Introduction
Hepatocellular carcinoma (HCC) is a common type of
primary liver cancer, the 5-year survival rate of which is poor
(1–3). Due to the high incidence and mortality
associated with HCC, it is necessary to explore novel prognostic
biomarkers and therapeutic targets for patients with HCC. With the
ongoing development of microarray and gene sequencing technologies,
bioinformatics approaches have been widely applied in numerous
research fields, including tumorigenesis and cancer progression
(4). Moreover, gene expression
profiles have been used to identify differentially expressed genes
(DEGs) associated with the prognosis of patients with HCC, and
these genes may be potential candidates for targeted treatment
(5).
Weighted gene co-expression network analysis (WGCNA)
is a widely used data mining method used especially for studying
biological networks based on high-throughput gene expression
profiles (6). In the present study,
DEGs were identified in The Cancer Genome Atlas-liver HCC
(TCGA-LIHC) and International Cancer Genome Consortium liver
cancer-RIKEN, Japan (ICGC LIRI-JP) cohorts. Subsequently, WGCNA was
used to screen meaningful modules and identify novel prognostic
biomarkers using least absolute shrinkage and selection operator
(LASSO) Cox regression. Furthermore, the prognostic prediction
value of the biomarkers was validated in patients with HCC.
Finally, a rarely reported gene, epoxide hydrolase 2 (EPHX2), was
selected for further investigation.
EPHX2 encodes for soluble epoxide hydrolase (sEH)
(7), an important enzyme in
endogenous lipid epoxide degradation, particularly the inactivation
of epoxyeicosatrienoic acids (EETs) (8). Cytochrome P450 (CYP) epoxygenases
convert arachidonic acid to EETs (9). Dysregulation of EPHX2 is associated
with the pathogenesis of various diseases, such as renal and
hepatic malignant neoplasms (10),
hypertension (11) and
hypercholesterolemia (12). The
Gene Ontology (GO) annotation of EPHX2 involves xenobiotic
metabolism, especially the hydrolysis of trans-substituted epoxides
(13); however, the detailed
functions of EPHX2 in the progression of HCC remain unclear. In the
present study, EPHX2 was identified as an independent prognostic
biomarker based on gene expression data. The mRNA expression levels
of EPHX2 were also evaluated in vitro. Furthermore, the
functions of EPHX2 were investigated using GO analysis, Kyoto
Encyclopedia of Genes and Genomes (KEGG) analysis and gene set
enrichment analysis (GSEA). The results suggested that
downregulation of EPHX2 was associated with tumor progression and
poor prognosis of HCC.
Materials and methods
Data collection and preprocessing
Gene expression profiles were obtained from TCGA
(https://portal.gdc.cancer.gov/repository/), ICGC
(https://dcc.icgc.org/releases/release_28/) (14) and Gene Expression Omnibus (GEO)
databases (https://www.ncbi.nlm.nih.gov/geo/) (15). For TCGA-LIHC cohort, gene expression
profiles produced by the Illumina HiSeq RNA-Seq platform were
downloaded from TCGA database and normalized using the variance
stabilizing transformation (VST) function in ‘DESeq2’ R package (R
Core Team; http://www.r-project.org) (16). Clinical data were extracted from
TCGA database and MSI scores were downloaded from cBioPortal
database (http://www.cbioportal.org/). For the
ICGC LIRI-JP cohort, gene expression profiles and clinical
information were downloaded from the ICGC database (17) and normalized using VST. Clinical
information about the tumor grade was available for only 212
patients. For the GSE14520 cohort, gene expression profiles were
downloaded from the GEO database and normalized using the limma
package (18). The gene expression
profiles of TCGA Pan-Cancer were downloaded from the UCSC database
(http://xena.ucsc.edu/) (19) and Student's t-test was used to
analyze EPHX2 expression between tumor and normal tissues in
different types of cancer.
Identification and validation of the
prognostic gene signature
‘Deseq2’ R package was used to screen the DEGs
between HCC and paired normal samples in TCGA-LIHC and ICGC LIRI-JP
cohorts (16). Adjusted P<0.05
and |log2fold change (FC)| >1 were set as the cut-off
thresholds. The results were presented in a volcano map using
‘ggplot2’ R package. The TCGA-LIHC cohort was set as the training
cohort, and the ICGC LIRI-JP cohort as the validation cohort. The
co-expression network of DEGs in TCGA-LIHC and ICGC LIRI-JP cohorts
was constructed based on TCGA-LIHC cohort using the R package
‘WGCNA’ (6). The soft-thresholding
power with a slope close to 1 and a scale-free R2 close
to 0.9 was selected to transform the adjacency matrix to a
topological overlap matrix. The soft-thresholding power was set as
7 (scale-free R2=0.91, slope=−1.49). Cut height was set
as 0.25 and the minimal module size was set as 30 for network
construction and module detection. The module with the highest
correlation with HCC was considered as the key module. Univariate
Cox proportional hazard regression analysis of those genes whose
gene significance (GS) >0.2 and module membership (MM) >0.6
was conducted to screen overall survival (OS)-associated genes.
Identified genes were further used to produce the prognostic
multiple-gene signature using the LASSO Cox regression with the
‘glmnet’ package in R (20,21). The following risk score formula was
used: Risk score (mRNA-based classifier) = sum of coefficients ×
expression levels of mRNA. The median risk score was used as a
cut-off value to divide the patients into high- and low-risk groups
for the prognostic prediction. The prognostic gene signature was
validated in the ICGC LIRI-JP cohort using the aforementioned
formula.
Oncomine database
The Oncomine database (https://www.oncomine.org) is an integrated online
cancer microarray database for DNA or RNA sequence analysis
(22). In the present study,
transcriptional expression of EPHX2 between cancer and matched
normal samples was obtained from the Oncomine database. EPHX2
levels in various cancer types were compared using Student's
t-test. The cut-off P-value and FC were as follows: P-value, 0.05;
FC, 1.5; gene rank, 10%; data type, mRNA. EPHX2 expression in HCC
was compared among the five datasets (15,23–25) by
Oncomine meta-analysis.
Kaplan-Meier plotter database
analysis
The Kaplan-Meier plotter database (http://kmplot.com/analysis/) is an online tool
containing gene expression and clinical data, which is commonly
used to evaluate the prognostic value of different genes among 21
types of cancer (26). The sources
of this database are GEO, TCGA and European Genome Phenome Archive.
The prognostic value of EPHX2 mRNA expression in pan-cancer
(n=7,489) including 21 different types of cancer was evaluated
using the Kaplan-Meier plotter database (http://kmplot.com/analysis/index.php?p=service&cancer=pancancer_rnaseq).
Patient samples were split into two groups by auto select best
cut-off. The log-rank P-value and hazard ratio (HR) with 95%
confidence intervals (CIs) were obtained.
GO and KEGG analyses
Spearman correlation analysis (R software 3.6.3) was
carried out to identify 500 genes closely correlated with EPHX2.
Functions of EPHX2 and 500 EPHX2-associated genes were investigated
using GO and KEGG analyses with the ‘clusterProfiler’ R package
(26). Adjusted P<0.05 was
considered as significant. GO terms and KEGG pathways were
presented using the R package ‘GOplot’ (27). GO analysis was based on three
factors, including biological process (BP), cellular component (CC)
and molecular function (MF), which could predict the functional
roles of EPHX2 and the 500 related genes. KEGG analysis was used to
identify the pathways associated with EPHX2 and the 500 related
genes.
GSEA
GSEA v4.0.3 (http://www.broad.mit.edu/gsea/) was used to analyze
the association between EPHX2 expression and biological pathways
(28). Pre-defined gene sets
(c2.cp.kegg.v7.0.symbols.gmt) were obtained from the Molecular
Signatures Database (MsigDB; http://software.broadinstitute.org/gsea/msigdb). The
patients in the TCGA-LIHC or ICGC LIRI-JP cohort were sorted into
high- and low-EPHX2 expression groups using median mRNA expression
levels of EPHX2. False discovery rate <0.25 and nominal
P<0.05 were set as the cut-off thresholds.
Patients and specimens
Two independent cohorts of patients with HCC were
used in the present study. For cohort one, tissue microarrays
(TMAs) containing 90 pairs of HCC and matched adjacent
non-cancerous tissue samples with complete clinical and follow-up
data were purchased from Shanghai Liao Ding Biotechnology Co., Ltd.
For cohort two, a total of 12 paired HCC and matched adjacent
non-cancerous tissue samples (distance from tumor margin, ≤3 cm;
cirrhosis tissue was excluded) were obtained from patients (age
range, 40–69 years; five male patients and seven female patients)
who were diagnosed with HCC at the Second Affiliated Hospital of
Chongqing Medical University (Chongqing, China) between January
2018 and December 2019. All patients did not undergo chemotherapy
or radiotherapy before surgery. After performing surgical
resection, tissue samples were immediately frozen and stored in
liquid nitrogen until further use. All cases were histologically
confirmed. The study protocol was approved by the Ethics Committee
of the Second Affiliated Hospital of Chongqing Medical University
(approval no. 2020-186). All patients provided written informed
consent.
Cell culture
Normal human hepatocytes MIHA and liver cancer cell
lines Huh7, HepG2, MHCC-97H and MHCC-97L were purchased from the
American Type Culture Collection. All cell lines were authenticated
by STR profiling. Cells were cultured using Dulbecco's modified
Eagle medium (Gibco; Thermo Fisher Scientific, Inc.) supplemented
with 10% heat-inactivated fetal bovine serum (Gibco; Thermo Fisher
Scientific, Inc.), penicillin (100 U/ml) and streptomycin (100
µg/ml; Beyotime Institute of Biotechnology) and maintained at 37°C
in a humidified incubator containing 5% CO2.
Reverse transcription- quantitative
polymerase chain reaction (RT-qPCR)
Total RNA was extracted from specimens in cohort two
or cells using TRIzol® reagent (Invitrogen; Thermo
Fisher Scientific, Inc.), and 1 µg RNA was reverse transcribed into
cDNA using PrimeScript RT Reagent kit with gDNA Eraser (Takara Bio,
Inc.) according to the manufacturer's introductions. Subsequently,
qPCR was carried out using SYBR Green PCR Master Mix (Takara Bio,
Inc.) according to the manufacturer's protocols. PCR amplification
was performed as follows: Initial denaturation at 95°C for 10 min,
followed by 35 cycles of a two-step PCR at 95°C for 14 sec and 60°C
for 1 min. The reaction volume was 10 µl. Relative gene expression
was normalized to GAPDH and calculated using the 2−ΔΔCq
method (29). The following primer
pairs were used: EPHX2, forward, 5′-CCTTCATACCAGCAAATCCCAACA-3′ and
reverse, 5′-TTCAGCCTCAGCCACTCCT-3′; GAPDH, forward,
5′-GATCATCAGCAATGCCTCCT-3′ and reverse,
5′-GAGTCCTTCCACGATACCAA-3′.
