Introduction
In 2012, Shalev et al (1) described a novel syndrome in two
unrelated consanguineous Arab families with multiple members
exhibiting severe short stature, skeletal dysplasia, unusual facial
features, brachydactyly and hypotrichosis. Shaheen et al
(2) and Sarig et al
(3) identified that the biallelic
variants of POC1 centriolar protein A (POC1A) cause short
stature, onychodysplasia, facial dysmorphism and hypotrichosis
(SOFT) syndrome. Prevalence of SOFT syndrome remains unclear. Thus
far, only 13 families, with 11 POC1A pathogenic variants
identified in 27 patients, have been reported worldwide (4). All of these families were sporadically
distributed in different countries (Table SI), including Saudi Arabia, Turkey,
Iran and Oman (Middle East); Spain, Monaco and Italy (Europe);
South Korea (East Asia); and Chile (South America).
Patients with SOFT syndrome have been reported to
exhibit a wide range of variable phenotypes, including severe pre-
and postnatal growth retardation, onychodysplasia, hypotrichosis,
facial deformities (such as long triangular face, prominent
forehead and pointed chin), skeletal abnormalities (such as short
long bones and irregular changes in metaphysis, short femoral neck,
and delayed ossification of carpal and vertebral bones), as well as
a poor response to recombinant human growth hormone (rhGH) therapy
(1,3,5–9). An
atypical type of SOFT syndrome has also been reported in two other
unrelated families, where affected members also show insulin
resistance caused by dyslipidemia (10,11).
Data regarding SOFT syndrome are limited due to the
scarcity and phenotypic diversity of this novel entity, and its
clinical identification remains a challenge. For this reason,
genotypic and phenotypic features of SOFT syndrome require further
investigation. In the present case report, a Chinese patient with
SOFT syndrome is presented. Exome sequencing identified compound
heterozygous variants of POC1A. Furthermore, follow-up
radiological imaging at various ages revealed several novel
phenotypic findings relating to SOFT syndrome.
Case report
General information
A female patient was referred to Hunan Children's
Hospital (Changsha, China) for growth retardation on February 11,
2019. The patient was born via spontaneous vaginal delivery at full
term and had a birth weight of 1.8 kg (<3rd percentile) and a
birth height of 40 cm (<3rd percentile). Her parents (both
Chinese Han) were healthy and from two unrelated families. Her
father was 36 years old and her mother was 35 years old. Her
father's height was 175 cm (50-75th percentile) and her mother's
height was 155 cm (10-25th percentile). When the patient was aged 7
years and 8 months, her height was 102.5 cm (<3rd percentile)
and her weight was 17.5 kg (<3rd percentile). She had an unusual
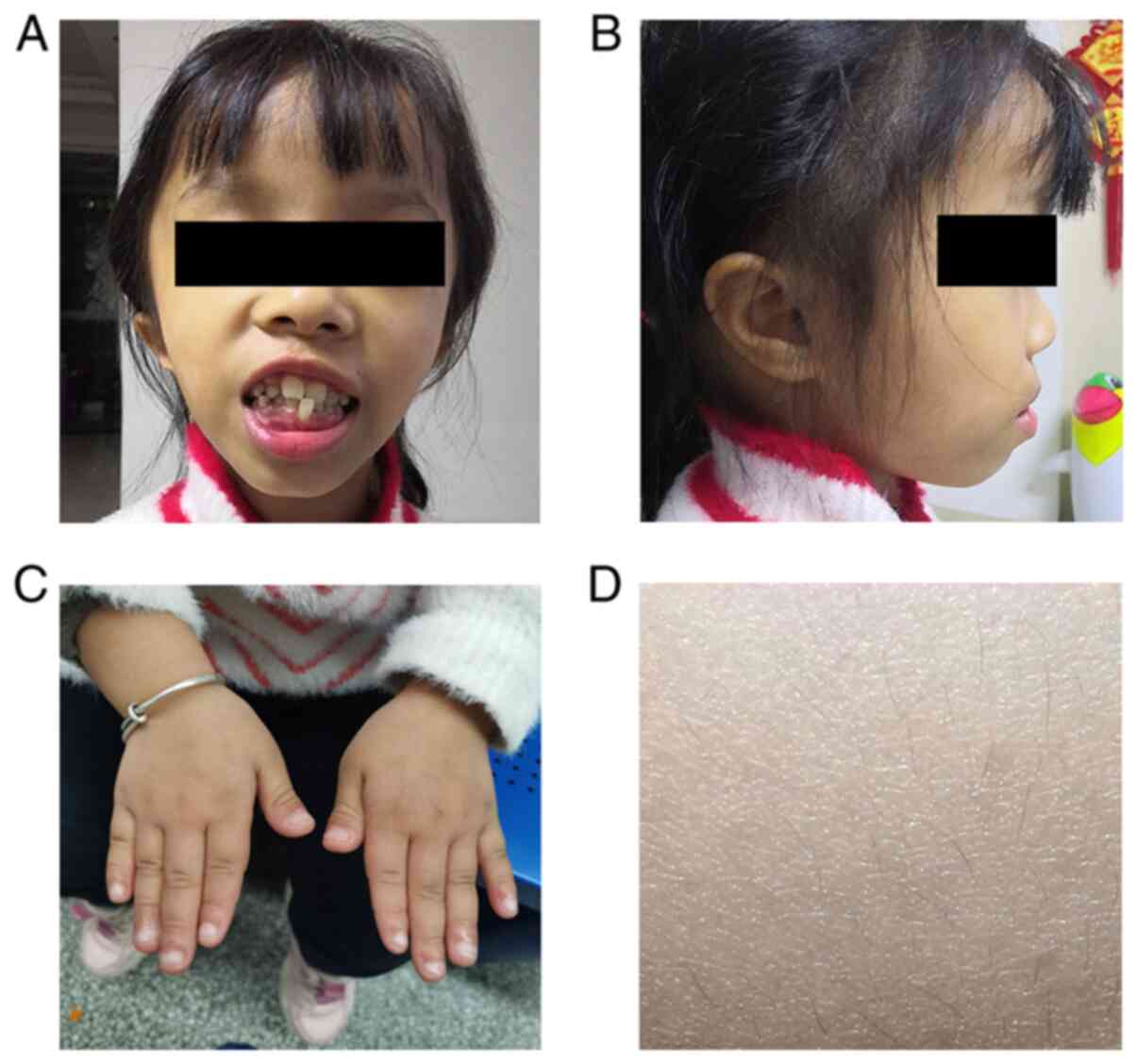
facial appearance, with a triangular face, prominent forehead and
irregularly positioned teeth (Fig. 1A
and B). Her fingers were stubby and with nail hypoplasia
(Fig. 1C), and her body hair was
sparse (Fig. 1D). Her intelligence
quotient was 78 according to the Chinese Wechsler Intelligence
Scale for Children (12). The
