Introduction
Acute kidney injury (AKI) describes acute renal
function decline, which results in >60% of patients requiring
intensive care (1). The etiology of
AKI can be multiple, and sepsis, which is defined as the presence
of any organ dysfunction that results from the deleterious response
of the host to infection, is a common offender. Indeed, the kidney
is one of the most commonly affected organs during sepsis and
sepsis-induced (SI)-AKI substantially contributes to the morbidity
and mortality of patients who suffer from sepsis (2). The most common cause of SI-AKI is
gram-negative bacterial infection, and lipopolysaccharide (LPS),
which is an important constituent of the outer membrane of
gram-negative bacteria, can be an important contributor to risk
associated with AKI (3). LPS can
activate Toll-like receptor 4 (TLR4) signaling and NF-κB, leading
to the generation of vital effectors, such as TNFα and IL-1β
(4). Excessive inflammatory
cytokine generation can be deleterious to the kidneys (5). Pathologically, SI-AKI involves the
presentation of peritubular endothelial dysfunction, tubular injury
and inflammatory cell infiltration (6–8).
Although the understanding of the pathophysiology of SI-AKI has
improved, its incidence remains high, ≤60% of patients with sepsis
have AKI (2), rendering SI-AKI a
frequent complication for those suffering from critical illnesses
(2). Therefore, effective
treatments for SI-AKI, in which LPS-induced SI-AKI constitutes the
majority cases, are urgently required.
TLR4 is a transmembrane protein with extracellular
leucine-rich repeats (9). The
primary ligand for binding TLR4 is LPS. Upon recognizing LPS, TLR4
triggers the IL-1 receptor-domain-containing adaptor-inducing
IFN-β- and myeloid differentiation primary response 88
(MyD88)-dependent signal, causing the production of proinflammatory
cytokines and type I interferon via activation of NF-κB, interferon
regulatory factor-3 and MAPK signaling pathways (10). The renal expression of TLR4 is
primarily located in the tubular epithelia, but it can also be
found in the glomeruli and vascular endothelia (5). It is possible that TLR4 may serve as a
potential therapeutic target for LPS-induced AKI and the use of a
monoclonal antibody (mAb) is a promising approach (5,11). The
monoclonal antibodies are frequently administered among patients
with inflammation-triggered diseases (11). In prior studies, humanized anti-TLR4
Fab fragments with gene-splicing were created using phage antibody
library technology and antibodies based on a prokaryotic vector
were successfully constructed, followed by the collection of
purified anti-TLR4 Fab antibodies (11,12).
It was also revealed that the produced anti-TLR4 mAb can
effectively counteract LPS-induced damage by blocking TLR4
signaling in macrophages (11,12).
In light of these findings, the protective effects against
LPS-related AKI exerted by the humanized anti-TLR4 mAb and the
associated mechanisms in mice were further investigated in the
present study.
Materials and methods
Reagents, diagnostic kits and
antibodies
The humanized anti-TLR4 mAb was provided by
Professor Jin Zhu (Nanjing Medical University, Nanjing, China;
patent no. ZL201410765623.8). The Escherichia coli 0111 B4
LPS was purchased from Sigma-Aldrich (Merck KGaA). The diagnostic
kit for serum creatinine and the test kit for blood urea nitrogen
(BUN) were purchased from Beckman Coulter, Inc. For detecting
IL-1β, IL-6 (Rockland Immunochemicals Inc. Mouse IL-1 beta
AccuSignal ELISA kit; cat. no. KOA0211 and Mouse IL-6 AccuSignal
ELISA kit; cat. no. KOA0226) and TNFα (Dakewe Bio-engineering Co.,
Ltd. Mouse TNF-α Precoated ELISA kit; cat. no. 1217202) ELISA kits
were used. For western blotting, the antibodies targeted against
Bax (cat. no. 14796), MyD88 (cat. no. 4283), phosphorylated (p)-IκB
(cat. no. 2859), IκB (cat. no. 4812), p-IKKα/β (cat. no. 2697),
p-p65 (cat. no. 3033), p65 (cat. no. 8242) and GAPDH (cat. no.
5174) were obtained from Cell Signaling Technology, Inc. The
antibody targeted against kidney injury molecule-1 (KIM-1) was
purchased from Novus Biologicals, LLC (cat. no. NBP1-76701) and the
antibody targeted against Bcl-2 (cat. no. ab182858) and IKKα/β
(cat. no. ab178870) was purchased from Abcam. The HRP-linked
polymer detection system was obtained from Shanghai Changdao
Biotechnology Co. Ltd.
Animal experiments: Group assignment
and treatment
Female C57BL/6 mice (9 weeks old, 18–22 g) were
obtained from Changzhou Cavens Lab Animal Co. Ltd. All animals were
acclimated for 7 days before experiment initiation, placed in a
12-h light/dark cycle at a temperature of 22±2°C in an
air-conditioned room, and given access to food and water ad
libitum. All procedures were carried out according to the
Guidelines for Laboratory Animal Care of the US National Institutes
of Health (13). The present study
was approved by the Research Ethics Committee of The Affiliated
Wuxi People's Hospital of Nanjing Medical University (approval no.
KS202089). All applicable international, national and/or
institutional guidelines for the care and use of animals were
followed. A total of 32 mice were randomly divided into four groups
(n=8 per group): i) Control; ii) LPS; iii) LPS + humanized
anti-TLR4 antibody (1 µg/g); and iv) LPS + humanized anti-TLR4
antibody (10 µg/g). The concentration of humanized anti-TLR4
antibody was selected according to the results of pre experiments.
To induce AKI, mice in the LPS group were intraperitoneally
administered 10 µg/g body weight of LPS dissolved in normal saline.
Mice in the LPS + humanized anti-TLR4 antibody groups received a
humanized anti-TLR4 antibody injection through the tail vein at 4 h
before the LPS challenge. Mice in the control group were
intraperitoneally administered 10 µg/g body weight of normal
saline. All mice were sacrificed at 24 h after LPS stimulation or
saline injection, and the blood and kidneys were collected.
Isoflurane inhalation anesthesia was used (induction concentration,
3%; maintenance concentration, 2%) and venous blood was collected
from the orbital sinus, followed by the removal of the bilateral
kidney. Then, mice were sacrificed by CO2 asphyxia (50%
CO2 replacement rate).
Serum biochemical and cytokine
analysis
The sera obtained from mice was isolated from total
blood by centrifugation (3,000 × g for 10 min at room temperature).
Subsequently, serum creatinine and blood urea nitrogen (BUN) of
serum were measured using commercially available kits. The levels
of serum IL-6, TNFα and IL-1β were determined using ELISA kits
according to manufacturer's protocols.
Renal histopathological
examination
Mice renal tissues were fixed with 10% formaldehyde,
embedded in paraffin, sectioned at 4 µm thick and stained with
hematoxylin and eosin (hematoxylin aqueous solution for 5 min,
alcohol eosin for 1–2 min at room temperature). Pathological
examination was performed under a light microscope (magnification,
×200) by two pathologists, who were blinded to the treatment. The
severities of renal tubuli that presented necrosis, brush border
loss, interstitial edema and tubular dilation were classified into
five categories: 0, none; 1, 0–20%; 2, 20–50%; 3, 50–70%; and 4,
>70% (4).
For the immunohistochemical (IHC) staining, the
paraffin-embedded kidney sections were deparaffinized and
rehydrated in a graded ethanol series. Using a microwave, the
antigen was recovered using sodium citrate solution (pH 6.0).
