Introduction
Ischemia-reperfusion (I-R) injury plays a
significant role in several human diseases, such as acute
myocardial infarction, stroke and multiorgan failure (1). Not surprisingly, I-R injury is the
most frequent cause of acute kidney injury with renal tubular
epithelial cells being extremely vulnerable due to their high
metabolic demands (2). Thus,
delineating the molecular mechanisms that govern I-R injury deems a
significant research issue, as it may lead to novel therapeutic
strategies.
Indoleamine 2,3-dioxygenase 1 (IDO) is a
rate-limiting enzyme that degrades tryptophan through the
kynurenine pathway. IDO initially engaged immunologists' attention
due to its immunomodulatory properties (3). IDO may activate two main pathways.
Tryptophan depletion by increasing the uncharged tryptophanyl-tRNA
activates the general control non-derepressible-2 kinase (GCN2K)
(4–6). In parallel, the produced kynurenine
activates the aryl-hydrocarbon receptor (AhR) (7,8).
However, the role of IDO seems to extend beyond the immune system.
Experimentally, in the mouse kidney, it has been shown that I-R
injury increases IDO expression, whereas IDO inhibition ameliorates
kidney injury and preserves renal function (9). Nevertheless, the exact molecular
mechanisms are still unknown. Also, in cultures of renal tubular
epithelial cells subjected to reoxygenation, cell death depends on
the activation of AhR (10). Since
kynurenine is a known endogenous activator of AhR (7), the aforementioned
reoxygenation-induced AhR activation might result from the
reoxygenation-induced IDO upregulation and the subsequent
kynurenine overproduction.
The present study evaluated the kinetics of IDO
expression and its effect on cell survival in primary renal
proximal tubular epithelial cells (RPTECs) subjected to I-R injury.
I-R injury consists of two consecutive but pathophysiologically
distinct phases. During ischemia, cell death ensues due to cell
energy collapse. However, the setting alters during reoxygenation,
as cell death results from overproduction of reactive oxygen
species (ROS) (1). Notably,
confirming the pathophysiological difference between the two phases
of I-R injury, previous studies showed that during ischemia, RPTECs
death ensues through apoptosis (11,12).
In contrast to this, reperfusion induces lipid peroxidation and
ferroptotic cell death (12–14).
To evaluate the effect of the two different phases
of I-R injury on IDO kinetics and how the latter may affect RPTECs
survival, the current study developed a proper cell culture system.
RPTECs were cultured under anoxia to simulate ischemia. To imitate
reperfusion, RPTECs were initially cultured under anoxia, then
washed, fresh culture medium was added and cells were cultured
under normoxic conditions. Whenever needed, the IDO inhibitor
1-DL-methyl-tryptophane (1-MT) (15), the AhR inhibitor CH223191 (16) or the ferroptosis inhibitor
α-tocopherol were used (17). The
IDO-triggered molecular pathways that may induce cell apoptosis
during anoxia or cell ferroptosis due to reoxygenation were
evaluated.
Materials and methods
Cell culture and imaging
Primary C57BL/6 mouse RPTECs (cat. no. C57-6015;
Cell Biologics, Inc.) were cultured in Complete Epithelial Cell
Medium/w kit, supplemented with epithelial cell growth supplement
(epithelial growth factor, insulin, transferrin, L-glutamine,
selenium, fetal bovine serum, and antibiotics) (cat. no. M6621;
Cell Biologics, Inc.). The aforementioned primary cells were
differentiated, well-characterized passage one RPTECs. Cells were
expanded in 75-cm2 flasks, and passage three cells were
used for the experiments.
Cells were seeded at a density of 10,000 cells per
well in 96-well plates or at a density of 300,000 cells per well in
6-well plates and incubated for 16 h before the onset of anoxic
conditions. To reduce oxygen levels to <1%, the GasPak™ EZ
Anaerobe Container System with Indicator (cat. no. 26001; BD
Biosciences) was used. Cells within the anaerobic container were
cultured at 37°C. These anoxic conditions simulated ischemia
(12).
An inverted microscope (Axiovert 40C; Carl Zeiss AG)
and a digital camera (3MP USB2.0 Microscope Digital Camera;
AmScope) with the related software (AmScope v. ×64, 3.7.3036;
AmScope) were used for cell imaging.
A reciprocal approach based on the cell imaging hard
end-point of anoxia- or reoxygenation-induced cell death was used
for selecting the appropriate time points. Cell imaging detected
that the time required for cell death of untreated cells due to
anoxia was 48 h. Onset of reoxygenation experiments was at half of
that time, which is after 24 h. Notably, cell imaging did not
detect a difference in confluency between the control cells and the
cells subjected to 24 h of anoxia.
In reoxygenation experiments, cells were washed with
Dulbecco's phosphate buffer saline (PBS) (Sigma-Aldrich; Merck
KGaA), supplemented with fresh culture medium, and placed at 37°C
in a humidified atmosphere containing 5% CO2. These conditions
imitate reperfusion (12). Cell
imaging detected that the time required for the death of untreated
cells due to reoxygenation was only 4 h.
As live cells are required to conduct reliable
experiments, the various parameters were evaluated at half of the
time needed for severe deterioration of untreated cells under
anoxia or reoxygenation. Thus, anoxia experiments were performed
after 24 h of anoxia and reoxygenation experiments after 2 h of
reoxygenation.
Whenever needed, cells were treated with 100 µM IDO
inhibitor 1-MT (Sigma-Aldrich; Merck KGaA), 3 µM AhR inhibitor
CH223191 (Sigma-Aldrich; Merck KGaA) or 100 µM ferroptosis
inhibitor α-tocopherol (Sigma-Aldrich; Merck KGaA). In the anoxia
experiments, such treatments started at the onset of the anoxic
conditions. In the reoxygenation experiments, such treatments
started at the beginning of the reoxygenation in previously
untreated cells that were subjected to 24 h of anoxia.
IDO mRNA level
Cells were cultured in 6-well plates (300,000 cells
per well) and were subjected or not to anoxia or reoxygenation.
Total cellular RNA was isolated from RPTECs using the
TRIzol® reagent (cat. no. 15596026; Invitrogen; Thermo
Fisher Scientific, Inc.) according to the manufacturer's
instructions. RNA concentration was measured on an
EnSpire® Multimode Plate Reader (PerkinElmer, Inc.), and
5 µg was used for first-strand cDNA synthesis using the
PrimeScript™ II Reverse Transcriptase (cat. no. 2690A; Takara Bio,
Inc.). RT was performed under the following conditions: 25°C for 5
min, 42°C for 60 min and 70°C for 15 min. The PCR platform used was
an Eppendorf Reaplex 4 MasterCycler (Eppendorf). The resultant cDNA
samples were subjected to 30 cycles of PCR amplification in the
presence of specific sense and antisense primers for mouse IDO and
glyceraldehyde 3-phosphate dehydrogenase (GAPDH) as an internal
control. The following thermocycling conditions were used: Initial
denaturation step at 94°C for 2 min; followed by 30 cycles of
annealing at 60°C for 50 sec, elongation at 72°C for 1 min and
denaturation at 94°C for 30 sec. The primer sequences used were as
follows: IDO sense, 5′-AGGATCCTTGAAGACCACCA-3′ and antisense,
5′-CCAATAGAGAGACGAGGAAG-3′ (398 bp); and GAPDH sense,
5′-GCCAAGGTCATCCATGACAACTTTGG-3′ and antisense,
5′-GCCTGCTTCACCACCTTCTTGATGTC-3′ (348 bp). Primers were obtained
from Eurofins Scientific. The amplified PCR products were
electrophoresed in a 1.0% agarose gel (cat. no. A9539;
Sigma-Aldrich; Merck KGaA) and stained with ethidium bromide (cat.
no. 1116150001; Sigma-Aldrich; Merck KGaA). Digital images were
obtained with a Kodak digital device (Kodak). Densitometric
analysis was performed using ImageJ software version 1.53f
(National Institutes of Health). These experiments were repeated
three times.
Cell death, ROS production, tryptophan
catabolism and kynurenine production
Besides cell imaging, cell death was also assessed
in cells cultured in 96-well plates (10,000 cells per well) by LDH
release using the Cytotox Non-Radioactive Cytotoxic Assay kit (cat.
no. G1780; Promega Corporation) according to the manufacturer's
protocol. The LDH release assay was performed to detect both
necrotic and apoptotic death (18).
Cell death was calculated using the following equation: Cell death
(%) = (LDH in the supernatant: total LDH) ×100. These experiments
were repeated six times.
ROS generation was evaluated in cells cultured in
96-well plates (10,000 cells per well). Once the incubation period
was over, 5 µM fluorogenic probe CellROX® Deep Red
Reagent (cat. no. C10422; Invitrogen; Thermo Fisher Scientific,
Inc.) was added for 30 min at 37°C. The cells were then washed with
PBS, and fluorescence signal intensity was measured on an
EnSpire® Multimode Plate Reader (PerkinElmer, Inc.).
These experiments were repeated six times.
Tryptophan catabolism and kynurenine production were
assessed by their concentration in the cell culture supernatant.
Cells were cultured in 6-well plates (300,000 cells per well). Once
the incubation period was over, tryptophan and kynurenine
concentrations were measured using the kynurenine/tryptophan ratio
ELISA pack (cat. no. ISE-2227; ImmuSmol). Limits of detections of
the kit for kynurenine and tryptophan are below 47.5 ng/ml and 1.2
µg/ml, respectively. These experiments were repeated six times.
Proteins of interest
Once anoxia or reoxygenation period was over, RPTECs
cultured in 6-well plates (300,000 cells per well) were lysed with
the T-PER tissue protein extraction reagent (Thermo Fisher
Scientific, Inc.) supplemented with protease (Sigma-Aldrich; Merck
KGaA) and phosphatase inhibitors (Roche Diagnostics). Bradford
assay (Sigma-Aldrich; Merck KGaA) was used for protein
quantification, and 10 μg from each protein extract was loaded per
lane and electrophoresed on an SDS-PAGE gel (4-12% Bis-Tris gels;
Thermo Fisher Scientific, Inc.) and transferred onto a
polyvinylidene fluoride (PVDF) membrane (Thermo Fisher Scientific,
Inc.). The LumiSensor Plus Chemiluminescent HRP Substrate kit
(GenScript) was used for the enhanced chemiluminescent detection of
the western blot bands. The Restore Western Blot Stripping Buffer
(Thermo Fisher Scientific, Inc.) was used for re-probing the PVDF
membranes. Densitometric analysis was performed with the ImageJ
software. These experiments were repeated four times.