patient underwent rhGH therapy for 6 months and her height
increased by 4.7 cm. Over the subsequent 3 months, her height
increased by only 0.5 cm and the rhGH therapy was discontinued.
No abnormalities were observed in laboratory tests,
which included complete blood count, urine analysis, vitamin D
level, blood sugar, thyroid function level, insulin-like growth
factor (IGF) binding protein 3, IGF1 and growth hormone stimulation
tests. Female patients with Turner syndrome (sex chromosome
aneuploidy) can exhibit a variety of developmental problems,
including short height; therefore, Giemsa (GTG) banding was
performed on the patient, but her karyotype was identified to be
normal (46 XX; Fig. S1A and B).
Ultrasound indicated left renal pelvis separation and mild
tricuspid regurgitation (data not shown).
Ethics statement
Parents/guardians of both the patient and the
matched control, as well as family members whose data are presented
in this case report, provided written informed consent for
publication of the data and associated images in the present case
report. This study and all the data described in the paper were
approved by the Ethics Committee of the Hunan Children's Hospital
(approval no. HCHLL-020-16).
GTG banding
Peripheral venous blood was collected in a
vacutainer vial containing sodium heparin. Slides were prepared
using standard cytogenetic methods. GTG banding at a 400- to a
550-band level was performed in accordance with the standard
laboratory protocol (13). Two
different cultures corresponding to two different series of the
slides from the sample were separately prepared and analyzed. At
least 40 metaphases were examined for the proband of the family.
For the second round of GTG banding evaluation, 100 metaphases were
evaluated for the proband.
Next generation sequencing
All three family members were subjected to exome
sequencing as described previously (13). Briefly, genomic DNA from three
family members was isolated from the peripheral blood using a DNA
Isolation Kit (Blood DNA Kit V2; cat. no. CW2553; CoWin
Biosciences). The genomic DNA was quantified using the Qubit™ dsDNA
HS Assay Kit (cat. no. Q32851; Invitrogen; Thermo Fisher
Scientific, Inc.). The DNA of each patient was fragmented into 180-
to 280-bp segments using a Covaris bath sonicator (duty cycle, 10%;
intensity, 5; cycles per burst, 200; 3 min for 25°C). A library was
prepared and captured with an Agilent SureSelect Human All Exon V6
kit (Agilent Technologies, Inc.). The quality-passed library was
sequenced on an Illumina HiSeq X Ten sequencing system (Illumina,
Inc.). BWA software (http://bio-bwa.sourceforge.net/) was used to align raw
data against the human reference genome (GRCh37/hg19).
Single-nucleotide polymorphisms (SNPs), small insertions/deletions
(indels) and quality recalibration were identified using GATK
software (Genome Analysis Toolkit; www.broadinstitute.org/gatk). All variants were
annotated using ANNOVAR
(annovar.openbioinformatics.org/en/latest/). A raw Binary Base Call
file was converted into a FASTQ file, 12 G bases were obtained for
each sample and the average yield was ~16.3 Gb with an error rate
of <0.1%.