Following antigen extraction and internal peroxidase quenching by
3% H2O2, the sections were incubated
overnight with Bax mAb (1:400), Bcl-2 mAb (1:500) and KIM-1
polyclonal antibody (1:100) at 4°C. Then the sections were
incubated with HRP-bonded second antibody (1:200) for 30 min and
stained with hematoxylin for 3 min at room temperature, followed by
the collection of four digital images of each non-overlapping
microscopic field of the renal cortex and medulla using an Eclipse
Ni light microscope (Nikon Corporation; magnification, ×200). The
positive stained areas of KIM-1, Bax and Bcl-2, based on the unit
area (magnification, ×200), were calculated as the percentage of
all examined areas using a digital image analysis program [ImageJ
V1.8.0.112 (National Institutes of Health); DS-Ri2 Special color
imaging system for microscope; Nikon Corporation)].
Western blotting
RIPA buffer (CoWin Biosciences) was used to extract
total protein from renal tissues. The BCA method was used to
determine the protein concentration. The total protein samples (30
µg) were separated via 12% SDS-PAGE and transferred onto PVDF
membranes, which were then blocked with 5% albumin from bovine
serum in TBS for 60 min at room temperature. The membranes were
incubated overnight at 4°C with primary antibodies targeted
against: MyD88 (1:1,000), p-p65 (1:1,000), p65 (1:1,000), p-IKKα/β
(1:1,000), IKKα/β (1:1,000), p-IκB (1:1,000), IκB (1:1,000), Bax
(1:1,000), KIM-1 (1:500), Bcl-2 (1:5,000) and GAPDH (1:2,000).
After incubation with HRP-labeled Goat Anti-Rabbit IgG (Beyotime
Institute of Biotechnology; cat. no. A0208; 1:1,000) for 60 min at
37°C, the bands were visualized using an enhanced chemiluminescence
reagent (Thermo Fisher Scientific, Inc.). Aperio Image Analysis
(Leica Microsystems GmbH; MAN-0013, revision G) was used to
semi-quantify protein expression. GAPDH was used as the loading
control. A total of eight samples were used for each analysis and
the analysis was repeated three times each. There were three
independent tests per experiment.
Reverse transcription-quantitative PCR
(RT-qPCR)
RT-qPCR was performed to determine the mRNA
expression levels of MyD88, IKKα/β, IκB, p65 and KIM-1 using
specific primers. Total RNA was extracted from renal tissues using
TRIzol® (Invitrogen; Thermo Fisher Scientific, Inc.).
Then, the RNA was reverse transcribed into cDNA using RevertAid
First Strand cDNA Synthesis kit (Thermo Fisher Scientific, Inc.;
cat. no. K1622) according to the manufacturer's protocol, followed
by qPCR using SYBR-Green qPCR Master Mix (Thermo Fisher Scientific,
Inc.) and the ABI PRISM 7300 sequence detection system (Applied
Biosystems; Thermo Fisher Scientific, Inc.) in a 384-well PCR
plate. The thermocycling conditions used for qPCR were as follows:
95°C for 10 min; 40 cycles of 95°C for 15 sec and 60°C for 45 sec;
followed by dissociation at 95°C for 15 sec, 60°C for 60 sec, 95°C
for 15 sec and 60°C for 15 sec to detect the melting temperature.
The primer sequences for each gene were: MyD88 forward,
5′-CCCCACTCGCAGTTTGTTG-3′ and reverse, 5′-GATGCCTCCCAGTTCCTTTG-3′;
IKKα/β forward, 5′-GAGACACGGAAGGCAACC-3′ and reverse,
5′-GAGACACGGAAGGCAACC-3′; IκB forward, 5′-GACTGACATTGTGGACCTGC-3′
and reverse, 5′-GACTGACATTGTGGACCTGC-3′; p65 forward,
5′-ATCTGTTTCCCCTCATCTTTCC-3′ and reverse,
5′-CAGCCTCATAGTAGCCATCCC-3′; KIM-1 forward,
5′-AATGGCACTGTGACATCCTC-3′ and reverse, 5′-GAGACACGGAAGGCAACC-3′;
GAPDH forward, 5′ CTGCCCAGAACATCATCC 3′ and reverse, 5′
CTCAGATGCCTGCTTCAC 3′. mRNA expression levels were normalized to
the internal reference gene GAPDH. The RT-PCR data were calculated
by using the 2−ΔΔCq method (14).
Statistical analysis
SPSS (version 18; SPSS, Inc.) was used for the
statistical analysis. Continuous data are presented as the mean ±
SEM. The differences among multiple treatment groups were analyzed
using one-way ANOVA followed by Tukey's post hoc test (parametric)
or the Kruskal-Wallis test followed by Dunn's post hoc test
(non-parametric). There were six samples in each group, and three
multiplex RT-PCR analyses were performed on one sample. A total of
eight samples for each western blot analysis run and western
blotting and all other experiments were repeated three times.
P<0.05 was considered to indicate a statistically significant
difference.
Results
Effects of the humanized anti-TLR4 mAb
on LPS-induced renal histopathology
The effect of the humanized anti-TLR4 mAb on
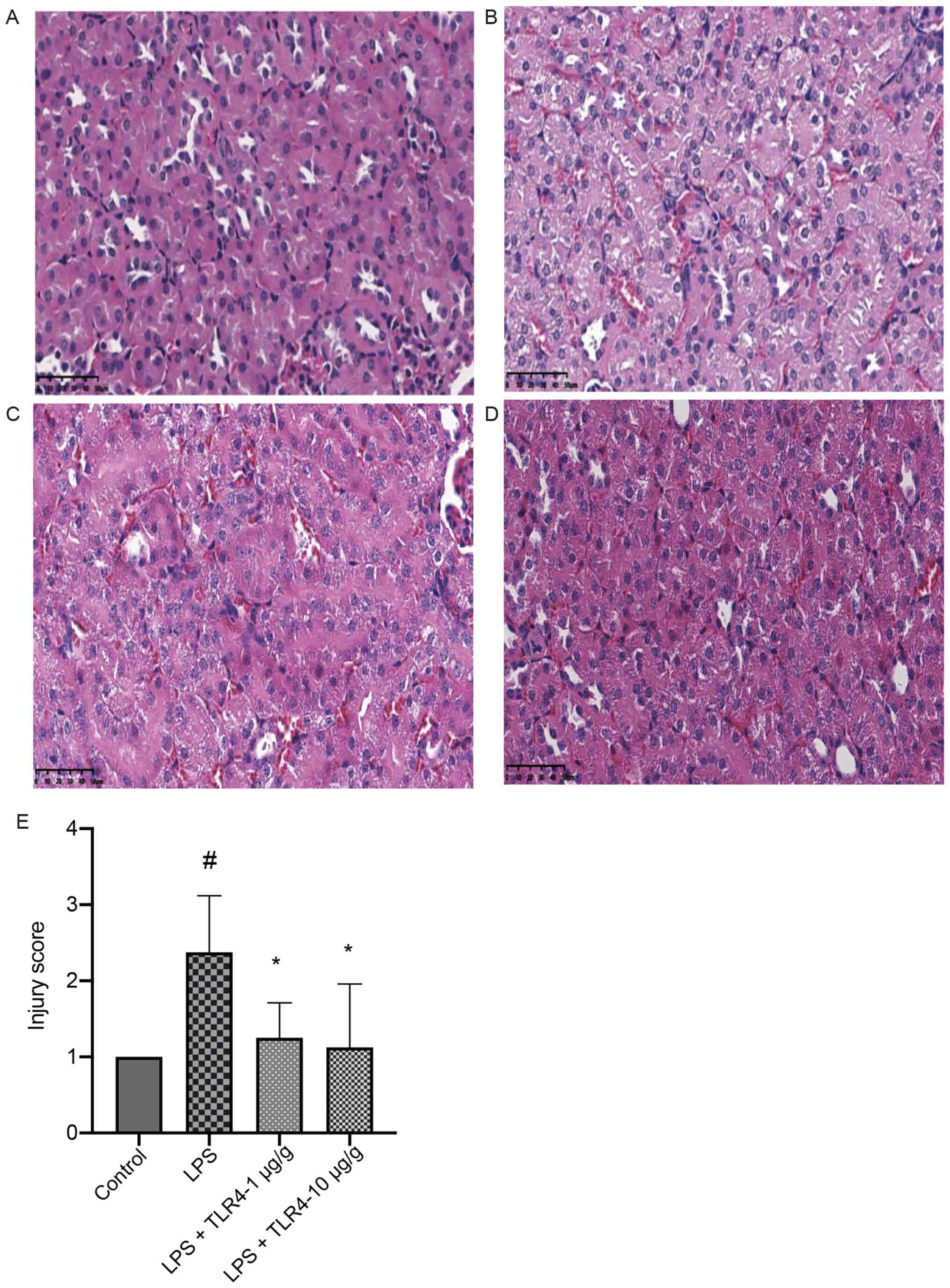
LPS-related AKI was first examined based on the renal pathology.
