Introduction
Calpainopathy, or limb-girdle muscular dystrophy
recessive 1 [LGMDR1; Online Mendelian Inheritance in Man
(OMIM):253600], previously known as limb-girdle muscular dystrophy
type 2A (LGMD2A), has an incidence of 1 in 100,000 and is the
commonest type of LGMD in Europe, accounting for approximately 30%
of muscular dystrophies. However, LGMDs are relatively rare in
China (1–3). The clinical manifestations of LGMDR1
are similar to those of other types of LGMDs, including progressive
weakness and muscle atrophy in the shoulder, pelvic girdle muscles
and proximal limb muscles. Notably, lower limb and body muscles are
most affected. Some patients present with an abnormal gait or
difficulty climbing stairs and lifting weights, accompanied by
aggravated locomotor abilities (4–6). The
phenotypic manifestations of LGMDR1 are highly heterogeneous and
can be inconsistent even within the same family. Symptoms and
physical signs may appear at any age and generally worsen over
time, although they remain mild in some cases (7,8). As
the pathophysiological mechanism underlying LGMDR1 is currently
unknown, no effective treatment is available for this disease.
Variants in calcium-activated neutral proteinase 3
(CAPN3; OMIM:114240) can cause LGMDR1 (autosomal recessive)
and limb-girdle muscular dystrophy dominant 4 (LGMDD4; LGMD1I;
OMIM:618129; autosomal dominant), the clinical manifestations of
which are similar except for its morbidity, inheritance patterns
and severity of symptoms (9).
LGMDR1 is the first disease for which the genetic cause has been
conclusively attributed to defective CAPN3 via specific
molecular biological effects (10).
To date, more than 500 different CAPN3 variants have been
reported in the Leiden muscular dystrophy database (https://databases.lovd.nl/shared/genes/CAPN3)
(11). The commonest type are
missense variants (~60%), which are mostly compound heterozygous
variants (12–14). The three different autosomal
recessive subtypes associated with CAPN3 variants can be
diagnosed based on the distribution of muscle weakness and age at
the time of onset: Shoulder- and limb-girdle psoas muscular
dystrophy (also known as Leiden-Mobius LGMD), shoulder and bone
LGMD (also known as Erb LGMD) and hyper-creatine kinase-emia, which
is typically observed in children or young adults (5). In most cases, the affected individuals
do not present muscular symptoms but exhibit elevated serum
creatine kinase (CK) levels (5,15).
CAPN3, localized on chromosome 15q15.1
(16–18), spans more than 138 kb of genomic DNA
with 24 exons and encodes a 94-kDa protease enzyme, calpain 3,
consisting of 821 amino acids (NM_000070.2) (19). Calpain 3 protein, a
calcium-dependent non-lysosomal neutral cysteine protease widely
found in the sarcomere structure, serves a role in muscle
regeneration, sarcolemmal repair, muscle assembly and remodeling,
as well as cytoskeleton regulation and calcium homeostasis
(20,21). The main structure of calpain 3
includes four functional domains, a cysteine protease (II) and
penta-EF-hand (PEF) (IV) domains, that respectively code for
cysteine protease and calcium-binding domains involved in
calcium-dependent enzyme activation (22,23).
Due to the high clinical and genetic heterogeneity
of LGMDs, it is challenging to distinguish different subtypes only
on the basis of clinical symptoms and physical signs (24–26).
As such, a large number of patients may be misdiagnosed in the
early stage. Thus, genetic testing is strongly recommended to offer
a reliable and conclusive differential diagnosis.
The present study described a 23-year-old man who
presented with consistently elevated serum CK as his only symptom
and was initially diagnosed with polymyositis. A strategy was
devised to couple next-generation sequencing (NGS) with Sanger
sequencing validation to identify and validate the disease-causing
variants in this proband. The results provided insight for further
research on the pathogenesis of LGMDs and to accelerate prenatal
diagnosis development.
Materials and methods
Subjects
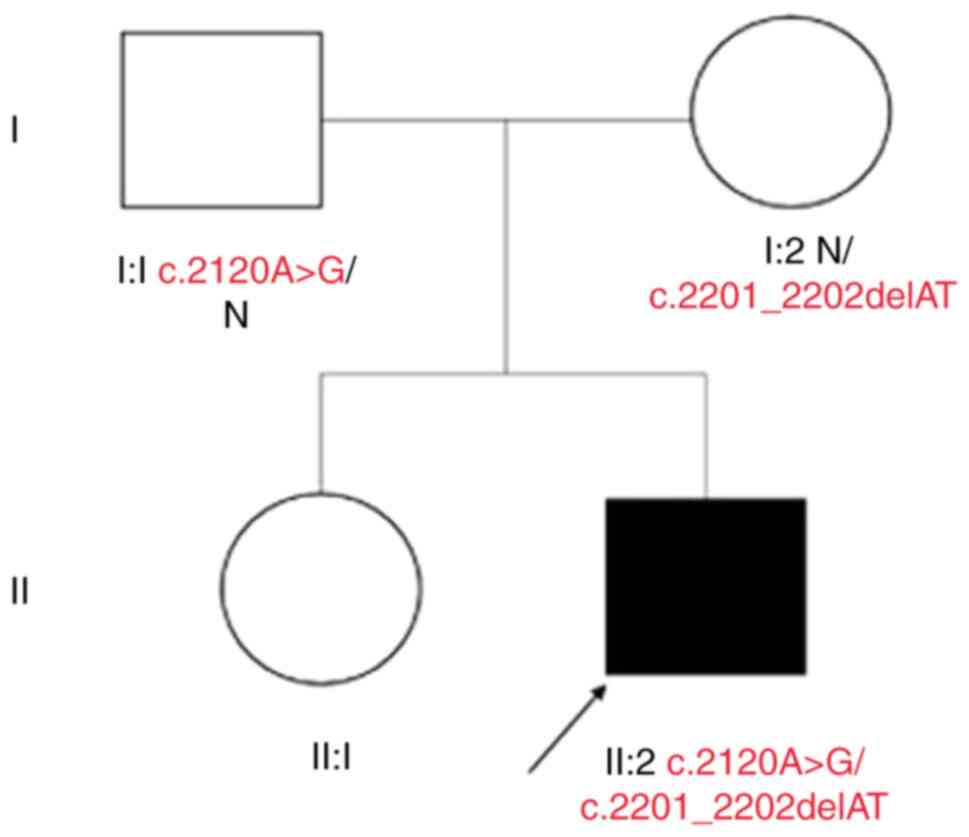
The proband (II:2), a 23-year-old male born to
healthy, non-consanguineous parents (Fig. 1), was admitted to the Affiliated
Hospital of Qingdao University due to consistently elevated,
although fluctuating, CK levels. Based on laboratory examinations,
the proband was initially diagnosed with polymyositis by the Immune
Department of the Affiliated Hospital of Qingdao University
(Shandong, China). However, a successive series of physical
examinations and auxiliary tests, including serum CK measurement,
electromyography, magnetic resonance imaging (MRI) and muscle
biopsy, were performed to complement and validate the presumptive
diagnosis. Control samples were randomly collected from patients
who presented to The Affiliated Hospital of Qingdao University
since August 2020. The control group comprised 200 healthy
individuals (age, 18–65 years) from the ethnic Han Chinese
population in Shandong Province. The male-to-female ratio was 1:1.