Sanger sequencing
Two pairs of primers were synthesized by BGI Group
to confirm the compound heterozygous variants of POC1A,
according to GRCh37/hg19. The primers were as follows:
chr3:52179936 forward, 5′-GGCCATCTCAGACCCATTTA-3′ and reverse,
5′-GAAAGGAGGTGTCTGGGTCA-3′; chr3:52159160 forward,
5′-TTCTGAGATGCAGCCATGAG-3′ and reverse, 5′-CCTGGACTTGTCCCTGTTGT-3′.
Polymerase chain reaction (PCR) amplification was performed using
the genomic DNA as a template in a Goldstar® PCR kit
(cat. no. CW0655M; CoWin Biosciences), according to the
manufacturer's protocols. Sanger sequencing was conducted using a
BigDye® Terminator v3.1 cycle sequencing kit (Applied
Biosystems; Thermo Fisher Scientific, Inc.) according to the
manufacturer's protocol. The amplified PCR products were purified
and then run on an Applied Biosystems™ 3500 series genetic analyzer
(Thermo Fisher Scientific, Inc.). The detailed protocol for the
primers is provided in Table I.
 | Table I.PCR conditions for amplification of
POC1 centriolar protein A exon 6 and exon 8 fragments. |
Table I.
PCR conditions for amplification of
POC1 centriolar protein A exon 6 and exon 8 fragments.
| Exon | Variant | Forward primer | Reverse primer | Product size
(bp) | Annealing temperature
(°C) |
|---|
| 6 |
c.593_605delGTGGGACGTGCAT | chr3:52159160-F | chr3:52159160-R | 530 | 62 |
| 8 | c.850_851insG | chr3:52179936-F | chr3:52179936-R | 351 | 60 |
Diagnostic protocol
The diagnostic protocol of SOFT syndrome can be
described as follows: i) Clinical signs by physical examination,
including short stature, onychodysplasia, facial deformities and
hypotrichosis (3,5,7); ii)
skeletal imaging changes by X-ray including short and thick long
bones with irregular changes in metaphysis, short femoral neck and
delayed ossification of carpal and vertebral bones (1,5,6); iii)