Western blotting
Total protein was extracted from tissue samples in
cohort two or cultured cells using ice-cold
radioimmunoprecipitation assay buffer (Beyotime Institute of
Biotechnology) supplemented with protease and phosphatase inhibitor
cocktails (Roche Diagnostics). Cell lysates were boiled at 100°C
for 10 min. Protein concentration was determined using the
bicinchoninic acid Protein Assay kit (Thermo Fisher Scientific,
Inc.). Equal amounts (40 µg) of protein samples were separated by
10% sodium dodecyl sulfate-polyacrylamide gel electrophoresis, and
further transferred to polyvinylidene difluoride membranes
(MilliporeSigma). Membranes were then blocked with 5% nonfat milk
in Tris-buffered saline-0.1% Tween at room temperature for 2 h and
incubated with primary antibodies against EPHX2 (1:1,000; cat. no.
10833-1-AP; Proteintech Group, Inc.) and β-actin (1:5,000; cat. no.
66009-1-lg; Proteintech Group, Inc.) at 4°C overnight.
Subsequently, the membrane was incubated with a horseradish
peroxidase-conjugated goat anti-rabbit (1:2,000; cat. no.
SA00001-2; Proteintech Group, Inc.) or goat anti-mouse (1:2,000;
cat. no. SA00001-1; Proteintech Group, Inc.) secondary antibodies
at room temperature for 1 h. Protein bands were visualized using an
enhanced chemiluminescence (ECL) detection kit (EMD Millipore). An
ECL Western blot analysis system (Bio-Rad Laboratories, Inc.) was
used to evaluate the bands. The intensity of protein bands was
semi-quantified using Image Lab software (Version 6.0.1; Bio-Rad
Laboratories, Inc.) and normalized to β-actin.
Immunohistochemistry (IHC)
The tissue sections on the TMAs (thickness, 4 µm)
were deparaffinized using xylene and rehydrated using alcohol.
Samples were then subjected to heat-induced antigen retrieval using
citrate buffer (0.01 M; pH 6.4) in a pressure cooker for 5 min, and
cooled to room temperature. Subsequently, the sections were treated
with 3% hydrogen peroxide for 10 min at room temperature to block
endogenous peroxidase activity, and incubated with goat serum
(Beijing Dingguo Changsheng Biotechnology Co., Ltd.) for 1 h at
room temperature to block nonspecific antibody binding and
incubated with an EPHX2 antibody (1:100; cat. no. 10833-1-AP;
Proteintech Group, Inc.) overnight at 4°C in a humidified chamber.
After incubation with the secondary goat anti-rabbit IgG
(horseradish peroxidase) antibody (1:200; cat. no. ab150077; Abcam)
for 30 min at room temperature, coloration with 3,3-diaminobenzidin
was performed for 10 min at room temperature. Subsequently, the
samples were counterstained with hematoxylin for 2 min at room
temperature, dehydrated in a gradient series of ethanol, and then
mounted with neutral gum. The stained tissue slices were analyzed
by two different pathologists blinded to patients' clinical
characteristics with a light microscope (BX53; Olympus
Corporation). The intensity of IHC staining was semi-quantified
using ImageJ software (Version 1.50i; National Institutes of
Health). IHC scores were calculated using the following formula:
IHC score = intensity score × percentage score of stained cells.
Staining intensity was scored from 0 to 3 (0, negative; 1, weak; 2,
moderate; 3, strong). The percentage of positively stained cells
was scored from 0 to 100. Therefore, the IHC score ranged from 0 to
300. The median IHC score was defined as the cut-off value for low
and high expression.
Statistical analyses
Data were presented as the means ± standard
deviation and statistical analyses were performed using R software
(Version 3.6.3; http://www.r-project.org). Wilcoxon matched-pairs test
or Student's t-test was performed to compare differences between
two groups, and One-way ANOVA followed by Bonferroni post-hoc test
was used to compare differences between multiple groups. The
χ2 test or Fisher's exact test was performed to
determine the association between EPHX2 expression and the clinical
characteristics. Kaplan-Meier survival analysis was carried out to
evaluate the prognostic value of gene signature and EPHX2, and the
log-rank test was performed to analyze significance. When the
survival curves crossed, the two-stage procedure was used for
significance analysis (30,31). Univariate and multivariate Cox
proportional hazard regression analyses were also carried out to
investigate the association between EPHX2 expression and OS.
Spearman correlation test was performed to identify
EPHX2-associated genes and assess the correlation between EPHX2
expression and microsatellite instability (MSI). Receiver operating
characteristic (ROC) analysis was used to determine the sensitivity
and specificity of survival prediction using the gene signature
risk score. The area under the curve (AUC) served as an indicator
of prognostic accuracy. All the experiments were performed at least
three times. P<0.05 was used to indicate a statistically
significant difference.
Results
WGCNA and key module
identification
Overall, 4,453 and 4,257 DEGs were identified in
TCGA-LIHC and ICGC LIRI-JP cohorts as presented in the volcano
plots (Fig. S1A and B). A total of
2,589 DEGs in both cohorts were selected for subsequent analyses
and presented in a Venn diagram (Fig.
S1C). Expression data of these 2,589 DEGs from TCGA-LIHC cohort
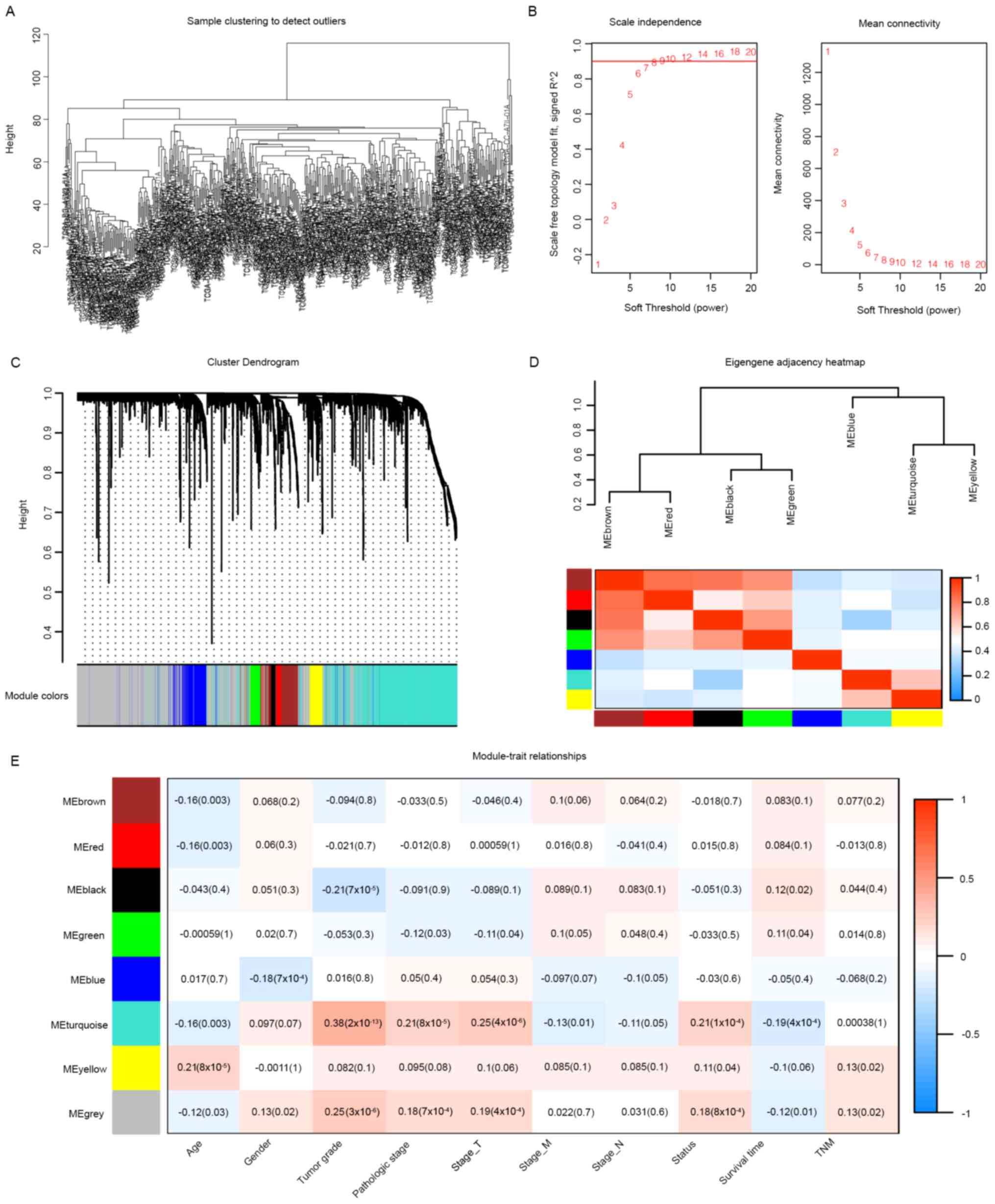
were used for WGCNA (Fig. 1). Eight
clustering modules (Fig. 1C and D)
were used to set the soft-threshold power as 7 (scale-free
R2=0.91, slope=−1.49; Figs.
1B and S2) and cut height as
0.25. The turquoise module was closely associated with clinical
traits, especially tumor grade (correlation coefficient=0.38,
P=2×10−13; Fig. 1E) and
was considered as the key module. Moreover, 251 associated genes
from the turquoise module were selected as novel candidates for
further analyses (GS>0.2 and MM>0.6). Subsequently, these
genes were subjected to univariate Cox regression analysis, and 239
genes were closely correlated with the OS (Table SI).
