approved the final
manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Glossary
Abbreviations
Abbreviations:
|
AA
|
arachidonic acid
|
|
ACSL4
|
acyl-CoA synthetase long-chain family
4
|
|
AdA
|
adrenoyl
|
|
ALA
|
alpha-lipoic acid
|
|
ALT
|
alanine aminotransferase
|
|
ASRK1
|
apoptosis signal-regulating kinase
1
|
|
ATP
|
adenosine triphosphate
|
|
BMP
|
bone morphogenetic protein
|
|
CAT
|
catalase, Ccl2, chemokine (C-C motif)
ligand 2
|
|
Cxcl1
|
chemokine (C-X-C motif) ligand 1
|
|
Cxcl2
|
chemokine (C-X-C motif) ligand 2
|
|
CYP
|
cytochrome P450
|
|
CVF
|
cobra venom factor
|
|
DISC
|
death-inducing signalling complex
|
|
DMT1
|
divalent metal transporter 1
|
|
ERK
|
extracellular signal-regulated
kinase
|
|
Fas
|
Fas cell surface death receptor
|
|
Fer-1
|
ferrostatin-1
|
|
FN
|
fibronectin
|
|
FINs
|
ferroptosis-inducing agents
|
|
GdCl3
|
gadolinium chloride
|
|
GP
|
glycogen phosphorylase
|
|
GPX4
|
glutathione peroxidase 4
|
|
GSH
|
glutathione
|
|
GSSG
|
oxidized glutathione
|
|
HSPB1
|
shock protein family B member 1
|
|
IL
|
interleukin
|
|
INOS
|
inducible nitric oxide synthase
|
|
IP
|
inositol phosphate
|
|
IPC
|
ischemic preconditioning
|
|
IRI
|
ischemia-reperfusion injury
|
|
JAK
|
Janus kinase
|
|
JNK
|
c-Jun N-terminal kinases
|
|
LOOH
|
alkyl radical
|
|
LOXs
|
lipoxygenases
|
|
LPCAT3
|
lysophosphatidylcholine
acyltransferase 3
|
|
LT
|
leukotrienes
|
|
MAPK
|
mitogen-activated protein kinase
|
|
MEK
|
mitogen-activated protein kinase
|
|
MLKL
|
mixed-lineage kinase domain-like
|
|
NADPH
|
nicotinamide adenine dinucleotide
phosphate
|
|
NF-κB
|
transcription factors nuclear factor
κB
|
|
NLRP3
|
pyrin domain containing 3
|
|
NOX
|
nitric oxide
|
|
PE-OOH
|
phosphatidylethanolamine-OOH
|
|
PHKG2
|
phosphorylase kinase G2
|
|
PMNs
|
polymorphonuclear neutrophils
|
|
PUFAs
|
polyunsaturated fatty acids
|
|
RCD
|
regulated cell death
|
|
RIPK1
|
receptor-interacting
serine/threonine-protein kinase 1
|
|
ROS
|
reactive oxygen species
|
|
RSL3
|
Ras-selective lethal small
molecule
|
|
SOD
|
superoxide dismutase
|
|
STAT3
|
signal transducer and activator of
transcription 3
|
|
STEAP3
|
six-transmembrane epithelial antigen
of prostate 3
|
|
TfR
|
transferrin receptor
|
|
TLR4
|
Toll-like receptor 4
|
|
TNF-α
|
tumor necrosis factor-α
|
|
TRAF-2
|
TNF-receptor-associated factor 2
|
References
|
1
|
Dickson KB and Zhou J: Role of reactive
oxygen species and iron in host defence against infection. Front
Biosci (Landmark Ed). 25:1600–1616. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Kagan VE, Mao G, Qu F, Angeli JP, Doll S,
Croix CS, Dar HH, Liu B, Tyurin VA, Ritov VB, et al: Oxidized
arachidonic and adrenic PEs navigate cells to ferroptosis. Nat Chem
Biol. 13:81–90. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Valko M, Jomova K, Rhodes CJ, Kuča K and
Musílek K: Redox- and non-redox-metal-induced formation of free
radicals and their role in human disease. Arch Toxicol. 90:1–37.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Cao JY and Dixon SJ: Mechanisms of
ferroptosis. Cell Mol Life Sci. 73:2195–2209. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Dixon SJ, Lemberg KM, Lamprecht MR, Skouta
R, Zaitsev EM, Gleason CE, Patel DN, Bauer AJ, Cantley AM, Yang WS,
et al: Ferroptosis: An iron-dependent form of nonapoptotic cell
death. Cell. 149:1060–1072. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Xie Y, Hou W, Song X, Yu Y, Huang J, Sun
X, Kang R and Tang D: Ferroptosis: Process and function. Cell Death
Differ. 23:369–379. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Macías-Rodríguez RU, Inzaugarat ME,
Ruiz-Margáin A, Nelson LJ, Trautwein C and Cubero FJ: Reclassifying
hepatic cell death during liver damage: Ferroptosis-A novel form of
non-apoptotic cell death? Int J Mol Sci. 21:16512020. View Article : Google Scholar
|
|
8
|
Yamada N, Karasawa T, Wakiya T, Sadatomo
A, Ito H, Kamata R, Watanabe S, Komada T, Kimura H, Sanada Y, et
al: Iron overload as a risk factor for hepatic ischemia-reperfusion
injury in liver transplantation: Potential role of ferroptosis. Am
J Transplant. 20:1606–1618. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Ploeg RJ, D'Alessandro AM, Knechtle SJ,
Stegall MD, Pirsch JD, Hoffmann RM, Sasaki T, Sollinger HW, Belzer
FO and Kalayoglu M: Risk factors for primary dysfunction after
liver transplantation-a multivariate analysis. Transplantation.