The proband and parents as well as individuals in the control group
signed informed consent forms prior to venous blood collection. The
present study was approved by the ethics committee of the
Affiliated Hospital of Qingdao University (approval no.
qdfy20203789) and conformed to the guidelines set forth by the
Declaration of Helsinki.
Genomic DNA preparation
From a three-person family, genomic DNA (gDNA) was
extracted from 200 µl of peripheral blood samples in the presence
of ethylenediaminetetraacetic acid as an anticoagulant with a DNA
Extraction kit (Qiagen GmbH) according to the manufacturer's
instructions. The optical density value of the gDNA was determined
with a Sim-100 ultramicro spectrophotometer (Thermo Fisher
Scientific, Inc.). gDNA samples were prepared as Illumina
sequencing libraries. Targeted sequencing was performed on the
proband samples (II:2). The pathogenic variant was validated by
Sanger sequencing in both proband and parental samples.
Whole-exome sequencing (WES) for
variant screening
WES technology was used to screen the mutated sites
in CAPN3 (NM_000070.2, NP_000061.1). WES was performed according to
the human exome capture protocol from Illumina's TruSeq Exome
Enrichment Guide (SureSelectXT Target Enrichment System for
Illumina Paired-End Sequencing Library, Agilent Technologies,
Inc.). The exome enrichment probe sets were constructed with the
Agilent Human All Exon 50 Mb Exome Enrichment kit and sequenced on
a HiSeq 4000 NGS platform (Illumina, Inc.). The captured gDNA
library was sequenced on the Illumina HiSeq 4000 platform and 200
(2×100) bp were generated from the final library fragment using V2
Reagent 1.8 software (Illumina, Inc.; data after June 22, 2011).
The average depth of the target area was 257.15 and the target
bases with coverage of at least 50× were 75.81%, 20× were 82.23%,
10× were 89.04%, 4× were 93.56% and 1× were 96.09%, respectively.
Following sequencing, low-quality variants were filtered out to
obtain clean reads. Paired-end sequence reads were mapped to the
human reference genome hg19 with Burrows-Wheeler Aligner (27). Genome Analysis Toolkit software
(https://gatk.broadinstitute.org/hc/en-us) (28) was used to identify single-nucleotide
polymorphisms and insertions or deletions. All identified variants
were submitted to ANNOVAR (version 2020Oct07) (29) for functional annotation and genetic
filtering. All information presented in the present study was
directly extracted from the reference data set or calculated in
batches of all variants. Common variants were excluded by
comparison with >1,000 exomes sequenced in our laboratory for
unrelated conditions and subsequently filtered with the dbSNP v137
(ncbi.nlm.nih.gov/snp/), 1000 Genomes Project
(ncbi.nlm.nih.gov/variation/tools/1000genomes/), National Heart,
Lung and Blood Institute, Exome Sequencing Project
(evs.gs.washington.edu/EVS/) and ExAC databases
(exac.broadinstitute.org). Variant filtration was based on the
following criteria: i) Excluding untranslated region 3 and 5
variants, non-coding RNA intron variants and intron and synonymous
variants; ii) excluding minor allele frequency >0.1 variants
(30); and iii) ≥50% of the harmful
variants in the bioinformatics software [PolyPhen-2
(genetics.bwh.harvard.edu/pph2/), SIFT
(sift.jcvi.org/www/SIFT_BLink_submit.html) and Mutation Taster
(mutationtaster.org/ChrPos.html)] were retained.
Sanger sequencing validation
The variations detected by WES were validated by
Sanger sequencing. Healthy controls (200) from the Han Chinese
ethnic population in Shandong Province were randomly selected for
validation. The male-to-female ratio was 1:1. The two pairs of
primers covering the variants were: Forward,
5′-ATCCTGCCCAAGCAAAAGTG-3′; reverse, 3′-GCCGGACTGGTCTGTGTCAT-5′ for
c.2120A>G in exon 20 (primer pair one) and forward,
5′-GGTAGGACAGCCCGGAGTCT-3′; reverse: 5′-TTGGGCCTGCCTTCTATTTTC-3′
for c.2201_2202delAT in exon 21 (primer pair two). Identical
amplification conditions for were used for both primer pairs in a
total volume of 25 µl containing 250 nM dNTPs, 100 ng of template
DNA, 0.5 mM of each primer and 1.25 units AmpliTaq Gold DNA
polymerase in 1X reaction buffer (10 mM Tris HCl, pH 8.3, 50 mM
KCl, 2.5 mM MgCl2). PCR was performed as follows:
initial denaturation at 94°C for 5 min, followed by 35 cycles
comprising denaturation at 94°C for 30 sec, annealing at 55°C for
primer pair one and 58°C for primer pair two for 60 sec and
extension at 72°C for 30 sec; after which a final extension was
conducted at 72°C for 10 min. Amplified PCR products were purified
and sequenced using appropriate PCR primers and a BigDye Terminator
Cycle Sequencing kit (Applied Biosystems; Thermo Fisher Scientific,
Inc.) and evaluated on an ABI 3730XL automated sequencer (Applied
Biosystems; Thermo Fisher Scientific, Inc.) for variant
analysis.
Bioinformatics analysis
Functional impacts of the variants were predicted
in silico using the following software. Multiple sequence
alignment was conducted to analyze gene sequence conservation. The
CAPN3 amino acid sequences from multiple species were obtained from
the UniProt website (http://www.uniprot.org). PolyPhen-2 (http://genetics.bwh.harvard.edu/pph2/)
and SIFT (http://sift.jcvi.org/www/SIFT_BLink_submit.html)
software were used to analyze and predict the function of the
identified variants.
The structure of a CAPN3 orthologue (PDB code 4OKH)
was used to predict the structural role of CAPN3 residues (31). Using the SWISS-MODEL program
(31), comparative modeling and
energy minimization methods were used to model the
three-dimensional structure of CAPN3 (32). The orthologous residues were
determined by alignment with the amino acid sequence of ClustalW-2.
PyMOL-1 (pymol.org/2/ V 2.4.1) was used to simulate and visualize
the three-dimensional protein structure of the wild-type (WT) and
mutant proteins.