molecular genetic test to identify POCA1 pathogenic variants
(2,3).
Genetic findings
Given the poor response to rhGH and the patient's
unusual facial appearance, exome sequencing was performed on all
three family members. Only one gene (POC1A; NM_015426.5)
with two variants (minor allele frequency <0.0001) remained in
the patient: i) c.850_851insG in exon 8, resulting in amino acid
changes p.Glu284Glyfs*9 and ii) c.593_605delGTGGGACGTGCAT (deletion
mutation) in exon 6, resulting in amino acid changes
p.Ser198Metfs*10. For these two variants, one came from her father,
and the other came from her mother (Fig. 2A-D). According to the American
College of Medical Genetics and Genomics guidelines (14), these two variants are pathogenic and
were confirmed through Sanger sequencing analysis (Fig. 2A-D).
Imaging observations
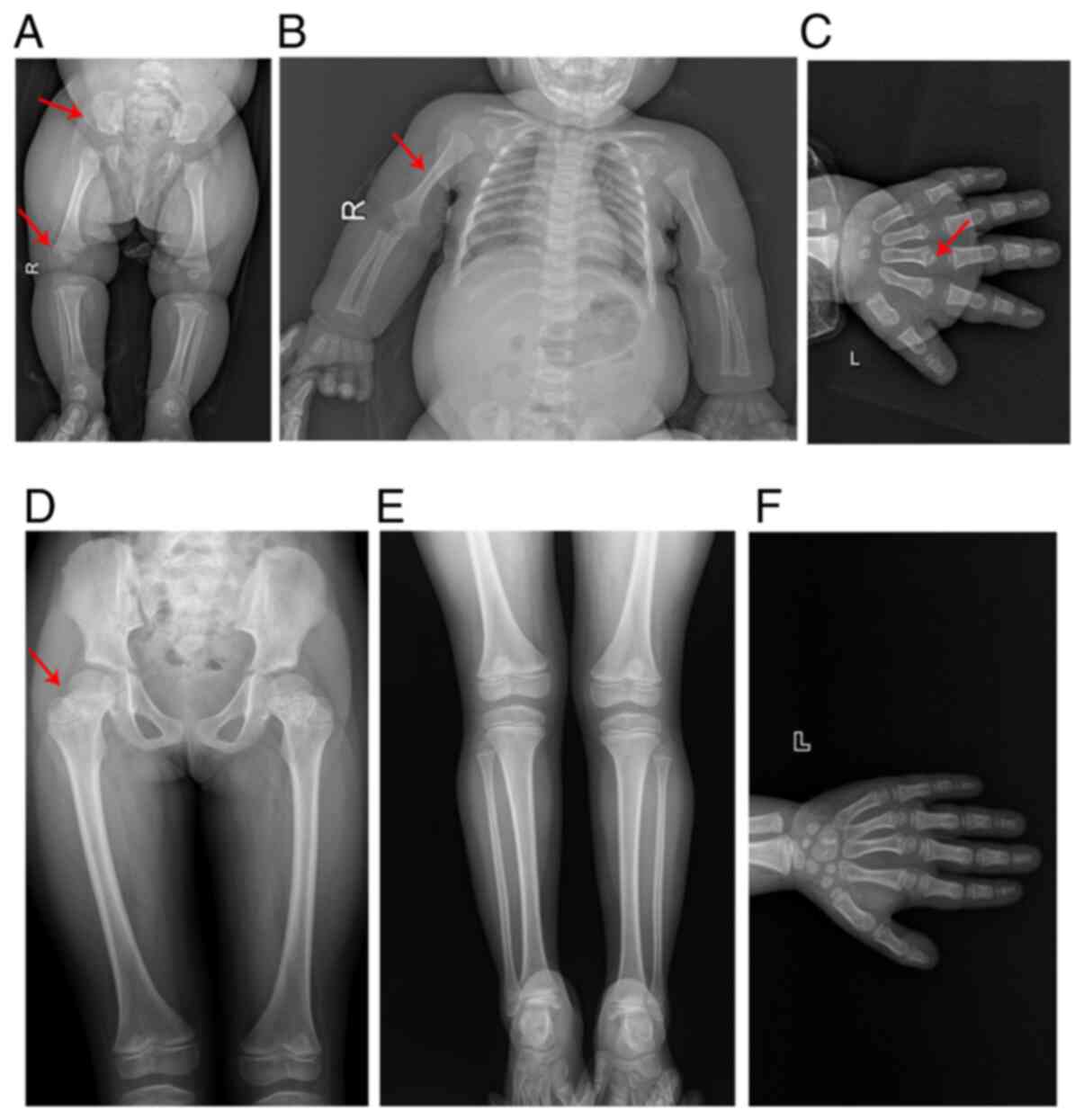
X-ray examinations were performed on the patient at
age 4 (Fig. 3A and B) and 12
(Fig. 3C) months. The images
demonstrated that the long bones of the limbs and the metacarpal
bones were thicker and shorter than those of an age- and
gender-matched control. Furthermore, the metaphysis of these bones
was widened, the ala of the ilium was square and the bottom of the
ilium was shortened (Fig. 3A-C).
However, when the patient was 7 years old (Fig. 3D), the dysplasia of these bones
became less apparent and the malformation in the femur bones
evolved into femur bones with a thicker and shorter femoral neck
(Fig. 3D-F). Meanwhile, it was
observed that when the patient was 6.3 years old, her carpal bone
age was ~5.1 years old, according to the Tanner-Whitehouse 3 method
(15) (Fig. 3F).
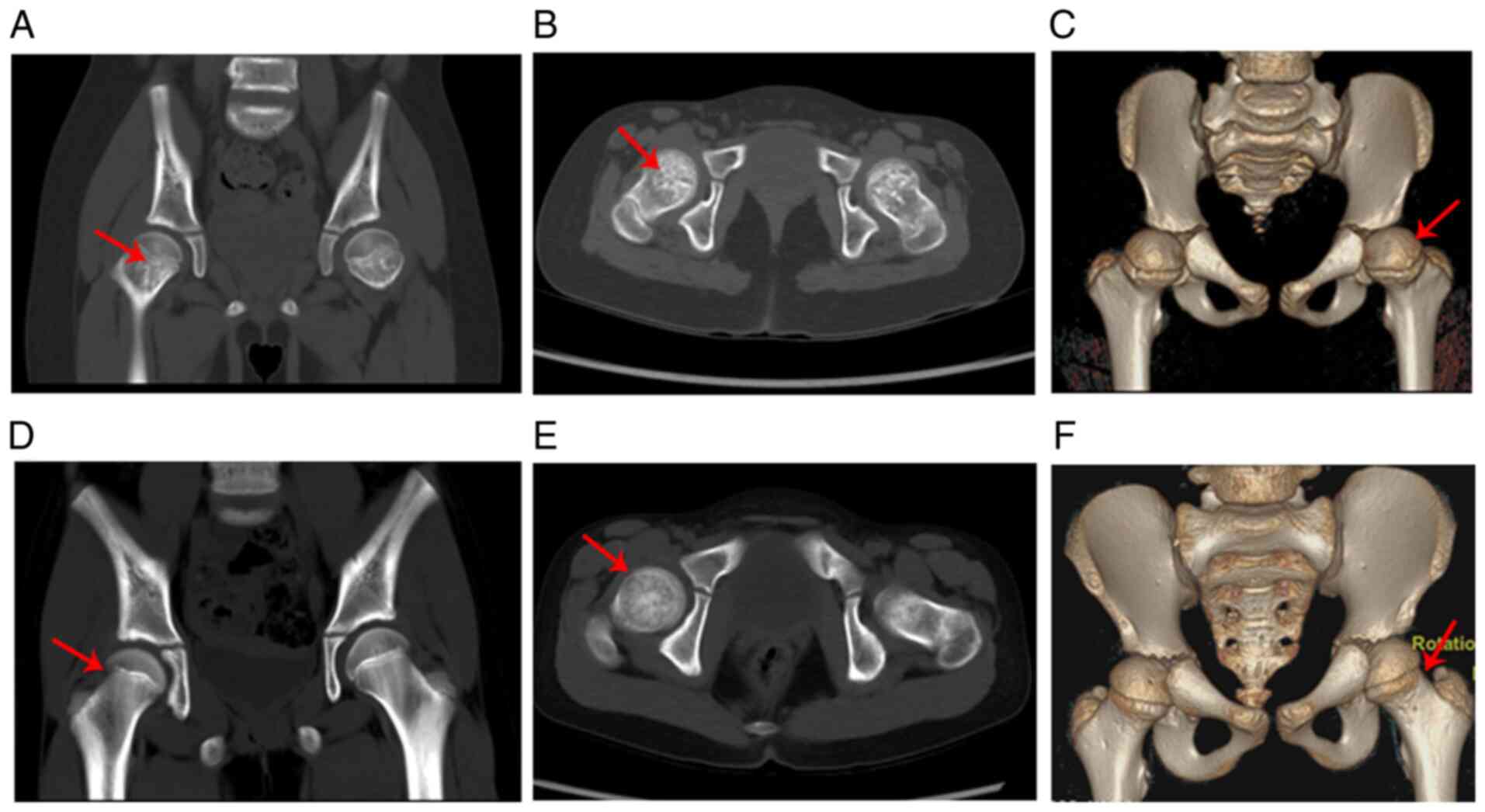
At the age of 7 years and 8 months, the patient was
subjected to spiral computed tomography (CT) and the results showed