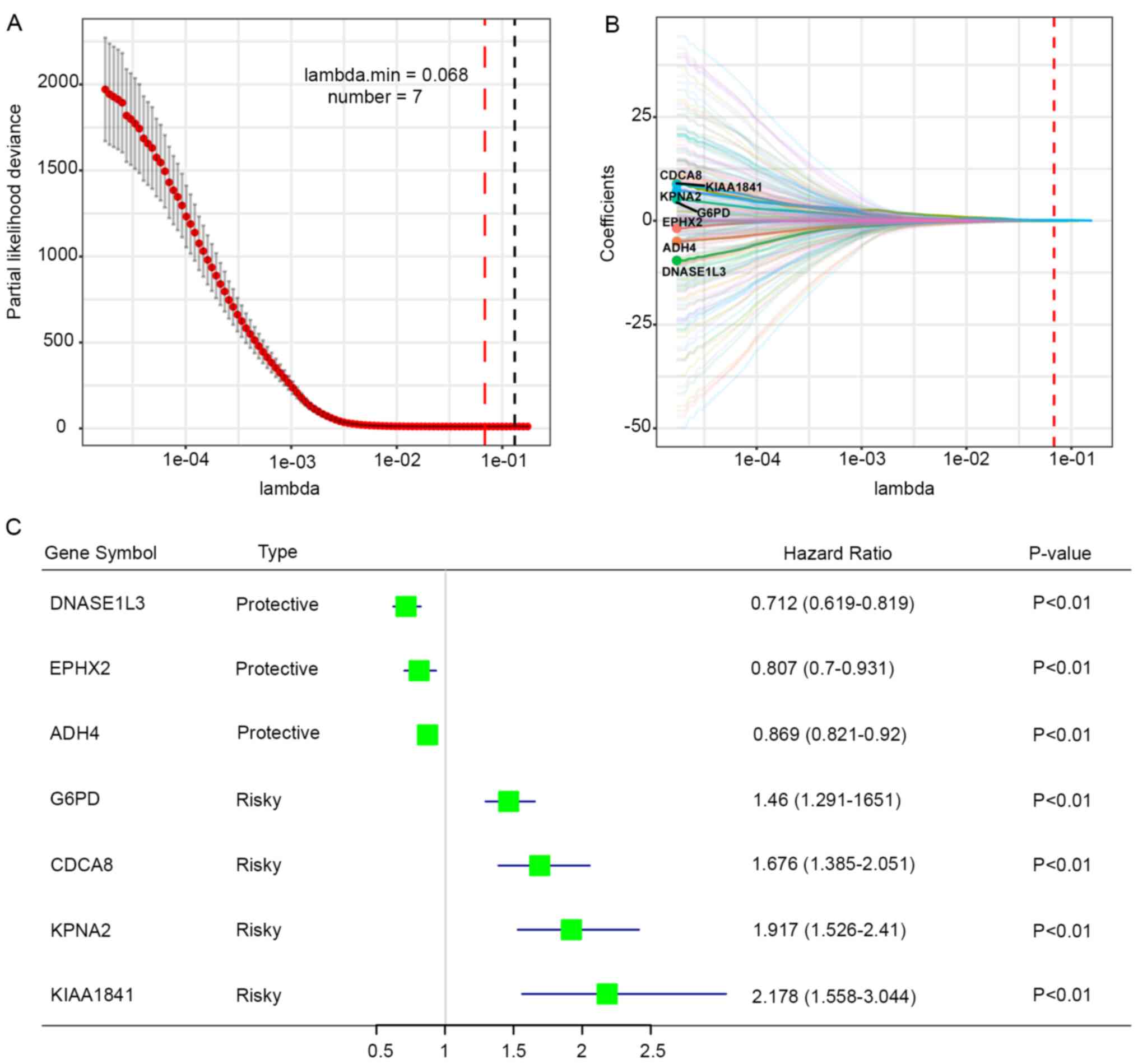
The aforementioned genes were further applied to the
LASSO Cox regression analysis and the regression coefficient of
each gene was calculated. The prognostic gene signature produced
the best performance when seven genes (DNASE1L3, EPHX2, ADH4, G6PD,
CDCA8, KPNA2 and KIAA1841) were included (Fig. 2A). Regression coefficients of these
genes were presented in Fig. 2B.
Three genes (DNASE1L3, EPHX2 and ADH4) with a HR<1 were
considered as protective genes, whereas four genes (G6PD, CDCA8,
KPNA2 and KIAA1841) with a HR>1 were considered as risk factors
(Fig. 2C). Risk score was
calculated for each patient using the following formula: Risk score
= (−0.0011 × DNASE1L3 expression) + (−0.0029 × EPHX2 expression) +
(−0.0281 × ADH4 expression) + (0.1459 × G6PD expression) + (0.0509
× CDCA8 expression) + (0.1594 × KPNA2 expression) + (0.0623 ×
KIAA1841 expression).
Validation of the prognostic gene
signature
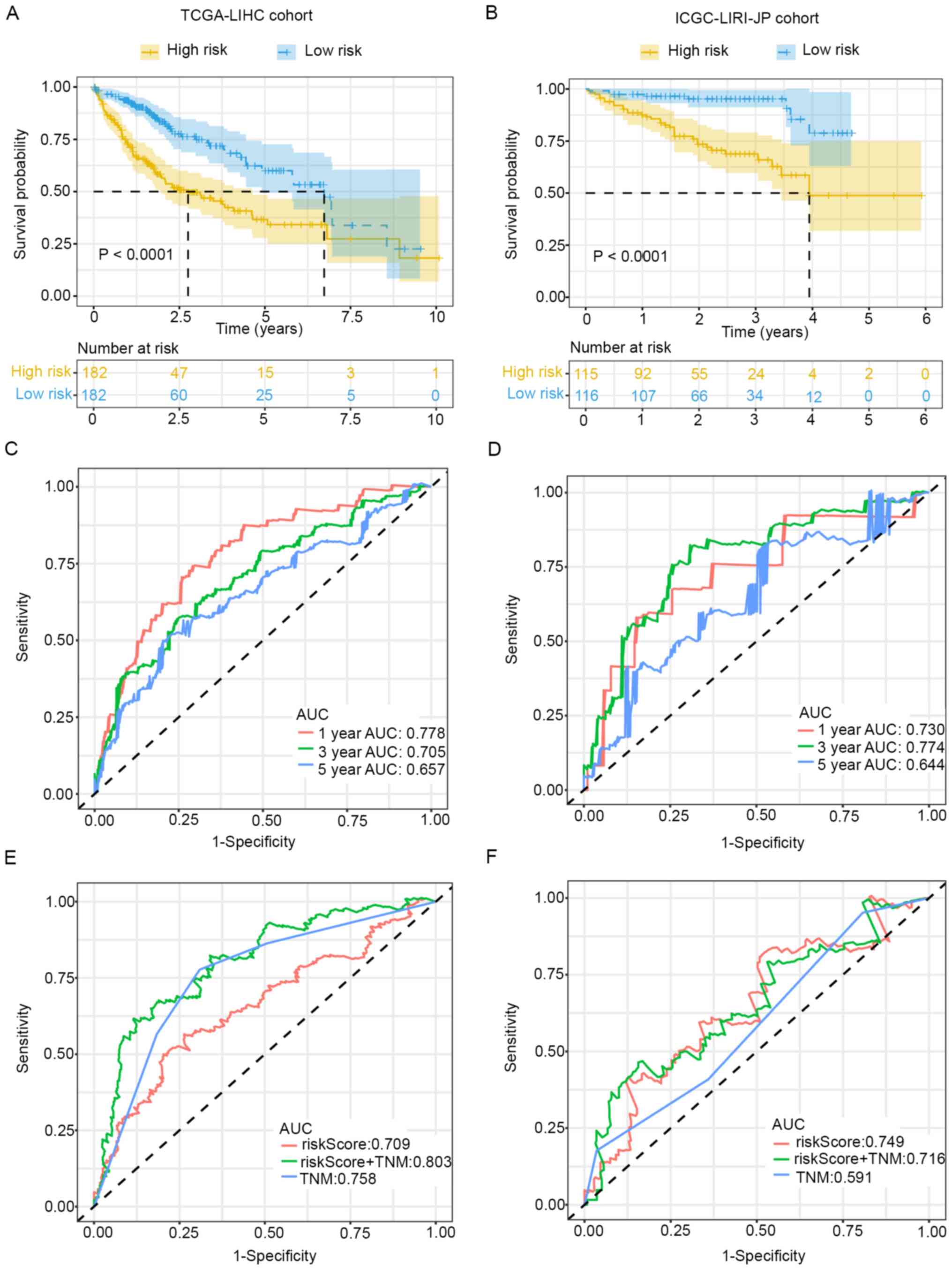
The prognostic prediction value of this seven-gene
signature was further validated. Kaplan-Meier survival analysis
indicated that patients with low-risk scores exhibited better
survival in the training (P<0.0001) and validation cohorts
(P<0.0001) (Fig. 3A and B). ROC
curve analysis was also conducted to evaluate the predictive
ability of the signature. Time-dependent ROC for OS at different
time points indicated that AUC values for 1-, 3- and 5-year OS were
0.778, 0.705 and 0.657 in the training cohort (Fig. 3C), and the values were 0.730, 0.774
and 0.644 in the validation cohort (Fig. 3D).