Results
Clinical features
The proband was a sporadic case with no related
symptoms manifesting in the parents. The proband was admitted to
the Affiliated Hospital of Qingdao University for consistently
elevated, although fluctuating, CK levels. The clinical course was
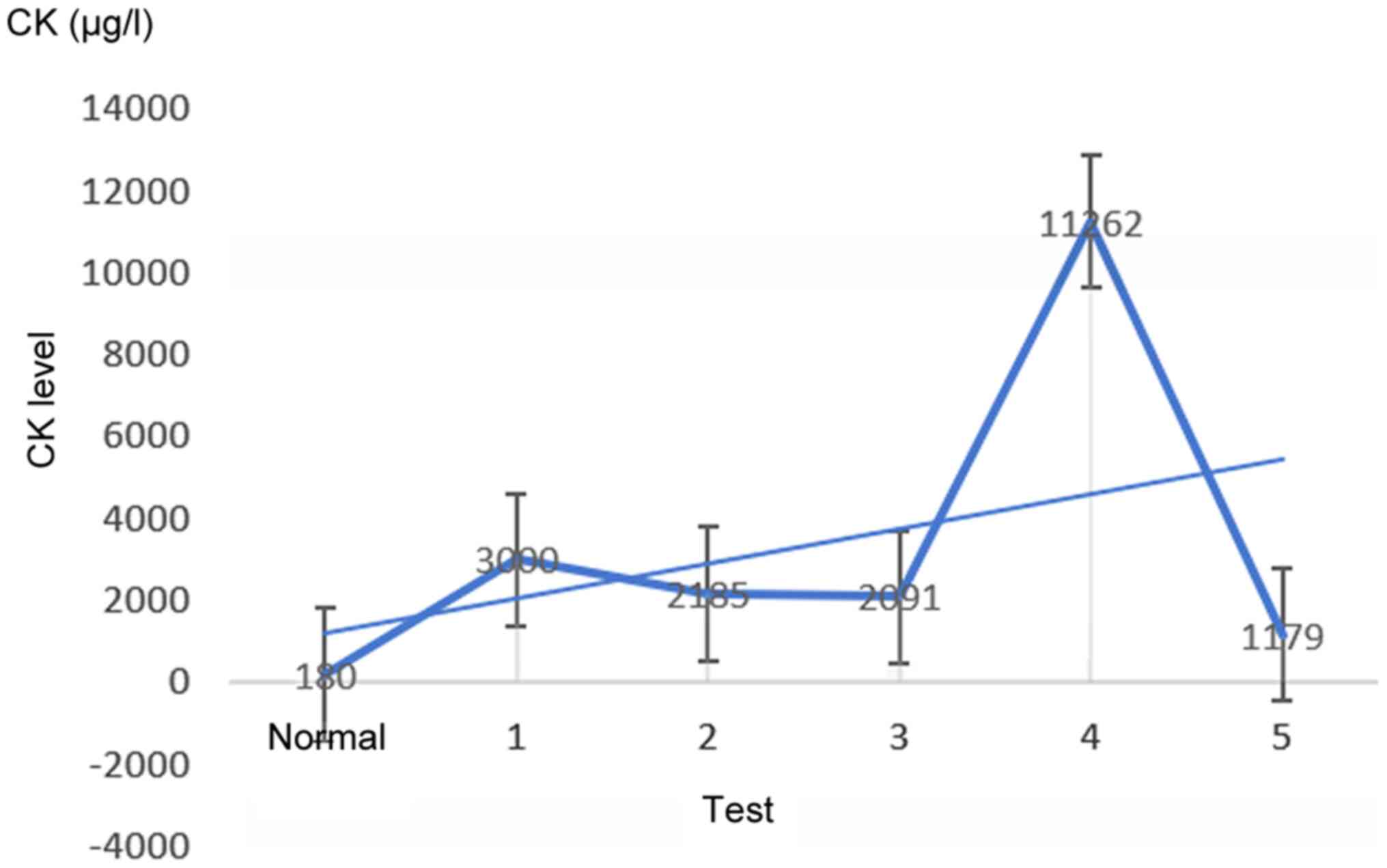
as follows: The proband was found to have elevated CK (3,000 µg/l;
physiological range 38–174 µg/l) when he visited the hospital for a
skin rash in 2015, without any muscle aches. Then, seven days
later, the patient was admitted to the Department of Immunology of
the hospital (CK: 2,185 µg/l) and was initially diagnosed as
possible polymyositis; thus, he was administered hormone combined
with immunosuppressive therapy. No evident therapeutic effect was
observed and his CK levels widely fluctuated during treatment.
Until 2020, the patient presented no abnormalities, a normal
walking posture and regular squatting and stair climbing abilities
and could lift heavy objects. As the elevated CK levels did not
affect his everyday life, this condition remained undiagnosed and
untreated. On June 1, 2020, for the first time, the patient felt
weakness in the left lower limb after several hours of driving. At
that time, the CK level was 2,091 µg/l; except for slightly uneven
shoulder blades, no muscular abnormalities were observed. The
muscle volume was normal and no evidently abnormal distribution was
evidenced by muscle MRI. However, 20 days later, the CK level was
11,262 µg/l for no apparent reasons. Following hormone therapy, the
CK level decreased to 1,179 µg/l. In addition to increased CK, the
patient also had increased total bilirubin (61.6 µmol/l) and direct
bilirubin (16.7 µmol/l). Since disease onset, the patient's weight
did not significantly vary and no family member showed similar
symptoms. Except for the persistently elevated CK level, the
patient did not show numbness, tingling, or pain in his hands or
feet and other general physical symptoms were normal, including
eating, urination, defecation and cranial nerve function.
The patient showed a normal cognitive function in
the neuropsychological test via the Minor Mental State Examination
(33). On physical examination, the
patient presented approximately average muscle strength and volume.
Muscle strength was graded according to the Medical Research
Council scale (34): Neck flexors
5-/5, neck extensors 5/5, bilateral shoulder abductors 5-/5, left
elbow flexors/extensors 5/5, right elbow flexors/extensors 5/5,
bilateral hip flexors/extensors, bilateral wrist flexors/extensors,
knee flexors/extensors and ankle dorsiflexors/plantar flexors 5-/5.
No extraocular, facial, or bulbar muscle weakness or significant
muscle atrophies were detected.
Laboratory examination showed a regular blood
routine, serum potassium, erythrocyte sedimentation rate and
C-reactive protein level. Markedly, the CK had been maintained at a
high-level, reaching a maximum of 11,262 µg/l, which is 50-fold
higher than the physiological level and independent of symptom
severity (Fig. 2). The CK-MB and
lactate dehydrogenase of the patient increased to 96 and 356 µg/l,
respectively, which may cause inflammatory myositis (35). Electromyography examination showed
no noticeable myopathy changes, including motor unit action
potentials with short duration, small amplitude and increased
polyphasic potentials. Muscle MRI did not show abnormal signs of
asymmetric muscle involvement. Muscle biopsy revealed a muscle
tissue morphology consistent with normal muscle tissue and the
results of immunohistochemical staining were normal. Genetic
testing was performed after LGMD was diagnosed.
