Introduction
Leukonychia is defined by a white discoloration of
the nail plate. The pathophysiology of the disease is not yet fully
understood; however, it can be classified into three categories
(true leukonychia, apparent leukonychia and pseudoleukonychia)
based on the site of origin (1).
True leukonychia can either be acquired or inherited. Acquired true
leukonychia is a result of alterations in the nail matrix due to
other medical conditions or external exposure (1). Hereditary leukonychia is extremely
rare and it may be considered as a benign isolated occurrence or
may be associated with a range of systemic diseases (2). Mutations in the phospholipase C δ 1
(PLCD1) gene have been identified as a major causative
factor in hereditary leukonychia (HL) (1). The PLCD1 gene maps to the short
arm of chromosome 3 (3p22.2), consists of 15 exons, and the gene
encodes two isoforms, 777 or 756 amino acids in length.
PLCD1 is the enzyme required for Ca2+ signal
transduction in a number of tissues, such as the foreskin,
keratinocytes, nail matrix and fibroblasts, which is significant in
the manifestation of HL (3).
The present study investigated a Chinese family with
HL, some of whom were diagnosed with koilonychia during their
childhood, as well as a sporadic patient who presented with HL.
Genome-wide linkage analysis revealed the linkage of the family to
chromosome 3 and Sanger sequencing of PLCD1 gene identified
one novel heterozygous missense mutation c.1384G>A (p.E462K).