Compared with the control group, the kidneys in the LPS group
displayed prominent pathologies and pretreatment with the humanized
anti-TLR4 mAb (1 µg/g and 10 µg/g) significantly improved such
renal injuries (Fig. 1).
Effects of the humanized anti-TLR4 mAb
on the renal function of LPS-treated mice
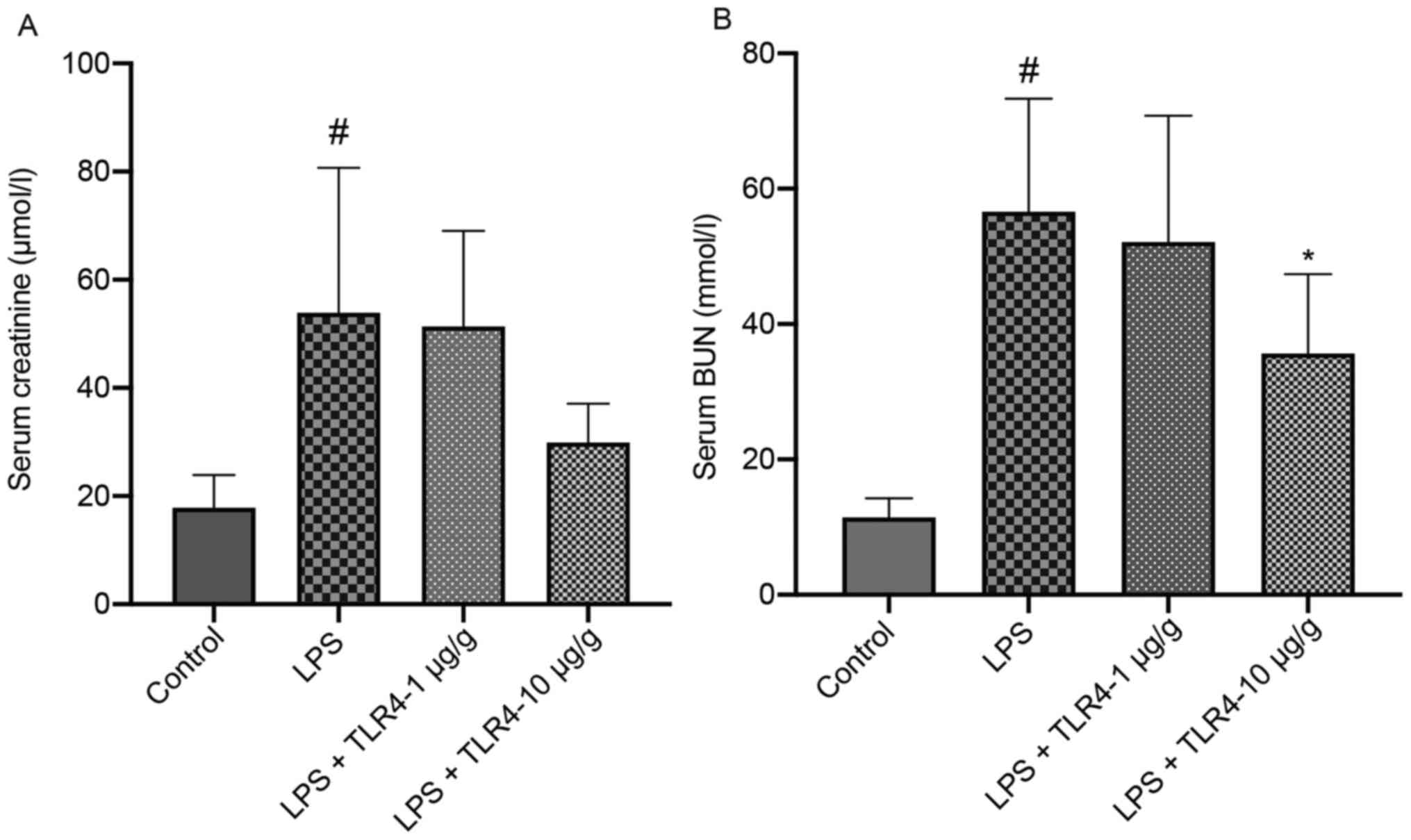
The serum BUN and creatinine levels were further
examined to evaluate the effect of the humanized anti-TLR4 mAb on
renal function. Compared with the control group, mice treated with
LPS displayed significantly elevated serum BUN and creatinine
levels, whereas LPS-induced alterations in BUN were significantly
attenuated by the administration of the humanized anti-TLR4 mAb (10
µg/g; Fig. 2). Serum creatinine
(log-transformed) was also attenuated by the administration of the
humanized anti-TLR4 mAb, but the difference was not
significant.
Effects of the humanized anti-TLR4 mAb
on LPS-induced alterations in IL-6, TNFα and IL-1 β levels based on
ELISAs
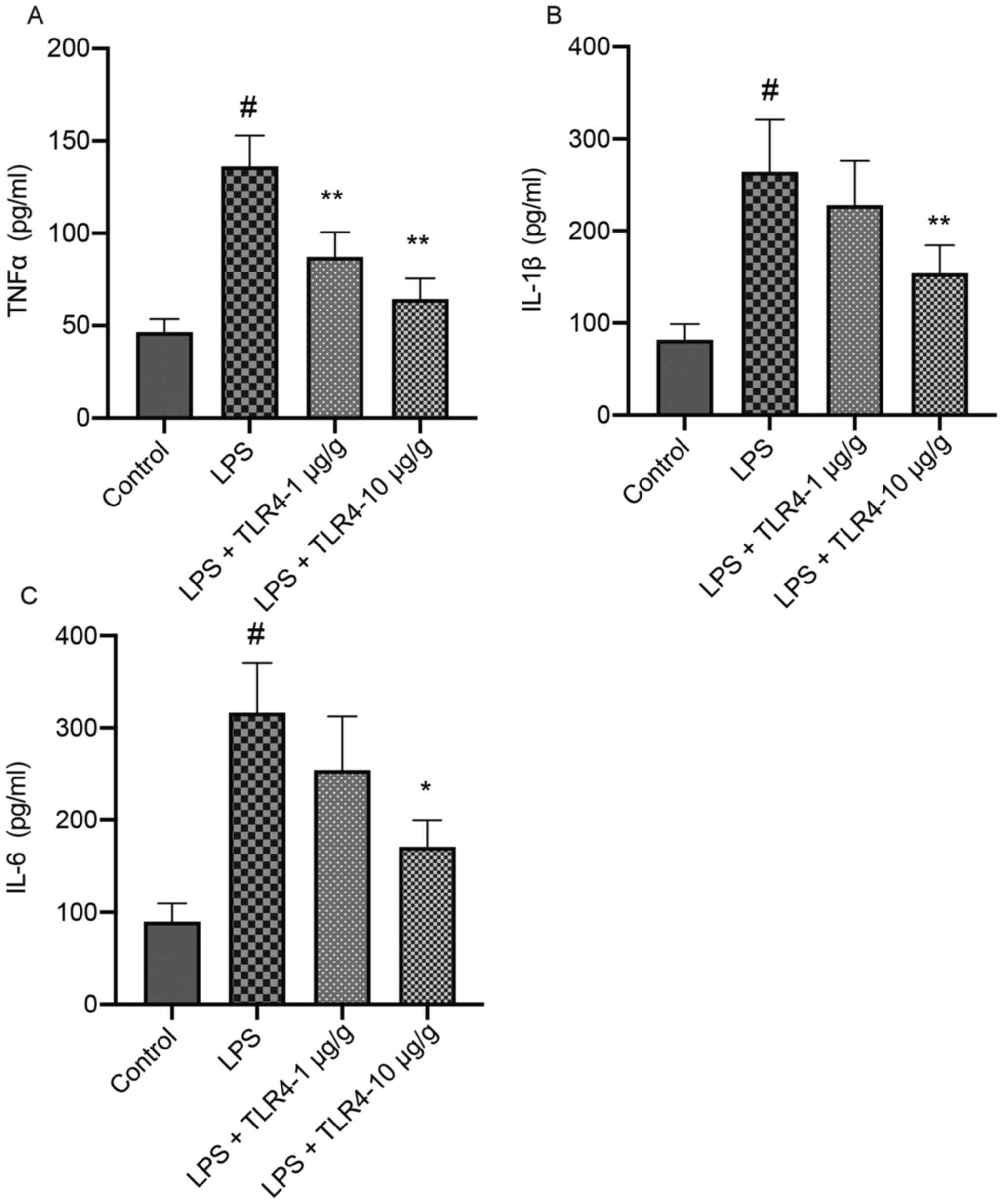
Compared with the control group, LPS significantly
increased the serum TNFα, IL-6 and IL-1β levels, which was
attenuated by pretreatment with the humanized anti-TLR4 mAb in a
dose-dependent manner (Fig. 3).