Genetic analysis
The average depth of sequencing was 257.15×; coupled
with the results of Sanger sequencing verification, high sequencing
reliability was obtained. WES data were filtered to exclude
non-genetic variants and then compared with the dbSNP and 1000 G
databases. Subsequently, two likely pathogenic compound
heterozygous variants in CAPN3 (NM_000070.2) (predicted by
bioinformatics analyses) were validated by Sanger sequencing in the
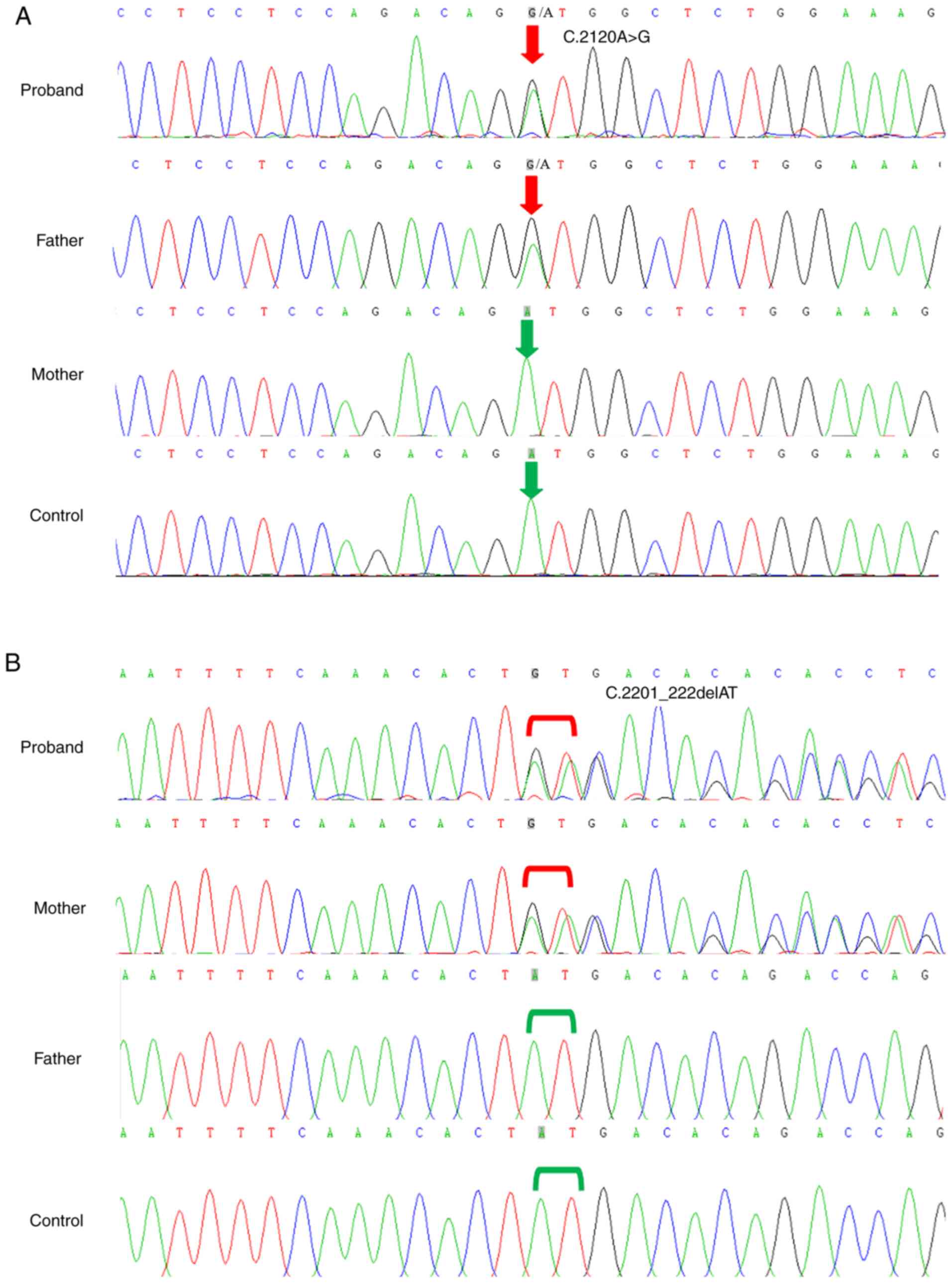
proband, a missense variant c.2120A>G/p.(Asp707Gly)
(NM_000070.2: c.2120A>G, NP_000061.1: p.(Asp707Gly)) and
deletion variant c.2201_2202delAT/p.(Tyr734*) [NM_000070.2:
c.2201_2202delAT, NP_000061.1: p.(Tyr734*)], inherited from his
father and mother, respectively.
The missense variant c.2120A>G/p.(Asp707Gly) in
exon 20 resulted in a single-nucleotide polymorphism (A-G) at site
2120 in the coding region of CAPN3 (NM_000070.2), which
introduced an aspartic acid residue that replaced a glycine in
codon 707 (Fig. 3A). This variant
was previously reported in multiple (>10) homozygous or compound
heterozygous patients with LGMDs (PM3-PVS) (36), which co-segregated within a family
(PP1-PM) (37). The normal
population database includes this variant; its frequency is 0.0148%
(42/282854, gnomAD; PM2); bioinformatics analysis software SIFT,
PolyPhen2 and Mutation Taster consistently predicted the variant to
be harmful (PP3). In addition, the variant is included in ClinVar
as a ‘pathogenic/suspected pathogenic variant’ (36). According to available evidence, the
variant is defined as pathogenic (PM3-PVS+PM2+PP1-PM+PP3) based on
the 2015 ACMG guidelines for sequence variant interpretation
(38).
A novel deletion variant
c.2201_2202delAT/p.(Tyr734*) occurred in exon 21 of CAPN3
(NM_000070.2), which resulted in a variant at site 734 of the
encoded protein that converted a tyrosine residue into a stop codon
(Fig. 3B), which may cause protein
truncation or activate nonsense-mediated CAPN3 mRNA
degradation, thereby affecting the function of the protein product
encoded by CAPN3 (PVS1). This variant was not detected in
the normal population database (PM2); in the trans-position, the
pathogenic variant c.2201_2202delAT/p.(Tyr734*) was detected (PM3).
According to the 2015 ACMG guidelines (38) for sequence variant interpretation,
this variant is defined as pathogenic (PVS1+PM2+PM3).
The probability of these variants occurring in the
population is extremely low and neither variant was not found in
200 identical ethnic healthy, unrelated controls from the Shandong
population with the WT genotype. Based on the data of the present
study and the characteristics of autosomal recessive inheritance,
it was found that the CAPN3 compound heterozygous variants
(c.2120A>G and c.2201_2202delAT) co-segregated with the LGMDR1
phenotypes in this three-person family.