Blots were incubated at 4°C for 16 h with each
primary antibody and for 30 min at room temperature with the
appropriate secondary antibody. Primary antibodies were specific
the following proteins: IDO (1:200; cat. no. sc-25809), GCN2K
(1:100; cat. no. sc-374609) (both from Santa Cruz Biotechnology,
Inc.), phosphorylated at Thr899 GCN2K (p-GCN2K; 1:1,000; cat. no.
ab75836; Abcam), eukaryotic translation initiation factor-2α
(eIF2α; 1:100; cat. no. sc-133132; Santa Cruz Biotechnology, Inc.),
p at Ser51 eIF2α (p-eIF2α; 1:1,000; cat. no. 9721; Cell Signaling
Technology, Inc.), activating transcription factor 4 (ATF4; 1:500;
cat. no. CSB-PA002272KA01HU), ATF3 (1:500; cat. no. CSB-PA020022)
(both Cusabio Technology LLC), C/EBP homologous protein (CHOP;
1:1,000; cat. no. 5554), p53 (1:1,000; cat. no. 2524), p at Ser15
p53 (p-p53; 1:1,000; cat. no. 9284), Bax (1:1,000; cat. no. 5023)
(all Cell Signaling Technology, Inc.), death receptor 5 (DR5;
1:500; cat. no. CSB-PA018500; Cusabio Technology LLC), activated
cleaved caspase-3 (CC3; 1:1,000; cat. no. ab13847; Abcam), AhR
(1:200; cat. no. sc-133088), cytochrome P450 family 1 subfamily A
polypeptide 1 (CYP1A1; 1:500; cat. no. sc-25304) (both Santa Cruz
Biotechnology, Inc.) and β-actin (1:2,500; cat. no. 4967; Cell
Signaling Technology, Inc.). Anti-mouse (1:1,000; cat. no. 7076) or
anti-rabbit (1:1,000, cat. no. 7074) (both Cell Signaling
Technology, Inc.) IgG HRP-conjugated secondary antibodies were
used.
Statistical analysis
Statistical analysis was performed with SPSS
software version 20 (IBM Corp.). The one-sample Kolmogorov-Smirnov
test verified that all variables were normally distributed except
the cell imaging results. An unpaired Student's t-test or one-way
ANOVA with Bonferroni's post hoc test were used for comparison of
means. For analyzing the cell imaging results, the Mann-Whitney U
test or the Kruskal-Wallis H test with Dunn's post hoc test were
used. Results are expressed as the mean ± SEM, and P<0.05 was
considered to indicate a statistically significant difference.
Western blotting results were normalized against β-actin and PCR
results were normalized against GAPDH.
Results
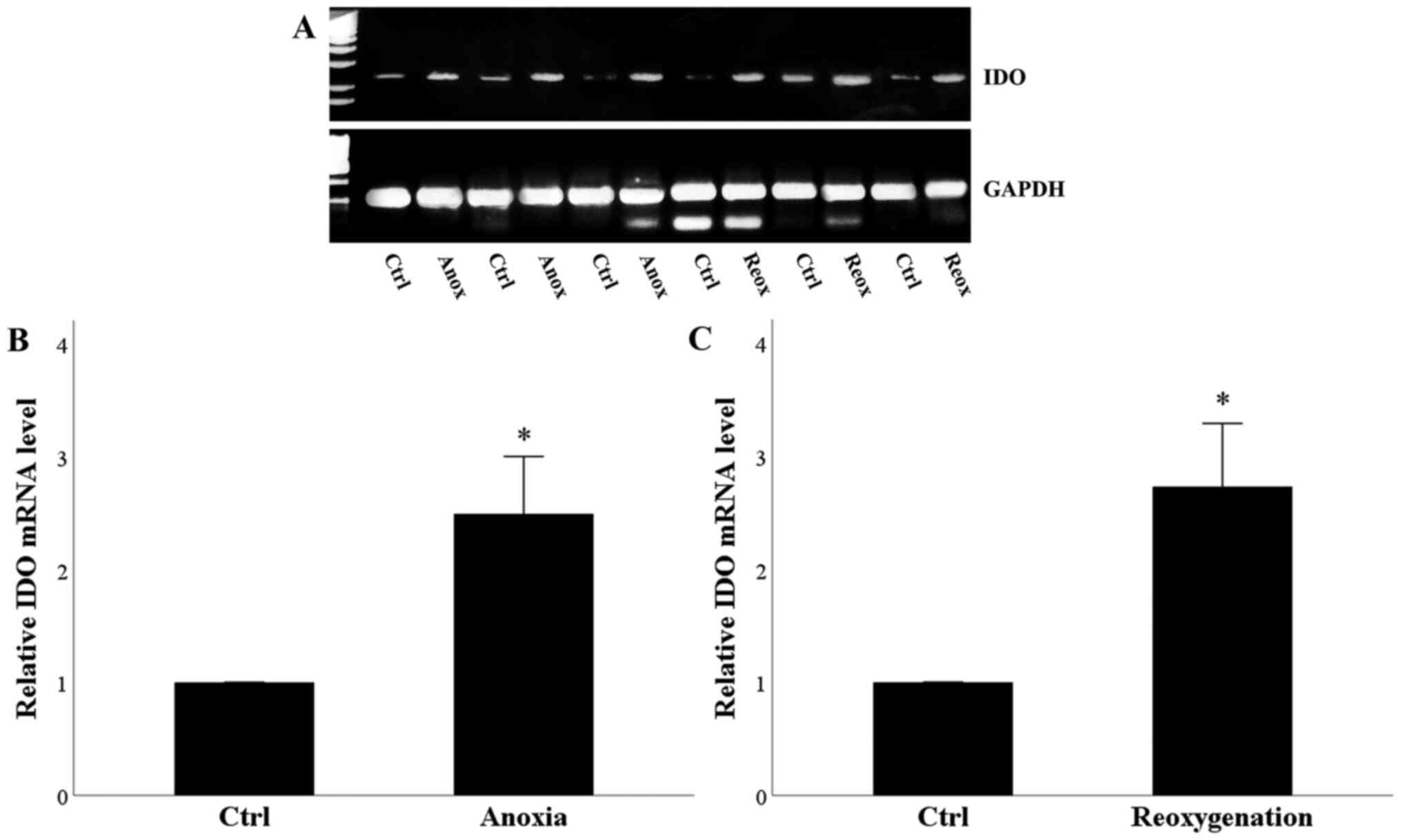
Anoxia or reoxygenation increases IDO
mRNA expression
RPTECs remained under normoxic conditions for 24 h
or subjected to 24 h of anoxia. Compared with the control cells,
anoxia increased IDO mRNA level significantly (Fig. 1A and B). Control RPTECs remained
under normoxic conditions for 24 h, washed and remained for another
2 h period under normoxia before mRNA extraction. Treated RPTECs
were subjected to 24 h of anoxia, and then washed and cultured for
another 2-h period under normoxic conditions. Compared with the
control cells, reoxygenation enhanced IDO mRNA level significantly
(Fig. 1A and C). Thus, both anoxia
and reoxygenation increased the mRNA expression of IDO.
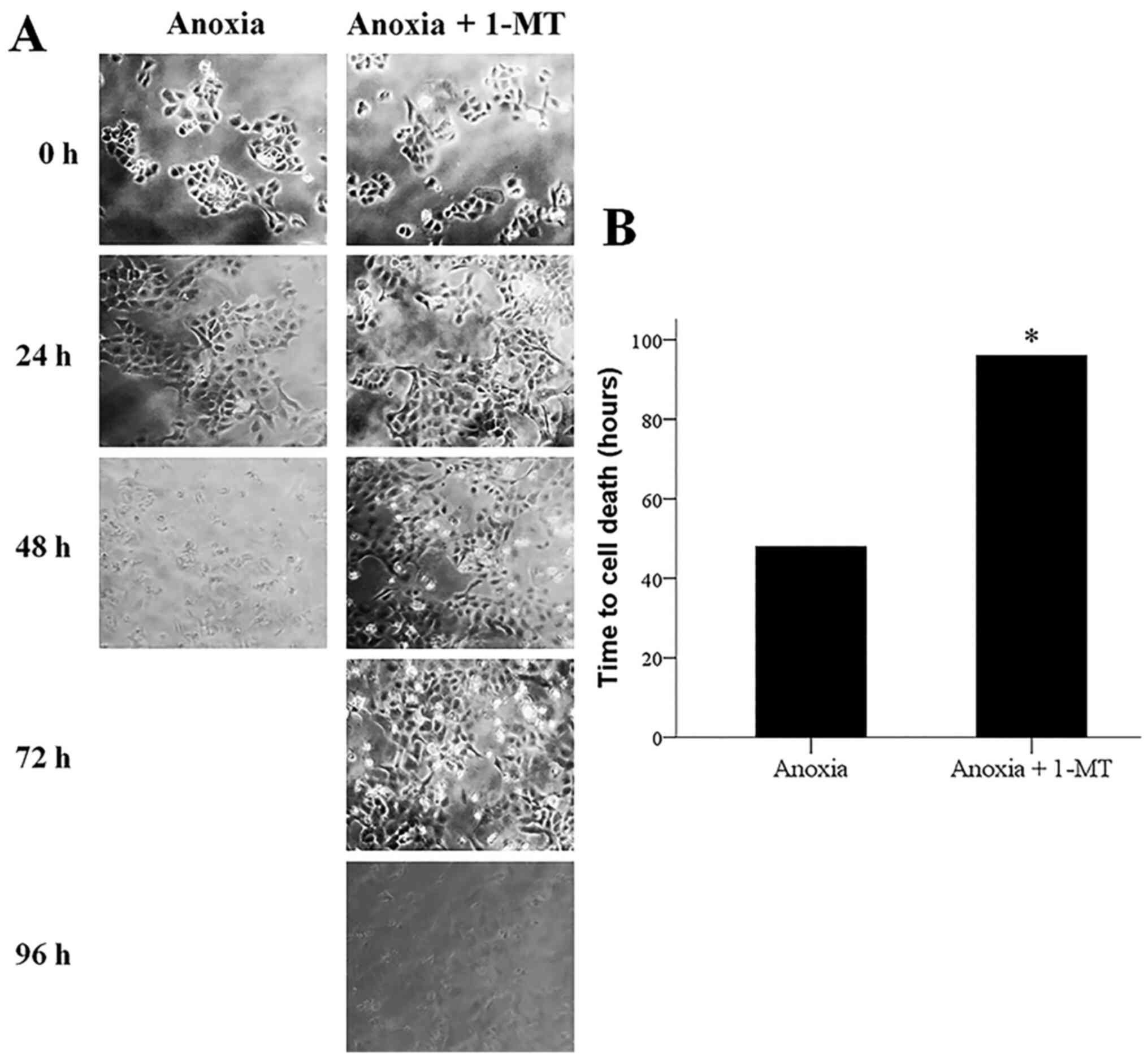
Inhibition of IDO prevents
anoxia-induced apoptosis
Cell imaging revealed that anoxia induced cell
death, whereas the IDO inhibitor 1-MT prevented anoxia-induced cell
death (Fig. 2A and B). Western
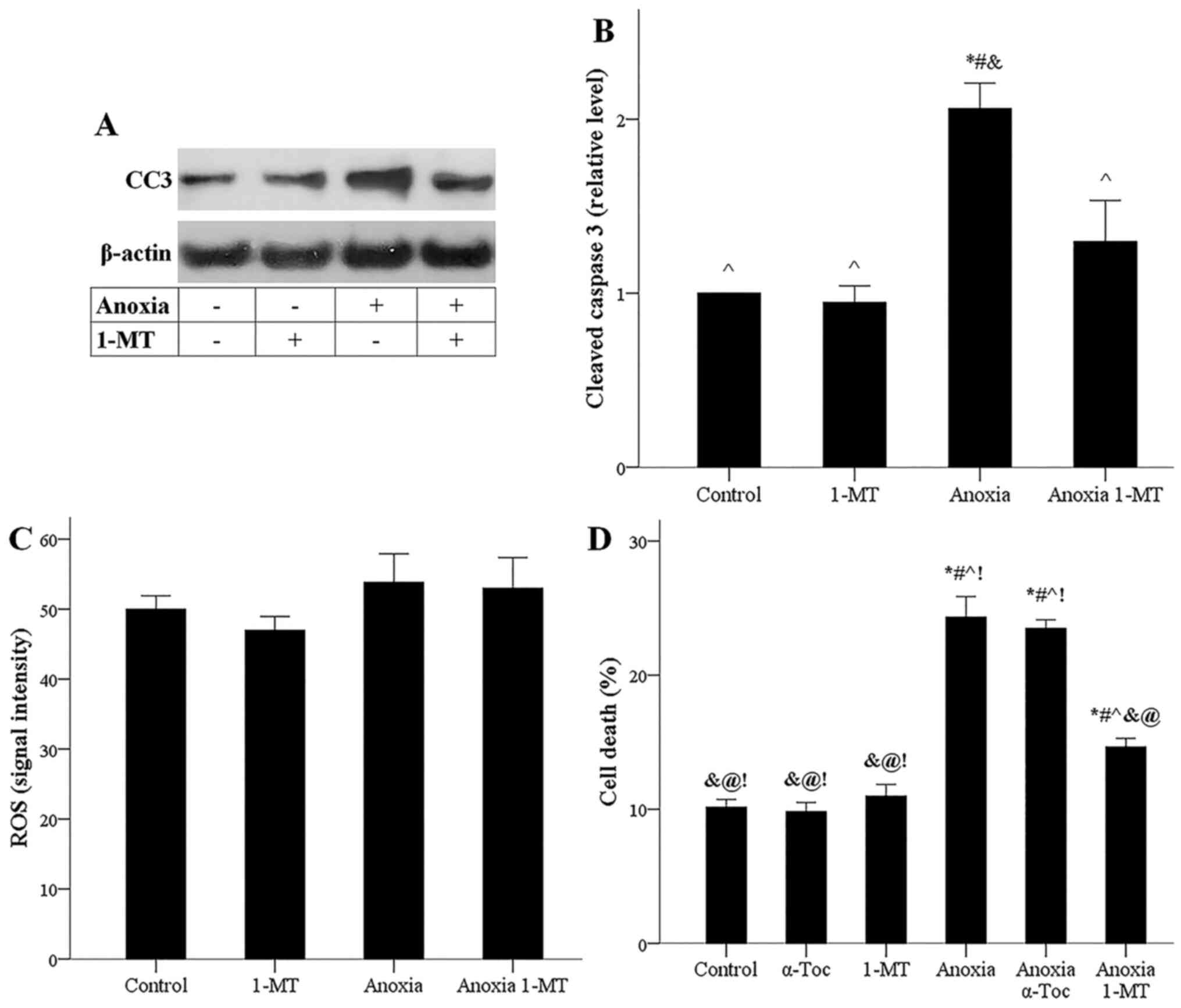