55:807–813. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Zhai Y, Petrowsky H, Hong JC, Busuttil RW
and Kupiec-Weglinski JW: Ischaemia-reperfusion injury in liver
transplantation-from bench to bedside. Nat Rev Gastroenterol
Hepatol. 10:79–89. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Ikeda T, Yanaga K, Kishikawa K, Kakizoe S,
Shimada M and Sugimachi K: Ischemic injury in liver
transplantation: Difference in injury sites between warm and cold
ischemia in rats. Hepatology. 16:454–461. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Cannistra M, Ruggiero M, Zullo A, Gallelli
G, Serafini S, Maria M, Naso A, Grande R, Serra R and Nardo B:
Hepatic ischemia reperfusion injury: A systematic review of
literature and the role of current drugs and biomarkers. Int J
Surg. 33 (Suppl 1):S57–S70. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Zhai Y, Busuttil RW and Kupiec-Weglinski
JW: Liver ischemia and reperfusion injury: New insights into
mechanisms of innate-adaptive immune-mediated tissue inflammation.
Am J Transplant. 11:1563–1569. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Duarte S, Shen XD, Fondevila C, Busuttil
RW and Coito AJ: Fibronectin-α4β1 interactions in hepatic cold
ischemia and reperfusion injury: Regulation of MMP-9 and MT1-MMP
via the p38 MAPK pathway. Am J Transplant. 12:2689–2699. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Decuypere JP, Ceulemans LJ, Agostinis P,
Monbaliu D, Naesens M, Pirenne J and Jochmans I: Autophagy and the
Kidney: Implications for ischemia-reperfusion injury and therapy.
Am J Kidney Dis. 66:699–709. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Pu T, Liao XH, Sun H, Guo H, Jiang X, Peng
JB, Zhang L and Liu Q: Augmenter of liver regeneration regulates
autophagy in renal ischemia-reperfusion injury via the AMPK/mTOR
pathway. Apoptosis. 22:955–969. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Zhong X, Xiao Q, Liu Z, Wang W, Lai CH,
Yang W, Yue P, Ye Q and Xiao J: TAK242 suppresses the TLR4
signaling pathway and ameliorates DCD liver IRI in rats. Mol Med
Rep. 20:2101–2110. 2019.PubMed/NCBI
|
|
18
|
Oliveira THC, Marques PE, Proost P and
Teixeira MMM: Neutrophils: A cornerstone of liver ischemia and
reperfusion injury. Lab Invest. 98:51–62. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Wang L, Zhang Z, Li M, Wang F, Jia Y,
Zhang F, Shao J, Chen A and Zheng S: P53-dependent induction of
ferroptosis is required for artemether to alleviate carbon
tetrachloride-induced liver fibrosis and hepatic stellate cell
activation. Iubmb Life. 71:45–56. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Li L, Tan J, Miao Y, Lei P and Zhang Q:
ROS and autophagy: Interactions and molecular regulatory
mechanisms. Cell Mol Neurobiol. 35:615–621. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Datta G, Fuller BJ and Davidson BR:
Molecular mechanisms of liver ischemia reperfusion injury: Insights
from transgenic knockout models. World J Gastroenterol.
19:1683–1698. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Papadopoulos D, Siempis T, Theodorakou E
and Tsoulfas G: Hepatic ischemia and reperfusion injury and trauma:
Current concepts. Arch Trauma Res. 2:63–70. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Dhanasekaran DN and Reddy EP: JNK
signaling in apoptosis. Oncogene. 27:6245–6251. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Wanner GA, Ertel W, Muller P, Hofer Y,
Leiderer R, Menger MD and Messmer K: Liver ischemia and reperfusion
induces a systemic inflammatory response through Kupffer cell
activation. Shock. 5:34–40. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Lentsch AB, Yoshidome H, Kato A, Warner
RL, Cheadle WG, Ward PA and Edwards MJ: Requirement for
interleukin-12 in the pathogenesis of warm hepatic
ischemia/reperfusion injury in mice. Hepatology. 30:1448–1453.
1999. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Jaeschke H: Reactive oxygen and mechanisms
of inflammatory liver injury. J Gastroenterol Hepatol. 15:718–724.