Multiple sequence alignment and
molecular structure modeling
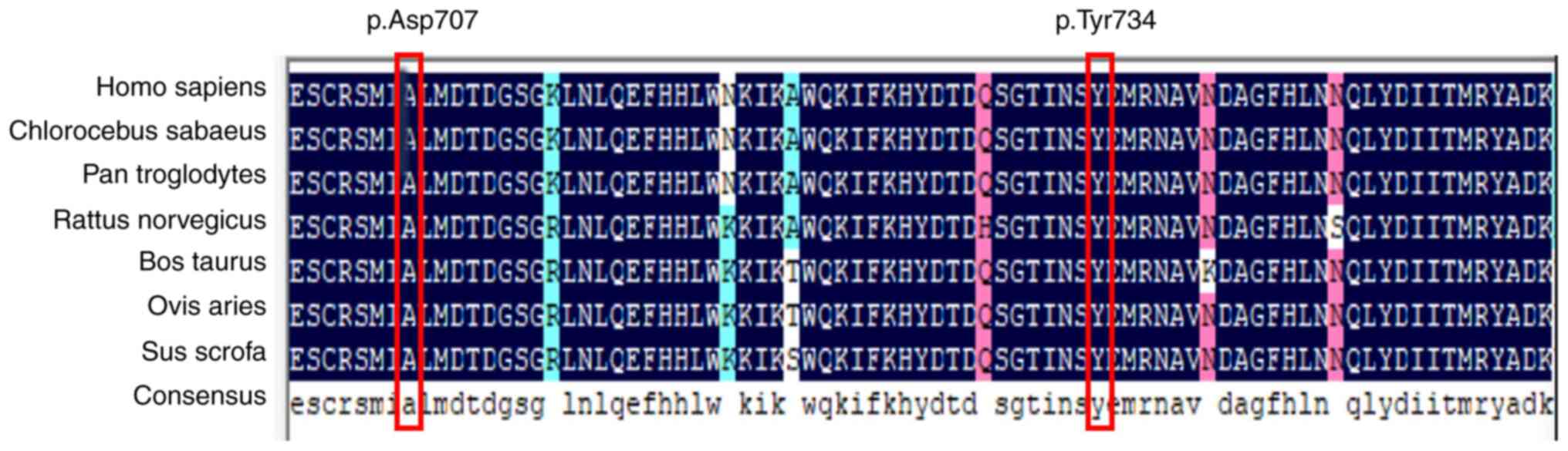
Multiple sequence alignment was performed to analyze
protein sequence conservation. This analysis of CAPN3 protein from
six species, including sheep, pig, cow, rat, chimpanzee and monkey,
showed that the aspartic acid and tyrosine residues at positions
707 and 734 of CAPN3 are highly conserved (Fig. 4).
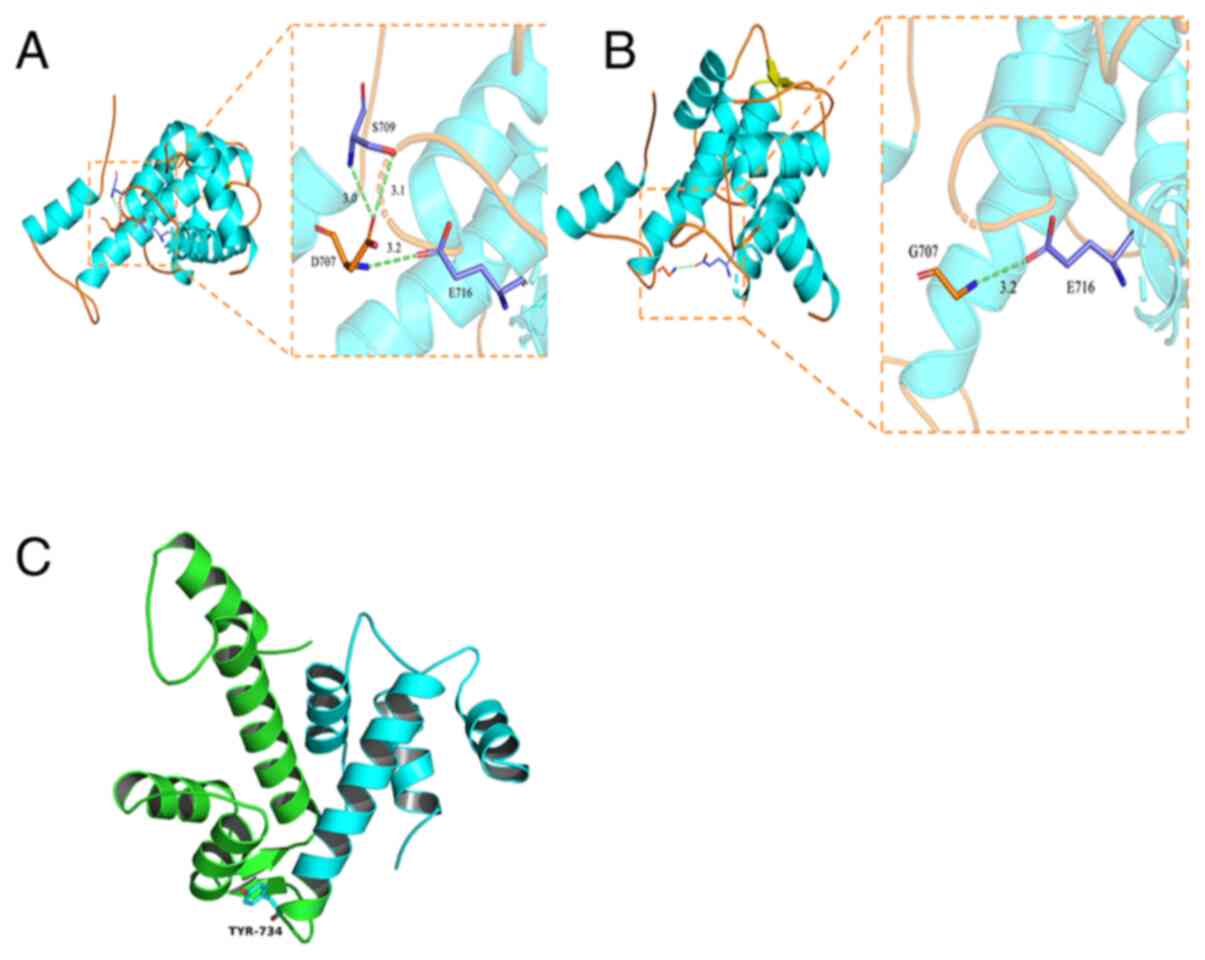
A homologous PDB protein sequence was downloaded
from the RCSB database (rcsb.org/) (PDB code 4OKH) and PyMOL-1
software was used to simulate the CAPN3 three-dimensional protein
structure (Fig. 5). Fig. 5A and B shows the interaction between
residue 707 (p.Asp707) with surrounding amino acids before and
after the variant. The aspartic acid at position 707 and serine at
709 formed two hydrogen bonds (indicated by green dotted lines, 3.0
and 3.1 represent the hydrogen bond distance between Asp707 and
surrounding residues) and glutamic acid at site 716 forms a
hydrogen bond (indicated by 3.2) with aspartic acid at position
707. Due to a point variant, the glycine at position 707 fails to
form hydrogen bonds with surrounding amino acid residues. Compared
with the WT sequence, the acid residue on position 707 lacks
interactions with serine residues, weakening the interaction
between the residue at position 707 with the surrounding amino
acids, affecting overall protein stability. Fig. 5C shows that the deletion due to the
tyrosine 734 nonsense variant resulted in a truncated protein and
functional impairment.