Further ROC curve analysis for OS within different
groups revealed that a combination of TNM stage and risk score
could improve the prognostic prediction ability in the training
cohort (Fig. 3E). However, the
predictive ability of risk score alone was much higher in the
validation cohort (Fig. 3F). In
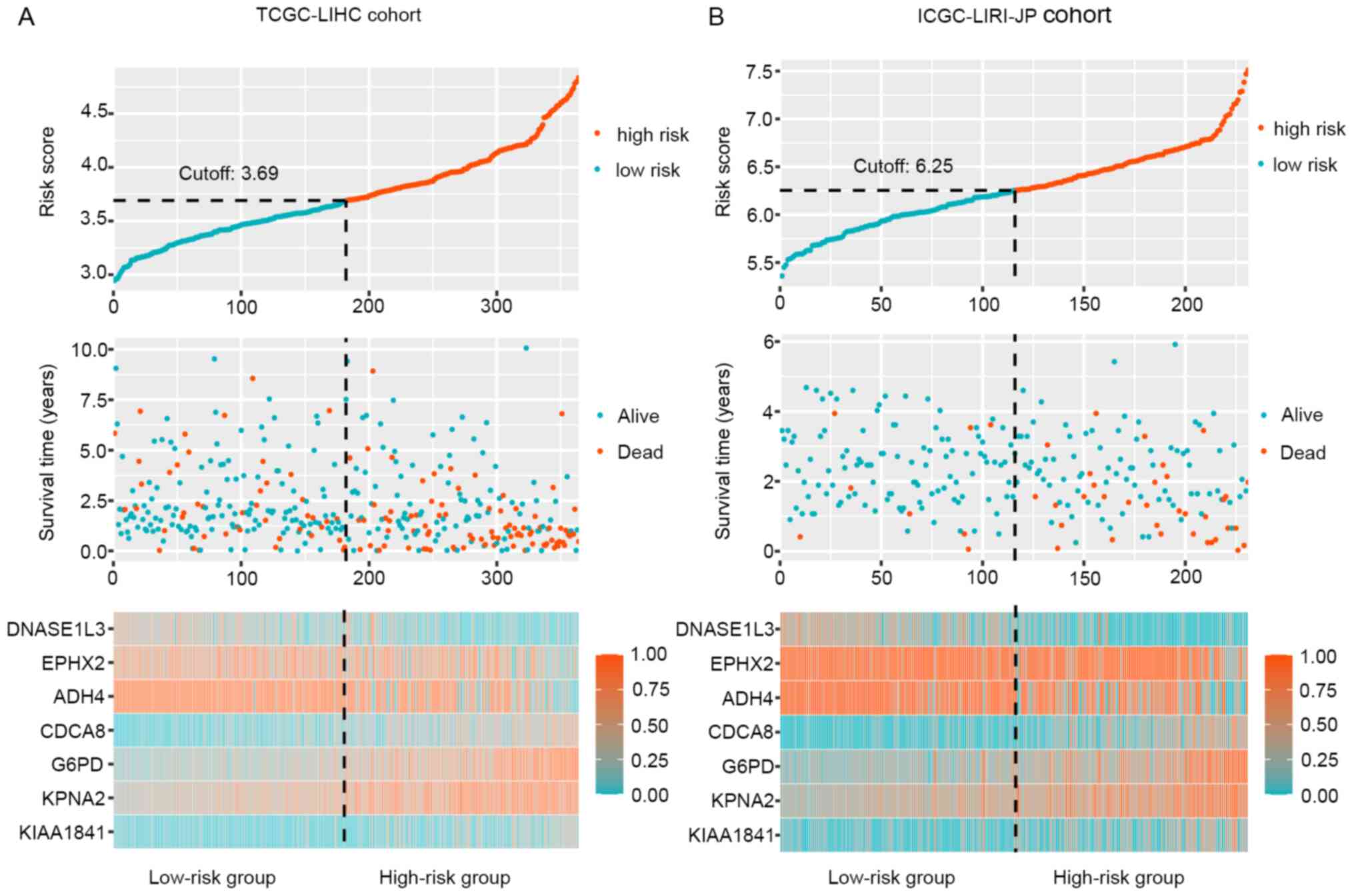
addition, the distribution of risk scores, survival time and
therapeutic outcomes in the training and validation cohorts are
presented in Fig. 4. The heat map
revealed upregulation of the four risk factors and downregulation
of the three protective genes in the high-risk group. In
conclusion, combination of this novel prognostic model with
conventional TNM staging might increase prognostic prediction
ability in HCC. Among these seven genes, EPHX2 was rarely reported
in HCC. Therefore, EPHX2 was selected for further validation.
mRNA expression levels of EPHX2 in
different types of cancer
The mRNA expression levels of EPHX2 in tumor and
normal tissues were compared in different types of cancer using
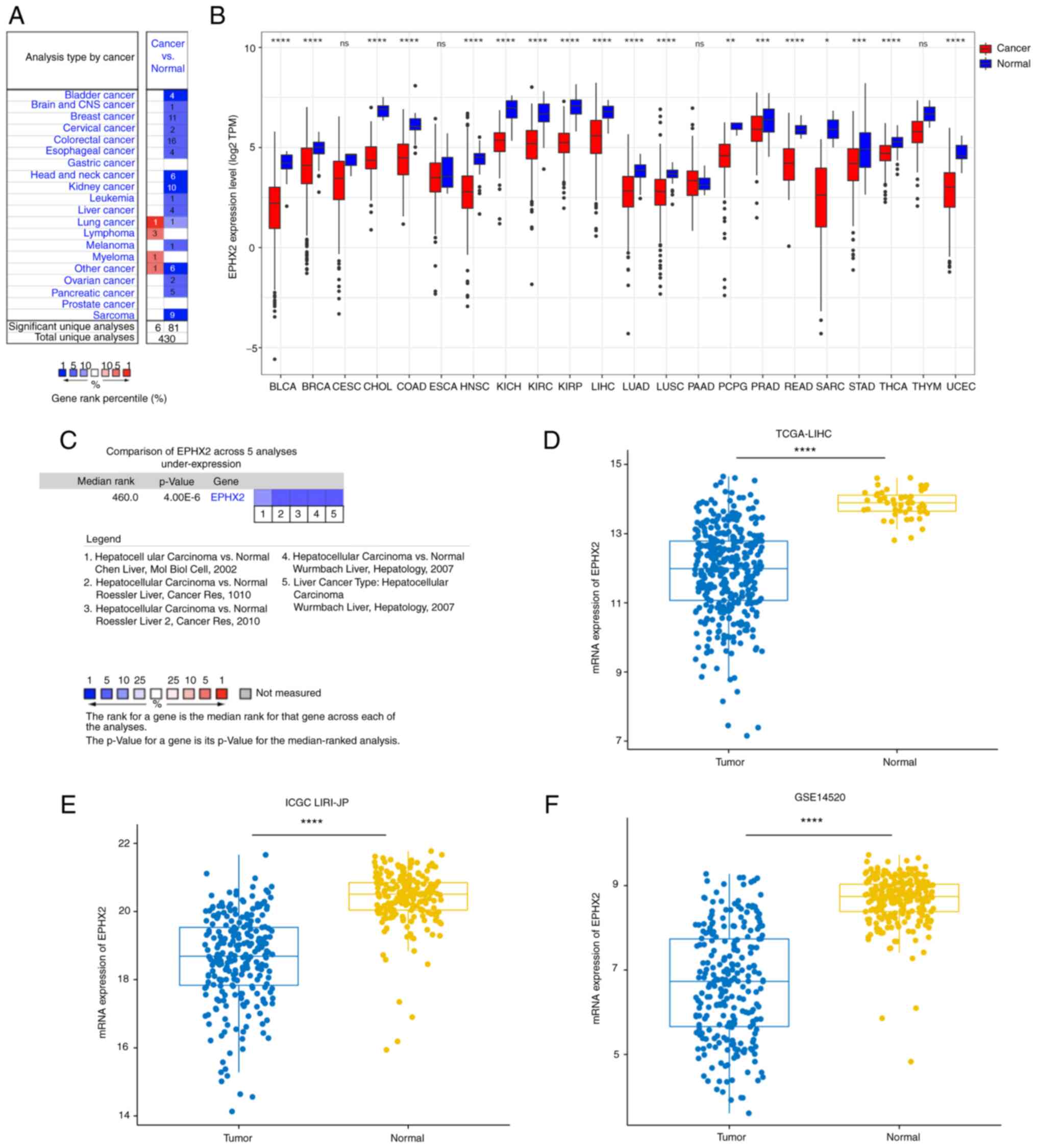
Oncomine and TCGA databases. In the Oncomine database, EPHX2
expression was decreased in numerous types of cancer, including
leukemia, melanoma, sarcoma, and esophageal, breast, kidney, liver,
colorectal, bladder, ovarian, cervical. brain and central nervous
system, head and neck, and pancreatic cancer (Fig. 5A). Moreover, EPHX2 expression in
cancer and normal specimens was compared using TCGA database
(Fig. 5B). Similarly, the
expression levels of EPHX2 were significantly reduced in tumor
samples, such as urothelial bladder carcinoma (BLCA), invasive
breast carcinoma, cholangiocarcinoma, colon adenocarcinoma, head
and neck squamous carcinoma (HNSC), kidney chromophobe, kidney
renal clear cell carcinoma (KIRC), kidney renal papillary cell
carcinoma (KIRP), LIHC, lung adenocarcinoma (LUAD), lung squamous
cell carcinoma (LUSC), pheochromocytoma and paraganglioma, prostate
adenocarcinoma, rectum adenocarcinoma, sarcoma, stomach
adenocarcinoma (STAD), thyroid carcinoma (THCA) and uterine corpus
endometrial carcinoma (UCEC). These results revealed downregulation
of EPHX2 in various types of cancer, which could be associated with
tumor progression.
Downregulation of EPHX2 in HCC
A comparison of EPHX2 expression levels in HCC from
various studies in the Oncomine database revealed downregulation of
EPHX2 expression in HCC tissues compared with those in adjacent
normal samples (Fig. 5C).