blotting showed that anoxia increased the level of activated CC3,
on which all the apoptotic pathways converge (19). Τhe IDO inhibitor 1-MT prevented
anoxia-induced CC3 upregulation (Fig.
3A and B).
Possibly due to lack of oxygen, anoxia did not
upregulate ROS generation in the presence or not of 1-MT (Fig. 3C). The LDH release assay confirmed
the results obtained by cell imaging, demonstrating that anoxia
induces cell death, whereas the IDO inhibitor 1-MT decreases
anoxia-induced cell death. Notably, the ferroptosis inhibitor
α-tocopherol did not affect anoxia-induced cell death, ensuring
that ferroptosis does not occur under anoxia (Fig. 3D). The latter is in accordance with
the aforementioned stable ROS levels since ROS overproduction is
required for ferroptosis (17).
Hence, during anoxia, cell death ensues through apoptosis, whereas
ferroptosis does not take place. Inhibition of IDO could prevent
anoxia-induced apoptotic cell death.
Anoxia upregulates IDO, depletes
tryptophan and activates GCN2K
Anoxia induced IDO overexpression, a result that was
not affected by the IDO inhibitor 1-MT (Fig. 4A and D). Tryptophan catabolism was
assessed by its concentration in the cell culture supernatant. The
results confirmed that anoxia-induced IDO-overexpression decreased
tryptophan concentration significantly, whereas 1-MT prevents
tryptophan depletion (Fig. 4E).
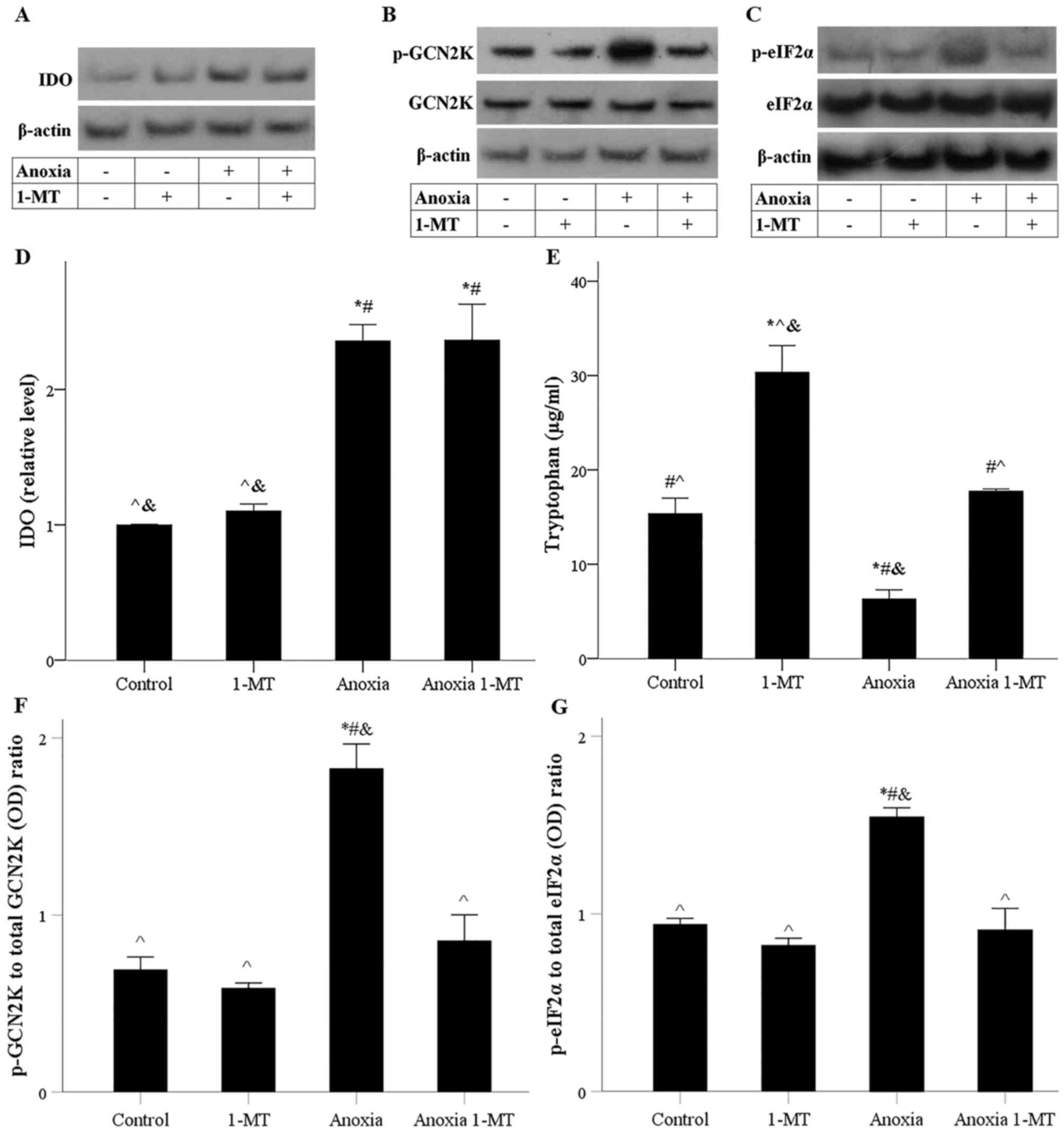 | Figure 4.Effect of anoxia in the presence or
absence of the IDO inhibitor 1-MT on IDO expression, tryptophan
catabolism, p-GCN2K and p-eIF2α levels. Representative western
blotting of (A) IDO, (B) p- and total GCN2K and (C) p- and total
eIF2α levels. (D) Semi-quantification of IDO protein levels. (E)
Tryptophan catabolism was assessed by its concentration in the cell
culture supernatant. (F) p-GCN2K/GCN2K and (G) p-eIF2α/eIF2α
ratios. *P<0.05 vs. control; #P<0.05 vs. control
with 1-MT; ^P<0.05 vs. anoxia;
&P<0.05 vs. anoxia with 1-MT. 1-MT,
1-DL-methyltryptophan; IDO, indoleamine 2,3-dioxygenase 1; p-,
phosphorylated; GCN2K, general control nonderepressible-2 kinase;
eIF2α, eukaryotic translation initiation factor 2α; OD, optical
density. |
Tryptophan depletion activated GCN2K as it was
assessed by the level of its activated phosphorylated form. As
expected, the absence of tryptophan depletion in cells treated with
1-MT prevented anoxia-induced GCN2K activation (Fig. 4B and F). Anoxia-induced GCN2K
activation led to increased phosphorylated eIF2α, a substrate of
the GCN2K (6). In 1-MT-treated
cells, the absence of GCN2K activation prevented anoxia-induced
eIF2α phosphorylation (Fig. 4C and
G). Therefore, anoxia increased the protein expression of IDO.
IDO could deplete tryptophan and activate GCN2K. 1-MT inhibited IDO
activity and prevented GCN2K activation.
IDO-induced GCN2 activation triggers
apoptotic pathways
Under anoxia, the p-eIF2α translational target ATF4
was upregulated, whereas 1-MT prevented anoxia-induced ATF4
upregulation (Fig. 5A and F). In
cells subjected to anoxia, the ATF4 transcriptional target CHOP
increased, while the IDO inhibitor 1-MT blocked anoxia-induced CHOP
upregulation (Fig. 5B and G).