Effects of the humanized anti-TLR4 mAb
on the expression of MyD88, IKKα/β, IκB, p65 and KIM-1 in renal
tissues based on RT-qPCR
The mRNA expression levels of MyD88, IKKα/β, IκB,
p65 and KIM-1 in the kidneys were significantly higher in the LPS
group compared with the control group (Fig. 4). LPS-induced increases in the
expression levels of MyD88, IKKα/β and IκB were significantly
attenuated by pretreatment with the humanized anti-TLR4 mAb (10
µg/g). Pretreatment with the humanized anti-TLR4 mAb decreased p65
and KIM-1 expression levels in LPS-treated mice, but the difference
was not significant.
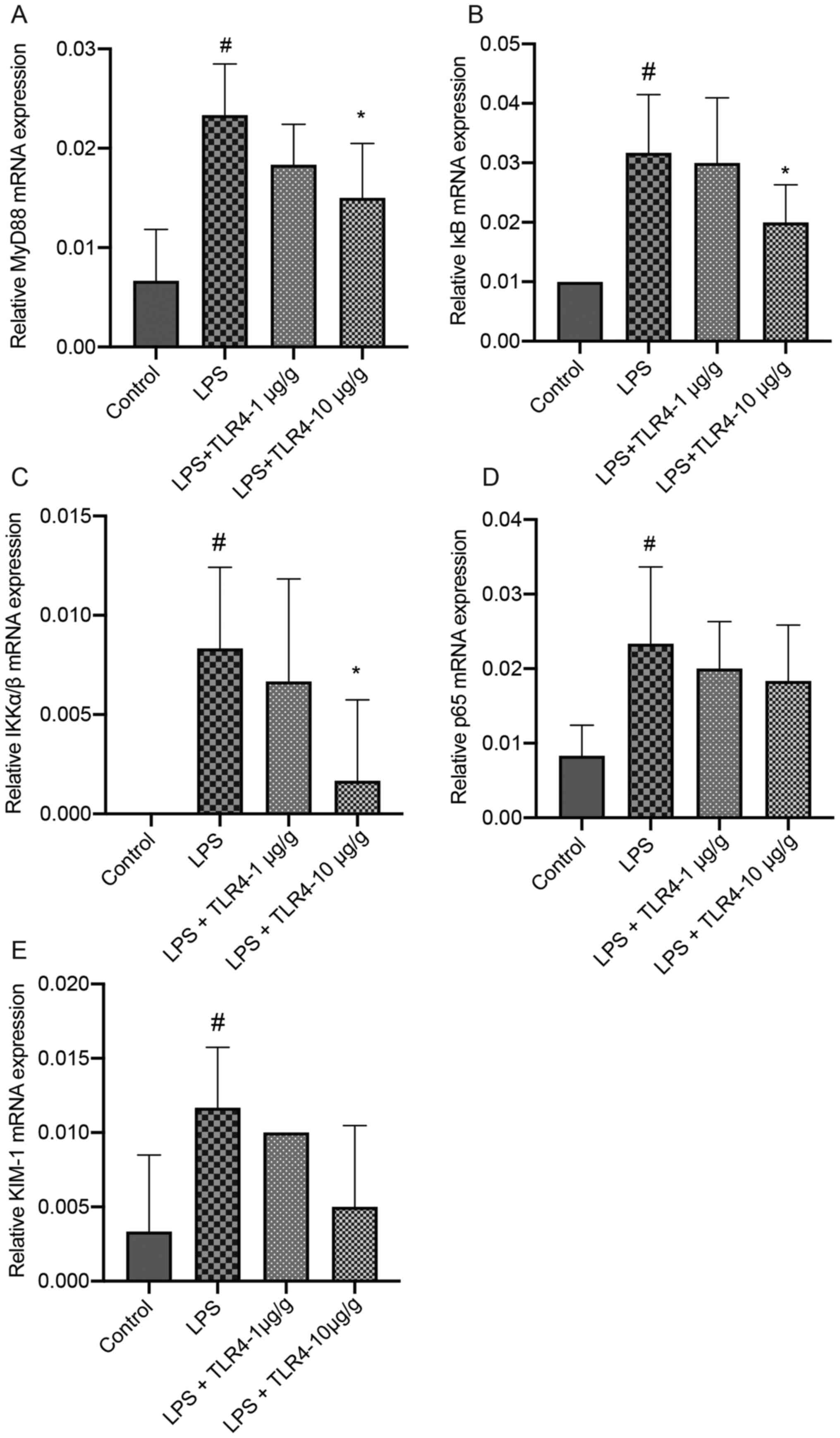 | Figure 4.Effects of the humanized anti-TLR4
mAb on the expression of MyD88, IκB, IKKα/β, p65 and KIM-1 in renal
tissues. The kidneys were collected at 24 h after LPS treatment.