Furthermore, EPHX2 expression was reduced in HCC specimens in
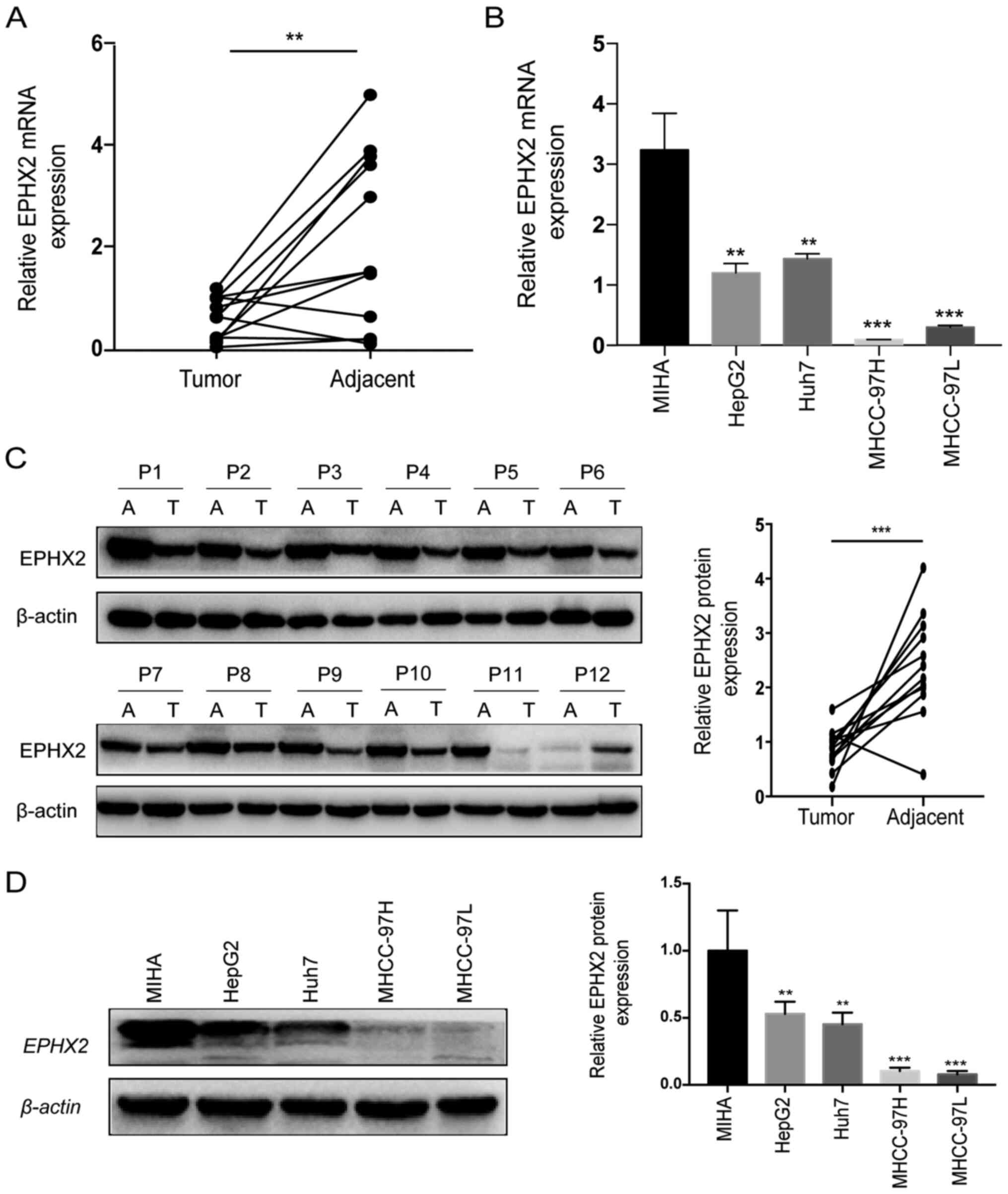
TCGA-LIHC, ICGC LIRI-JP and GSE14520 cohorts (Fig. 5D-F). Furthermore, RT-qPCR and
western blotting were performed to evaluate the mRNA and protein
expression levels of EPHX2 in 12 paired HCC and normal samples. The
expression levels of EPHX2 were significantly decreased in HCC
tissues compared with those in adjacent non-cancerous tissues
(Fig. 6A). As presented in Fig. 6C, the protein expression levels of
EPHX2 were also markedly reduced in HCC samples. Furthermore, EPHX2
expression was detected in normal hepatocytes and HCC cells. The
mRNA and protein expression levels of EPHX2 were markedly decreased
in HCC cells compared with those in normal hepatocytes (Fig. 6B and D). These findings revealed the
downregulation of EPHX2 in HCC.
Association of EPHX2 and
clinicopathological traits in HCC
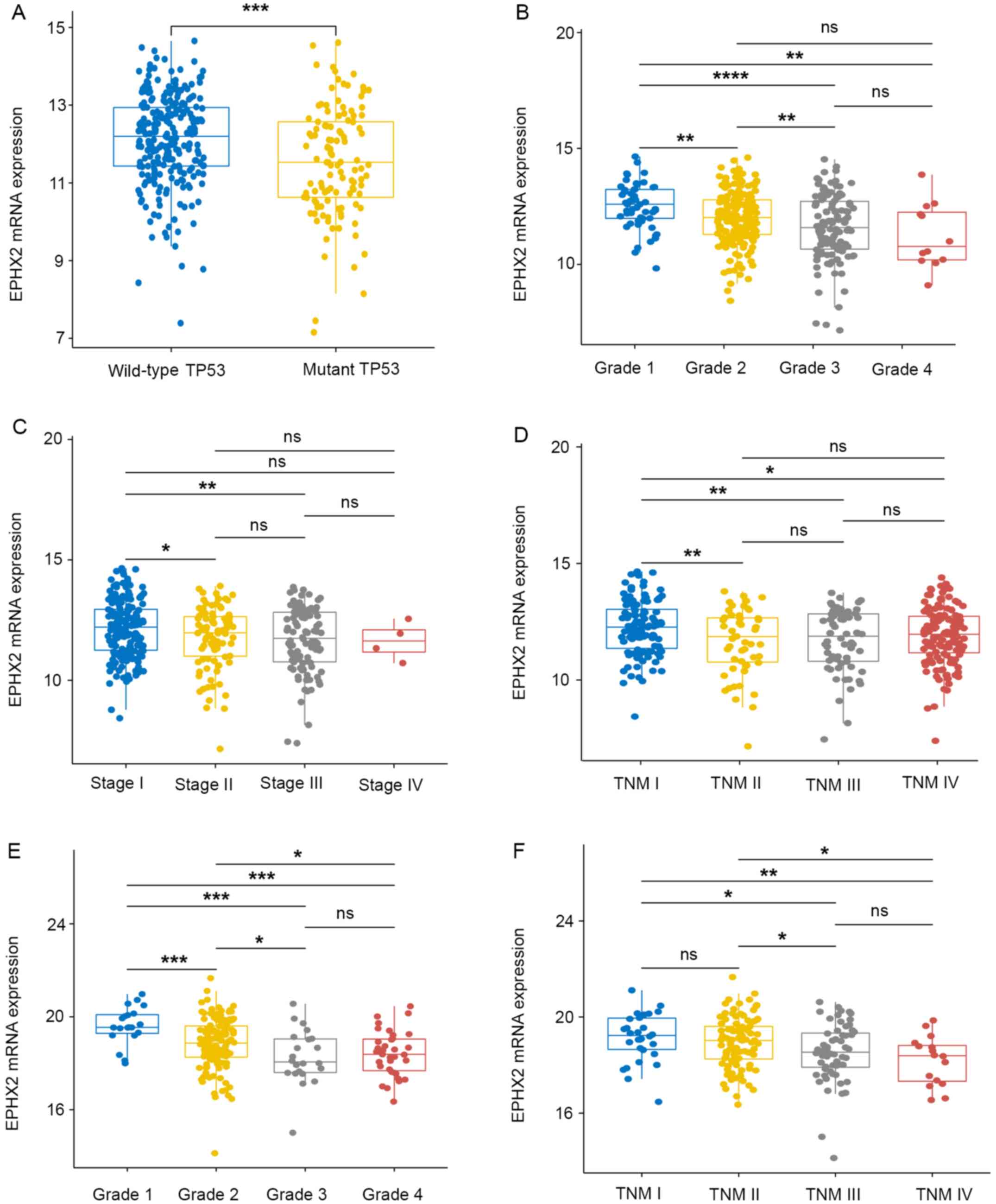
The association between EPHX2 expression and
clinicopathological traits in patients with HCC was further
determined. The mRNA expression levels of EPHX2 were evaluated in
different subgroups in TCGA-LIHC cohort, including with or without
TP53 mutations, various tumor grades and pathological/TNM stages
(Fig. 7A-D). EPHX2 expression was
also examined in subgroups in the ICGC LIRI-JP cohort, including
various tumor grades and TNM stages (Fig. 7E and F). In TCGA LIHC cohort, the
lowest mRNA expression levels of EPHX2 were detected in patients
with TP53 mutations, grade 4 and TNM stage III, whereas in the ICGC
LIRI-JP cohort, the lowest expression levels were found in grade 3
and TNM stage IV (Fig. 7). Due to
the limited number of stage IV patients (only four HCC patients
were at stage IV), no significant difference was observed between
EPHX2 expression in stage IV and any other stages in TCGA-LIHC
cohort. However, the mRNA expression of EPHX2 in pathological stage
III was significantly lower than that in stage I and II. These
results indicated that patients with HCC with TP53 mutations and at
advanced tumor grade and pathological/TNM stage exhibited lower
EPHX2 expression. The present study also assessed the correlation
between EPHX2 expression and microsatellite instability (MSI); the
results demonstrated that increased EPHX2 expression was associated
with decreased MSI in LIHC (R=−0.12; P=0.027; Fig. S3). Furthermore, clinicopathological
analysis suggested that downregulation of EPHX2 was associated with
advanced tumor grade and female sex in TCGA-LIHC cohort, while in
the ICGC LIRI-JP cohort, this was associated with higher TNM stage
and tumor grade (Table SII). These
results indicated that higher EPHX2 expression was detected in
patients with early-stage HCC, whereas patients at the advanced
stages exhibited lower EPHX2 levels.
EPHX2 is a potential tumor suppressor
gene
Kaplan-Meier plotter database was used to identify
the cancer types whose prognostic values were related to EPHX2
expression in OS and recurrence-free survival (RFS). The results
indicated that patients with higher EPHX2 levels exhibited better
OS prognosis in eight types of cancer: LIHC (HR=0.57; 95%
CI=0.4–0.81; P=0.0013), LUAD (HR=0.59; 95% CI=0.41–0.84; P=0.0034),
cervical squamous cell carcinoma (CESC; HR=0.42; 95% CI=0.26–0.68;
P=2.5×10−4), HNSC (HR=0.61; 95% CI=0436-0.86; P=0.0048),
KIRC (HR=0.44; 95% CI=0.31–0.63; P=4.5×10-6), KIRP (HR=0.42; 95%
CI=0.23–0.76; P=0.003), pancreatic ductal adenocarcinoma (PDAC;
HR=0.53; 95% CI= 0.34–0.82; P=0.0041) and UCEC (HR=1.71; 95%
CI=1.03–2.84; P=0.036) (Fig. S4).
Moreover, eight types of cancer exhibited improved RFS prognosis
when EPHX2 was highly expressed; the results indicated that
patients with higher EPHX2 levels exhibited lower recurrence rates
in BLCA, KIRP, LUAD, LUSC, PDAC, STAD and THCA (Fig. S5). These findings indicated that
EPHX2 expression could be associated with the prognosis of
different types of cancer, and it may be a putative survival
predictor for patients with HCC.