Moreover, another novel mutation c.770G>A (p.R257H) in the
sporadic case of leukonychia was detected.
Case report
Clinical characteristics of the
patients
A family consisting of 6 affected and 10 unaffected
individuals, and 1 sporadic patient with white-colored nails from
birth were enrolled in the present study. Informed consent was
obtained from all study participants prior to their inclusion in
the study. The present study was approved by the Institutional
Ethical Committee of Shanghai Skin Disease Hospital and was
performed in accordance with the guidelines of the Declaration of
Helsinki (4). The family had no
consanguineous marriages and the findings were consistent with an
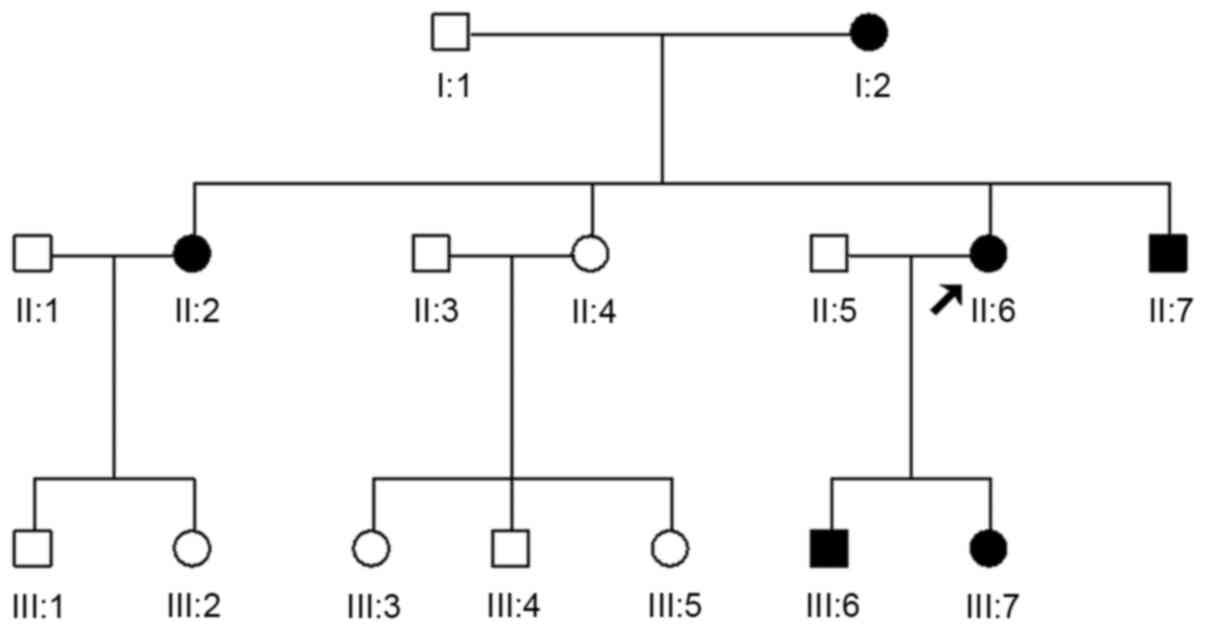
autosomal dominant mode of inheritance of the disease (Fig. 1). The proband was an 8-year-old boy
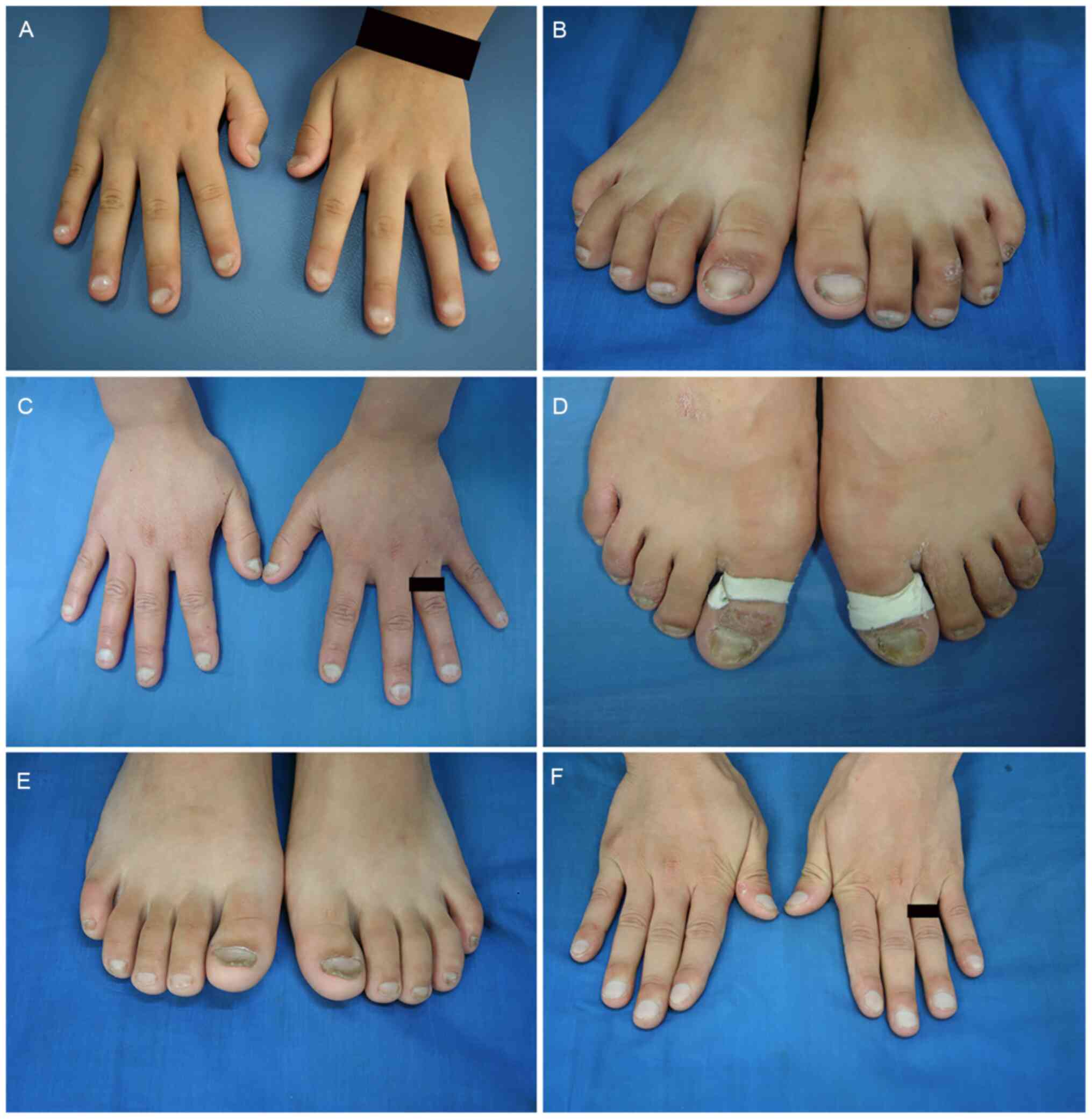
born with white-colored soft nails. The nails became flat or
spoon-shaped with the passing of time. Upon examination, his
fingernails were quite thin with a concave surface, and the distal
edges of some of the nail plates were rough and hyperpigmented,
particularly those on both thumbs. The toenails had minor lesions
along with varying degrees of browning at the distal edges
(Fig. 2A and B). A white
discoloration of the nail plates was present since birth. Upon
applying pressure over the nail plates, no fading of the whiteness
was observed. A skin, hair and systemic examination revealed no
abnormal findings. Furthermore, no evidence of any associated