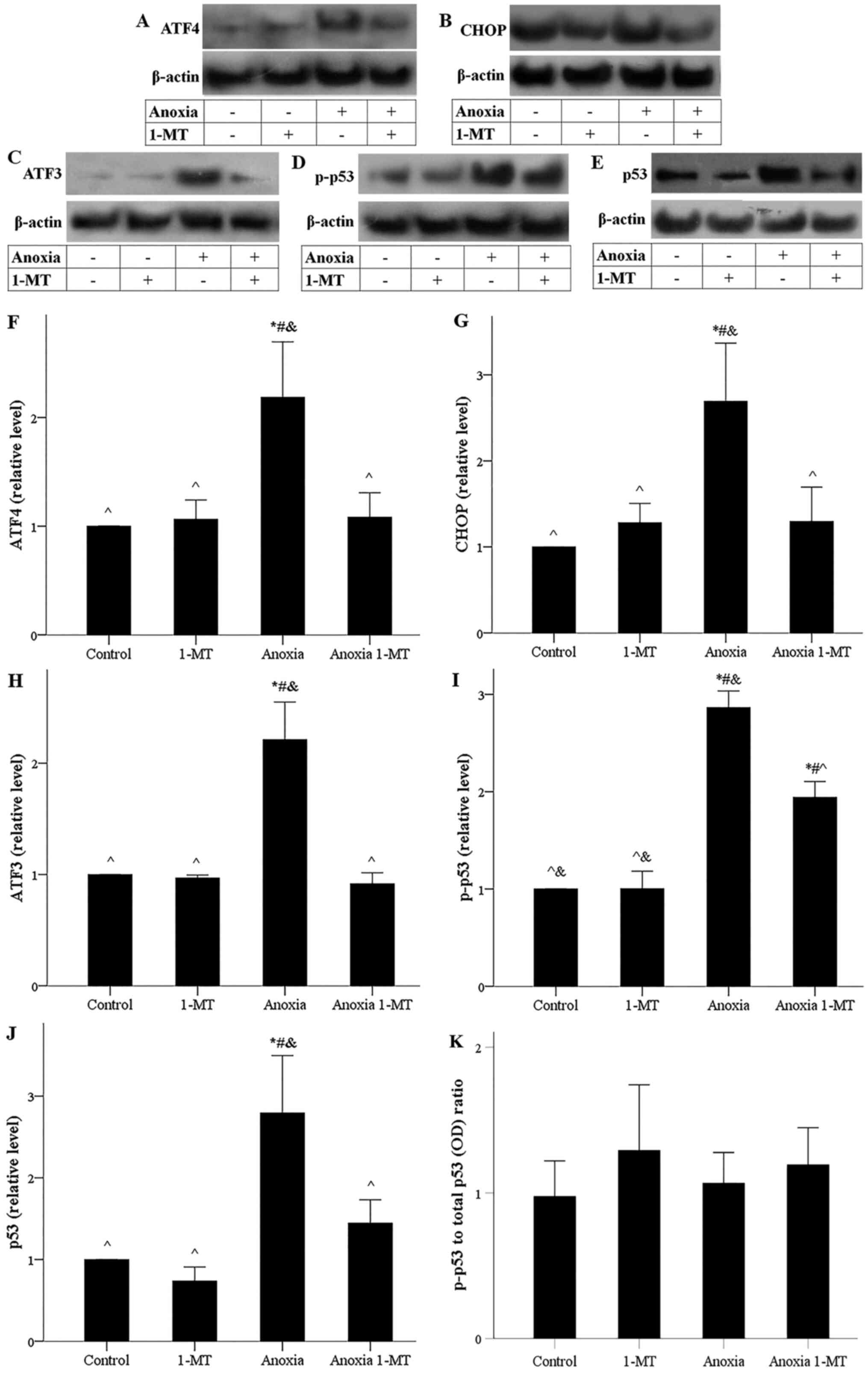 | Figure 5.Effect of anoxia in the presence or
absence of the IDO inhibitor 1-MT on ATF4, CHOP, ATF3, p-p53 and
p53 levels. Representative western blots for the levels of (A)
ATF4, (B) CHOP, (C) ATF3, (D) p-p53 and (E) p53.
Semi-quantification of (F) ATF4, (G) CHOP, (H) ATF3, (I) p-p53 and
(J) p53 protein levels. (K) p-p53/total p53 ratio. *P<0.05 vs.
control; #P<0.05 vs. control with 1-MT;
^P<0.05 vs. anoxia; &P<0.05 vs.
anoxia with 1-MT. 1-MT, 1-DL-methyltryptophan; IDO, indoleamine
2,3-dioxygenase 1; ATF4, activating transcription factor 4; CHOP;
C/EBP homologous protein; ATF4, activating transcription factor 3;
p-, phosphorylated; OD, optical density. |
Under anoxia, ATF3, a transcriptional target of
ATF4, expression increased, whereas 1-MT prevented anoxia-induced
ATF3 upregulation (Fig. 5C and H).
Anoxia induced phosphorylation of p53, an effect that was reduced
significantly by 1-MT (Fig. 5D and
I). In cells subjected to anoxia, the increase of ATF3 and
p-p53 was accompanied by p53 upregulation, while 1-MT prevented
anoxia-induced p53 upregulation (Fig.
5E and J). The p-p53 to total p53 ratio remained stable under
all conditions (Fig. 5K),
indicating that phosphorylation of p53 at Ser15 controls total p53
level.
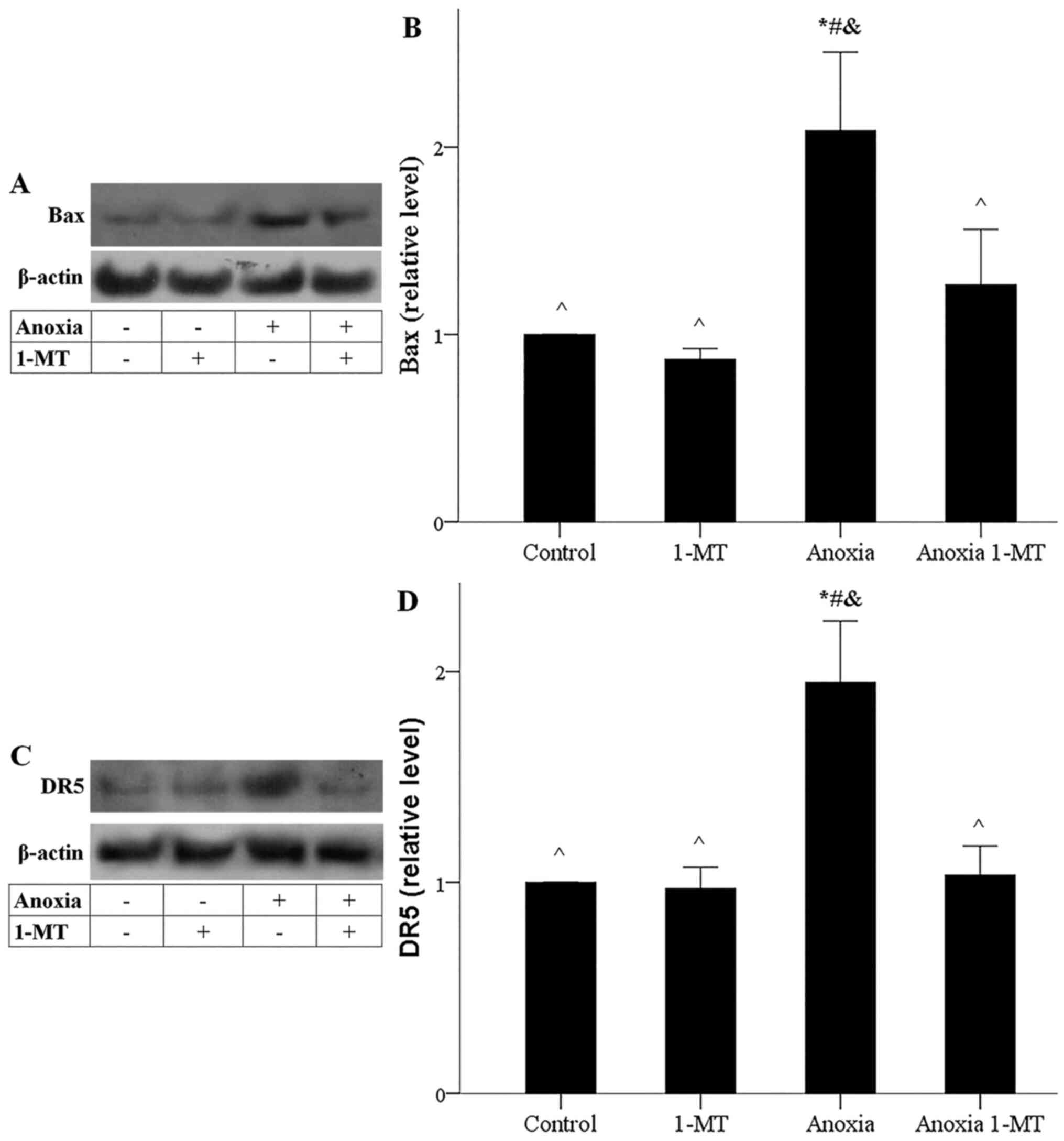
Anoxia-induced increase of CHOP and p53 was
accompanied by elevated levels of their transcriptional target Bax.
1-MT inhibited anoxia-induced Bax upregulation (Fig. 6A and B). Also, anoxia-induced
increase of CHOP and p53 was accompanied by enhanced expression of
their transcriptional target DR5. 1-MT blocked anoxia-induced DR5
upregulation as well (Fig. 6C and
D). Thus, anoxia-induced GCN2K activation could trigger the
activation of apoptotic pathways, whereas inhibition of IDO
prevented the activation of the aforementioned apoptotic
pathways.