EPHX2 is an independent prognostic
biomarker in HCC
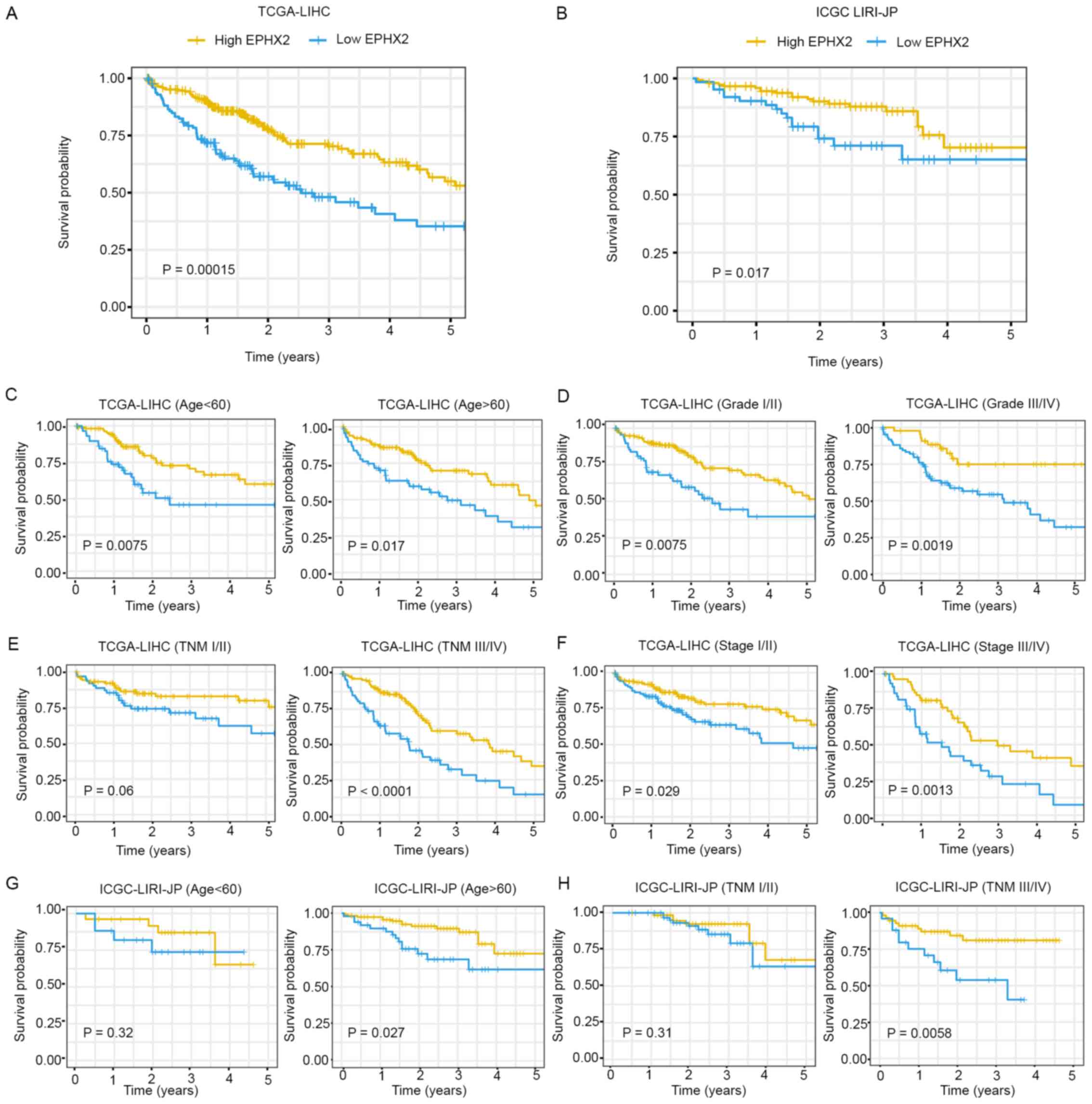
The prognostic value of EPHX2 in HCC was further
evaluated. Kaplan-Meier survival curves indicated that patients
with higher EPHX2 levels exhibited better prognosis in TCGA-LIHC
(P=1.5×10−4) and ICGC LIRI-JP (P=0.017) cohorts
(Fig. 8A and B). Furthermore,
Kaplan-Meier analysis of OS was performed in patients with HCC
according to age, tumor grade, TNM stage and pathologic stage in
the TCGA-LIHC cohort, and age and TNM stage in the ICGC LIRI-JP
cohort, respectively. In TCGA-LIHC cohort, EPHX2 expression
affected OS rates in the different age, tumor grade, pathological
stage and TNM stage III/IV subgroups (P<0.05), but not in the
TNM stage I/II subgroup (P=0.06) (Fig.
8C-F). In the ICGC LIRI-JP cohort, patients with higher EPHX2
expression exhibited better OS in the age >60 years (P=0.027)
and TNM stage III/IV (P=0.0058) subgroups, but not in the age
<60 years (P=0.32) and TNM stage I/II (P=0.31) subgroups
(Fig. 8G and H).
The independent prognostic value of EPHX2 expression
with regard to OS was determined in TCGA-LIHC and ICGC LIRI-JP
cohorts. In the univariate analysis, patients with HCC with higher
pathological stage, TNM stage and lower EPHX2 levels exhibited
poorer OS in TCGA-LIHC cohort, and patients with lower EPHX2
expression also exhibited poorer OS in the ICGC LIRI-JP cohort
(Table SIII). In the multivariate
analysis, improved OS was detected in patients with HCC with higher
EPHX2 levels in TCGA-LIHC cohort (Table SII; HR=0.8433, 95%
CI=0.7442–0.9555, P=0.0075); whereas, no significant value was
identified in ICGC LIRI-JP cohort. Taken together, the mRNA
expression levels of EPHX2 were associated with prognosis in HCC,
and EPHX2 expression may be a promising survival predictor for
patients with HCC.
Functional enrichment analysis for
EPHX2 in HCC
To explore the detailed functions of EPHX2, GO
analysis, KEGG analysis and GSEA were carried out. Firstly, A total
of 500 genes significantly correlated with EPHX2 were screened by
Spearman correlation test. Then, functions of EPHX2 and the 500
associated genes were analyzed using GO and KEGG analyses in
TCGA-LIHC and ICGC LIRI-JP cohorts. As presented in Fig. 9A and C, commonly enriched BPs in
both cohorts were involved in metabolic reprogramming, such as
‘organic acid catabolic process’, ‘small molecule catabolic
process’, ‘fatty acid metabolic process’, ‘carboxylic acid
catabolic process’, ‘alpha-amino acid metabolic process’ and
‘cellular amino acid metabolic process’; CCs, such as ‘peroxisome’,
‘mitochondrial matrix’, ‘microbody part’, ‘microbody’, ‘peroxisomal
matrix’ and ‘peroxisomal part’. MFs, such as ‘oxidoreductase
activity, acting on CH-OH group of donors’, ‘coenzyme binding’,
‘iron ion binding’, ‘vitamin binding’ and ‘oxidoreductase activity
and acting on paired donors’. In KEGG analysis, enriched pathways,
including ‘peroxisome’, ‘carbon metabolism’, ‘complement and
coagulation cascades’, ‘valine, leucine and isoleucine degradation’
and ‘propanoate metabolism’, were associated with the functions of
EPHX2 and the 500 associated genes in HCC (Fig. 9B and D).
To identify the biological pathways associated with
EPHX2, GSEA was further performed to compare the high- and
low-EPHX2 expression groups in TCGA-LIHC and ICGC LIRI-JP cohorts.
In general, 33 and 33 KEGG pathways were enriched in the high-EPHX2
group in TCGA-LIHC and ICGC LIRI-JP cohorts, respectively; whereas,
only one significantly enriched pathway was identified in the
low-EPHX2 group in TCGA-LIHC cohort (Table SIV). Elevated EPHX2 levels were
significantly associated with numerous metabolic pathways in both
cohorts, such as ‘PPAR signaling pathway’, ‘tyrosine metabolism,
‘propanoate metabolism’, ‘histidine metabolism’, ‘valine, leucine
and isoleucine degradation’, ‘retinal metabolism’, ‘primary bile
acid biosynthesis’, and ‘fatty acid metabolism’ (Fig. 9E). Furthermore, low EPHX2 expression
was negatively associated with ‘olfactory transduction pathway’.
These results indicated that EPHX2 was involved in catabolic
processes and peroxisome metabolism in HCC, and it might be
associated with metabolic reprogramming in HCC.
Validation of clinical and prognostic
value of EPHX2 in TMAs
Finally, clinical and prognostic value of EPHX2 was
validated in TMAs with complete clinical and follow-up data. Those
eight patients whose missing area of tissue section was >50%
were excluded. Protein expression levels of EPHX2 were determined
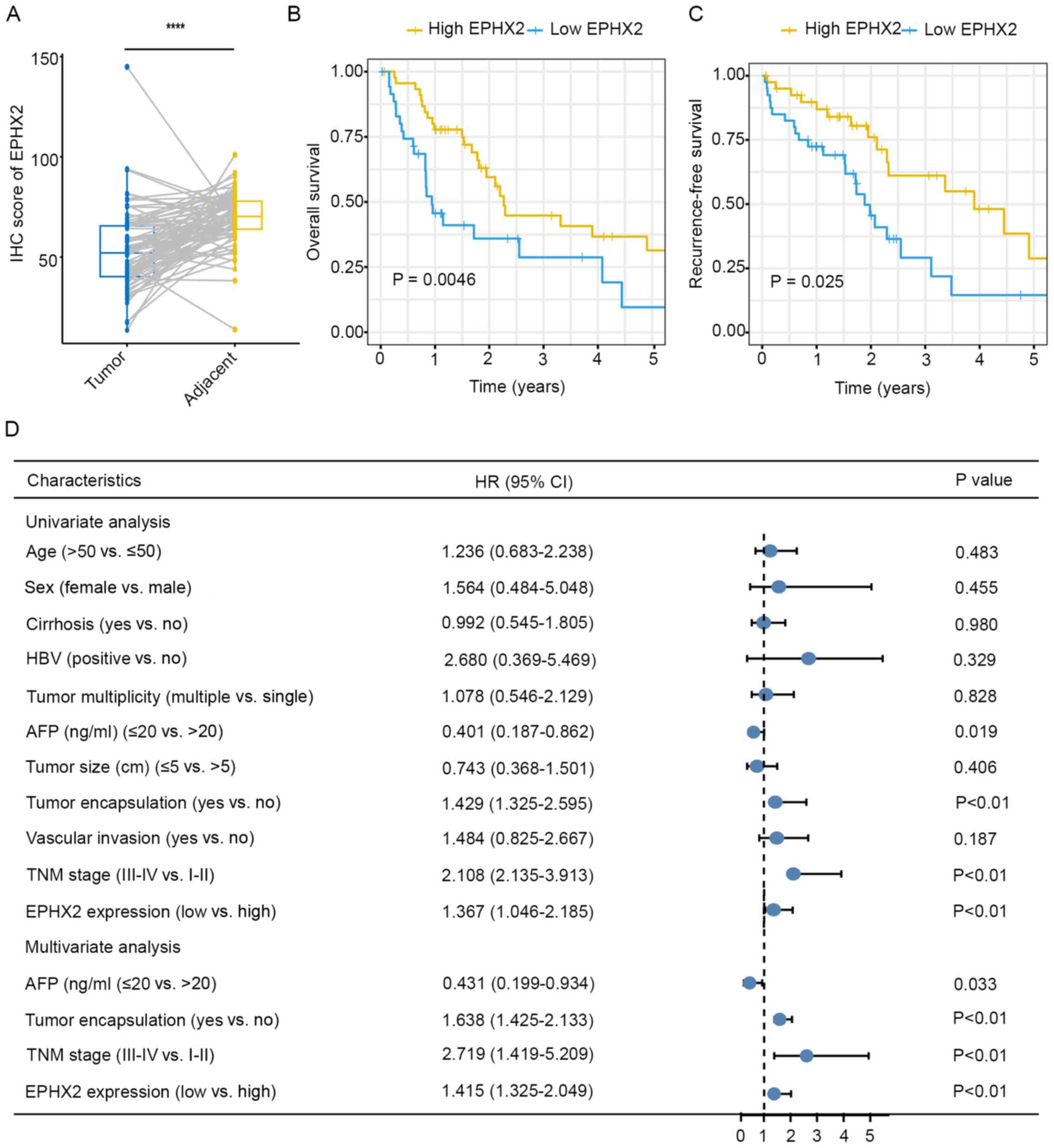
in TMAs using IHC, and the results revealed downregulation of EPHX2
in HCC tissues (Figs. S6 and
10A). Kaplan-Meier survival
analysis suggested that the high EPHX2 group exhibited better OS
(P=0.0046; Fig. 10B) and lower
recurrence (P=0.025; Fig. 10C).