conditions was found. Routine laboratory test results were normal.
A direct microscopy test of fungus from the fingernails and
toenails yielded negative results. Serum levels of iron, zinc and
Ca2+ were normal. Oral treatment with Theragran Junior
did not improve the condition. Upon examination of the other
members of the family, 6 patients with leukonychia, including 2
males and 4 females with similar spoon-shaped nails during their
childhood (out of 15 members of 3 generations) were identified
(Fig. 2C-F). The changes in the
nails of all patients were similar; however, certain differences
were observed. The similarities were that the spoon-shaped
appearances of the nails had improved to varying degrees, and the
brown color of the distal nails had a tendency of centripetal
growth with age. The differences lied in the appearance of the
individual patients' nails. The nails of one of the affected
members (II:7) only exhibited spoon-shaped changes without browning
during childhood, and they became normal during adulthood. However,
the browning of the proband's nails was more evident.
Linkage of hereditary leukonychia to
chromosome 3
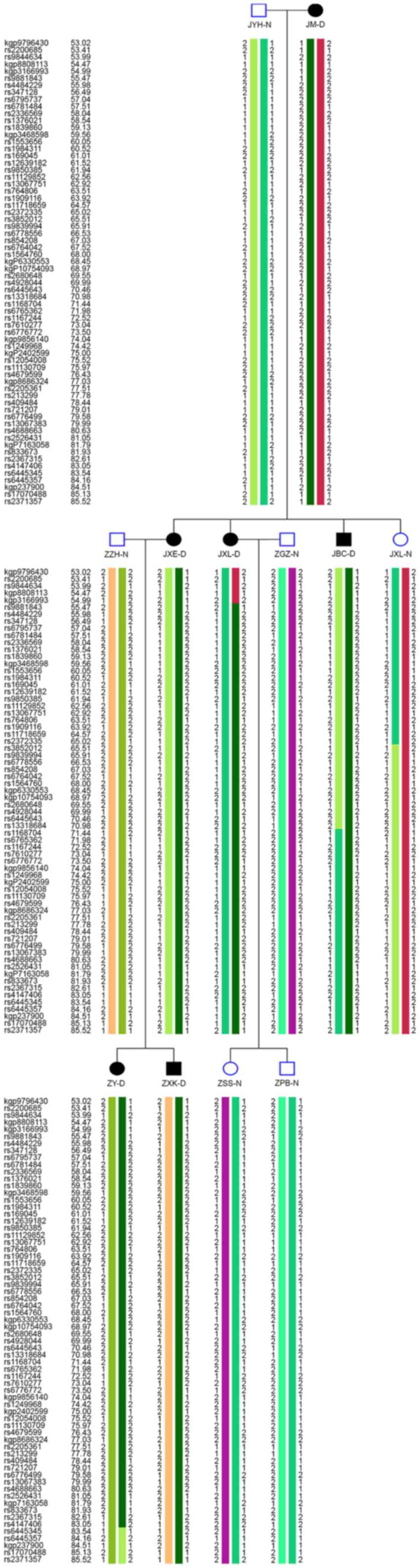
Owing to the typical features of koilonychia in the
patients in the present study, an Illumina Infinium HumanLinkage-24
panel (Illumina, Inc.) was used to genotype 12 individuals from the
family (I:1, I:2, II:1, II:2, II:4, II:5, II:6, II:7, III:1, III:2,
III:6 and III:7) (Fig. 3).