Subsequently, reverse transcription-quantitative PCR was performed
to assess the expression levels of (A) MyD88, (B) IκB, (C) IKKα/β,
(D) p65 and (E) KIM-1. #P<0.05 vs. control;
*P<0.05 vs. LPS. TLR4, Toll-like receptor 4; mAb, monoclonal
antibody; MyD88, myeloid differentiation primary response 88;
KIM-1, kidney injury molecule-1; LPS, lipopolysaccharide. |
Effects of the humanized anti-TLR4 mAb
on the expression of Bax, Bcl-2 and KIM-1 in renal tissues based on
IHC staining
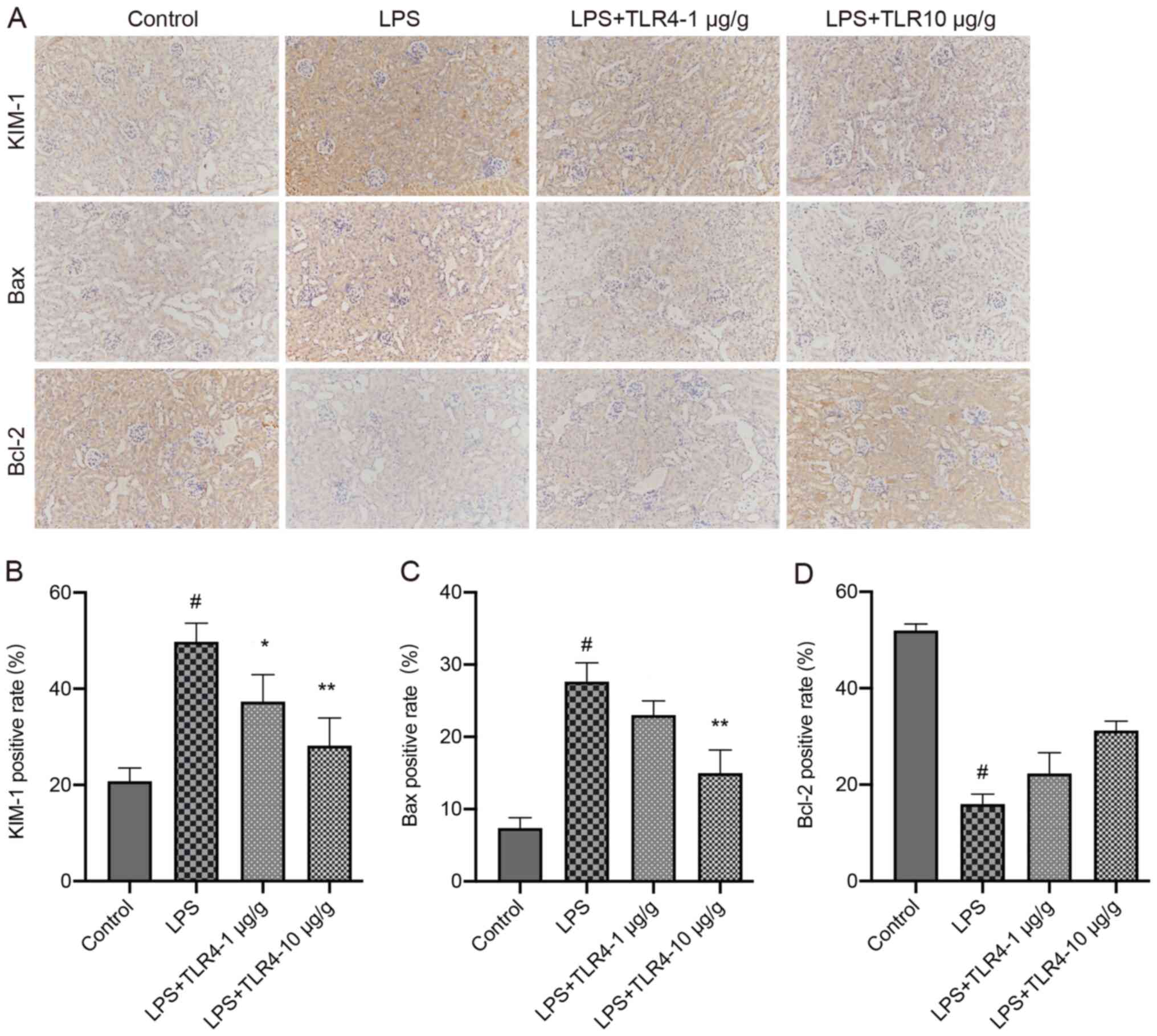
The percentage of positively stained areas of KIM-1
and Bax in the LPS group were significantly increased compared with
the control group (Fig. 5).
However, pretreatment with the humanized anti-TLR4 mAb
significantly decreased KIM-1 and Bax expression compared with the
LPS group. In the LPS group, the positively stained areas of Bcl-2
were significantly decreased compared with the control group,
whereas pretreatment with the humanized anti-TLR4 mAb increased
Bcl-2 positive staining, but the difference was not
significant.
Effects of the humanized anti-TLR4 mAb
on the expression of MyD88, p-IKKα/β, p-IκB and p-p65 in renal
tissues based on western blotting
Compared with the control group, LPS treatment
significantly increased the expression levels of MyD88, p-IKKα/β,
p-IκB and p-p65 in renal tissues (Fig.
6). LPS-induced increases in the expression of MyD88, p-IKKα/β,
p-p65 and p-IκB were significantly attenuated by pretreatment with
the humanized anti-TLR4 mAb.
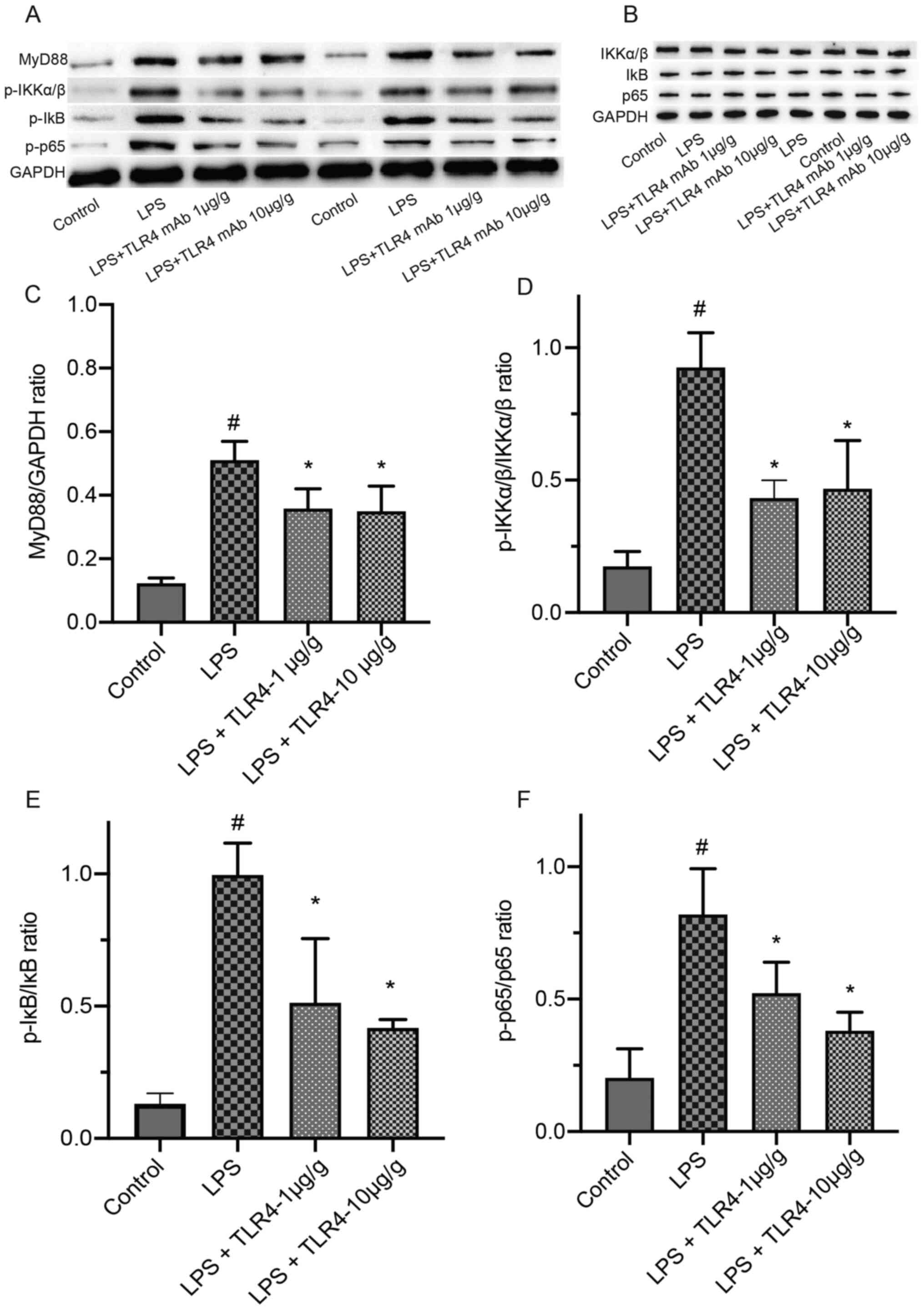 | Figure 6.Effects of the humanized anti-TLR4
mAb on the expression of MyD88, p-IKKα/β, p-IκB, p-p65, IKKα/β, IκB
and p65 in renal tissues by western blotting. The renal tissues
were collected after LPS treatment for 24 h. (A) The expression of
MyD88, p-IKKα/β, p-IκB and p-p65 and (B) IKKα/β, IκB and p65
Quantification of (C) MyD88, (D) p-IKKα/β/IKKα/β ratio (E)
p-IκB/IκB ratio and (F) p-p65/p65 ratio expression levels.