Inhibition of IDO prevents
reoxygenation-induced ferroptosis
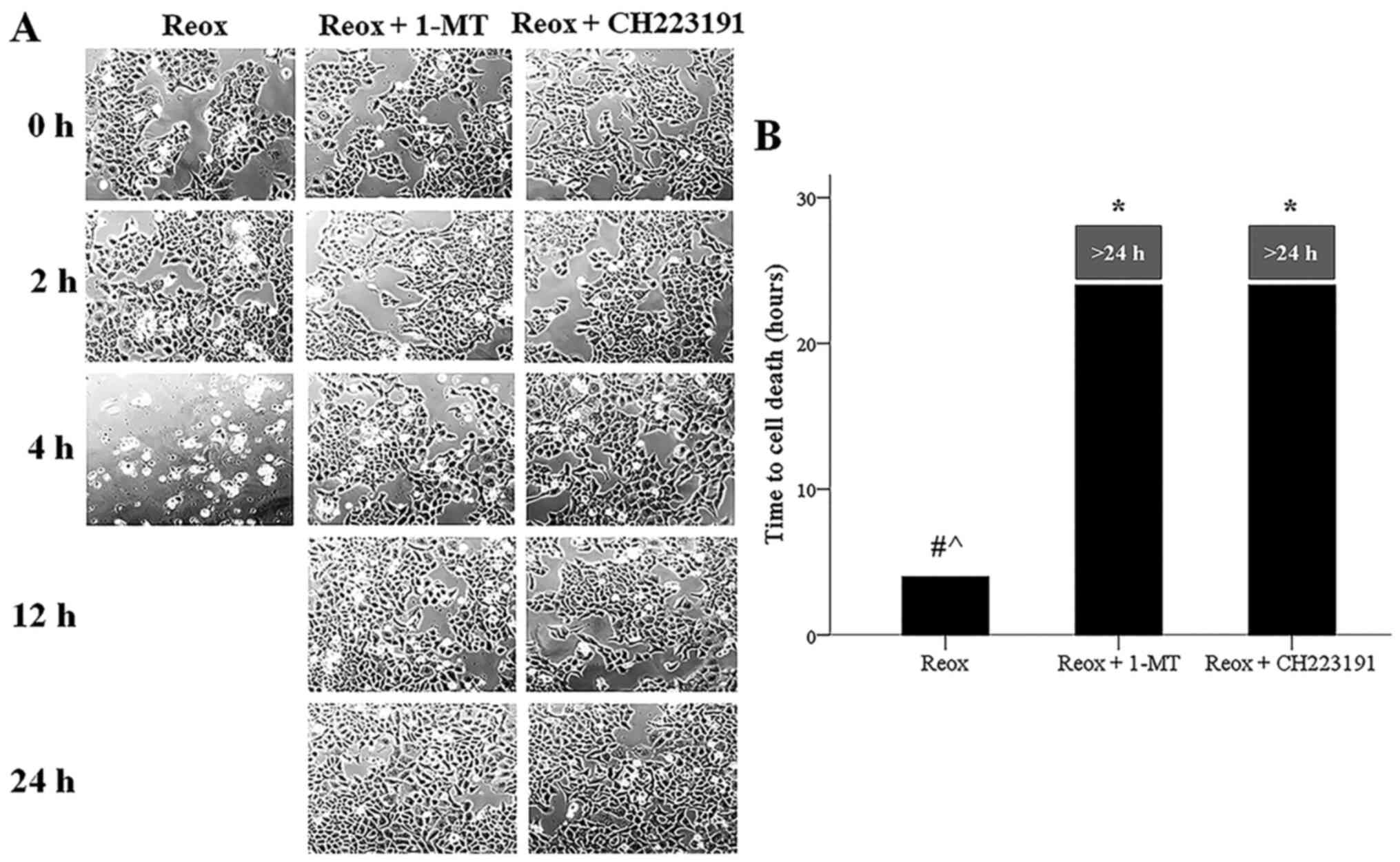
Cell imaging showed that reoxygenation induced cell
death, while both 1-MT and the AhR inhibitor CH223191 prevented
reoxygenation-induced cell death (Fig.
7A and B). Meanwhile, irrespective of the presence of 1-MT or
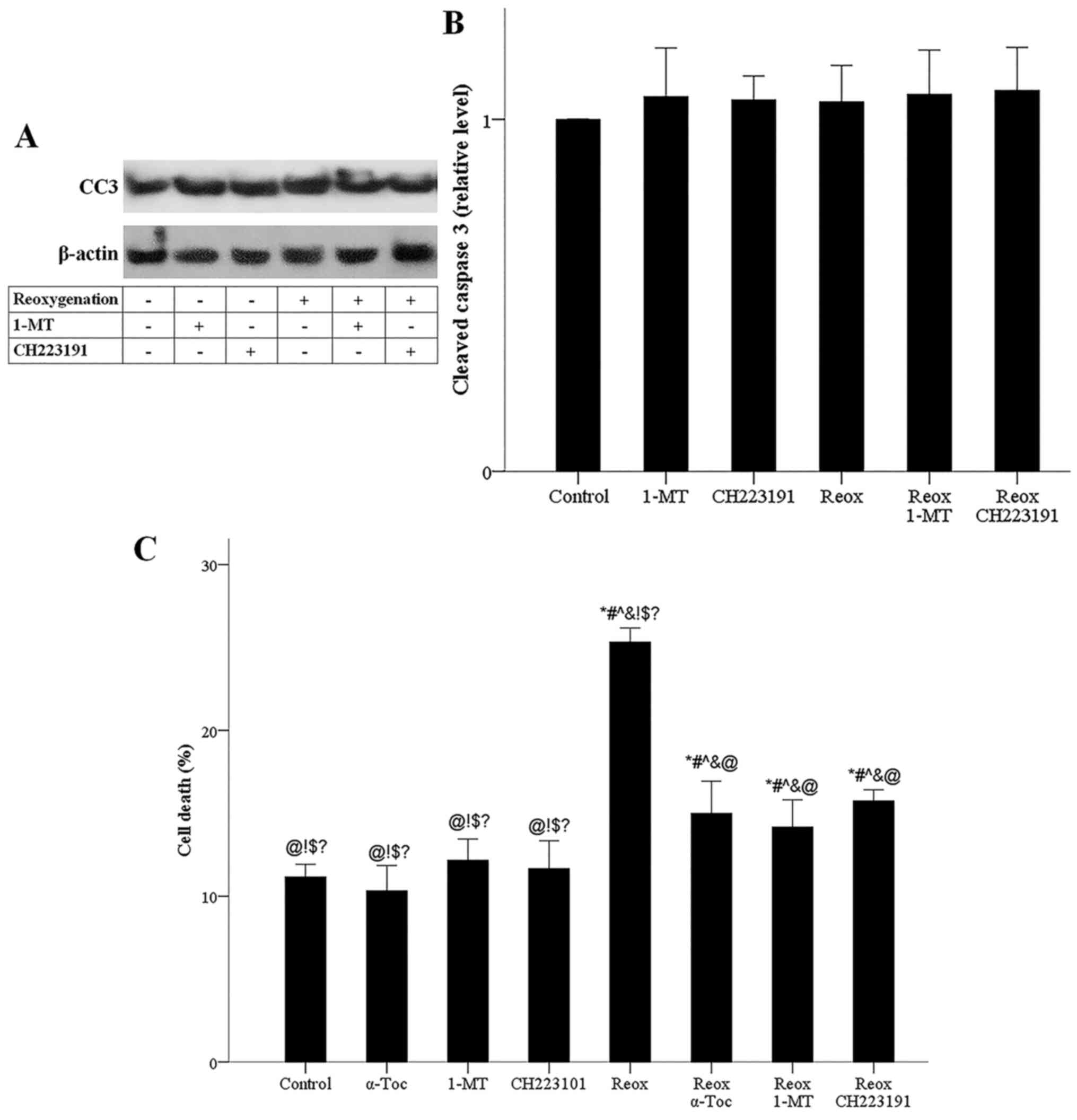
CH223191, reoxygenation did not affect the level of activated CC3,
indicating that under reoxygenation, apoptosis does not take place
(Fig. 8A and B).
The LDH release assay confirmed the results obtained
by cell imaging results, since it detected that reoxygenation
induces cell death, while 1-MT prevented reoxygenation-induced cell
death. CH223191 also prevented reoxygenation-induced cell death,
indicating that, under reoxygenation, cell death is mediated by
AhR. Notably, the ferroptosis inhibitor α-tocopherol blocked
reoxygenation-induced cell death, confirming the ferroptotic nature
of cell death under reoxygenation (Fig.
8C). Therefore, reoxygenation induced ferroptotic cell death,
whereas apoptosis did not take place, and the results indicated
that ferroptosis was mediated by IDO and AhR.
Reoxygenation upregulates IDO,
increases kynurenine and triggers AhR-induced ROS generation
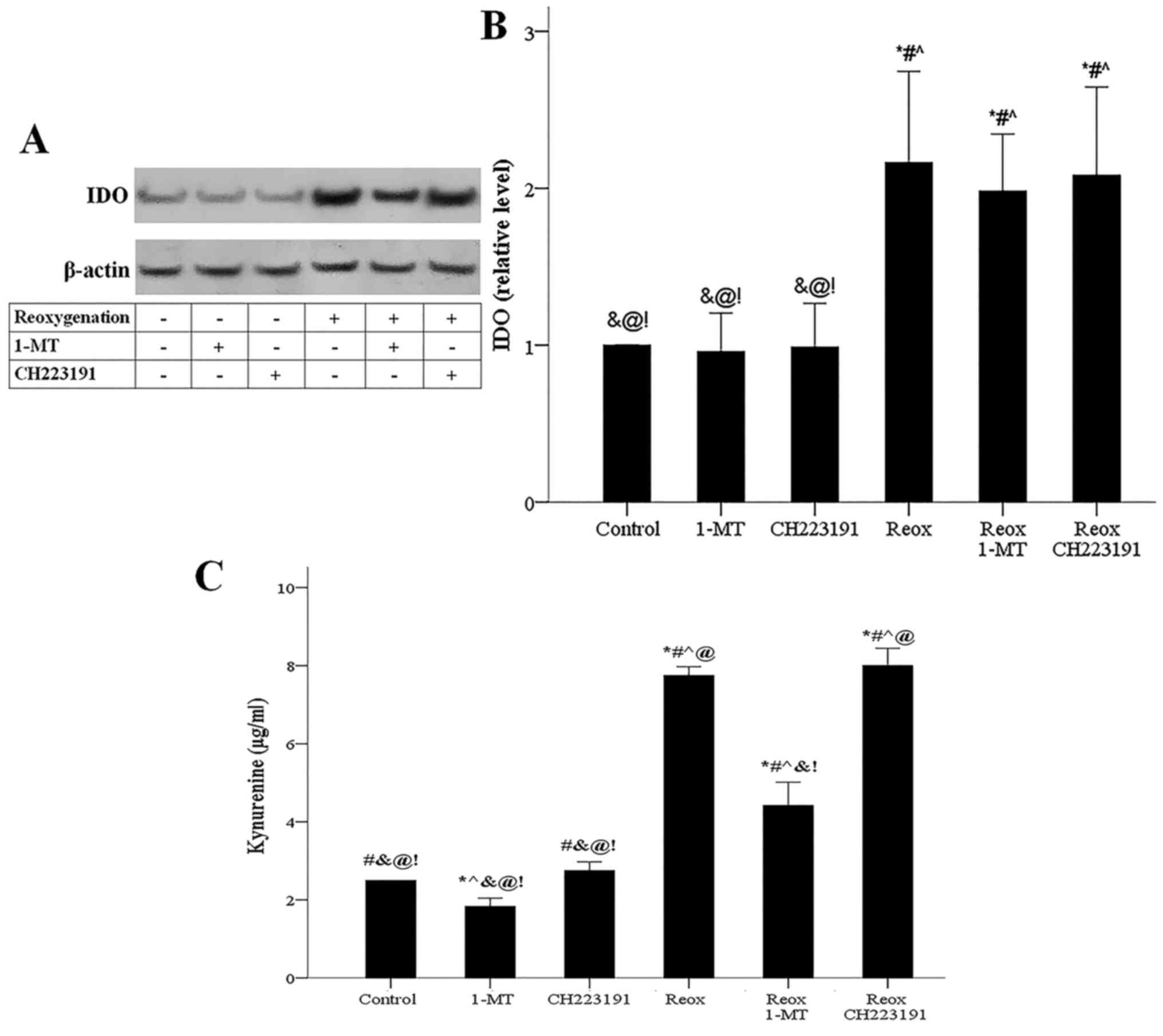
Reoxygenation upregulated IDO expression, while
neither 1-MT nor CH223191 affected IDO (Fig. 9A and B). In cells subjected to
reoxygenation, IDO-overexpression was accompanied by increased
kynurenine production assessed by its concentration in the cell
culture supernatant. Inhibition of IDO mediated by 1-MT decreased
reoxygenation-induced kynurenine-overproduction, while the AhR
inhibitor CH223191 did not affect kynurenine levels (Fig. 9C).