Genome-wide linkage analysis was performed with a total of 6,518
single nucleotide polymorphisms (SNP) markers; their average
genetic and physical distances were 442 kb and 0.55 cM. In the
linkage analysis, SNPs with a call rate <90%, monomorphic SNPs
and non-Mendelian transmitted markers were removed; a total number
of 45,716 informative autosomal SNPs remained. Multipoint
parametric linkage analyses were performed with the MERLIN program
version 1.1.2 (5). A fully
penetrant autosomal-dominant model was used with a rare disease
frequency of 0.0001. Critical recombination events of the pedigree
members were also determined through haplotype construction in
MERLIN.
Genome-wide linkage analysis in the family revealed
that the multipoint Log of Odds (LOD) scores of the third
chromosome were >2.0, indicating linkages to this chromosome. No
other locus suggestive of linkage was detected. Recombination
events were observed in individuals II:3 and III:1. These
recombination events defined that the physical distance of the
susceptibility region was a 28.56 cM interval (54.98–83.54 cM
between kgp3166993 and rs6445345). All affected individuals shared
a common haplotype across this disease interval.
Sequencing analysis
One known pathogenic gene for leukonychia was
located in the mapped region (6).
Primers were designed for all 15 coding exons of PLCD1
(RefSeq accession no. NM_006225). The QIAquick PCR Purification kit
(Qiagen, Inc.) was used to purify the PCR products and PLCD1
was sequenced with an ABI PRISM 3730 automated sequencer (Applied
Biosystems; Thermo Fisher Scientific, Inc.). Sequence comparisons
and analysis were performed using the Phred-Phrap-Consed program
version 12.0 (5). A total of 10
templates were sequenced per subject. The accession number of the
reference sequence used for the identification of mutations/SNPs is
NM_001130964.2. The software used for mutation/SNP analysis in the
present study was Chromas 233 (5).
A total of 100 unrelated population-matched control peripheral
blood samples were sequenced to exclude the possibility of
identification of the polymorphism in the PLCD1 gene. The
unrelated matched control samples used in the study were collected
from volunteers who had come to the hospital for physical
examination between January and March in 2013. The median age of
100 unrelated population was 37.5. The age range was 23–61. Among
them, 58 individuals were male and 42 individuals were female. All
data were cited from //asia.ensembl.org/index.html.