Discussion
The present study observed a sporadic male case of
LGMDR1 and identified two compound heterozygous variants in
CAPN3, namely c.2120A>G/p. (Asp707Gly) and
c.2201_2202delAT/p.(Tyr734*), which co-segregated with the LGMDR1
phenotypes in the proband's family. The genetic analysis of this
family showed that the parents of this proband were obligate
heterozygotes, as evidenced by Sanger sequencing and carried one
CAPN3 pathogenic variant each. Therefore, it was considered that
this proband had an autosomal recessive inheritance (AR-LGMDR1).
Each sibling of this proband had a 25% possibility of being
affected at conception, a 50% possibility of being an asymptomatic
carrier and a 25% possibility of being unaffected but not a
carrier. In silico analysis of these CAPN3 variants
revealed all deleterious results according to the ACMG guidelines
for sequence variant interpretation. Additionally, the heterozygous
compound variants were absent from 200 healthy, unrelated controls
from the same ethnic group in the Shandong population with WT
genotypes, suggesting they are pathogenic variants responsible for
the LGMDR1 phenotypes.
Genetic analysis revealed a novel frameshift variant
c.2201_2202delAT at the heterozygous state in exon 21 and a known
missense variant (c.2120A>G) in exon 20 of CAPN3
(36). Notably, both parents were
carriers of pathogenic variants, as evidenced by Sanger sequencing,
without related neuromuscular weakness or atrophies and normal CK
levels. Therefore, it was considered that this proband possessed
autosomal recessive inheritance. The CAPN3 missense variant
p.(Asp707Gly) reportedly causes autosomal recessive inheritance,
leading to severe and progressive clinical features and typical and
evident muscular weakness and atrophies (11,36,37,39).
Missense CAPN3 variants not only affect calpain 3 enzymatic
activity, but also affect binding between protein molecules,
thereby affecting protein integrity (20). Notably, previous studies showed that
CAPN3 variants can also cause the autosomal dominant pattern
of LGMDD4 and deletion variants c.643_663del21, c.598_612del15, as
well as missense variant c.1333G>A (8,40,41)
can trigger similarly mild clinical features in the case of LGMDR1.
Nevertheless, in the present study, the newly identified
c.2201_2202delAT deletion variant contributed to the autosomal
recessive LGMDR1 pattern because of genetic heterogeneity. Although
this novel deletion created a truncated calpain 3 protein, the
prognosis for LGMDR1 is unpredictable depending on whether the
mutated site is located in a critically functional area (42).
It is well-known that missense variants in
CAPN3 are the most common type and frequently occur in the
cysteine protease and PEF domain of calpain 3. The PEF domain binds
four Ca2+ ions per protomer through EF1, EF2, EF3 and
EF5 and three Ca2+-binding EF-hands are concentrated
near the protease core, which may facilitate calpain 3
homodimerization and calcium ion binding and radically alter the
local charge within the dimer during Ca2+ signaling
(20,32). The two CAPN3 variants
identified in the present study are respectively located in EF2 and
EF3 of the PEF domain and may significantly change the 3D structure
and disrupt the conformational stability of domain IV, thereby
affecting the transmission of calcium signals.
Hyper-creatine kinase-emia refers to a clinically
asymptomatic condition in which high serum CK levels are
accidentally found during physical examination. CK levels may
fluctuate, typically 5–80-fold above physiological levels. Among
the reported cases, the serum CK level is elevated in almost all
patients with hyper-creatine kinase-emia, showing values more than
10-fold higher than the normal level but has no apparent
relationship with disease severity (4,43).
Fanin et al (44) found that
6 of 58 patients with CAPN3 variants presented with
hyper-creatine kinase-emia and were asymptomatic, whereas only one
was heterozygous for the c.550delA variant. A cohort study showed a
frequency of 1.9% in German patients with LGMD who carried the
deletion variant c.550delA in CAPN3, exhibiting sustained and
isolated hyper-creatine kinase-emia (45). Notably, the calpainopathy-related
hyper-creatine kinase-emia in the present study was diagnosed at
the age of 18 years based on elevated CK levels but without
myopathic symptoms. However, genetic analysis revealed CAPN3
variants. Notably, few patients with hyper-creatine kinase-emia are
asymptomatic or present only mild muscle weakness, indicating that
very mild, late-onset calpainopathy phenotypes occurred in the
proband in the present study.
Notably, some patients with normal CK levels present
myopathy-related symptoms. Furthermore, the clinical phenotype of
LGMDR1 is highly heterogeneous. The clinical phenotypes may
completely differ, even between siblings with the same variant. In
addition, the lack of a definitive correlation between gene variant
sites and variant types (23,46,47)
further hinders accurate diagnoses. Thus, the severity and
prognosis cannot be predicted based on the variant type and
location alone (46). Severe cases
may suffer disabilities during adolescence, which seriously affects
motor abilities and eventually leads to the patient requiring a
wheelchair; mild cases only present with mild myasthenia,
hyper-creatine kinase-emia, or pseudometabolic myopathy in
adulthood (42). Simple
hyper-creatine kinase-emia may be either a mild form of LGMDR1 or
the preclinical stage of LGMDR1. The patient can be asymptomatic
for many years but progressive weakness symptoms can appear over
time. The high clinical heterogeneity of this disease is
remarkable. Therefore, clinically asymptomatic hyper-creatine
kinase-emia should be carefully investigated.
By performing muscular biopsies, Fanin et al
(13) found that the pathological
score of muscle tissue and degrees of fiber regeneration and
degeneration in patients with LGMDR1 may be related to disease
development. However, in the present case study, the male proband
presented normal results. However, the pathogenesis of muscle
changes caused by CAPN3 variants requires further
investigation.
In summary, the present study determined the genetic
etiology of LGMDR1 in a family with CAPN3 variants and
performed genetic counseling for the proband, suggesting a genetic
explanation for the simple hyper-creatine kinase-emia. The present
study expanded the current clinical and genetic spectrum of LGMDR1,
providing useful insights for further research on the pathogenesis
of LGMDs and accelerating the development of prenatal
diagnosis.
Acknowledgments
Not applicable.
Funding
The present study was supported by grants from the
National Natural Science Foundation (NSFC; grant no. 81371499) and
National Key Research and Development Program of China (grant nos.
2016YFC1000306 and 2005DKA32408).
Availability of data and materials
The datasets used and/or analyzed during the current
study have been submitted to the ClinVar database [accession
numbers VCV000468648.8 (https://www.ncbi.nlm.nih.gov/clinvar/variation/468648/)
and VCV000992898.1 (https://www.ncbi.nlm.nih.gov/clinvar/variation/992898/)].