that the bone density of the femoral neck was uneven (patchy high
or low density) and that the femoral neck was thicker and shorter
than those of the age- and gender-matched control (Fig. 4A-F). The three-dimensional
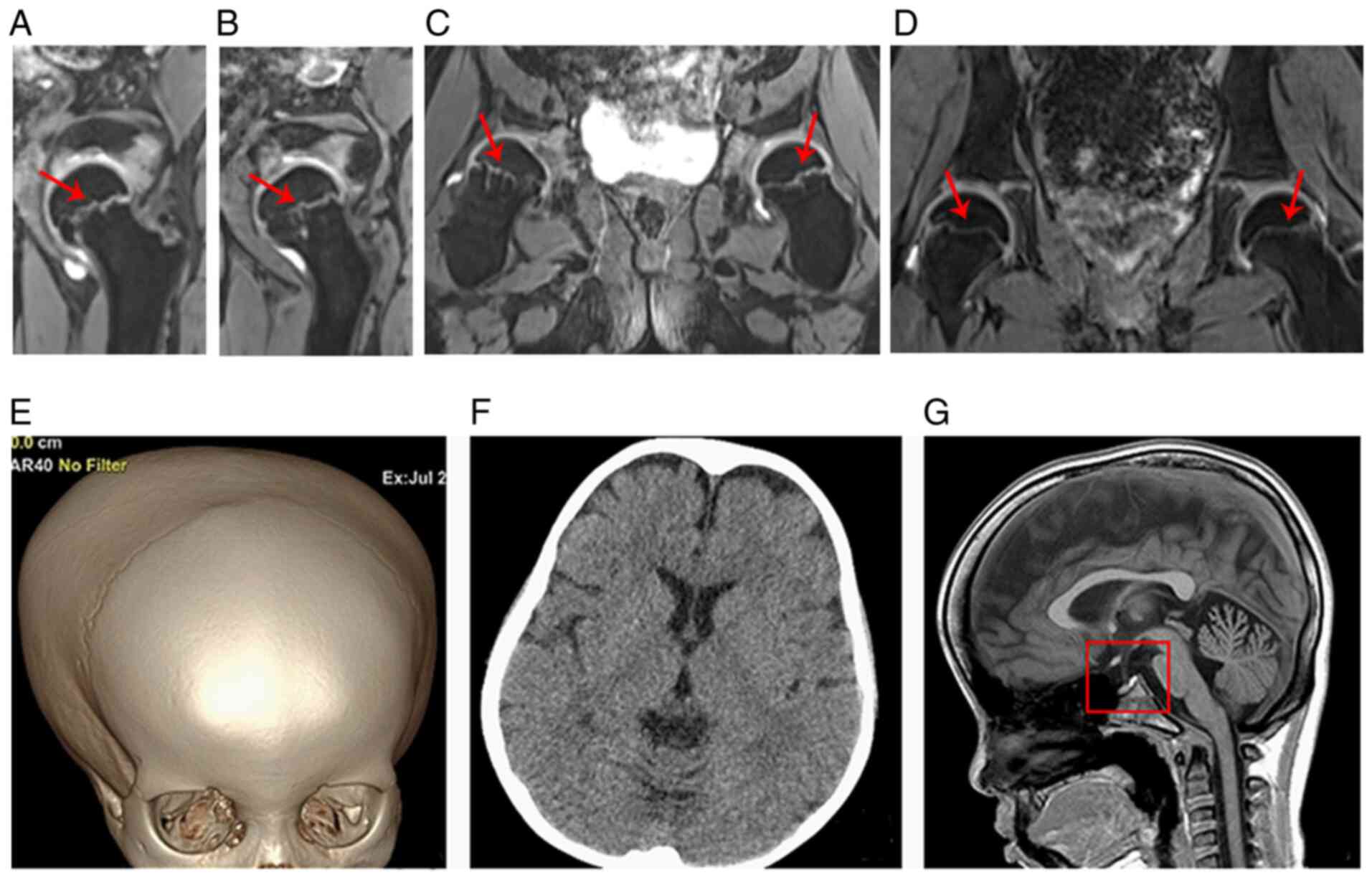
double-echo steady state with water excitation MRI of the hip
further revealed that the thickness of the proximal femoral
epiphyseal plate was uneven and that the metaphysis of the proximal
femur showed a stripe-like hyperintensity (such hyperintensity was
continuous to the adjacent epiphyseal plate) (Fig. 5A-C). Fig. 5D shows the normal hip MRI of the
age- and sex-matched control in coronal view. The three-dimensional
volume rendering of the cranium identified the disproportionate
cerebral and facial cranium as trigonocephaly, and the brain CT
scan showed that the bilateral frontal extracerebral space was
widened (Fig. 5E and F). The brain
MRI indicated that the sella was flat and exhibited hook-like
changes (Fig. 5G).
Discussion
Patients with SOFT syndrome exhibit numerous
overlapping clinical features with several other syndromes, such as
3M syndrome, Russell-Silver syndrome and Mulibrey nanism. These
clinical features include: i) Severe prenatal and postnatal growth
retardation; ii) facial dysmorphism, including a triangular-shaped
face and prominent forehead; and iii) normal intelligence (5). SOFT syndrome was diagnosed by the
following features in the present case report: i) The patient had
distinctive features, such as onychodysplasia, hypotrichosis and
variable skeletal manifestations, including short long bones and
irregular changes in metaphysis, which are consistent with the
phenotypes of SOFT syndrome; ii) exome sequencing data did not
reveal any rare variants on genes, as identified in 3M syndrome
(CUL7, OBSL1, CCDC8) (16),