Identification of mutations in the
PLCD1 gene
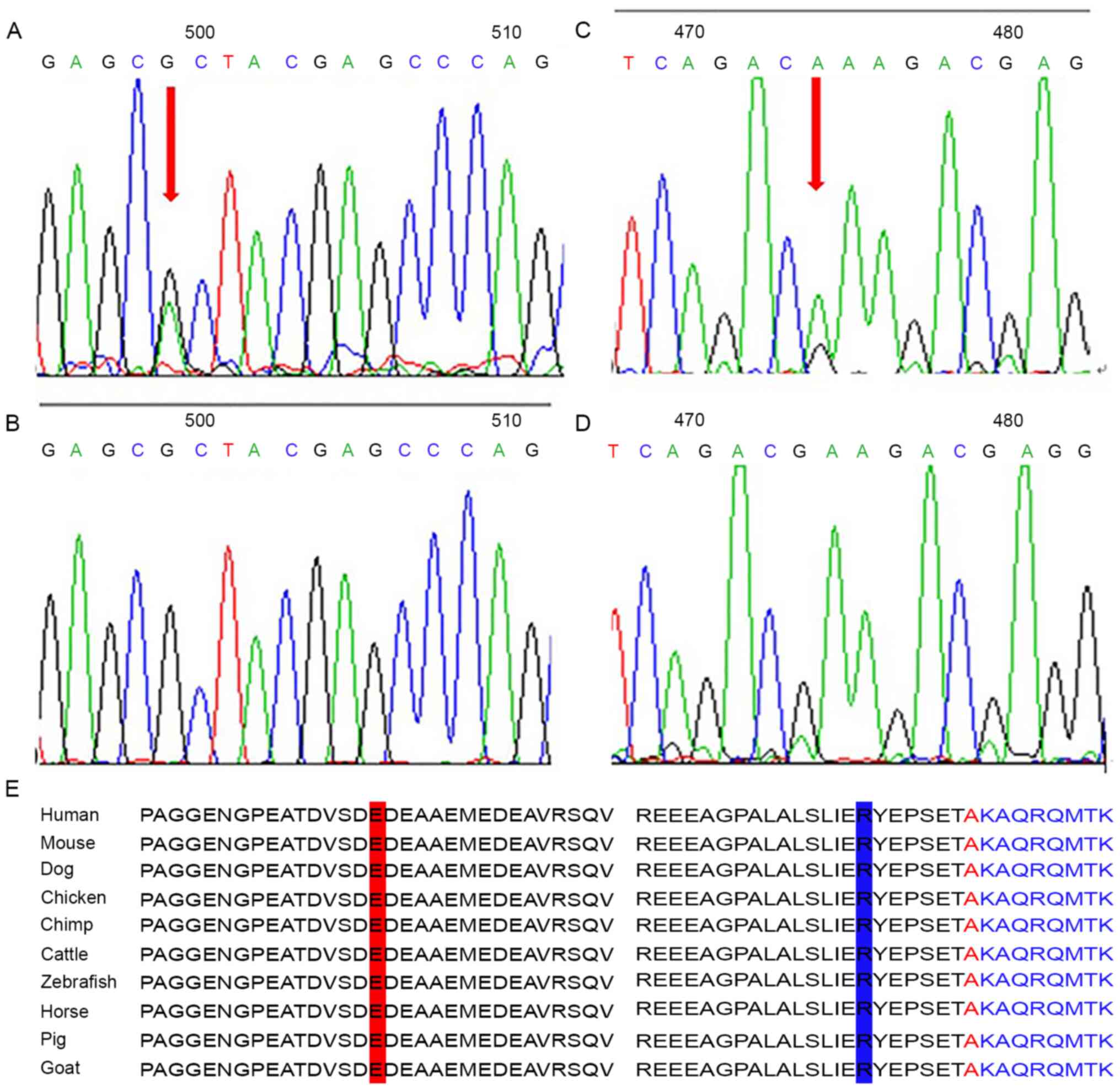
Direct sequencing of PLCD1 revealed a
heterozygous missense mutation involving a G to A transition at
nucleotide position 1384 (c.1384G>A) of the gene in the Chinese
pedigree patients (Fig. 4C and D).