#P<0.01 vs. control; *P<0.01 vs. LPS. TLR4,
Toll-like receptor 4; mAb, monoclonal antibody; MyD88, myeloid
differentiation primary response 88; p, phosphorylated; LPS,
lipopolysaccharide. |
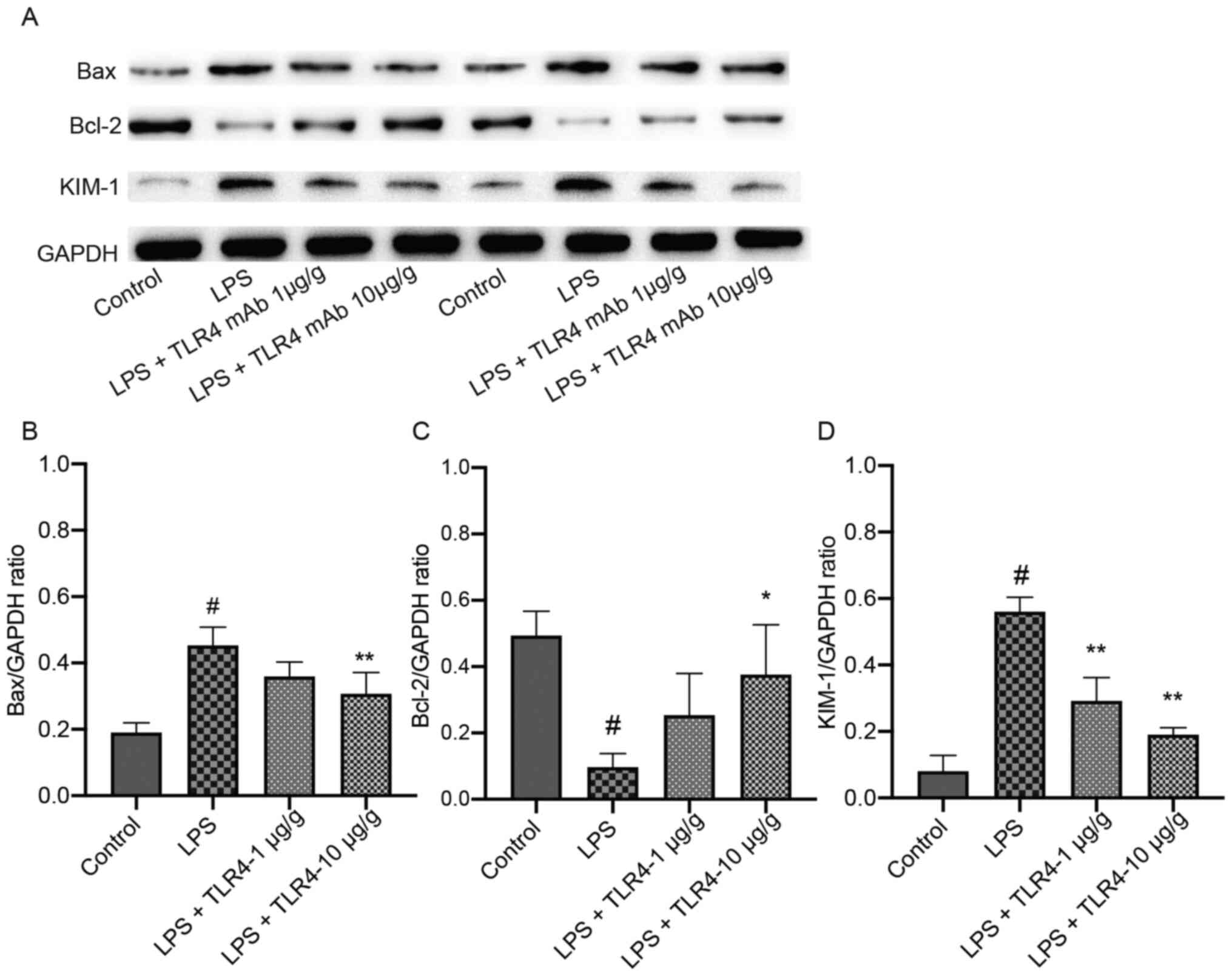
Effects of the humanized anti-TLR4 mAb
on the expression of Bax, Bcl-2 and KIM-1 in renal tissues based on
western blotting
The expression levels of KIM-1 and Bax were
significantly higher in the LPS group compared with the control
group. The expression of these proteins significantly declined
after pretreatment with the humanized anti-TLR4 mAb (Fig. 7). Furthermore, Bcl-2 expression in
the LPS group was significantly decreased compared with the control
group, but pretreatment with the humanized anti-TLR4 mAb (10 µg/g)
significantly increased Bcl-2 expression levels in LPS-treated
mice.
Discussion
The occurrence of AKI is associated with a higher
risk of developing complications that involve various organs and
systems (15). Among in-patients,
>5% are at risk of manifesting AKI, which increases the risk of
mortality and the subsequent development of chronic kidney disease
(16). Among those that require
intensive care, >50% have AKI, the severity of which is closely
associated with an increase in hospitalization mortality (17). At present, the management plan for
patients with SI-AKI consists primarily of infection control,
transfusion and optimal resuscitation, the use of vasoactive drugs
and the prompt initiation of renal replacement therapy (2). However, patients with SI-AKI continue
to suffer from high mortality. Therefore, novel therapeutics for
effective management are urgently required. Previous studies
reported that the humanized anti-TLR4 mAb can effectively
ameliorate LPS-triggered inflammatory responses in macrophages by
blocking the TLR4 signal (11,12).
In the present study, the effect of the humanized anti-TLR4 mAb on
LPS-induced AKI mice was investigated and it was shown that LPS
induced significant AKI in treated mice, as indicated by elevated
serum creatinine, BUN and KIM-1, which is an early biomarker of AKI
(18), expression levels. It was
further revealed that, compared with the LPS group, the
administration of the humanized anti-TLR4 mAb downregulated KIM-1
expression at the mRNA, protein and tissue level, although this was
not significant at the mRNA level. Administration of the humanized
anti-TLR4 mAb was accompanied by improved renal histopathological
changes and downregulated serum BUN and creatinine levels in
LPS-treated mice. Collectively, the results of the present study
indicated that the humanized anti-TLR4 mAb may alleviate the
severity of LPS-induced AKI in animal models.
Since male mice have been found to be more
aggressive prior to the experiments, the differences in animal
behavior might alter experimental results. Hence, female mice were
selected in the present study. The periods of adolescence,
middle-age and old age of mice are 60–90 days, 120–180 days and
>180 days after birth, respectively. It was found that
6-month-old mice became old with weaker immunity, displayed a
declined physical status, thinner hair and slower reactions
(19), and had poorer liver and
kidney function. Renal aging is associated with alterations in
renal morphology and functional decline (20). The glomerular filtration rate of
C57BL/6 mice also decreases with age (21). The literature suggests that most
reports of LPS-induced AKI involve mice aged 8–12 weeks (15,22).
Therefore, the present study used 9-week-old mice after a 7-day
acclimation period.