Russell-Silver syndrome (GRB10, YWHAE) (17,18)
and Mulibrey nanism (TRIM37) (19); iii) exome sequencing identified two
compound heterozygous variants of POCA1, which is the
causative gene of SOFT syndrome.
POC1 consists of POC1A and POC1B (20). POC1A (located on 3p21.2)
encodes POC1 centriolar protein A (2), which plays an important role in the
early steps of centriole duplication and in the later phases of
centriole length control, ensuring centriole integrity and proper
mitotic spindle formation (20).
POC1A and POC1B are conserved proteins, including an N-terminal
WD40 domain, which likely forms a seven-bladed β-propeller and a
C-terminal coiled-coil, which contains a highly conserved sequence
(3,20,21).
All of the POCA1 pathogenic variants reported to date
disrupt the WD40 domains (5–7,9,21).
Consequently, the disruption interrupts the normal function of POC1
proteins and then causes the centrosome to fail to assemble;
consequently, mitotic spindles form abnormally (5–7). In
addition, the POC1A variants also cause the aberrant
distribution of centrosomal microtubules (3,7).
Centrosomal microtubules play an essential role in proper Golgi
assembly and trafficking, and several studies have shown a
connection between the abnormal function of the Golgi and syndromes
that feature bone and skin defects (3,7).
Therefore, POC1A plays an important role in the formation of
bones and skin, and the dysfunction of centrosome formation may be
the main molecular mechanism of SOFT syndrome (3). In the present study, according to the
variants position at the nucleic acid level, the predicted amino
acid changes in two POC1A variants are truncating from the
WD40 domains. Therefore both variants of the present study may
cause dysfunction of the centrosome, and the interruption of Golgi
assembly and trafficking.