2000. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Fan C, Zwacka RM and Engelhardt JF:
Therapeutic approaches for ischemia/reperfusion injury in the
liver. J Mol Med (Berl). 77:577–592. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Bauer M and Bauer I: Heme oxygenase-1:
Redox regulation and role in the hepatic response to oxidative
stress. Antioxid Redox Signal. 4:749–758. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Rensing H, Jaeschke H, Bauer I, Patau C,
Datene V, Pannen BH and Bauer M: Differential activation pattern of
redox-sensitive transcription factors and stress-inducible dilator
systems heme oxygenase-1 and inducible nitric oxide synthase in
hemorrhagic and endotoxic shock. Crit Care Med. 29:1962–1971. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Jaeschke H, Ho YS, Fisher MA, Lawson JA
and Farhood A: Glutathione peroxidase-deficient mice are more
susceptible to neutrophil-mediated hepatic parenchymal cell injury
during endotoxemia: Importance of an intracellular oxidant stress.
Hepatology. 29:443–450. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Essani NA, Fisher MA and Jaeschke H:
Inhibition of NF-kappa B activation by dimethyl sulfoxide
correlates with suppression of TNF-alpha formation, reduced ICAM-1
gene transcription, and protection against endotoxin-induced liver
injury. Shock. 7:90–96. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Li JM and Shah AM: Differential
NADPH-versus NADH-dependent superoxide production by phagocyte-type
endothelial cell NADPH oxidase. Cardiovasc Res. 52:477–486. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Ozaki M, Deshpande SS, Angkeow P, Bellan
J, Lowenstein CJ, Dinauer MC, Goldschmidt-Clermont PJ and Irani K:
Inhibition of the Rac1 GTPase protects against nonlethal
ischemia/reperfusion-induced necrosis and apoptosis in vivo. FASEB
J. 14:418–429. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Li Y, Qian L and Yuan J: Small molecule
probes for cellular death machines. Curr Opin Chem Biol. 39:74–82.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Degterev A and Linkermann A: Generation of
small molecules to interfere with regulated necrosis. Cell Mol Life
Sci. 73:2251–2267. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Dixon SJ and Stockwell BR: The role of
iron and reactive oxygen species in cell death. Nat Chem Biol.
10:9–17. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Pignatello JJ, Oliveros E and MacKay A:
Advanced oxidation processes for organic contaminant destruction
based on the Fenton reaction and related chemistry. J Crit Rev
Environmental Sci Technol. 36:1–84. 2006. View Article : Google Scholar
|
|
38
|
Wang H, An P, Xie E, Wu Q, Fang X, Gao H,
Zhang Z, Li Y, Wang X, Zhang J, et al: Characterization of
ferroptosis in murine models of hemochromatosis. Hepatology.
66:449–465. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Sun X, Ou Z, Xie M, Kang R, Fan Y, Niu X,
Wang H, Cao L and Tang D: HSPB1 as a novel regulator of ferroptotic
cancer cell death. Oncogene. 34:5617–5625. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Chen H, Zheng C, Zhang Y, Chang YZ, Qian
ZM and Shen X: Heat shock protein 27 downregulates the transferrin
receptor 1-mediated iron uptake. Int J Biochem Cell Biol.
38:1402–1416. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Arrigo AP, Virot S, Chaufour S, Firdaus W,
Kretz-Remy C and Diaz-Latoud C: Hsp27 consolidates intracellular
redox homeostasis by upholding glutathione in its reduced form and
by decreasing iron intracellular levels. Antioxid Redox Signal.
7:414–422. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Lei P, Bai T and Sun Y: Mechanisms of
ferroptosis and relations with regulated cell death: A review.
Front Physiol. 10:1392019. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Krieg P and Furstenberger G: The role of
lipoxygenases in epidermis. Biochim Biophys Acta. 1841:390–400.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Haeggstrom JZ and Funk CD: Lipoxygenase
and leukotriene pathways: Biochemistry, biology, and roles in
disease. Chem Rev. 111:5866–5898. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Choi J, Chon JK, Kim S and Shin W:
Conformational flexibility in mammalian 15S-lipoxygenase:
Reinterpretation of the crystallographic data. Proteins.
70:1023–1032. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Shintoku R, Takigawa Y, Yamada K, Kubota
C, Yoshimoto Y, Takeuchi T, Koshiishi I and Torii S:
Lipoxygenase-mediated generation of lipid peroxides enhances
ferroptosis induced by erastin and RSL3. Cancer Sci. 108:2187–2194.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Yang WS, Kim KJ, Gaschler MM, Patel M,
Shchepinov MS and Stockwell BR: Peroxidation of polyunsaturated
fatty acids by lipoxygenases drives ferroptosis. Proc Natl Acad Sci
USA. 113:E4966–E4975. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Reddy PH and Beal MF: Are mitochondria
critical in the pathogenesis of Alzheimer's disease? Brain Res
Brain Res Rev. 49:618–632. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Ademowo OS, Dias HKI, Burton DGA and
Griffiths HR: Lipid (per) oxidation in mitochondria: An emerging
target in the ageing process? Biogerontology. 18:859–879. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Wong-Ekkabut J, Xu Z, Triampo W, Tang IM,
Tieleman DP and Monticelli L: Effect of lipid peroxidation on the
properties of lipid bilayers: A molecular dynamics study. Biophys
J. 93:4225–4236. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Simoncini C, Orsucci D, Caldarazzo Ienco
E, Siciliano G, Bonuccelli U and Mancuso M: Alzheimer's
pathogenesis and its link to the mitochondrion. Oxid Med Cell
Longev. 2015:8039422015. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Eckl PM, Ortner A and Esterbauer H:
Genotoxic properties of 4-hydroxyalkenals and analogous aldehydes.