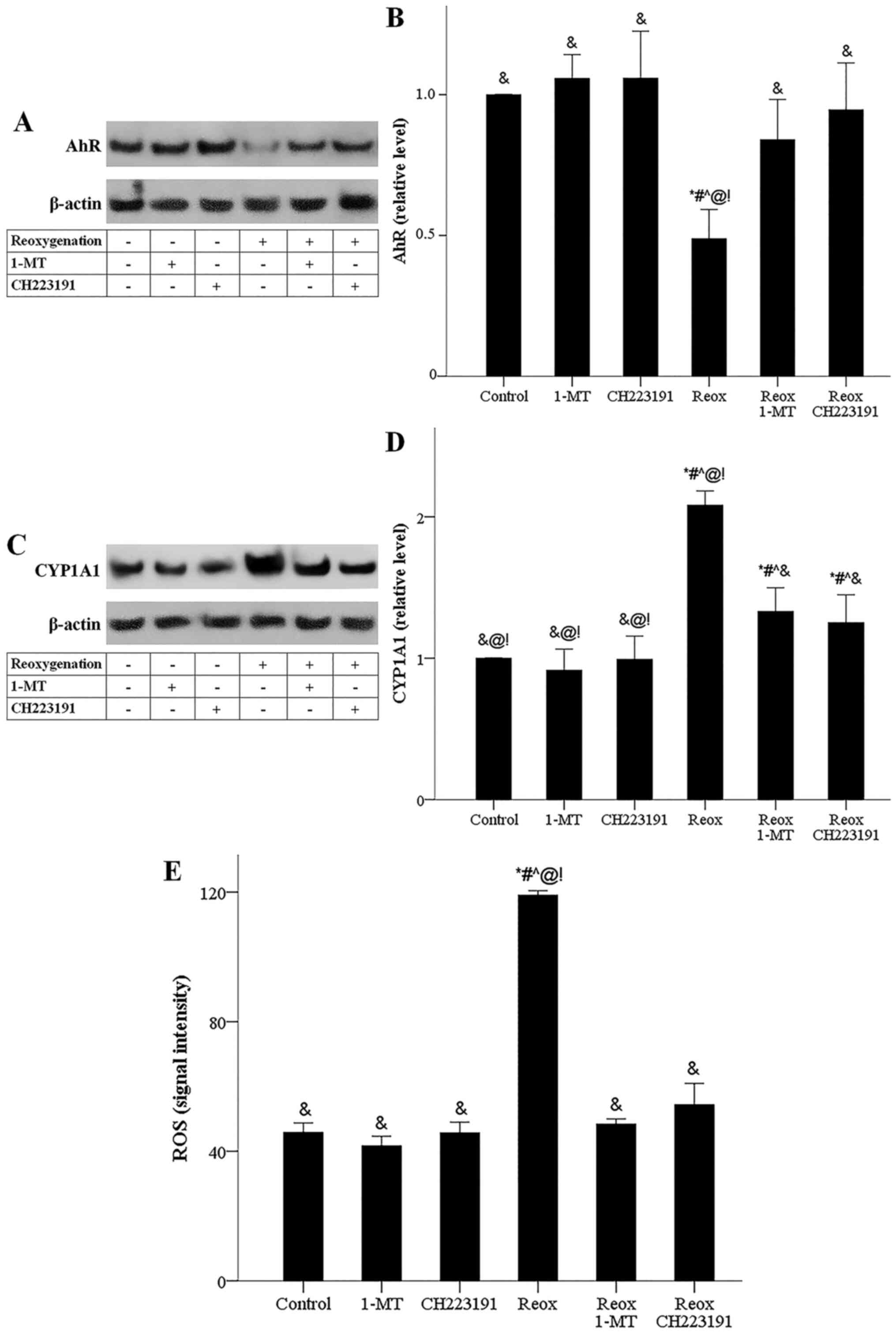
The reoxygenation-induced kynurenine-overproduction
was accompanied by a reduction in AhR levels. Reduction in AhR
levels reflects the activation status of the receptor as only the
activated form of AhR is targeted by proteasomal degradation
leading to decreased AhR cellular levels (20,21).
Under the reoxygenation conditions, both 1-MT, which by inhibiting
IDO prevents kynurenine production (7,8,15), and
CH223192, which inhibits AhR directly (16), prevented AhR activation (Fig. 10A and B). In cells subjected to
reoxygenation, activated AhR led to an enhanced expression of its
transcriptional target, CYP1A1. 1-MT decreased
reoxygenation-induced CYP1A1-overexpression possibly by reducing
kynurenine-induced AhR activation. As expected, the AhR inhibitor
CH223191 also decreased reoxygenation-induced CYP1A1-overexpression
(Fig. 10C and D). In cells
subjected to reoxygenation, CYP1A1 upregulation was accompanied by
RO-overproduction, a prerequisite for ferroptotic cell death
(17). Both 1-MT and CH223191
blocked reoxygenation-induced ROS-overproduction (Fig. 10E).
Discussion
Triggered by experimental data that showed
upregulation of IDO in a model of kidney I-R injury and a
beneficial effect of IDO inhibition (9), the present study evaluated IDO
kinetics and the subsequent molecular pathways that may affect cell
survival under anoxia or reoxygenation in RPTECs.
In accordance with a previous study (12), the current results showed that
anoxia induced apoptotic and not ferroptotic cell death as anoxia
did not increase ROS generation, which is a prerequisite for
ferroptosis (17). Anoxia increased
IDO mRNA and protein expression. The subsequent tryptophan
depletion led to autophosphorylation and activation of the GCN2K,
likely due to increased uncharged tryptophanyl-tRNA (6). GCN2K phosphorylated its substrate
eIF2α, which is known to alter the cellular translational program
(6). In accordance with previous
studies (6,22), p-eIF2α enhanced ATF4, which in turn
upregulated the expression of its transcriptional target CHOP. CHOP
is known to upregulate pro-apoptotic factors involved in both the
intrinsic and extrinsic apoptotic pathways (22). Two pro-apoptotic factors were also
evaluated in the present study. Anoxia-induced increased expression
of CHOP, Bax and DR5. By increasing mitochondrial membrane
permeability, Bax contributes to the intrinsic apoptotic pathway
(19,22). DR5, also known as TNF-related
apoptosis-inducing ligand receptor 2, sensitizes cells to the
extrinsic apoptotic pathway (22).
The role of IDO in sensitizing cultured RPTECs subjected to I-R
injury to the extrinsic apoptotic pathway has been detected
previously (23). Eventually, the
present study observed that anoxia causes apoptotic cell death by
activating the caspase-3, in which all the apoptotic pathways
converge (19). The fact that
anoxia-induced IDO upregulation is responsible for the
aforementioned molecular events was evaluated and confirmed using
the IDO inhibitor 1-MT, which inhibited all the described pathway
components.
Another pro-apoptotic factor that plays a
significant role in I-R-induced apoptosis is transcription factor
p53. Silencing of p53 with siRNA attenuates ischemic acute kidney
injury (24). In accordance with a
previous study (22), the present
study reported that anoxia-induced ATF4-upregulation is accompanied
by increased ATF3 expression. ATF3 upregulates p53 by increasing
its gene transcription or by inhibiting p53 proteasomal degradation
through direct interaction with p53 or with the mouse double minute
2 homolog (MDM2) (25–27). Indeed, in the current study, anoxia
upregulated p53. p53 contributes to the observed increase of Bax
and DR5 expression since p53 transcribes several pro-apoptotic
genes, including the Bax and DR5 (28,29).
1-MT also inhibited all the aforementioned pathway components,
confirming that anoxia-induced IDO-overexpression is responsible
for p53 upregulation.
Another common mechanism of p53 upregulation
involves its phosphorylation at Ser15 by the ataxia-telangiectasia
mutated (ATM)/ataxia-telangiectasia and Rad3-related protein (ATR)
complex. This phosphorylation leads to p53 dissociation from the
MDM2, saving p53 from proteasomal degradation (29). The present study demonstrated that
anoxia enhanced p53 phosphorylation, while 1-MT significantly
decreased this phosphorylation. A previous study reported that
ATM/ATR forms a complex with aminoacyl-tRNA synthetase-interacting
multifunctional protein-3/p18 (AIMP3/p18) in the nucleus in order
to phosphorylate p53 (30).
However, AIMP3/p18 remains in the cytoplasm in a complex with
methionyl-tRNA synthetase (MRS). Activated GCN2K phosphorylates
MRS, allowing AIMP3/p18 release. In its turn AIMP3/p18 translocates
into the nucleus and interacts with ATM/ATR (31). Collectively, the pathways involved
in anoxia-induced IDO-mediated apoptosis are depicted in Fig. 11.
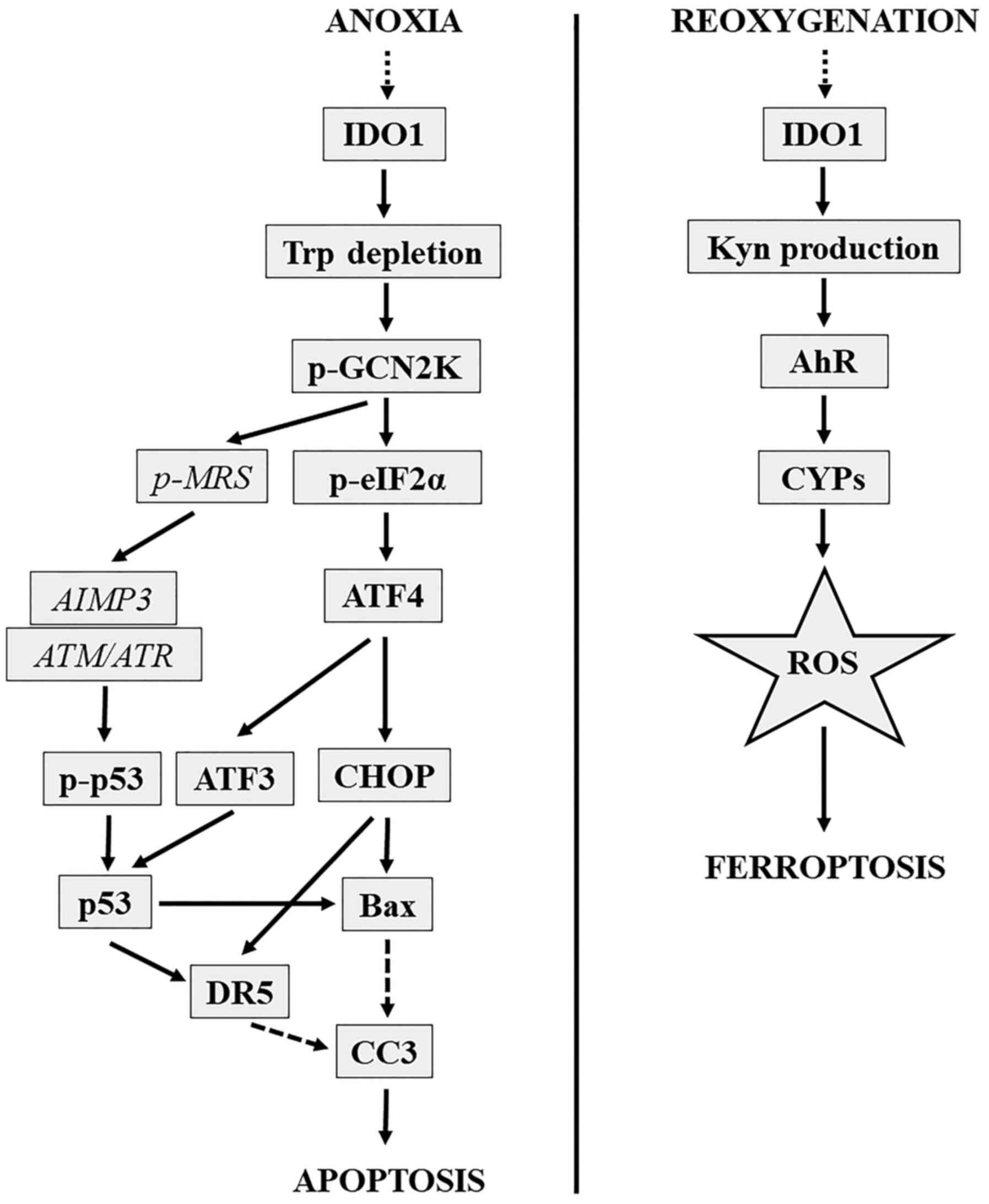 | Figure 11.IDO-mediated anoxia-induced apoptosis
and reoxygenation-induced ferroptosis molecular pathways.