Further clinicopathological analysis in the TMAs cohort indicated
that EPHX2 expression was associated with tumor encapsulation,
tumor multiplicity, vascular invasion and TNM stage (Table I). In addition, univariate Cox
regression analysis revealed that lower EPHX2 levels were
associated with poorer OS (HR=1.367; 95% CI=1.046–2.185; P<0.01;
Fig. 10D) and higher recurrence
(HR=1.101; 95% CI=1.042–1.365; P=0.01; Fig. S7). In addition, multivariate Cox
regression analysis suggested that EPHX2 was an independent
prediction indicator for OS (HR=1.415; 95% CI =1.325–2.049;
P<0.01; Fig. 10D). These
results revealed the clinical and prognostic value of EPHX2 in HCC
and suggested that EPHX2 could be an independent prognostic
biomarker for HCC.
 | Table I.Association between EPHX2 expression
and clinicopathological features. |
Table I.
Association between EPHX2 expression
and clinicopathological features.
|
| EPHX2 |
|
|---|
|
|
|
|
|---|
| Variables | All cases
(n=82) | Low expression
(n=41) | High expression
(n=41) | Fishers exact test
or χ2 P-value |
|---|
| Age, years, n
(%) |
|
|
| 1.000 |
|
≤50 | 48 (58.5) | 24 (58.5) | 24 (58.5) |
|
|
>50 | 34 (41.5) | 17 (41.5) | 17 (41.5) |
|
| Sex, n (%) |
|
|
| 1.000 |
|
Female | 8 (9.8) | 4 (9.8) | 4 (9.8) |
|
|
Male | 74 (90.2) | 37 (90.2) | 37 (90.2) |
|
| Cirrhosis, n
(%) |
|
|
| 0.822 |
| No | 49 (59.8) | 24 (58.5) | 25 (61.0) |
|
|
Yes | 33 (40.2) | 17 (41.5) | 16 (39.0) |
|
| HBV, n (%) |
|
|
| 1.000 |
|
Negative | 4 (4.9) | 2 (4.9) | 2 (4.9) |
|
|
Positive | 78 (95.1) | 39 (95.1) | 39 (95.1) |
|
| Tumor multiplicity,
n (%) |
|
|
| 0.021 |
|
Single | 62 (75.6) | 26 (63.4) | 36 (87.8) |
|
|
Multiple | 20 (24.4) | 15 (36.6) | 5 (12.2) |
|
| α-fetoprotein
(ng/ml), n (%) |
|
|
| 0.809 |
|
≤20 | 24 (29.3) | 13 (31.7) | 11 (26.8) |
|
|
>20 | 58 (70.7) | 28 (68.3) | 30 (73.2) |
|
| Tumor size (cm), n
(%) |
|
|
| 0.770 |
| ≤5 | 68 (82.9) | 33 (80.5) | 35 (85.4) |
|
|
>5 | 14 (17.1) | 8 (19.5) | 6 (14.6) |
|
| Tumor
encapsulation, n (%) |
|
|
| 0.006 |
|
Complete | 29 (35.4) | 8 (19.5) | 21 (51.2) |
|
|
None | 53 (64.6) | 33 (80.5) | 20 (48.8) |
|
| Vascular invasion,
n (%) |
|
|
| 0.021 |
| No | 53 (64.6) | 21 (51.2) | 32 (78.0) |
|
|
Yes | 29 (35.4) | 20 (48.8) | 9 (22.0) |
|
| TNM stage, n
(%) |
|
|
| 0.015 |
|
III–IV | 44 (53.7) | 28 (68.3) | 16 (39.0) |
|
|
I–II | 38 (46.3) | 13 (31.7) | 25 (61.0) |
|
Discussion
HCC is a common type of cancer associated with high
morbidity and mortality, and the therapeutic outcome is poor for
patients at advanced or metastatic stages. The pathogenesis of HCC
involves genomic mutation, environmental intervention, modulation
of molecular pathways involved in hepatocarcinogenesis and tumor
progression (32). Targeted
therapies have been used to improve the survival of patients with
advanced HCC (33–36); however, it is still necessary to
improve the OS of patients with HCC, thus novel biomarkers should
be identified. In the present study, notable modules correlated
with clinical traits were identified using WGCNA, and a gene
signature associated with OS was developed by LASSO. Furthermore,
its performance for prognostic prediction was validated in
TCGA-LIHC and ICGC LIRI-JP cohorts. In consistence with the present
findings, Zhang et al (37)
revealed that EPHX2, together with five other genes, was associated
with OS in patients with HCC based on TCGA data (37). In the present study, the seven genes
in the signature were EPHX2, KPNA2, KIAA1841, G6PD, CDCA8, ADH4 and
DNASE1L3. Among these genes, EPHX2 was dysregulated in numerous
types of cancer, including HCC, and was associated with cancer
prognosis based on bioinformatics analysis, suggesting that it
might serve an essential role in tumor progression. Since EPHX2 has
rarely been reported in HCC, to the best of our knowledge, it was
selected for further validation in the present study.
EPHX2 encodes sEH, which is expressed in various
human malignant neoplasms, including HCC (10). Downregulation of EPHX2 was confirmed
in HCC tissues and cells in the present study, and its
downregulation was associated with shorter OS and RFS. Furthermore,
EPHX2 was identified as an independent prognostic biomarker for OS
in patients with HCC. Moreover, clinicopathological analysis
suggested that downregulation of EPHX2 was associated with advanced
tumor grade/TNM stage and poor prognosis in patients with HCC,
suggesting that patients with early-stage HCC could exhibit higher
EPHX2 expression. Finally, the clinical and prognostic value of
EPHX2 was evaluated in TMAs with complete clinical and follow-up
data. Functional analysis revealed that EPHX2 was closely
associated with ‘complement/coagulation cascade’,
‘peroxisome/carbon metabolism’, ‘CYP’, ‘catabolic processes of
carboxylic acids’, ‘small molecules’, ‘fatty acids’, ‘organic
acids’ and other metabolic pathways, suggesting that EPHX2 was
closely associated with metabolic reprogramming in HCC.
It has been reported that CYP2J2 expression may be
increased in HCC tissues compared with that in normal controls
(38). EPHX2 protein catalyzes the
hydrolysis of EETs, which are the major products synthesized from
arachidonic acids by CYP (9).
Further studies have also indicated that the addition of EETs or
overexpression of CYP2J2 could promote cell proliferation in human
malignant neoplasms, including HCC (39,40).
CYP epoxygenases and the epoxide metabolites have also been
reported to induce proliferation/metastasis and trigger
angiogenesis in various types of cancer (40). Conversely, inhibitors of CYP2J2
could suppress the growth of tumor cells with high CYP2J2 levels,
such as HCC, breast and lung cancer cells (41). The studies of sEH in solid tumors
have indicated that dual inhibition of sEH and cyclooxygenase-2 may
suppress tumor growth in HCC and lung cancer (40,41).
In addition, inhibition of EPHX2 could result in the accumulation
of EETs, consequently promoting tumor growth and metastasis in
patients with HCC (40). These
findings suggested that EPHX2 may be considered a prognostic
biomarker and therapeutic target in HCC, and it may exert important
roles in the progression of HCC. Further research, including in
vitro and in vivo studies, are required to validate the
anti-oncogenic role of EPHX2 in HCC and to investigate the
underlying molecular mechanisms of EPHX2-modulated metabolic
reprogramming in HCC.
In summary, a seven-gene signature was constructed
and validated, which was correlated with the development of HCC.
Moreover, a rarely reported gene, EPHX2, was selected and
downregulation of EPHX2 was confirmed in HCC. In addition, higher
EPHX2 expression was detected in patients with early-stage HCC.
Furthermore, patients with higher EPHX2 levels exhibited better
prognosis, thus EPHX2 could be an independent prognostic biomarker
for OS of patients with HCC. Additionally, functional enrichment
analyses revealed that EPHX2 expression was associated with
metabolic processes and peroxisomal components, suggesting that
EPHX2 could be involved in metabolic reprogramming of HCC. These
data indicated that downregulation of EPHX2 might be associated
with the progression and poor prognosis of HCC, and EPHX2 could be
a novel therapeutic approach for targeted treatment of patients
with HCC.
Supplementary Material
Supporting Data
Supporting Data
Acknowledgements
Not applicable.
Funding
The present study was supported by China
Mega-Project for Infectious Diseases (grant no.
2017ZX10203202004).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
ZM and LQ initiated and designed the study. KZ, YB,
SL and HC performed the analyses and interpreted the data. KZ, LK
and QL carried out the experiments. KZ, SL and HC produced the
figures and tables. KZ, YB and HC drafted the paper. ZM, LQ and LL
reviewed the manuscript. ZM and KZ confirm the authenticity of all
the raw data. All authors read and approved the final
manuscript.