Mutat Res. 290:183–192. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Siems W and Grune T: Intracellular
metabolism of 4-hydroxynonenal. Mol Aspects Med. 24:167–175. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Stockwell BR, Friedmann AJ, Bayir H, Bush
AI, Conrad M, Dixon SJ, Fulda S, Gascon S, Hatzios SK, Kagan VE, et
al: Ferroptosis: A regulated cell death nexus linking metabolism,
redox biology, and disease. Cell. 171:273–285. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Tuo QZ, Lei P, Jackman KA, Li XL, Xiong H,
Li XL, Liuyang ZY, Roisman L, Zhang ST, Ayton S, et al:
Tau-mediated iron export prevents ferroptotic damage after ischemic
stroke. Mol Psychiatry. 22:1520–1530. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Skouta R, Dixon SJ, Wang J, Dunn DE, Orman
M, Shimada K, Rosenberg PA, Lo DC, Weinberg JM, Linkermann A and
Stockwell BR: Ferrostatins inhibit oxidative lipid damage and cell
death in diverse disease models. J Am Chem Soc. 136:4551–4556.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Friedmann AJ, Schneider M, Proneth B,
Tyurina YY, Tyurin VA, Hammond VJ, Herbach N, Aichler M, Walch A,
Eggenhofer E, et al: Inactivation of the ferroptosis regulator Gpx4
triggers acute renal failure in mice. Nat Cell Biol. 16:1180–1191.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Netea MG, van de Veerdonk FL, van der Meer
JW, Dinarello CA and Joosten LA: Inflammasome-independent
regulation of IL-1-family cytokines. Annu Rev Immunol. 33:49–77.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Kiziltas S: Toll-like receptors in
pathophysiology of liver diseases. World J Hepatol. 8:1354–1369.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Usui F, Shirasuna K, Kimura H, Tatsumi K,
Kawashima A, Karasawa T, Yoshimura K, Aoki H, Tsutsui H, Noda T, et
al: Inflammasome activation by mitochondrial oxidative stress in
macrophages leads to the development of angiotensin II-induced
aortic aneurysm. Arterioscler Thromb Vasc Biol. 35:127–136. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Usui F, Shirasuna K, Kimura H, Tatsumi K,
Kawashima A, Karasawa T, Hida S, Sagara J, Taniguchi S and
Takahashi M: Critical role of caspase-1 in vascular inflammation
and development of atherosclerosis in Western diet-fed
apolipoprotein E-deficient mice. Biochem Biophys Res Commun.
425:162–168. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Kawaguchi M, Takahashi M, Hata T, Kashima
Y, Usui F, Morimoto H, Izawa A, Takahashi Y, Masumoto J, Koyama J,
et al: Inflammasome activation of cardiac fibroblasts is essential
for myocardial ischemia/reperfusion injury. Circulation.
123:594–604. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Sadatomo A, Inoue Y, Ito H, Karasawa T,
Kimura H, Watanabe S, Mizushina Y, Nakamura J, Kamata R, Kasahara
T, et al: Interaction of neutrophils with macrophages promotes
IL-1β maturation and contributes to hepatic ischemia-reperfusion
injury. J Immunol. 199:3306–3315. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Skaar EP: The battle for iron between
bacterial pathogens and their vertebrate hosts. PLoS Pathog.
6:e10009492010. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Wang L, Harrington L, Trebicka E, Shi HN,
Kagan JC, Hong CC, Lin HY, Babitt JL and Cherayil BJ: Selective
modulation of TLR4-activated inflammatory responses by altered iron
homeostasis in mice. J Clin Invest. 119:3322–3328. 2009.PubMed/NCBI
|
|
66
|
Weiss G, Werner-Felmayer G, Werner ER,
Grunewald K, Wachter H and Hentze MW: Iron regulates nitric oxide
synthase activity by controlling nuclear transcription. J Exp Med.
180:969–976. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Bubici C, Papa S, Dean K and Franzoso G:
Mutual cross-talk between reactive oxygen species and nuclear
factor-kappa B: Molecular basis and biological significance.
Oncogene. 25:6731–6748. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Ganz T and Nemeth E: Hepcidin and iron
homeostasis. Biochim Biophys Acta. 1823:1434–1443. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Wunderer F, Traeger L, Sigurslid HH,
Meybohm P, Bloch DB and Malhotra R: The role of hepcidin and iron
homeostasis in atherosclerosis. Pharmacol Res. 153:1046642020.