IDO-mediated anoxia-induced apoptotic molecular pathway is depicted
on the left. IDO-mediated reoxygenation-induced ferroptotic
molecular pathway is depicted on the right. AIMP3/p18,
aminoacyl-tRNA synthetase-interacting multifunctional
protein-3/p18; AhR, aryl-hydrocarbon receptor; ATF3, activating
transcription factor 3; ATF4, activating transcription factor 4;
ATM/ATR, ataxia-telangiectasia mutated/ataxia-telangiectasia and
Rad3 related protein complex; CHOP, C/EBP homologous protein; CC3,
cleaved caspase-3; CYP1A1, cytochrome P450 family 1 subfamily A
polypeptide 1; DR5, death receptor 5; IDO, indoleamine
2,3-dioxygenase 1; Kyn, kynurenine; p-, phosphorylated; eIF2a,
eukaryotic translation initiation factor-2α; GCN2K, general control
nonderepressible-2 kinase; MRS, methionyl-tRNA synthetase; p53,
p53; ROS, reactive oxygen species; Trp, tryptophan. |
The exact mechanism that induces IDO expression
under anoxic conditions was not evaluated in the present study.
However, in dendritic cells, anoxia induces ATP release into the
extracellular space where it is converted to adenosine. Free
adenosine induces IDO expression through the adenosine A3 receptor
(32). Also, extracellular ATP can
upregulate IDO in mesenchymal cells directly through purinergic
receptors (33). Since ATP release
is a typical response to various types of stress and in numerous
cell types (34), the possibility
of extracellular ATP-induced IDO expression in RPTECs subjected to
I-R deserves evaluation in future studies.
The present study reported that reoxygenation
induces ferroptotic and not apoptotic cell death, which is in
accordance with previous studies (12–14).
ROS-overproduction is a prerequisite for ferroptotic cell death
(17). Notably, RPTECs may be
particularly vulnerable to ROS since they fail to upregulate
certain anti-oxidant defense mechanisms during reoxygenation
(35). A source of cellular ROS is
the cytochrome P450 superfamily (CYPs) enzymes, which produce ROS
during the oxidation of their substrates (36). Experimental models of heart or liver
I-R injury show that inhibition of CYPs decreases ROS production
and organ dysfunction (37,38). Certain CYPs, particularly CYP1A1,
CYP1A2 and CYP1B1, are transcriptional targets of AhR (39). Notably, in experimental models of
lung or heart I-R injury, inhibition of AhR was beneficial
(40,41). Also, a recent study has shown that
in the context of RPTECs, the primary source of ROS for
reoxygenation-induced ferroptosis are the CYPs, which are
upregulated due to AhR activation (10). AhR is activated by various exogenous
and endogenous ligands with kynurenine, a product of tryptophan
catabolism by IDO, being one of the endogenous AhR ligands
(7,42).
In the present study, reoxygenation upregulated IDO
mRNA level and protein expression. As expected, the subsequent
kynurenine production activated AhR. It should be noted that when
AhR is activated, it becomes vulnerable to proteasomal degradation
resulting in lower total cellular levels (20,21).
The reoxygenation-induced AhR activation was also confirmed by the
increase of the AhR transcriptional target CYP1A1.
CYP1A1-upregulation was accompanied by ROS-overproduction, which
resulted in ferroptotic cell death. To establish the role of IDO in
the aforementioned pathway, the IDO inhibitor 1-MT was added, which
suppressed reoxygenation-induced kynurenine production, AhR
activation, CYP1A1 expression, ROS generation and eventually cell
ferroptosis. The AhR inhibitor CH223191 blocked
reoxygenation-induced AhR activation, CYP1A1 expression, ROS
generation and ultimately cell ferroptosis. The latter confirms
that, in cells subjected to reoxygenation, IDO-upregulation induces
cell ferroptosis through the AhR pathway. Collectively, the
molecular pathway involved in the reoxygenation-induced
IDO-mediated ferroptosis is depicted in Fig. 11.
From a teleological perspective, the upregulation of
IDO under anoxia- or reoxygenation-induced stress should be part of
an adaptive mechanism aiming to protect the cell from the noxious
insult. As with most adaptive mechanisms, depending on the
intensity or the duration of the noxious insult, there may be
limits in the ability of IDO to protect the cell. There are
numerous such paradigms of adaptive responses, such as the
genotoxic stress response, the endoplasmic reticulum stress
response, the amino acid deprivation stress, autophagy or the
ferroptotic mechanism (6,22,43,44).
These attempt to restore cellular homeostasis against various
stressors. However, if the insult is too intense or lasts too long,
the same adaptive responses eventually lead to cell death (6,22,43,44).
It seems that RPTECs are so vulnerable to anoxia and reoxygenation
that at the time points used in the present study that they
committed to death. It should be noted that the time points
selected for the current experiments were based on the time needed
for cell death. It is possible that a shorter exposure of RPTECs to
anoxia had been used, then a protective role of IDO would be
revealed. It was demonstrated that IDO upregulated p53.
Hypothetically, a shorter time of exposure to anoxia may have
resulted in p53-induced p21-upregulation, which has an
anti-apoptotic effect (45). For
instance, albeit in other cell types, a study showed that under
hypoxic conditions, activation of GCN2K upregulates p53, resulting
in cell cycle arrest through p21-overexpression. However, p21
downregulates Bax, ultimately reducing apoptosis (46). On the contrary, at the time points
used in the present study, p53 induced Bax and DR5 expression and
eventually apoptosis. Certainly, due to the complexity of the
pathways evaluated in the current study, this subject deserves
further evaluation. However, in clinical practice, the ischemic
insult that may cause acute kidney injury usually lasts longer than
the time applied in the present study.
The lack of in vivo verification of the
experiments is a limitation of the current study. However, the
in vitro approach allowed us to apply strict experimental
conditions and evaluate the two pathophysiologically distinct
phases of I-R injury, ischemia and reperfusion, separately. Thus,
the present study could be considered as a starting point for
further in vivo studies.
Besides further clarifying the molecular mechanisms
involved in I-R injury, another important finding of the present
study is that both ischemia and reperfusion share a common feature;
IDO upregulation. This raises the opportunity to intervene in both
phases of I-R injury at once with a single therapy. Notably,
efforts to interfere with I-R injury by altering tryptophan levels
are feasible by administering tryptophan or applying a
tryptophan-free diet. Tryptophan is an essential amino acid not
synthesized by human cells, and its concentration is the lowest
among all the amino acids. In humans, a 2-day low tryptophan intake
results in tryptophan depletion (47). However, according to the current
results, tryptophan supplementation is expected to alleviate
apoptosis during the ischemic phase by decreasing GCN2K activation.
During the reperfusion phase, tryptophan supplementation is
expected to worsen ferroptosis by increasing kynurenine production
and AhR activation. On the other hand, tryptophan depletion is
expected to ameliorate ferroptosis during reperfusion and increase
apoptosis during ischemia. Thus, inhibition of IDO seems to be a
more reliable approach for attenuating I-R injury. Of note, various
IDO inhibitors have already been developed and tested in human
clinical trials for cancer immunotherapy (48).
In conclusion, in RPTECs, both anoxia and
reoxygenation upregulate IDO, which in turn induces GCN2K-mediated
apoptosis and AhR-mediated ferroptosis, respectively. The
inhibition of IDO may prove a useful therapeutic strategy for
preventing or attenuating I-R injury.
Acknowledgements
Not applicable.
Funding
No funding was received.
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
TE designed the study. GP and TE performed the
experiments, and collected the data. TE and GP confirm the
authenticity of all raw data. TE interpreted the data with help
from GP, SG, VL and IS. TE, GP, SG, VL and IS analyzed the results.
TE wrote the manuscript with help from GP. All authors read and
approved the final manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Wu MY, Yiang GT, Liao WT, Tsai AP, Cheng
YL, Cheng PW, Li CY and Li CJ: Current mechanistic concepts in
ischemia and reperfusion injury. Cell Physiol Biochem.
46:1650–1667. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Bonventre JV and Yang L: Cellular
pathophysiology of ischemic acute kidney injury. J Clin Invest.
121:4210–4221. 2011. View
Article : Google Scholar : PubMed/NCBI
|
|
3
|
King NJ and Thomas SR: Molecules in focus:
Indoleamine 2,3-dioxygenase. Int J Biochem Cell Biol. 39:2167–2172.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Munn DH, Zhou M, Attwood JT, Bondarev I,
Conway SJ, Marshall B, Brown C and Mellor AL: Prevention of
allogeneic fetal rejection by tryptophan catabolism. Science.