Authors' contributions
CZ and LX were responsible for data curation; CZ and
XZ performed formal analysis; SL and FC were responsible for
funding acquisition; DL and LX performed clinical investigation. SL
and FC were responsible for project administration; CZ, LX, XZ, DL,
SL and FC acquired materials; SL and FC supervised the present
study. CZ and LX confirm the authenticity of all the raw data. CZ
wrote the original draft; and CZ, SL and FC wrote, reviewed and
edited the manuscript. All authors read and approved the final
manuscript.
Ethics approval and consent to
participate
The present study was approved by the ethics
committee of the Affiliated Hospital of Qingdao University
(approval no. qdfy20203789) and was conducted according to the
Declaration of Helsinki.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Glossary
Abbreviations
Abbreviations:
|
CAPN3
|
calcium-activated neutral proteinase
3
|
|
CK
|
creatine kinase
|
|
LGMD
|
limb-girdle muscular dystrophies
|
|
LGMD2A
|
limb-girdle muscular dystrophy type
2A
|
|
LGMDR1
|
limb-girdle muscular dystrophy
recessive 1
|
|
PEF
|
penta-EF-hand
|
|
WT
|
wild-type
|
|
MRI
|
magnetic resonance imaging
|
|
NGS
|
next-generation sequencing
|
|
WES
|
whole-exome sequencing
|
References
|
1
|
Wicklund MP: The limb-girdle muscular
dystrophies. Continuum (Minneap Minn). 25:1599–1618.
2019.PubMed/NCBI
|
|
2
|
Nallamilli BRR, Chakravorty S, Kesari A,
Tanner A, Ankala A, Schneider T, da Silva C, Beadling R, Alexander
JJ, Askree SH, et al: Genetic landscape and novel disease
mechanisms from a large LGMD cohort of 4656 patients. Ann Clin
Transl Neurol. 5:1574–1587. 2018. View
Article : Google Scholar : PubMed/NCBI
|
|
3
|
Oliveira Santos M, Ninitas P and Conceição
I: Severe limb-girdle muscular dystrophy 2A in two young siblings
from Guinea-Bissau associated with a novel null homozygous mutation
in CAPN3 gene. Neuromuscul Disord. 28:1003–1005. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Bushby KM: The limb-girdle muscular
dystrophies-multiple genes, multiple mechanisms. Hum Mol Genet.
8:1875–1882. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Angelini C and Fanin M: Calpainopathy.
GeneReviews®. Adam MP, Ardinger HH, Pagon RA and Wallace
SE: University of Washington; Seattle, WA: 2005
|
|
6
|
Straub V, Murphy A and Udd B; LGMD
workshop study group, : 229th ENMC international workshop: Limb
girdle muscular dystrophies - Nomenclature and reformed
classification Naarden, the Netherlands, 17–19 March 2017.
Neuromuscul Disord. 28:702–710. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Landires I, Núñez-Samudio V, Fernandez J,
Sarria C, Villareal V, Córdoba F, Apráez-Ippolito G, Martínez S,
Vidal OM, Vélez JI, et al: Calpainopathy: Description of a novel
mutation and clinical presentation with early severe contractures.
Genes (Basel). 11:E1292020. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Martinez-Thompson JM, Niu Z, Tracy JA,
Moore SA, Swenson A, Wieben ED and Milone M: Autosomal dominant
calpainopathy due to heterozygous CAPN3 C.643_663del21. Muscle
Nerve. 57:679–683. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Richard I, Hogrel JY, Stockholm D, Payan
CA, Fougerousse F, Eymard B, Mignard C, Lopez de Munain A, Fardeau
M and Urtizberea JA; Calpainopathy Study Group, : Natural history
of LGMD2A for delineating outcome measures in clinical trials. Ann
Clin Transl Neurol. 3:248–265. 2016. View
Article : Google Scholar : PubMed/NCBI
|
|
10
|
Richard I, Broux O, Allamand V,
Fougerousse F, Chiannilkulchai N, Bourg N, Brenguier L, Devaud C,
Pasturaud P, Roudaut C, et al: Mutations in the proteolytic enzyme
calpain 3 cause limb-girdle muscular dystrophy type 2A. Cell.
81:27–40. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Leiden Database, . https://databases.lovd.nl/shared/genes/CAPN3September
2–2020
|
|
12
|
Park HJ, Jang H, Lee JH, Shin HY, Cho SR,
Park KD, Bang D, Lee MG, Kim SM, Lee JH, et al: Clinical and
pathological heterogeneity of korean patients with CAPN3 mutations.
Yonsei Med J. 57:173–179. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Fanin M, Nascimbeni AC and Angelini C:
Gender difference in limb-girdle muscular dystrophy: A muscle fiber
morphometric study in 101 patients. Clin Neuropathol. 33:179–185.
2014. View
Article : Google Scholar : PubMed/NCBI
|
|
14
|
Fanin M and Angelini C: Progress and
challenges in diagnosis of dysferlinopathy. Muscle Nerve.
54:821–835. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Angelini C, Nardetto L, Borsato C, Padoan
R, Fanin M, Nascimbeni AC and Tasca E: The clinical course of
calpainopathy (LGMD2A) and dysferlinopathy (LGMD2B). Neurol Res.
32:41–46. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Duguez S, Bartoli M and Richard I: Calpain
3: A key regulator of the sarcomere? FEBS J. 273:3427–3436. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Pantoja-Melendez CA, Miranda-Duarte A,
Roque-Ramirez B and Zenteno JC: Epidemiological and molecular
characterization of a Mexican population isolate with high
prevalence of limb-girdle muscular dystrophy type 2A due to a novel
calpain-3 mutation. PLoS One. 12:e01702802017. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Gallardo E, Saenz A and Illa I:
Limb-girdle muscular dystrophy 2A. Handb Clin Neurol. 101:97–110.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Ono Y, Ojima K, Shinkai-Ouchi F, Hata S
and Sorimachi H: An eccentric calpain, CAPN3/p94/calpain-3.
Biochimie. 122:169–187. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Lasa-Elgarresta J, Mosqueira-Martín L,
Naldaiz-Gastesi N, Sáenz A, López de Munain A and
Vallejo-Illarramendi A: Calcium mechanisms in limb-girdle muscular
dystrophy with CAPN3 mutations. Int J Mol Sci. 20:E45482019.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Hauerslev S, Sveen ML, Duno M, Angelini C,
Vissing J and Krag TO: Calpain 3 is important for muscle
regeneration: Evidence from patients with limb girdle muscular
dystrophies. BMC Musculoskelet Disord. 13:432012. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Maki M: Structures and functions of
penta-EF-hand calcium-binding proteins and their interacting
partners: Enigmatic relationships between ALG-2 and calpain-7.
Biosci Biotechnol Biochem. 84:651–660. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Sáenz A, Leturcq F, Cobo AM, Poza JJ,
Ferrer X, Otaegui D, Camaño P, Urtasun M, Vílchez J,
Gutiérrez-Rivas E, et al: LGMD2A: Genotype-phenotype correlations
based on a large mutational survey on the calpain 3 gene. Brain.