View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Yang WS, SriRamaratnam R, Welsch ME,
Shimada K, Skouta R, Viswanathan VS, Cheah JH, Clemons PA, Shamji
AF, Clish CB, et al: Regulation of ferroptotic cancer cell death by
GPX4. Cell. 156:317–331. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Jornot L, Petersen H and Junod AF:
Hydrogen peroxide-induced DNA damage is independent of nuclear
calcium but dependent on redox-active ions. Biochem J. 335:85–94.
1998. View Article : Google Scholar : PubMed/NCBI
|
|
72
|
Mills EM, Takeda K, Yu ZX, Ferrans V,
Katagiri Y, Jiang H, Lavigne MC, Leto TL and Guroff G: Nerve growth
factor treatment prevents the increase in superoxide produced by
epidermal growth factor in PC12 cells. J Biol Chem.
273:22165–22168. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
73
|
Hurst R, Bao Y, Jemth P, Mannervik B and
Williamson G: Phospholipid hydroperoxide glutathione peroxidase
activity of rat class theta glutathione transferase T2-2. Biochem
Soc Trans. 25:S5591997. View Article : Google Scholar : PubMed/NCBI
|
|
74
|
Yin GY, Yin YF and He XF: Effect of
Zhuchun pill on immunity and endocrine function of elderly with
kidney-yang deficiency. Zhongguo Zhong Xi Yi Jie He Za Zhi.
15:601–603. 1995.(In Chinese). PubMed/NCBI
|
|
75
|
Lee YJ, Galoforo SS, Berns CM, Chen JC,
Davis BH, Sim JE, Corry PM and Spitz DR: Glucose
deprivation-induced cytotoxicity and alterations in
mitogen-activated protein kinase activation are mediated by
oxidative stress in multidrug-resistant human breast carcinoma
cells. J Biol Chem. 273:5294–5299. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
76
|
Bae YS, Kang SW, Seo MS, Baines IC, Tekle
E, Chock PB and Rhee SG: Epidermal growth factor (EGF)-induced
generation of hydrogen peroxide. Role in EGF receptor-mediated
tyrosine phosphorylation. J Biol Chem. 272:217–221. 1997.
View Article : Google Scholar : PubMed/NCBI
|
|
77
|
Jaeschke H and Farhood A: Neutrophil and
Kupffer cell-induced oxidant stress and ischemia-reperfusion injury
in rat liver. Am J Physiol. 260:G355–G362. 1991.PubMed/NCBI
|
|
78
|
Straatsburg IH, Boermeester MA, Wolbink
GJ, van Gulik TM, Gouma DJ, Frederiks WM and Hack CE: Complement
activation induced by ischemia-reperfusion in humans: A study in
patients undergoing partial hepatectomy. J Hepatol. 32:783–791.
2000. View Article : Google Scholar : PubMed/NCBI
|
|
79
|
Bajt ML, Farhood A and Jaeschke H: Effects
of CXC chemokines on neutrophil activation and sequestration in
hepatic vasculature. Am J Physiol Gastrointest Liver Physiol.
281:G1188–G1195. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
80
|
Witthaut R, Farhood A, Smith CW and
Jaeschke H: Complement and tumor necrosis factor-alpha contribute
to Mac-1 (CD11b/CD18) up-regulation and systemic neutrophil
activation during endotoxemia in vivo. J Leukoc Biol. 55:105–111.
1994. View Article : Google Scholar : PubMed/NCBI
|
|
81
|
Jaeschke H, Farhood A, Bautista AP,
Spolarics Z and Spitzer JJ: Complement activates Kupffer cells and
neutrophils during reperfusion after hepatic ischemia. Am J
Physiol. 264:G801–G809. 1993.PubMed/NCBI
|
|
82
|
Yeh CG, Marsh HJ Jr, Carson GR, Berman L,
Concino MF, Scesney SM, Kuestner RE, Skibbens R, Donahue KA and Ip
SH: Recombinant soluble human complement receptor type 1 inhibits
inflammation in the reversed passive arthus reaction in rats. J
Immunol. 146:250–256. 1991.PubMed/NCBI
|
|
83
|
Weisman HF, Bartow T, Leppo MK, Marsh HJ,
Carson GR, Concino MF, Boyle MP, Roux KH, Weisfeldt ML and Fearon
DT: Soluble human complement receptor type 1: In vivo inhibitor of
complement suppressing post-ischemic myocardial inflammation and
necrosis. Science. 249:146–151. 1990. View Article : Google Scholar : PubMed/NCBI
|
|
84
|
Rymsa B, Wang JF and de Groot H:
O2−. Release by activated Kupffer cells upon
hypoxia-reoxygenation. Am J Physiol. 261:G602–G607. 1991.PubMed/NCBI
|
|
85
|
Marnett LJ: Lipid peroxidation-DNA damage
by malondialdehyde. Mutat Res. 424:83–95. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
86
|
Arslan M, Metin CF, Kucuk A, Ozturk L and
Yaylak F: Dexmedetomidine protects against lipid peroxidation and
erythrocyte deformability alterations in experimental hepatic
ischemia reperfusion injury. Libyan J Med. 7:2012.doi:
10.3402/ljm.v7i0.18185. View Article : Google Scholar : PubMed/NCBI
|
|
87
|
Kuypers FA: Red cell membrane damage. J
Heart Valve Dis. 7:387–395. 1998.PubMed/NCBI
|
|
88
|
Sivilotti ML: Oxidant stress and
haemolysis of the human erythrocyte. Toxicol Rev. 23:169–188. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
89
|
Therond P, Bonnefont-Rousselot D,
Davit-Spraul A, Conti M and Legrand A: Biomarkers of oxidative
stress: An analytical approach. Curr Opin Clin Nutr Metab Care.