Shalev et al (1) investigated eight patients with SOFT
syndrome in two families. Of these patients, three (ages 8, 15 and
25 years) received the skeletal imaging testing. Such three
patients exhibited a severe to mild level of epiphyseal and
metaphyseal changes in the long bones (metaphyseal changes are
obvious on the 8-year-old patient, but are mild on other older
patients) (1). In the present
study, the metaphysis abnormalities of the long bones and the
hypoplasia of the ilium were clearly observed when the patient was
4- and 12-months-old, respectively. However, the dysplasia of these
bones in the same patient was alleviated (or even disappeared) with
aging. A skeletal X-ray of the patient at 7 years old identified
that the metaphysis of the femur and metacarpal were becoming
nearly normal. Moreover, the ilium dysplasia became less evident at
this age. Therefore, similar to the X-ray examination results
described by Shalev et al (1), the present results indicated that the
metaphyseal dysplasia of the patient with SOFT syndrome was
alleviated with aging. Investigating the underlying mechanism of
the alleviation of metaphyseal and ilium dysplasia with age may
provide insights into therapeutic strategies for SOFT syndrome in
future.
Currently, the underlying mechanism of POC1A
mutations in skeletal deformities remains unclear. In animal models
with POC1A defects, certain studies implicated cell
proliferation defects of chondrocytes in growth plates (5,22).
However, to the best of our knowledge, such proliferation defects
have not been observed in humans. In the present study, the hip MRI
revealed that the metaphysis of the femoral neck had a
cartilaginous stripe signal and this finding was novel. This result
indicated that an abnormal signal in the femoral neck may be the
result of tissue imaging of immature chondrocytes during
endochondral ossification. These imaging features provided evidence
that the variant of POC1A was associated with the defect in
the proliferation of chondrocytes in the growth plate; as a result,
the growth of endochondral ossification was arrested. However, the
detailed mechanism remains unclear; therefore, further studies are
required to elucidate the function of POC1A in endochondral
ossification.
In conclusion, the present case report described a
Chinese patient with SOFT syndrome caused by two novel POC1A
pathogenic variants, expanding the mutational and clinical spectrum
of SOFT syndrome.
Supplementary Material
Supporting Data
Acknowledgements
Not applicable.
Funding
No funding was received.
Availability of data and materials
The datasets generated and/or analyzed during the
current study are available in the National Center for
Biotechnology Information (NCBI) ClinVar repository, (https://www.ncbi.nlm.nih.gov/clinvar/variation/996335/;
http://www.ncbi.nlm.nih.gov/clinvar/variation/996336/).
The raw sequencing data generated and/or analyzed during the
current study are not publicly available due to local government
ethical restrictions and to protect the privacy of the family, but
are available from the corresponding author on reasonable
request.
Authors' contributions
YZ performed sample collection and clinical
diagnosis, and reviewed and edited the manuscript. SL, YZ, YY, SH
and WH wrote and edited the original manuscript. YY provided
genetic guidance, helped to analyze the sequencing findings and
reviewed the manuscript. SH oversaw imaging. YZ, WH and SL
undertook sample collection and clinical data collection. All
authors have read and approved the final manuscript. YZ and YY
confirm the authenticity of all the raw data.
Ethics approval and consent to
participate
The present study was approved by the Ethics
Committee of Hunan Children's Hospital (Changsha, China).
Patient consent for publication
Prior to participating in this study, written
informed consent was obtained from all three family members.
Parents/guardians of both the patient and the matched control, as
well as members of the family included in the present study,
provided written informed consent for publication of the data and
associated images in the present case report.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Shalev SA, Spiegel R and Borochowitz ZU: A
distinctive autosomal recessive syndrome of severe disproportionate
short stature with short long bones, brachydactyly, and
hypotrichosis in two consanguineous Arab families. Eur J Med Genet.
55:256–264. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Shaheen R, Faqeih E, Shamseldin HE, Noche
RR, Sunker A, Alshammari MJ, Al-Sheddi T, Adly N, Al-Dosari MS,
Megason SG, et al: POC1A truncation mutation causes a ciliopathy in
humans characterized by primordial dwarfism. Am J Hum Genet.
91:330–336. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Sarig O, Nahum S, Rapaport D, Rapaport D,
Ishida-Yamamoto A, Fuchs-Telem D, Qiaoli L, Cohen-Katsenelson K,
Spiegel R, Nousbeck J, et al: Short stature, onychodysplasia,
facial dysmorphism, and hypotrichosis syndrome is caused by a POC1A
mutation. Am J Hum Genet. 91:337–342. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Al-Kindi A, Al-Shehhi M, Westenberger A,
Beetz C, Scott P, Brandau O, Abbasi-Moheb L, Yüksel Z, Bauer P,
Rolfs A and Grüning NM: A novel POC1A variant in an alternatively
spliced exon causes classic SOFT syndrome: Clinical presentation of
seven patients. J Hum Genet. 65:193–197. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Koparir A, Karatas OF, Yuceturk B, Yuksel
B, Bayrak AO, Gerdan OF, Sagiroglu MS, Gezdirici A, Kirimtay K,
Selcuk E, et al: Novel POC1A mutation in primordial dwarfism
reveals new insights for centriole biogenesis. Hum Mol Genet.