128:732–742. 2005. View Article : Google Scholar
|
|
24
|
Toral-Ojeda I, Aldanondo G,
Lasa-Elgarresta J, Lasa-Fernández H, Fernández-Torrón R, López de
Munain A and Vallejo-Illarramendi A: Calpain 3 deficiency affects
SERCA expression and function in the skeletal muscle. Expert Rev
Mol Med. 18:e72016. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Taveau M, Bourg N, Sillon G, Roudaut C,
Bartoli M and Richard I: Calpain 3 is activated through autolysis
within the active site and lyses sarcomeric and sarcolemmal
components. Mol Cell Biol. 23:9127–9135. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Taghizadeh E, Rezaee M, Barreto GE and
Sahebkar A: Prevalence, pathological mechanisms, and genetic basis
of limb-girdle muscular dystrophies: A review. J Cell Physiol.
234:7874–7884. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Li H and Durbin R: Fast and accurate
long-read alignment with Burrows-Wheeler transform. Bioinformatics.
26:589–595. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
McKenna A, Hanna M, Banks E, Sivachenko A,
Cibulskis K, Kernytsky A, Garimella K, Altshuler D, Gabriel S, Daly
M, et al: The Genome Analysis Tool kit: A MapReduce framework for
analyzing next-generation DNA sequencing data. Genome Res.
20:1297–1303. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Wang K, Li M and Hakonarson H: ANNOVAR:
Functional annotation of genetic variants from high-throughput
sequencing data. Nucleic Acids Res. 38:e1642010. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Tran KT, Le VS, Bui HTP, Do DH, Ly HTT,
Nguyen HT, Dao LTM, Nguyen TH, Vu DM, Ha LT, et al: Genetic
landscape of autism spectrum disorder in Vietnamese children. Sci
Rep. 10:50342020. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Schwede T, Kopp J, Guex N and Peitsch MC:
SWISS-MODEL: An automated protein homology-modeling server. Nucleic
Acids Res. 31:3381–3385. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Partha SK, Ravulapalli R, Allingham JS,
Campbell RL and Davies PL: Crystal structure of calpain-3
penta-EF-hand (PEF) domain - a homodimerized PEF family member with
calcium bound at the fifth EF-hand. FEBS J. 281:3138–3149. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Arevalo-Rodriguez I, Smailagic N, Roqué I
Figuls M, Ciapponi A, Sanchez-Perez E, Giannakou A, Pedraza OL,
Bonfill Cosp X and Cullum S: Mini-Mental State Examination (MMSE)
for the detection of Alzheimer's disease and other dementias in
people with mild cognitive impairment (MCI). Cochrane Database Syst
Rev. 2015:CD0107832015.PubMed/NCBI
|
|
34
|
Compston A: Aids to the investigation of
peripheral nerve injuries. Medical Research Council: Nerve Injuries
Research Committee. His Majesty's Stationery Office: 1942; pp. 48
(iii) and 74 figures and 7 diagrams; with aids to the examination
of the peripheral nervous system. By Michael O'Brien for the
Guarantors of Brain. Saunders Elsevier: 2010; pp. [8] 64 and 94
Figures, . Brain. 133:2838–2844. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Rasmussen LH, Madsen HN and Ladefoged SD:
Creatine phosphokinase MB and lactate dehydrogenase isoenzyme 1 in
polymyositis. Scand J Rheumatol. 14:427–430. 1985. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Minami N, Nishino I, Kobayashi O, Ikezoe
K, Goto Y and Nonaka I: Mutations of calpain 3 gene in patients
with sporadic limb-girdle muscular dystrophy in Japan. J Neurol
Sci. 171:31–37. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Park HJ, Jang H, Kim JH, Lee JH, Shin HY,
Kim SM, Park KD, Yim SV, Lee JH and Choi YC: Discovery of
pathogenic variants in a large Korean cohort of inherited muscular
disorders. Clin Genet. 91:403–410. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Richards S, Aziz N, Bale S, Bick D, Das S,
Gastier-Foster J, Grody WW, Hegde M, Lyon E, Spector E, et al ACMG
Laboratory Quality Assurance Committee, : Standards and guidelines
for the interpretation of sequence variants: A joint consensus
recommendation of the American College of Medical Genetics and
Genomics and the Association for Molecular Pathology. Genet Med.
17:405–424. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Ten Dam L, Frankhuizen WS, Linssen WHJP,
Straathof CS, Niks EH, Faber K, Fock A, Kuks JB, Brusse E, de Coo
R, et al: Autosomal recessive limb-girdle and Miyoshi muscular
dystrophies in the Netherlands: The clinical and molecular spectrum
of 244 patients. Clin Genet. 96:126–133. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Cerino M, Campana-Salort E, Salvi A,
Cintas P, Renard D, Juntas Morales R, Tard C, Leturcq F, Stojkovic
T, Bonello-Palot N, et al: Novel CAPN3 variant associated with an
autosomal dominant calpainopathy. Neuropathol Appl Neurobiol.
46:564–578. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Vissing J, Barresi R, Witting N, Van
Ghelue M, Gammelgaard L, Bindoff LA, Straub V, Lochmüller H, Hudson
J, Wahl CM, et al: A heterozygous 21-bp deletion in CAPN3 causes
dominantly inherited limb girdle muscular dystrophy. Brain.
139:2154–2163. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Urtasun M, Sáenz A, Roudaut C, Poza JJ,
Urtizberea JA, Cobo AM, Richard I, García Bragado F, Leturcq F,
Kaplan JC, et al: Limb-girdle muscular dystrophy in Guipúzcoa
(Basque Country, Spain). Brain. 121:1735–1747. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Fanin M and Angelini C: Protein and
genetic diagnosis of limb girdle muscular dystrophy type 2A: The
yield and the pitfalls. Muscle Nerve. 52:163–173. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Fanin M, Fulizio L, Nascimbeni AC,
Spinazzi M, Piluso G, Ventriglia VM, Ruzza G, Siciliano G, Trevisan
CP, Politano L, et al: Molecular diagnosis in LGMD2A: Mutation
analysis or protein testing? Hum Mutat. 24:52–62. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Hanisch F, Müller CR, Grimm D, Xue L,
Traufeller K, Merkenschlager A, Zierz S and Deschauer M: Frequency
of calpain-3 c.550delA mutation in limb girdle muscular dystrophy
type 2 and isolated hyperCKemia in German patients. Clin
Neuropathol. 26:157–163. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Fardeau M, Hillaire D, Mignard C, Feingold
N, Feingold J, Mignard D, de Ubeda B, Collin H, Tome FM, Richard I,
et al: Juvenile limb-girdle muscular dystrophy. Clinical,
histopathological and genetic data from a small community living in
the Reunion Island. Brain. 119:295–308. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Yu M, Zheng Y, Jin S, Gang Q, Wang Q, Yu
P, Lv H, Zhang W, Yuan Y and Wang Z: Mutational spectrum of Chinese
LGMD patients by targeted next-generation sequencing. PLoS One.
12:e01753432017. View Article : Google Scholar : PubMed/NCBI
|