3:373–384. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
90
|
Stahl W, Junghans A, de Boer B, Driomina
ES, Briviba K and Sies H: Carotenoid mixtures protect multilamellar
liposomes against oxidative damage: Synergistic effects of lycopene
and lutein. FEBS Lett. 427:305–308. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
91
|
Beaudeux JL, Gardes-Albert M, Delattre J,
Legrand A, Rousselet F and Peynet J: Resistance of lipoprotein(a)
to lipid peroxidation induced by oxygenated free radicals produced
by gamma radiolysis: A comparison with low-density lipoprotein.
Biochem J. 314:277–284. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
92
|
Jankowska R, Passowicz-Muszynska E, Banas
T, Marcinkowska A and Medrala W: The influence of vitamin A on
production of oxygen free radicals and activity of granulocyte
catalase in patients with chronic bronchitis. Pneumonol Alergol
Pol. 62:628–633. 1994.(In Polish). PubMed/NCBI
|
|
93
|
Malhi H, Guicciardi ME and Gores GJ:
Hepatocyte death: A clear and present danger. Physiol Rev.
90:1165–1194. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
94
|
Kischkel FC, Hellbardt S, Behrmann I,
Germer M, Pawlita M, Krammer PH and Peter ME:
Cytotoxicity-dependent APO-1 (Fas/CD95)-associated proteins form a
death-inducing signaling complex (DISC) with the receptor. EMBO.
14:5579–5588. 1995. View Article : Google Scholar
|
|
95
|
Stefan JR and Guy SS: The apoptosome:
Signalling platform of cell death. Nat Rev Mol Cell Bio. 8:405–413.
2007. View Article : Google Scholar
|
|
96
|
Yong-Ling PO, Douglas RG, Zhenyue H and
Tak WM: Cytochrome c: Functions beyond respiration. Nat Rev Mol
Cell Bio. 9:532–542. 2008. View Article : Google Scholar
|
|
97
|
Nicholas SW, Vishva D and Avi A: Death
receptor signal transducers: Nodes of coordination in immune
signaling networks. Nat Immunol. 10:348–355. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
98
|
Susan E: Apoptosis: A review of programmed
cell death. Toxicol Pathol. 35:495–516. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
99
|
Krysko DV, Vanden BT, D'Herde K and
Vandenabeele P: Apoptosis and necrosis: Detection, discrimination
and phagocytosis. Methods. 44:205–221. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
100
|
Ricardo W, Andrew O, Helen MB and Douglas
RG: Necroptosis in development, inflammation and disease. Nat Rev
Mol Cell Biol. 18:127–136. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
101
|
Ueno T and Komatsu M: Autophagy in the
liver: Functions in health and disease. Nat Rev Gastroenterol
Hepatol. 14:170–184. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
102
|
Pierre ER, Dominique CH, Richard M, Claire
F, Gérard F, Didier L, Éric OD, Pierre B, Dominique V and Fran OD:
Acute liver cell damage in patients with anorexia nervosa: A
possible role of starvation-induced hepatocyte autophagy.
Gastroenterology. 135:840–848.e1-e3. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
103
|
Donna D and Sharad K: Autophagy-dependent
cell death. Cell Death Differ. 26:605–616. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
104
|
Santana-Codina N and Mancias JD: The role
of NCOA4-mediated ferritinophagy in health and disease.
Pharmaceuticals (Basel). 11:1142018. View Article : Google Scholar
|
|
105
|
Hou W, Xie Y, Song X, Sun X, Lotze MT, Zeh
HJ, Kang R and Tang D: Autophagy promotes ferroptosis by
degradation of ferritin. Autophagy. 12:1425–1428. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
106
|
Veitch K, Maisin L and Hue L:
Trimetazidine effects on the damage to mitochondrial functions
caused by ischemia and reperfusion. Am J Cardiol. 76:B25–B30. 1995.
View Article : Google Scholar
|
|
107
|
Guarnieri C and Muscari C: Effect of
trimetazidine on mitochondrial function and oxidative damage during
reperfusion of ischemic hypertrophied rat myocardium. Pharmacology.