281:1191–1193. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Eleftheriadis T, Pissas G, Antoniadi G,
Spanoulis A, Liakopoulos V and Stefanidis I: Indoleamine
2,3-dioxygenase increases p53 levels in alloreactive human T cells,
and both indoleamine 2,3-dioxygenase and p53 suppress glucose
uptake, glycolysis and proliferation. Int Immunol. 26:673–684.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Castilho BA, Shanmugam R, Silva RC, Ramesh
R, Himme BM and Sattlegger E: Keeping the eIF2 alpha kinase Gcn2 in
check. Biochim Biophys Acta. 1843:1948–1968. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Mezrich JD, Fechner JH, Zhang X, Johnson
BP, Burlingham WJ and Bradfield CA: An interaction between
kynurenine and the aryl hydrocarbon receptor can generate
regulatory T cells. J Immunol. 185:3190–3198. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Eleftheriadis T, Pissas G, Liakopoulos V
and Stefanidis I: IDO decreases glycolysis and glutaminolysis by
activating GCN2K, while it increases fatty acid oxidation by
activating AhR, thus preserving CD4+ T cell survival and
proliferation. Int J Mol Med. 42:557–568. 2018.PubMed/NCBI
|
|
9
|
Mohib K, Wang S, Guan Q, Mellor AL, Sun H,
Du C and Jevnikar AM: Indoleamine 2,3-dioxygenase expression
promotes renal ischemia-reperfusion injury. Am J Physiol Renal
Physiol. 295:F226–F234. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Eleftheriadis T, Pissas G, Filippidis G,
Liakopoulos V and Stefanidis I: Reoxygenation induces reactive
oxygen species production and ferroptosis in renal tubular
epithelial cells by activating aryl hydrocarbon receptor. Mol Med
Rep. Nov 10–2020.(Epub ahead of print). doi:
10.3892/mmr.2020.11679. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Khan S, Cleveland RP, Koch CJ and
Schelling JR: Hypoxia induces renal tubular epithelial cell
apoptosis in chronic renal disease. Lab Invest. 79:1089–1099.
1999.PubMed/NCBI
|
|
12
|
Eleftheriadis T, Pissas G, Antoniadi G,
Liakopoulos V and Stefanidis I: cell death patterns due to warm
ischemia or reperfusion in renal tubular epithelial cells
originating from human, mouse, or the native hibernator hamster.
Biology (Basel). 7:72018.PubMed/NCBI
|
|
13
|
Eleftheriadis T, Pissas G, Liakopoulos V
and Stefanidis I: Factors that may protect the native hibernator
syrian hamster renal tubular epithelial cells from ferroptosis due
to warm anoxia-reoxygenation. Biology (Basel). 8:82019.
|
|
14
|
Linkermann A, Skouta R, Himmerkus N, Mulay
SR, Dewitz C, De Zen F, Prokai A, Zuchtriegel G, Krombach F, Welz
PS, et al: Synchronized renal tubular cell death involves
ferroptosis. Proc Natl Acad Sci USA. 111:16836–16841. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Jia L, Schweikart K, Tomaszewski J, Page
JG, Noker PE, Buhrow SA, Reid JM, Ames MM and Munn DH: Toxicology
and pharmacokinetics of 1-methyl-d-tryptophan: Absence of toxicity
due to saturating absorption. Food Chem Toxicol. 46:203–211. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Kim SH, Henry EC, Kim DK, Kim YH, Shin KJ,
Han MS, Lee TG, Kang JK, Gasiewicz TA, Ryu SH, et al: Novel
compound 2-methyl-2H-pyrazole-3-carboxylic acid
(2-methyl-4-o-tolylazo-phenyl)-amide (CH-223191) prevents
2,3,7,8-TCDD-induced toxicity by antagonizing the aryl hydrocarbon
receptor. Mol Pharmacol. 69:1871–1878. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Stockwell BR, Friedmann Angeli JP, Bayir
H, Bush AI, Conrad M, Dixon SJ, Fulda S, Gascón S, Hatzios SK,
Kagan VE, et al: Ferroptosis: A regulated cell death nexus linking
metabolism, redox biology, and disease. Cell. 171:273–285. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Lobner D: Comparison of the LDH and MTT
assays for quantifying cell death: Validity for neuronal apoptosis?
J Neurosci Methods. 96:147–152. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Fadeel B and Orrenius S: Apoptosis: A
basic biological phenomenon with wide-ranging implications in human
disease. J Intern Med. 258:479–517. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Pollenz RS: The mechanism of AH receptor
protein down-regulation (degradation) and its impact on AH
receptor-mediated gene regulation. Chem Biol Interact. 141:41–61.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Yang SC, Wu CH, Tu YK, Huang SY and Chou
PC: Exposure to 2,3,7,8-tetrachlorodibenzo-p-dioxin increases the
activation of aryl hydrocarbon receptor and is associated with the
aggressiveness of osteosarcoma MG-63 osteoblast-like cells. Oncol
Lett. 16:3849–3857. 2018.PubMed/NCBI
|
|
22
|
Hu H, Tian M, Ding C and Yu S: The C/EBP
homologous protein (CHOP) transcription factor functions in
endoplasmic reticulum stress-induced apoptosis and microbial
infection. Front Immunol. 9:30832019. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Mohib K, Guan Q, Diao H, Du C and Jevnikar
AM: Proapoptotic activity of indoleamine 2,3-dioxygenase expressed
in renal tubular epithelial cells. Am J Physiol Renal Physiol.
293:F801–F812. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Molitoris BA, Dagher PC, Sandoval RM,
Campos SB, Ashush H, Fridman E, Brafman A, Faerman A, Atkinson SJ,
Thompson JD, et al: siRNA targeted to p53 attenuates ischemic and
cisplatin-induced acute kidney injury. J Am Soc Nephrol.
20:1754–1764. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
You Z, Xu J, Li B, Ye H, Chen L, Liu Y and
Xiong X: The mechanism of ATF3 repression of epithelial-mesenchymal
transition and suppression of cell viability in cholangiocarcinoma
via p53 signal pathway. J Cell Mol Med. 23:2184–2193. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Wang Z, He Y, Deng W, Lang L, Yang H, Jin
B, Kolhe R, Ding HF, Zhang J, Hai T, et al: Atf3 deficiency
promotes genome instability and spontaneous tumorigenesis in mice.
Oncogene. 37:18–27. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Li X, Guo M, Cai L, Du T, Liu Y, Ding HF,
Wang H, Zhang J, Chen X and Yan C: Competitive ubiquitination
activates the tumor suppressor p53. Cell Death Differ.
27:1807–1818. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Taketani K, Kawauchi J, Tanaka-Okamoto M,
Ishizaki H, Tanaka Y, Sakai T, Miyoshi J, Maehara Y and Kitajima S:
Key role of ATF3 in p53-dependent DR5 induction upon DNA damage of
human colon cancer cells. Oncogene. 31:2210–2221. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Aubrey BJ, Kelly GL, Janic A, Herold MJ
and Strasser A: How does p53 induce apoptosis and how does this
relate to p53-mediated tumour suppression? Cell Death Differ.
25:104–113. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Park BJ, Kang JW, Lee SW, Choi SJ, Shin
YK, Ahn YH, Choi YH, Choi D, Lee KS and Kim S: The
haploinsufficient tumor suppressor p18 upregulates p53 via
interactions with ATM/ATR. Cell. 120:209–221. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Kwon NH, Kang T, Lee JY, Kim HH, Kim HR,
Hong J, Oh YS, Han JM, Ku MJ, Lee SY, et al: Dual role of
methionyl-tRNA synthetase in the regulation of translation and
tumor suppressor activity of aminoacyl-tRNA synthetase-interacting
multifunctional protein-3. Proc Natl Acad Sci USA. 108:19635–19640.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Song X, Zhang Y, Zhang L, Song W and Shi
L: Hypoxia enhances indoleamine 2,3-dioxygenase production in
dendritic cells. Oncotarget. 9:11572–11580. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Lotfi R, Steppe L, Hang R, Rojewski M,
Massold M, Jahrsdörfer B and Schrezenmeier H: ATP promotes
immunosuppressive capacities of mesenchymal stromal cells by
enhancing the expression of indoleamine dioxygenase. Immun Inflamm
Dis. 6:448–455. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Dosch M, Gerber J, Jebbawi F and Beldi G:
Mechanisms of ATP release by inflammatory cells. Int J Mol Sci.
19:192018. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Eleftheriadis T, Pissas G, Nikolaou E,
Filippidis G, Liakopoulos V and Stefanidis I: Mistimed H2S
upregulation, Nrf2 activation and antioxidant proteins levels in
renal tubular epithelial cells subjected to anoxia and
reoxygenation. Biomed Rep. 13:32020.PubMed/NCBI
|
|
36
|
Hrycay EG and Bandiera SM: Monooxygenase,
peroxidase and peroxygenase properties and reaction mechanisms of
cytochrome P450 Enzymes. Adv Exp Med Biol. 851:1–61. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Ishihara Y, Sekine M, Nakazawa M and
Shimamoto N: Suppression of myocardial ischemia-reperfusion injury
by inhibitors of cytochrome P450 in rats. Eur J Pharmacol.
611:64–71. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Shaik IH and Mehvar R: Effects of
cytochrome p450 inhibition by cimetidine on the warm hepatic
ischemia-reperfusion injury in rats. J Surg Res. 159:680–688. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Nebert DW, Dalton TP, Okey AB and Gonzalez
FJ: Role of aryl hydrocarbon receptor-mediated induction of the
CYP1 enzymes in environmental toxicity and cancer. J Biol Chem.
279:23847–23850. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Cuartero MI, Ballesteros I, de la Parra J,
Harkin AL, Abautret-Daly A, Sherwin E, Fernández-Salguero P, Corbí
AL, Lizasoain I and Moro MA: L-kynurenine/aryl hydrocarbon receptor
pathway mediates brain damage after experimental stroke.
Circulation. 130:2040–2051. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Couroucli XI, Liang YW, Jiang W, Barrios R
and Moorthy B: Attenuation of oxygen-induced abnormal lung
maturation in rats by retinoic acid: Possible role of cytochrome
P4501A enzymes. J Pharmacol Exp Ther. 317:946–954. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Stejskalova L, Dvorak Z and Pavek P:
Endogenous and exogenous ligands of aryl hydrocarbon receptor:
Current state of art. Curr Drug Metab. 12:198–212. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Bartek J and Lukas J: DNA damage
checkpoints: From initiation to recovery or adaptation. Curr Opin
Cell Biol. 19:238–245. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Galluzzi L, Bravo-San Pedro JM, Vitale I,
Aaronson SA, Abrams JM, Adam D, Alnemri ES, Altucci L, Andrews D,
Annicchiarico-Petruzzelli M, et al: Essential versus accessory
aspects of cell death: Recommendations of the NCCD 2015. Cell Death
Differ. 22:58–73. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Mirzayans R, Andrais B, Kumar P and Murray
D: Significance of wild-type p53 signaling in suppressing apoptosis
in response to chemical genotoxic agents: Impact on chemotherapy
outcome. Int J Mol Sci. 18:9282017. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Liu Y, László C, Liu Y, Liu W, Chen X,
Evans SC and Wu S: Regulation of G(1) arrest and apoptosis in
hypoxia by PERK and GCN2-mediated eIF2α phosphorylation. Neoplasia.
12:61–68. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Ellenbogen MA, Young SN, Dean P, Palmour
RM and Benkelfat C: Mood response to acute tryptophan depletion in
healthy volunteers: Sex differences and temporal stability.
Neuropsychopharmacology. 15:465–474. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Le Naour J, Galluzzi L, Zitvogel L,
Kroemer G and Vacchelli E: Trial watch: IDO inhibitors in cancer
therapy. OncoImmunology. 9:17776252020. View Article : Google Scholar : PubMed/NCBI
|