The induction of inflammatory cytokines triggered by
LPS serves an important role in the pathogenesis of AKI (23). It has been previously shown that the
serum levels of TNFα, IL-1β and IL-6 significantly increase during
episodes of LPS-induced AKI and that the inhibition of these
cytokines may alleviate such injury (24). A previous study revealed that the
humanized anti-TLR4 mAb can lower serum TNFα, IL-1 and IL-6 levels
and inhibit the expression of these cytokines in macrophages
stimulated by LPS (11). The
present study further showed that the humanized anti-TLR4 mAb
decreased the serum TNFα, IL-1β and IL-6 levels in mice with
LPS-induced AKI in a dose-dependent manner, suggesting that
inhibition of these inflammatory cytokines might be partially
responsible for the therapeutic effect of the humanized anti-TLR4
mAb against LPS-induced AKI.
TLR4 signaling is the primary pathway that mediates
renal inflammation (25). LPS
induces the release of inflammatory cytokines by activating TLR4,
which accounts for the subsequent development of renal injuries
(26). NF-κB is the main effector
that regulates the expression of several inflammatory cytokines
(27). By activating the TLR4/NF-κB
signaling pathway, LPS can enhance the production of TNFα, IL-1β
and IL-6 (15). It has been
previously shown that the humanized anti-TLR4 mAb can inhibit
LPS-induced NF-κB activation in macrophages (11,12).
Previous study have shown that TLR4 may serve as a potential
therapeutic target during AKI (5).
The anti-inflammatory mechanism underlying the humanized anti-TLR4
mAb was examined and the results demonstrated that the humanized
anti-TLR4 mAb (10 µg/g) downregulated LPS-induced elevations in the
mRNA expression levels of MyD88, IKKα/β and IκB. Furthermore, the
protein expression levels of MyD88, p-IKKα/β, p-IκB and p-p65 were
significantly lowered by pretreatment with the humanized anti-TLR4
mAb in LPS-treated mice. The present findings suggested that the
humanized anti-TLR4 mAb reduced the release of circulating
cytokines during LPS-induced AKI via suppressing TLR4/NF-κB
signaling.
Apoptosis also serves an important role in the
pathogenesis of SI-AKI (28).
Inflammatory exudates in injured kidneys induce apoptosis and
promote renal epithelial loss, which is a characteristic of AKI
(29,30). The primary mechanisms of renal
tubular cell apoptosis are the activation of Bax and the inhibition
of Bcl-2. The present study revealed that, compared with the
control group, LPS exposure significantly increased Bax expression,
but significantly decreased Bcl-2 expression, of which both effects
were significantly reversed by pretreatment with the humanized
anti-TLR4 mAb (10 µg/g). Therefore, it was hypothesized that the
humanized anti-TLR4 mAb may be able to protect against AKI by
reducing the expression of apoptosis-related proteins.
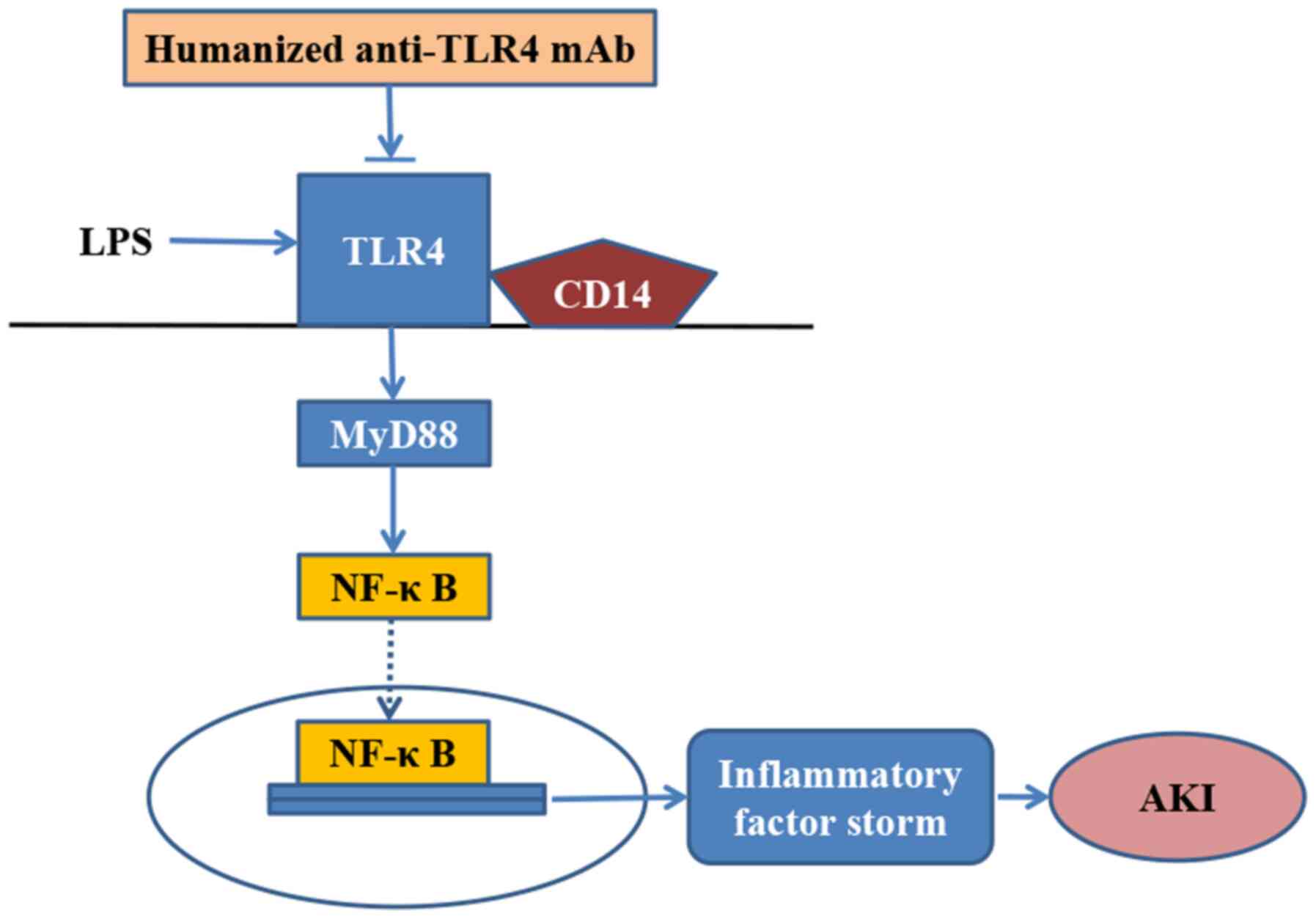
Overall, the present study revealed that the
humanized anti-TLR4 mAb exhibited a strong anti-inflammatory effect
during episodes of LPS-related AKI in mice. In addition, it
demonstrated that the humanized anti-TLR4 mAb further inhibited the
activation of NF-κB during LPS-related AKI (Fig. 8). Therefore, the humanized anti-TLR4
mAb may serve as a potential therapeutic for the management of
SI-AKI in the future.
Acknowledgements
Not applicable.
Funding
The present study was supported by the Scientific
Research Project of Wuxi Health Committee (grant no. Z201914), the
Scientific Research Projects of Jiangsu Provincial Health
Commission (grant no. LGY201801), the Scientific Research Project
of Wuxi People's Hospital (grant nos. RKA201804 and RKA201805) and
the Wuxi Medical Leadership Talent and Innovation Team (grant no.
CXTDJS001).
Availability of data and materials
The datasets generated and analyzed in the present
study are available from the corresponding author on reasonable
request.
Authors' contributions
JZ, CX and LW conceived and designed the experiment.
QZ, MW and YZ conducted the experiments. XL and LW analyzed the
data. QZ and LW wrote the manuscript. JZ, CX, QZ and LW confirm the
authenticity of all the raw data. All authors have read and
approved the final manuscript.