46:324–331. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
108
|
Miotto G, Rossetto M, Di Paolo ML, Orian
L, Venerando R, Roveri A, Vuckovic AM, Bosello TV, Zaccarin M,
Zennaro L, et al: Insight into the mechanism of ferroptosis
inhibition by ferrostatin-1. Redox Biol. 28:1013282020. View Article : Google Scholar : PubMed/NCBI
|
|
109
|
Feng Y, Madungwe NB, Imam AA, Tombo N and
Bopassa JC: Liproxstatin-1 protects the mouse myocardium against
ischemia/reperfusion injury by decreasing VDAC1 levels and
restoring GPX4 levels. Biochem Biophys Res Commun. 520:606–611.
2019. View Article : Google Scholar : PubMed/NCBI
|
|
110
|
Giakoustidis D, Papageorgiou G, Iliadis S,
Giakoustidis A, Kostopoulou E, Kontos N, Botsoglou E, Tsantilas D,
Papanikolaou V and Takoudas D: The protective effect of
alpha-tocopherol and GdCl3 against hepatic ischemia/reperfusion
injury. Surg Today. 36:450–456. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
111
|
Masaki H, Okano Y, Ochiai Y, Obayashi K,
Akamatsu H and Sakurai H: Alpha-tocopherol increases the
intracellular glutathione level in HaCaT keratinocytes. Free Radic
Res. 36:705–709. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
112
|
Lee WY and Lee SM: Protective effects of
alpha-tocopherol and ischemic preconditioning on hepatic
reperfusion injury. Arch Pharm Res. 28:1392–1399. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
113
|
Giakoustidis D, Papageorgiou G, Iliadis S,
Kontos N, Kostopoulou E, Papachrestou A, Tsantilas D, Spyridis C,
Takoudas D, Botsoglou N, et al: Intramuscular administration of
very high dose of alpha-tocopherol protects liver from severe
ischemia/reperfusion injury. World J Surg. 26:872–877. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
114
|
Gondolesi GE, Lausada N, Schinella G,
Semplici AM, Vidal MS, Luna GC, Toledo J, de Buschiazzo PM and
Raimondi JC: Reduction of ischemia-reperfusion injury in
parenchymal and nonparenchymal liver cells by donor treatment with
DL-alpha-tocopherol prior to organ harvest. Transplant Proc.
34:1086–1091. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
115
|
Ruttinger D, Vollmar B, Wanner GA and
Messmer K: In vivo assessment of hepatic alterations following
gadolinium chloride-induced Kupffer cell blockade. J Hepatol.
25:960–967. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
116
|
Cerwenka H, Khoschsorur G, Bacher H,
Werkgartner G, El-Shabrawi A, Quehenberger F, Rabl H and Mischinger
HJ: Normothermic liver ischemia and antioxidant treatment during
hepatic resections. Free Radic Res. 30:463–469. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
117
|
Kohen R and Nyska A: Oxidation of
biological systems: Oxidative stress phenomena, antioxidants, redox
reactions, and methods for their quantification. Toxicol Pathol.
30:620–650. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
118
|
Packer JE, Slater TF and Willson RL:
Direct observation of a free radical interaction between vitamin E
and vitamin C. Nature. 278:737–738. 1979. View Article : Google Scholar : PubMed/NCBI
|
|
119
|
Lee WY, Lee JS and Lee SM: Protective
effects of combined ischemic preconditioning and ascorbic acid on
mitochondrial injury in hepatic ischemia/reperfusion. J Surg Res.
142:45–52. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
120
|
Seo MY and Lee SM: Protective effect of
low dose of ascorbic acid on hepatobiliary function in hepatic
ischemia/reperfusion in rats. J Hepatol. 36:72–77. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
121
|
Rabl H, Khoschsorur G and Petek W:
Antioxidative vitamin treatment: Effect on lipid peroxidation and
limb swelling after revascularization operations. World J Surg.
19:738–744. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
122
|
Bilzer M and Lauterburg BH: Effects of
hypochlorous acid and chloramines on vascular resistance, cell
integrity, and biliary glutathione disulfide in the perfused rat
liver: Modulation by glutathione. J Hepatol. 13:84–89. 1991.
View Article : Google Scholar : PubMed/NCBI
|
|
123
|
Winterbourn CC and Metodiewa D: The
reaction of superoxide with reduced glutathione. Arch Biochem
Biophys. 314:284–290. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
124
|
Schauer RJ, Gerbes AL, Vonier D, Meissner
H, Michl P, Leiderer R, Schildberg FW, Messmer K and Bilzer M:
Glutathione protects the rat liver against reperfusion injury after
prolonged warm ischemia. Ann Surg. 239:220–231. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
125
|
Cotgreave IA: N-acetylcysteine:
Pharmacological considerations and experimental and clinical
applications. Adv Pharmacol. 38:205–227. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
126
|
Dulundu E, Ozel Y, Topaloglu U, Sehirli O,
Ercan F, Gedik N and Sener G: Alpha-lipoic acid protects against
hepatic ischemia-reperfusion injury in rats. Pharmacology.