This resulted in the substitution of a codon for glutamic acid at
amino acid position 462 to lysine (p.E462K). The other heterozygous
missense mutation detected in exon 5 in one sporadic case of
leukonychia was a G to A transition at nucleotide 770 (c.770
G>A) (Fig. 4A and B), which
resulted in the substitution of a codon for arginine at amino acid
position 257 to histidine (p.R257H). To exclude the possibility
that the two mutations do not represent non-pathogenic
polymorphisms, the Polyphen2 (polymorphism phenotyping version 2
(genetics.bwh.harvard.edu/pph2) tool
was used to assess the effects of these variations on the function
of the corresponding protein. It was found that the c.1384G>A
and c.770 G>A mutations in the PLCD1 gene were both
pathogenic. The Polyphen2 score for the c.1384G>A mutation was
0.984 (close to 1), while that for c.770 G>A was 0.885 (close to
1). PolyPhen scores were interpreted as follows: Benign, 0.00 to
0.20; possibly damaging, 0.20 to 0.85; and probably damaging, 0.85
to 1.00. In addition, a panel of 200 unaffected, unrelated and
ethnically matched control individuals were screened, and these
mutations were not identified. These findings indicate that both
mutations are not SNPs. The mutation p.E462K detected in the
Chinese pedigree patients and p.R257H detected in the sporadic case
of leukonychia have been shown to be highly conserved during
evolution amongst dog, zebrafish, horse, mouse, chicken, chimp,
cattle, pig and goat (Fig. 4E), and
may be critical for the function of the protein.
Discussion
HL is a rare nail pathology that causes partial or
total whitening of the nail plate. Histological analysis indicates
hyperkeratosis and parakeratosis from the nail bed to the nail
plate, and abnormal cornified cells in the nail plate (1,7). In
the present study, the clinical manifestations of the Chinese HL
family were not particularly typical, with some patients presenting
with unnoticeable leukonychia, but obvious koilonychia. Detailed
analysis revealed that all the affected members had koilonychia in
their childhood. This had misled the initial diagnosis as
hereditary koilonychia and genome-wide linkage analysis was used to
identify the pathogenic genes of koilonychia. Interestingly, it was
revealed that the multipoint LOD scores of the third chromosome
were >2.0, indicating linkages to this chromosome. One known
pathogenic gene for leukonychia, PLCD1, is present in this
chromosome (8). A novel
heterozygous missense mutation in the PLCD1 gene was then
identified by direct sequencing, which suggested that the accurate
diagnosis should be hereditary leukonychia. Another mutation in the
PLCD1 gene was also identified in a sporadic case, which
confirmed the final diagnosis.
After reviewing the literature related to hereditary
leukonychia, it was found that mutations in five genes are linked
to the pathogenesis of HL (9).
Different genes may be associated with distinct clinical phenotypes
of HL. In particular, mutations in PLCD1 are predominantly
reported in a large fraction of patients with HL. The PLCD1
gene consists of 15 exons, spanning 22.17 kb of genomic DNA on
chromosome 3p22.2. PLCD1 encodes an enzyme that mediates the
regulatory signaling of energy metabolism, Ca2+
homeostasis and intracellular movement (3,10,11).
Kiuru et al (8) discovered
five mutations in the PLCD1 on chromosome 3p22.2 in both
autosomal dominant and autosomal recessive HL. Of these, three
mutations, c.1309C>T (p.R437X), c.1792-10del-TGTAGTGGCC and
c.1055G>A (p.A285GfsX70), underlie the autosomal recessive form
of leuconychia, whereas the other two mutations, c.1720C>T
(p.A574T) and c.625T>C (p.C209R), are responsible for the
autosomal dominant form of the disease. It appears that the
recessive form is caused by a premature termination codon mutation,
and the autosomal dominant form by a missense mutation; one
possibility would be that the missense mutations exert a
dominant-negative effect on the wild-type allele with a complete
loss of function. Previously, Nomikos et al (12) determined significant divergent
enzymatic activities for leukonychia-linked mutant PLCD1,
confirming that mutations in PLCD1 cause HL. In the present
study, two novel heterozygous missense mutations, c.1384G>A
(p.E462K) and c.770 G>A (p.R257H), were identified in the family
with HL and one sporadic patient, respectively. Both of the
mutations are pathogenic and lead to the substitution of amino
acids, which was also confirmed by Polyphen2 (0.984 for
c.1384G>A and 0.885 for c.770 G>A). Moreover, it was found
that the clinical manifestations of leukonychia concomitant
koilonychias did not merely occur in the family described herein.