Ethics approval and consent to
participate
The present study was approved by the Ethics
Committee of the Second Affiliated Hospital of Chongqing Medical
University (approval no. 2020-186). All patients provided written
informed consent.
Patient consent for publication
Not applicable.
Competing interests
The authors declared that they have no competing
interests.
References
|
1
|
Bray F, Ferlay J, Soerjomataram I, Siegel
RL, Torre LA and Jemal A: Global cancer statistics 2018: GLOBOCAN
estimates of incidence and mortality worldwide for 36 cancers in
185 countries. CA Cancer J Clin. 68:394–424. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Jemal A, Ward EM, Johnson CJ, Cronin KA,
Ma J, Ryerson B, Mariotto A, Lake AJ, Wilson R, Sherman RL, et al:
Annual report to the Nation on the status of cancer, 1975–2014,
featuring survival. J Natl Cancer Inst. 109:djx0302017. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Fitzmaurice C, Abate D, Abbasi N,
Abbastabar H, Abd-Allah F, Abdel-Rahman O, Abdelalim A, Abdoli A,
Abdollahpour I, Abdulle AS, et al Global Burden of Disease Cancer
Collaboration, : Global, Regional, and National Cancer Incidence,
Mortality, Years of Life Lost, Years Lived With Disability, and
Disability-Adjusted Life-Years for 29 Cancer Groups, 1990 to 2017:
A systematic analysis for the global burden of disease study. JAMA
Oncol. 5:1749–1768. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Sia D, Villanueva A, Friedman SL and
Llovet JM: Liver cancer cell of origin, molecular class, and
effects on patient prognosis. Gastroenterology. 152:745–761. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Li X, Xu W, Kang W, Wong SH, Wang M, Zhou
Y, Fang X, Zhang X, Yang H, Wong CH, et al: Genomic analysis of
liver cancer unveils novel driver genes and distinct prognostic
features. Theranostics. 8:1740–1751. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Langfelder P and Horvath S: WGCNA: An R
package for weighted correlation network analysis. BMC
Bioinformatics. 9:5592008. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Larsson C, White I, Johansson C, Stark A
and Meijer J: Localization of the human soluble epoxide hydrolase
gene (EPHX2) to chromosomal region 8p21-p12. Hum Genet. 95:356–358.
1995. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Spector AA: Arachidonic acid cytochrome
P450 epoxygenase pathway. J Lipid Res. 50 (Suppl 1):S52–56. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Spector AA, Fang X, Snyder GD and
Weintraub NL: Epoxyeicosatrienoic acids (EETs): Metabolism and
biochemical function. Prog Lipid Res. 43:55–90. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Enayetallah AE, French RA and Grant DF:
Distribution of soluble epoxide hydrolase, cytochrome P450 2C8, 2C9
and 2J2 in human malignant neoplasms. J Mol Histol. 37:133–141.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Dreisbach AW, Japa S, Sigel A, Parenti MB,
Hess AE, Srinouanprachanh SL, Rettie AE, Kim H, Farin FM and Hamm
LL: The Prevalence of CYP2C8, 2C9, 2J2, and soluble epoxide
hydrolase polymorphisms in African Americans with hypertension. Am
J Hypertens. 18:1276–1281. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Enayetallah AE and Grant DF: Effects of
human soluble epoxide hydrolase polymorphisms on isoprenoid
phosphate hydrolysis. Biochem Biophys Res Commun. 341:254–260.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Decker M, Arand M and Cronin A: Mammalian
epoxide hydrolases in xenobiotic metabolism and signalling. Arch
Toxicol. 83:297–318. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Zhang J, Bajari R, Andric D, Gerthoffert
F, Lepsa A, Nahal-Bose H, Stein LD and Ferretti V: The
international cancer genome consortium data portal. Nat Biotechnol.
37:367–369. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Roessler S, Jia HL, Budhu A, Forgues M, Ye
QH, Lee JS, Thorgeirsson SS, Sun Z, Tang ZY, Qin LX, et al: A
unique metastasis gene signature enables prediction of tumor
relapse in early-stage hepatocellular carcinoma patients. Cancer
Res. 70:10202–10212. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Love MI, Huber W and Anders S: Moderated
estimation of fold change and dispersion for RNA-seq data with
DESeq2. Genome Biol. 15:5502014. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Fujimoto A, Furuta M, Totoki Y, Tsunoda T,
Kato M, Shiraishi Y, Tanaka H, Taniguchi H, Kawakami Y, Ueno M, et
al: Whole-genome mutational landscape and characterization of
noncoding and structural mutations in liver cancer. Nat Genet.
48:500–509. 2016. View
Article : Google Scholar : PubMed/NCBI
|
|
18
|
Ritchie ME, Phipson B, Wu D, Hu Y, Law CW,
Shi W and Smyth GK: limma powers differential expression analyses
for RNA-sequencing and microarray studies. Nucleic Acids Res.
43:e472015. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Goldman MJ, Craft B, Hastie M, Repečka K,
McDade F, Kamath A, Banerjee A, Luo Y, Rogers D, Brooks AN, et al:
Visualizing and interpreting cancer genomics data via the Xena
platform. Nat Biotechnol. 38:675–678. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Friedman J, Hastie T and Tibshirani R:
Regularization paths for generalized linear models via coordinate
descent. J Stat Softw. 33:1–22. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Sauerbrei W, Royston P and Binder H:
Selection of important variables and determination of functional
form for continuous predictors in multivariable model building.
Stat Med. 26:5512–5528. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Rhodes DR, Yu J, Shanker K, Deshpande N,
Varambally R, Ghosh D, Barrette T, Pandey A and Chinnaiyan AM:
ONCOMINE: A cancer microarray database and integrated data-mining
platform. Neoplasia. 6:1–6. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Chen X, Cheung ST, So S, Fan ST, Barry C,
Higgins J, Lai KM, Ji J, Dudoit S, Ng IO, et al: Gene expression
patterns in human liver cancers. Mol Biol Cell. 13:1929–1939. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Wurmbach E, Chen YB, Khitrov G, Zhang W,
Roayaie S, Schwartz M, Fiel I, Thung S, Mazzaferro V, Bruix J, et
al: Genome-wide molecular profiles of HCV-induced dysplasia and
hepatocellular carcinoma. Hepatology. 45:938–947. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Woo HG, Lee JH, Yoon JH, Kim CY, Lee HS,
Jang JJ, Yi NJ, Suh KS, Lee KU, Park ES, et al: Identification of a
cholangiocarcinoma-like gene expression trait in hepatocellular
carcinoma. Cancer Res. 70:3034–3041. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Yu G, Wang LG, Han Y and He QY:
clusterProfiler: An R package for comparing biological themes among
gene clusters. OMICS. 16:284–287. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Walter W, Sánchez-Cabo F and Ricote M:
GOplot: An R package for visually combining expression data with
functional analysis. Bioinformatics. 31:2912–2914. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Subramanian A, Tamayo P, Mootha VK,
Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub
TR, Lander ES, et al: Gene set enrichment analysis: A
knowledge-based approach for interpreting genome-wide expression
profiles. Proc Natl Acad Sci USA. 102:15545–15550. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Li H, Han D, Hou Y, Chen H and Chen Z:
Statistical inference methods for two crossing survival curves: A
comparison of methods. PLoS One. 10:e01167742015. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Qiu P and Sheng J: A two-stage procedure
for comparing hazard rate functions. J R Stat Soc Series B Stat
Methodol. 70:191–208. 2008.
|
|
32
|
Llovet JM, Montal R, Sia D and Finn RS:
Molecular therapies and precision medicine for hepatocellular
carcinoma. Nat Rev Clin Oncol. 15:599–616. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Llovet JM, Ricci S, Mazzaferro V, Hilgard
P, Gane E, Blanc JF, de Oliveira AC, Santoro A, Raoul JL, Forner A,
et al SHARP Investigators Study Group, : Sorafenib in advanced
hepatocellular carcinoma. N Engl J Med. 359:378–390. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Kudo M, Finn RS, Qin S, Han KH, Ikeda K,
Piscaglia F, Baron A, Park JW, Han G, Jassem J, et al: Lenvatinib
versus sorafenib in first-line treatment of patients with
unresectable hepatocellular carcinoma: A randomised phase 3
non-inferiority trial. Lancet. 391:1163–1173. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Bruix J, Qin S, Merle P, Granito A, Huang
YH, Bodoky G, Pracht M, Yokosuka O, Rosmorduc O, Breder V, et al
RESORCE Investigators, : Regorafenib for patients with
hepatocellular carcinoma who progressed on sorafenib treatment
(RESORCE): A randomised, double-blind, placebo-controlled, phase 3
trial. Lancet. 389:56–66. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Abou-Alfa GK, Meyer T, Cheng AL,
El-Khoueiry AB, Rimassa L, Ryoo BY, Cicin I, Merle P, Chen Y, Park
JW, et al: Cabozantinib in Patients with Advanced and Progressing
Hepatocellular Carcinoma. N Engl J Med. 379:54–63. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Zhang R, Ye J, Huang H and Du X: Mining
featured biomarkers associated with vascular invasion in HCC by
bioinformatics analysis with TCGA RNA sequencing data. Biomed
Pharmacother. 118:1092742019. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Xu X, Zhang XA and Wang DW: The roles of
CYP450 epoxygenases and metabolites, epoxyeicosatrienoic acids, in
cardiovascular and malignant diseases. Adv Drug Deliv Rev.
63:597–609. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Jiang JG, Chen CL, Card JW, Yang S, Chen
JX, Fu XN, Ning YG, Xiao X, Zeldin DC and Wang DW: Cytochrome P450
2J2 promotes the neoplastic phenotype of carcinoma cells and is
up-regulated in human tumors. Cancer Res. 65:4707–4715. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Panigrahy D, Greene ER, Pozzi A, Wang DW
and Zeldin DC: EET signaling in cancer. Cancer Metastasis Rev.
30:525–540. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Chen C, Wei X, Rao X, Wu J, Yang S, Chen
F, Ma D, Zhou J, Dackor RT, Zeldin DC, et al: Cytochrome P450 2J2
is highly expressed in hematologic malignant diseases and promotes
tumor cell growth. J Pharmacol Exp Ther. 336:344–355. 2011.
View Article : Google Scholar : PubMed/NCBI
|