79:163–170. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
127
|
Müller C, Dunschede F, Koch E, Vollmar AM
and Kiemer AK: Alpha-lipoic acid preconditioning reduces
ischemia-reperfusion injury of the rat liver via the PI3-kinase/Akt
pathway. Am J Physiol Gastrointest Liver Physiol. 285:G769–G778.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
128
|
Dunschede F, Erbes K, Kircher A,
Westermann S, Seifert J, Schad A, Oliver K, Kiemer AK and Theodor
J: Reduction of ischemia reperfusion injury after liver resection
and hepatic inflow occlusion by alpha-lipoic acid in humans. World
J Gastroenterol. 12:6812–6817. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
129
|
Kagan VE, Shvedova A, Serbinova E, Khan S,
Swanson C, Powell R and Packer L: Dihydrolipoic acid-a universal
antioxidant both in the membrane and in the aqueous phase.
Reduction of peroxyl, ascorbyl and chromanoxyl radicals. Biochem
Pharmacol. 44:1637–1649. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
130
|
Wang B, Xu H, Li J, Gao HM, Xing YH, Lin
Z, Li HJ, Wang YQ and Cao SH: Complement depletion with cobra venom
factor alleviates acute hepatic injury induced by
ischemiareperfusion. Mol Med Rep. 18:4523–4529. 2018.PubMed/NCBI
|
|
131
|
Mao YF, Yu QH, Zheng XF, Liu K, Liang WQ,
Wang YW, Deng XM and Jiang L: Pre-treatment with Cobra venom factor
alleviates acute lung injury induced by intestinal
ischemia-reperfusion in rats. Eur Rev Med Pharmacol Sci.
17:2207–2217. 2013.PubMed/NCBI
|
|
132
|
Vogel CW, Finnegan PW and Fritzinger DC:
Humanized cobra venom factor: Structure, activity, and therapeutic
efficacy in preclinical disease models. Mol Immunol. 61:191–203.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
133
|
Vogel CW and Fritzinger DC: Cobra venom
factor: Structure, function, and humanization for therapeutic
complement depletion. Toxicon. 56:1198–1222. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
134
|
Liu Y, Wang W, Li Y, Xiao Y, Cheng J and
Jia J: The 5-lipoxygenase inhibitor zileuton confers
neuroprotection against glutamate oxidative damage by inhibiting
ferroptosis. Biol Pharm Bull. 38:1234–1239. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
135
|
Kukan M, Vajdova K, Horecky J, Nagyova A,
Mehendale HM and Trnovec T: Effects of blockade of Kupffer cells by
gadolinium chloride on hepatobiliary function in cold
ischemia-reperfusion injury of rat liver. Hepatology. 26:1250–1257.
1997. View Article : Google Scholar : PubMed/NCBI
|
|
136
|
Cutrin JC, Boveris A, Zingaro B, Corvetti
G and Poli G: In situ determination by surface chemiluminescence of
temporal relationships between evolving warm ischemia-reperfusion
injury in rat liver and phagocyte activation and recruitment.
Hepatology. 31:622–632. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
137
|
Sindram D, Porte RJ, Hoffman MR, Bentley
RC and Clavien PA: Synergism between platelets and leukocytes in
inducing endothelial cell apoptosis in the cold ischemic rat liver:
A Kupffer cell-mediated injury. FASEB J. 15:1230–1232. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
138
|
Vajdova K, Smrekova R, Kukan M, Jakubovsky
J, van Rooijen N, Horecky J, Lutterova M and Wsolova L:
Endotoxin-induced aggravation of preservation-reperfusion injury of
rat liver and its modulation. J Hepatol. 32:112–120. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
139
|
Giakoustidis DE, Iliadis S, Tsantilas D,
Papageorgiou G, Kontos N, Kostopoulou E, Botsoglou NA, Gerasimidis
T and Dimitriadou A: Blockade of Kupffer cells by gadolinium
chloride reduces lipid peroxidation and protects liver from
ischemia/reperfusion injury. Hepatogastroenterology. 50:1587–1592.
2003.PubMed/NCBIPubMed/NCBI
|
|
140
|
Bremer C, Bradford BU, Hunt KJ, Knecht KT,
Connor HD, Mason RP and Thurman RG: Role of Kupffer cells in the
pathogenesis of hepatic reperfusion injury. Am J Physiol.
267:G630–G636. 1994.PubMed/NCBI
|
|
141
|
Dixon LJ, Barnes M, Tang H, Pritchard MT
and Nagy LE: Kupffer cells in the liver. Compr Physiol. 3:785–797.
2013.PubMed/NCBI
|
|
142
|
Callery MP, Kamei T and Flye MW: Kupffer
cell blockade increases mortality during intra-abdominal sepsis
despite improving systemic immunity. Arch Surg. 125:36–41. 1990.
View Article : Google Scholar : PubMed/NCBI
|