The condition has also been reported in few cases of Chinese and
Pakistani families (9,13–15).
Thus, this phenomena may not be a result of ethnic differences.
As per current knowledge, familial koilonychia is
another type of rare genodermatosis, which differs completely from
HL (15). To the best of our
knowledge, there are no reports of hereditary koilonychia with
leukonychia to date. It is inherited in an autosomal dominant
pattern with no predilection for sex. It may present at birth or
within the first few years of life. Nails are typically thin and
flat, developing degrees of concavity over time. In young children,
koilonychia of the toenails is commonly transient and idiopathic
(7). A previous literature review
revealed that mutations in PLCD1 only cause leukonychia
(16). Although it has been proven
through several proteomic studies that PLCD1 is abundant in
the human nail matrix, the mechanisms by which PLCD1
mutations result in leukonychia remain unknown (17), and the lack of functional
verification in the present study may be considered a limitation.
The present study was not able to determine whether the
PLCD1 gene was involved in the occurrence of hereditary
koilonychia. There are no studies which have identified any
pathogenic genes associated with familial koilonychia. Moreover, a
family of 5 generations with a syndrome of leukonychia, koilonychia
and multiple pilar cysts segregating as an autosomal dominant trait
was reported by Mutoh et al (18). No specific pathogenic genes
associated with the aforementioned syndrome were identified
however. The syndrome was ruled out since the family presented
herein did not have any other comorbidities.
In summary, HL is an extremely rare condition with
multiple clinical manifestations. The present study describes a
Chinese family with atypical characteristics of HL. The present
study not only provides novel insights for the accurate diagnosis
of HL and familial koilonychia, but also highlights the crucial
significance of genetic diagnosis. Furthermore, the findings
further confirm that mutations in PLCD1 underlie HL and add
another two pathogenic variants to the PLCD1 mutation
spectrum causing leukonychia, particularly in a Chinese Han
pedigree.
Acknowledgements
Not applicable.
Funding
This study was funded by grants from National
Natural Science Foundation of China (grant nos. 81471930 and
81472796).
Availability of data and materials
The raw Sanger sequencing data have been deposited
at China Nucleotide Sequence Archive (accession no. CNP0001358). It
is a usual practice in our group that the data is not published for
2 years. The datasets used during the present study are available
from the corresponding author upon reasonable request.
Authors' contributions
SS, MS and GZ designed the study, wrote the
protocol, collected the data, performed statistical analyses and
contributed to writing the manuscript. UK helped with data
collection, study design and coordinated the study. ML and XW
participated in the study design and helped to critically revise
the manuscript. SS and GZ were responsible for confirming the
authenticity of the raw data. All authors read and approved the
final manuscript.
Ethics approval and consent to
participate
Informed consent was obtained from all study
participants prior to their inclusion in the study. The present
study was approved by the Institutional Ethical Committee of
Shanghai Skin Disease Hospital and was performed in accordance with
the guidelines of the Declaration of Helsinki.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Pakornphadungsit K, Suchonwanit P,
Sriphojanart T and Chayavichitsilp P: Hereditary leukonychia
totalis: A case report and review of the literature. Case Rep
Dermatol. 10:82–88. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
De D and Handa S: Hereditary leukonychia
totalis. Indian J Dermatol Venereol Leprol. 73:355–357. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Kim H, Kim JY, Kim KA, Lim Y, Kim YH, Huh
PW, Lee KH, Han H, Wang YP and Rha HK: Identification of the
elements regulating the expression of the phospholipase C delta1.