24:5378–5387. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Ko JM, Jung S, Seo J, Shin CH, Cheong HI,
Choi M, Kim OH and Cho TJ: SOFT syndrome caused by compound
heterozygous mutations of POC1A and its skeletal manifestation. J
Hum Genet. 61:561–564. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Barraza-García J, Iván Rivera-Pedroza C,
Salamanca L, Belinchón A, López-González V, Sentchordi-Montané L,
del Pozo Á, Santos-Simarro F, Campos-Barros Á, Lapunzina P, et al:
Two novel POC1A mutations in the primordial dwarfism, SOFT
syndrome: Clinical homogeneity but also unreported malformations.
Am J Med Genet A. 170A:210–216. 2016. View Article : Google Scholar
|
|
8
|
Saida K, Silva S, Solar B, Fujita A,
Hamanaka K, Mitsuhashi S, Koshimizu E, Mizuguchi T, Miyatake S,
Takata A, et al: SOFT syndrome in a patient from Chile. Am J Med
Genet A. 179:338–340. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Mostofizadeh N, Gheidarloo M, Hashemipour
M and Dehkordi EH: SOFT syndrome: The first case in Iran. Adv
Biomed Res. 7:1282018. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Chen JH, Segni M, Payne F, Huang-Doran I,
Sleigh A, Adams C; UK10K Consortium, ; Savage DB, O'Rahilly S,
Semple RK and Barroso I: Truncation of POC1A associated with short
stature and extreme insulin resistance. J Mol Endocrinol.
55:147–158. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Giorgio E, Rubino E, Bruselles A, Pizzi S,
Rainero I, Duca S, Sirchia F, Pasini B, Tartaglia M and Brusco A: A
syndromic extreme insulin resistance caused by biallelic POC1A
mutations in exon 10. Eur J Endocrinol. 177:K21–K27. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Gong Y and Cai T: China revised the
Webster's intelligence scale for children. Chin J Clin Psychol.
2:1–6. 1994.PubMed/NCBI
|
|
13
|
Yang Y, Zheng Y, Li W, Li L, Tu M, Zhao L,
Mei H, Zhu G and Zhu Y: SMAD6 is frequently mutated in nonsyndromic
radioulnar synostosis. Genet Med. 21:2577–2585. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Richards S, Aziz N, Bale S, Bick D, Das S,
Gastier-Foster J, Grody WW, Hegde M, Lyon E, Spector E, et al:
Standards and guidelines for the interpretation of sequence
variants: A joint consensus recommendation of the American college
of medical genetics and genomics and the association for molecular
pathology. Genet Med. 17:405–424. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Tanner JM, Healy MJR, Goldstein H and
Cameron N: Assessment of skeletal maturity and prediction of adult
height (TW3 method). London: Saunders; pp. 1–48. 2001
|
|
16
|
Hu L, Wang X, Jin T, Han Y, Liu J, Jiang
M, Yan S, Fu X, An B and Huang S: Identification of two CUL7
variants in two Chinese families with 3-M syndrome by whole-exome
sequencing. J Clin Lab Anal. 34:e232652020. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Yoshihashi H, Maeyama K, Kosaki R, Ogata
T, Tsukahara M, Goto Y, Hata J, Matsuo N, Smith RJ and Kosaki K:
Imprinting of human GRB10 and its mutations in two patients with
russell-silver syndrome. Am J Hum Genet. 67:476–482. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Ho AC, Liu AP, Lun KS, Tang WF, Chan KY,
Lau EY, Tang MH, Tan TY and Chung BH: A newborn with a 790 kb
chromosome 17p13.3 microduplication presenting with aortic
stenosis, microcephaly and dysmorphic facial features-is cardiac
assessment necessary for all patients with 17p13.3
microduplication? Eur J Med Genet. 55:758–762. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Avela K, Lipsanen-Nyman M, Idänheimo N,
Seemanová E, Rosengren S, Mäkelä TP, Perheentupa J, Chapelle AD and
Lehesjoki AE: Gene encoding a new RING-B-box-Coiled-coil protein is
mutated in mulibrey nanism. Nat Genet. 25:298–301. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Venoux M, Tait X, Hames RS, Straatman KR,
Woodland HR and Fry AM: Poc1A and Poc1B act together in human cells
to ensure centriole integrity. J Cell Sci. 126:163–175. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Keller LC, Geimer S, Romijn E, Yates J
III, Zamora I and Marshall WF: Molecular architecture of the
centriole proteome: The conserved WD40 domain protein POC1 is
required for centriole duplication and length control. Mol Biol
Cell. 20:1150–1166. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Geister KA, Brinkmeier ML, Burgess DL,
Cavalcoli J, Cheung LY, Oatley JM, Oatley M, Wendt J and Camper SA:
Poc1a, a component of the centriole and cilia, causes skeletal
dysplasia and male infertility: A mousemodel: American Society of
Human Genetics Conference. San Diogo, California, USA: 2014
|