Ethics approval and consent to
participate
The present study was approved by the Research
Ethics Committee of The Affiliated Wuxi People's Hospital of
Nanjing Medical University (approval no. KS202089). All applicable
international, national and/or institutional guidelines for the
care and use of animals were followed.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Glossary
Abbreviations
Abbreviations:
|
AKI
|
acute kidney injury
|
|
BUN
|
blood urea nitrogen
|
|
KIM-1
|
kidney injury molecule-1
|
|
LPS
|
lipopolysaccharide
|
|
mAb
|
monoclonal antibody
|
|
SI-AKI
|
sepsis-induced AKI
|
|
TLR4
|
Toll-like receptor 4
|
References
|
1
|
Shu B, Feng Y, Gui Y, Lu Q, Wei W, Xue X,
Sun X, He W, Yang J and Dai C: Blockade of CD38 diminishes
lipopolysaccharide-induced macrophage classical activation and
acute kidney injury involving NF-κB signaling suppression. Cell
Signal. 42:249–258. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Poston JT and Koyner JL: Sepsis associated
acute kidney injury. BMJ. 364:k48912019. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Kuzmich NN, Sivak KV, Chubarev VN, Porozov
YB, Savateeva-Lyubimova TN and Peri F: TLR4 signaling pathway
modulators as potential therapeutics in inflammation and sepsis.
Vaccines (Basel). 5:342017. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Song J, Fan HJ, Li H, Ding H, Lv Q and Hou
SK: Zingerone ameliorates lipopolysaccharide-induced acute kidney
injury by inhibiting Toll-like receptor 4 signaling pathway. Eur J
Pharmacol. 772:108–114. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Anderberg SB, Luther T and Frithiof R:
Physiological aspects of Toll-like receptor 4 activation in
sepsis-induced acute kidney injury. Acta Physiol (Oxf).
219:573–588. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Tran M, Tam D, Bardia A, Bhasin M, Rowe
GC, Kher A, Zsengeller ZK, Akhavan-Sharif MR, Khankin EV,
Saintgeniez M, et al: PGC-1α promotes recovery after acute kidney
injury during systemic inflammation in mice. J Clin Invest.
121:4003–4014. 2011. View
Article : Google Scholar : PubMed/NCBI
|
|
7
|
Takasu O, Gaut JP, Watanabe E, To K,
Fagley RE, Sato B, Jarman S, Efimov IR, Janks DL, Srivastava A, et
al: Mechanisms of cardiac and renal dysfunction in patients dying
of sepsis. Am J Respir Crit Care Med. 187:509–517. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Jung YJ, Lee AS, Nguyen-Thanh T, Kim D,
Kang KP, Lee S, Park SK and Kim W: SIRT2 regulates LPS-induced
renal tubular CXCL2 and CCL2 expression. J Am Soc Nephrol.
26:1549–1560. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Kim HM, Park BS, Kim J-I, Kim SE, Lee J,
Oh SC, Enkhbayar P, Matsushima N, Lee H, Yoo OJ, et al: Crystal
structure of the TLR4-MD-2 complex with bound endotoxin antagonist
Eritoran. Cell. 130:906–917. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Akira S, Uematsu S and Takeuchi O:
Pathogen recognition and innate immunity. Cell. 124:783–801. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Cai B, Wang M, Zhu X, Xu J, Zheng W, Zhang
Y, Zheng F, Feng Z and Zhu J: The fab fragment of a humanized
anti-toll like receptor 4 (TLR4) monoclonal antibody reduces the
lipopolysaccharide response via TLR4 in mouse macrophage. Int J Mol
Sci. 16:25502–25515. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Wang M, Zheng W, Zhu X, Xu J, Cai B, Zhang
Y, Zheng F, Zhou L, Yang Z, Zhang X, et al: A human anti-toll like
receptor 4 Fab fragment inhibits lipopolysaccharide-induced
pro-inflammatory cytokines production in macrophages. PLoS One.
11:e01468562016. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Care NRCU and Animals AUOL; Guide for the
Care and Use of Laboratory Animals, : The National Academies
Collection: Reports funded by National Institutes of Health.
National Academies Press; Washington, DC: 2011
|
|
14
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) Method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Ye H-Y, Jin J, Jin L-W, Chen Y, Zhou Z-H
and Li Z-Y: Chlorogenic acid attenuates lipopolysaccharide-induced
acute kidney injury by inhibiting TLR4/NF-κB signal pathway.
Inflammation. 40:523–529. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Wonnacott A, Meran S, Amphlett B, Talabani
B and Phillips A: Epidemiology and outcomes in community-acquired
versus hospital-acquired AKI. Clin J Am Soc Nephrol. 9:1007–1014.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Hoste EA, Bagshaw SM, Bellomo R, Cely CM,
Colman R, Cruz DN, Edipidis K, Forni LG, Gomersall CD, Govil D, et
al: Epidemiology of acute kidney injury in critically ill patients:
The multinational AKI-EPI study. Intensive Care Med. 41:1411–1423.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Song J, Yu J, Prayogo GW, Cao W, Wu Y, Jia
Z and Zhang A: Understanding kidney injury molecule 1: A novel
immune factor in kidney pathophysiology. Am J Transl Res.
11:1219–1229. 2019.PubMed/NCBI
|
|
19
|
Qin C: Medical laboratory zoology.
People's Health Publishing House. 51–57. 2015.
|
|
20
|
Zhou XJ, Rakheja D, Yu X, Saxena R, Vaziri
ND and Silva FG: The aging kidney. Kidney Int. 74:710–720. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Hackbarth H and Harrison DE: Changes with
age in renal function and morphology in C57BL/6, CBA/HT6, and
B6CBAF1 mice. J Gerontol. 37:540–547. 1982. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Chen L, Yang S, Zumbrun EE, Guan H,
Nagarkatti PS and Nagarkatti M: Resveratrol attenuates
lipopolysaccharide-induced acute kidney injury by suppressing
inflammation driven by macrophages. Mol Nutr Food Res. 59:853–864.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Mortensen J, Shames B, Johnson CP and
Nilakantan V: MnTMPyP, a superoxide dismutase/catalase mimetic,
decreases inflammatory indices in ischemic acute kidney injury.
Inflamm Res. 60:299–307. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Xu D, Chen M, Ren X, Ren X and Wu Y:
Leonurine ameliorates LPS-induced acute kidney injury via
suppressing ROS-mediated NF-κB signaling pathway. Fitoterapia.
97:148–155. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Cunningham PN, Wang Y, Guo R, He G and
Quigg RJ: Role of Toll-like receptor 4 in endotoxin-induced acute
renal failure. J Immunol. 172:2629–2635. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Castoldi A, Braga TT, Correa-Costa M,
Aguiar CF, Bassi EJ, Correa-Silva R, Elias RM, Salvador F,
Moraes-Vieira PM, Cenedeze MA, et al: TLR2, TLR4 and the MYD88
signaling pathway are crucial for neutrophil migration in acute
kidney injury induced by sepsis. PLoS One. 7:e375842012. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Blackwell TS and Christman JW: The role of
nuclear factor-κ B in cytokine gene regulation. Am J Respir Cell
Mol Biol. 17:3–9. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Lee S-Y, Lee Y-S, Choi H-M, Ko YS, Lee HY,
Jo SK, Cho WY and Kim HK: Distinct pathophysiologic mechanisms of
septic acute kidney injury: Role of immune suppression and renal
tubular cell apoptosis in murine model of septic acute kidney
injury. Crit Care Med. 40:2997–3006. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Peerapornratana S, Manrique-Caballero CL,
Gómez H and Kellum JA: Acute kidney injury from sepsis: Current
concepts, epidemiology, pathophysiology, prevention and treatment.
Kidney Int. 96:1083–1099. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Gao L, Wu W-F, Dong L, Ren G-L, Li H-D,
Yang Q, Li X-F, Xu T, Li Z, Wu B-M, et al: Protocatechuic aldehyde
attenuates cisplatin-induced acute kidney injury by suppressing
nox-mediated oxidative stress and renal inflammation. Front
Pharmacol. 7:4792016. View Article : Google Scholar : PubMed/NCBI
|