Mol Cells. 14:29–34. 2002.PubMed/NCBI
|
|
4
|
Gensure RC, Mäkitie O, Barclay C, Chan C,
Depalma SR, Bastepe M, Abuzahra H, Couper R, Mundlos S, Sillence D,
et al: A novel COL1A1 mutation in infantile cortical hyperostosis
(Caffey disease) expands the spectrum of collagen-related
disorders. J Clin Invest. 115:1250–1257. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Zhong W, Pan Y, Shao Y, Yang Y, Yu B and
Lin Z: Atypical presentation of dyschromatosis universalis
hereditaria with a novel ABCB6 mutation. Clin Exp Dermatol.
44:e58–e60. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Mir H, Khan S, Arif MS, Ali G, Wali A,
Ansar M and Ahmad W: Mutations in the gene phospholipase C, delta-1
(PLCD1) underlying hereditary leukonychia. Eur J Dermatol.
22:736–739. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Fawcett RS, Linford S and Stulberg DL:
Nail abnormalities: Clues to systemic disease. Am Fam Physician.
69:1417–1424. 2004.PubMed/NCBI
|
|
8
|
Kiuru M, Kurban M, Itoh M, Petukhova L,
Shimomura Y, Wajid M and Christiano AM: Hereditary leukonychia, or
porcelain nails, resulting from mutations in PLCD1. Am J Human
Genetics. 88:839–844. 2011. View Article : Google Scholar
|
|
9
|
Xue K, Zheng Y, Shen C and Cui Y:
Identification of a novel PLCD1 mutation in Chinese Han pedigree
with hereditary leukonychia and koilonychia. J Cosmet Dermatol.
18:912–915. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Song JJ, Liu Q, Li Y, Yang ZS, Yang L,
Xiang TX, Ren GS and Chen JB: Epigenetic inactivation of PLCD1 in
chronic myeloid leukemia. Int J Mol Med. 30:179–184.
2012.PubMed/NCBI
|
|
11
|
Xiang T, Li L, Fan Y, Jiang Y, Ying Y,
Putti TC, Tao Q and Ren G: PLCD1 is a functional tumor suppressor
inducing G(2)/M arrest and frequently methylated in breast cancer.
Cancer Biol Ther. 10:520–527. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Nomikos M, Thanassoulas A, Beck K,
Theodoridou M, Kew J, Kashir J, Calver BL, Matthews E, Rizkallah P,
Sideratou Z, Nounesis G and Lai FA: Mutations in PLCδ1 associated
with hereditary leukonychia display divergent PIP2 hydrolytic
function. FEBS J. 283:4502–4514. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Brown PJ, Padgett JK and English JC III:
Sporadic congenital leukonychia with partial phenotype expression.
Cutis. 66:117–119. 2000.PubMed/NCBI
|
|
14
|
Khan T, Khan M, Yousaf A, Khan S, Naeem M,
Shah A, Murtaza G, Ali A, Jabeen N, Hussain HM, et al: Whole exome
sequencing identifies a novel dominant missense mutation underlying
leukonychia in a Pakistani family. J Hum Genet. 63:1071–1076. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Kwon NH, Kim JE, Cho BK, Jeong EG and Park
HJ: Sporadic congenital leukonychia with koilonychia. Int J
Dermatol. 51:1400–1402. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Walker J, Baran R, Vélez N and Jellinek N:
Koilonychia: An update on pathophysiology, differential diagnosis
and clinical relevance. J Eur Acad Dermatol Venereology.
30:1985–1991. 2016. View Article : Google Scholar
|
|
17
|
Rice RH, Xia YX, Alvarado RJ and Phinney
BS: Proteomic analysis of human nail plate. J Proteome Res.
9:6752–6758. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Mutoh M, Niiyama S, Nishikawa S, Oharaseki
T and Mukai H: A syndrome of leukonychia, koilonychia and multiple
pilar cysts. Acta Derm Venereol. 95:249–250. 2015. View Article : Google Scholar : PubMed/NCBI
|