Introduction
Acute lung injury (ALI) is a life-threatening lung
disease that can lead to refractory hypoxemia and respiratory
failure. Previous studies demonstrated that the age-adjusted
incidence of ALI was 86.2 per 100,000 person-years and that the
in-hospital mortality rate was 38.5% (1). Sepsis is a life-threatening disease
and may cause systemic inflammatory response syndrome and multiple
organ dysfunction, leading to high mortality among patients in the
intensive care unit (2,3). The lung is one of the most vulnerable
target organs during sepsis, and septic patients often have ALI,
which in turn facilitates the development of sepsis and multiple
organ failure via causing gas exchange impairment and intractable
hypoxemia (4–6). The pathogenesis of ALI involves the
death of the pulmonary endothelium and epithelium, autophagic
disorder and damage to the alveolar-capillary barrier. Thus,
inflammation and oxidative stress have indispensable roles in the
development of ALI (7–9). During ALI, leukocytes (e.g.,
macrophages and neutrophils) penetrate the lung interstitium and
release substantial proinflammatory cytokines to amplify the
inflammatory response. These leukocytes also increase the
production of reactive oxygen species (ROS) and induce oxidative
damage to lung cells. Additionally, excessive ROS levels activate
NACHT, LRR and PYD domain-containing protein 3 (NLRP3)
inflammasome, thereby accelerating the maturation and release of
proinflammatory cytokines (10–13).
Hence, it is reasonable to target inflammation and oxidative stress
to develop therapeutic strategies against ALI.
AMP-activated protein kinase (AMPK) has been
identified as an energy sensor in eukaryotic cells and has multiple
biological functions, including anti-inflammatory and antioxidant
capacities (14–17). Zhao et al (18) previously found that AMPK activation
attenuated nuclear factor-κB (NF-κB) transcription activity via
increasing TANK-binding kinase 1 phosphorylation, thereby
preventing adipose tissue inflammation. AMPK also suppressed NLRP3
inflammasome activation and alleviated lipopolysaccharide
(LPS)-induced ALI in mice (7,19).
Nuclear factor erythroid-2 related factor 2 (NRF2) functions as a
redox-sensitive transcription factor and is required for the
synthesis of various antioxidant enzymes, including superoxide
dismutase (SOD), catalase (CAT) and glutathione peroxidase (GPx)
(20–22). AMPK works upstream of NRF2, and
AMPK-mediated NRF2 activation protects against the development of
LPS-induced ALI (7,23,24).
These findings distinctly define AMPK as a strategic cellular
target for the treatment of ALI.
MicroRNAs (miRNA/miR) are a group of endogenous
short noncoding RNAs that act as negative gene regulators via
directly binding to the 3′-untranslated regions (UTRs) of
downstream messenger RNAs (25–27).
Numerous miRNAs have been proven to be essential for the
pathogenesis of ALI (28).
miR-351-5p was initially identified as a myogenesis-associated
miRNA and is responsible for skeletal muscle development by
targeting the phosphorylation kinase signaling cascade (29). Previous results demonstrated that an
miR-351-5p agomir increased inflammation, oxidative stress and
apoptosis, thereby aggravating intestinal ischemia/reperfusion
injury, whereas inhibiting miR-351-5p, either by antagomir or
dioscin, markedly ameliorated intestinal ischemia/reperfusion
injury in mice (30–32). In addition, da Silva et al
(33) determined that miR-351-5p
repressed PTEN expression and was essential for establishing a
proinflammatory environment in the H9c2 cell line. The aim of the
present study was to investigate the role and potential mechanisms
of miR-351-5p in ALI.
Materials and methods
Reagents and antibodies
LPS (from Escherichia coli O111:B4; cat. no.
L2360), H89 [a specific protein kinase A (PKA inhibitor); cat. no.
B1427] and the selective adenylate cyclase (AC) inhibitor
2′,5′-dideoxyadenosine (DDA; cat. no. D7408) were purchased from
Sigma-Aldrich (Merck KGaA). A lactate dehydrogenase (LDH) assay kit
(cat. no. ab102526), myeloperoxidase (MPO) ELISA kit (cat. no.
ab155458), malondialdehyde (MDA) assay kit (cat. no. ab118970),
protein carbonyl content assay kit (cat. no. ab126287), total
antioxidant capacity (TAOC) assay kit (cat. no. ab65329), total SOD
activity assay kit (cat. no. ab65354), CAT activity assay kit (cat.
no. ab83464), GPx assay kit (cat. no. ab102530), reduced
glutathione (GSH) assay kit (cat. no. ab235670), tumor necrosis
factor- α (TNF-α) ELISA kit (cat. no. ab208348), interleukin (IL-)
1β ELISA kit (cat. no. ab197742), IL-18 ELISA kit (cat. no.
ab216165), cAMP assay kit (cat. no. ab65355) and protein kinase A
(PKA) activity assay kit (cat. no. ab139435) were obtained from
Abcam. Compound C (CC; cat. no. S7840), a selective AMPK inhibitor,
was purchased from Selleck Chemicals. The Pierce BCA protein assay
kit (cat. no. 23227) and 2′,7′-dichlorofluorescin diacetate
(DCFH-DA; cat. no. C2938) were purchased from Thermo Fisher
Scientific, Inc. The TransAM® NRF2 kit (cat. no. 50296)
was obtained from Active Motif, Inc. miR-351-5p agomir (cat. no.
miR40000609-4-5), antagomir (cat. no. miR30000609-4-5) and their
negative controls (NC) were synthesized by Guangzhou RiboBio Co.,
Ltd. For western blotting, the following primary antibodies were
used at a dilution of 1:1,000: Anti-NRF2 (cat. no. ab137550;
Abcam), anti-GAPDH (cat. no. ab8245; Abcam), anti-NLRP3 (cat. no.
ab214185; Abcam), anti-caspase-1 p10 (cat. no. sc-56036; Santa Cruz
Biotechnology, Inc.), anti-phosphorylated (p-) AMPK (cat. no. 2535;
Cell Signaling Technology, Inc.) and anti-total (t-) AMPK (cat. no.
2603P; Cell Signaling Technology, Inc.).
Mice and treatments
A total of 180 male C57BL/6 mice (8–10 weeks old and
23–28 g weight) were purchased from Huafukang Bioscience Co., Ltd.,
and bred in a specific pathogen-free environment (25±2°C, 50±5%
humidity, 12-h light/dark cycle) with free access to food and
water. LPS is the major constituent of the outer membrane of
gram-negative bacteria and has emerged as a clinically relevant
model for ALI. In the present study, LPS was used to establish a
septic ALI model as previously described (7). ALI was induced through a single
intratracheal injection of LPS (5 mg/kg) dissolved in 50 µl sterile
saline for 12 h, while the control mice were treated with 50 µl
sterile saline intratracheally (7).
Arterial blood gas analysis, bronchoalveolar lavage fluid (BALF)
analysis, pulmonary edema, respiratory function measurement and
tissue injury biomarkers were detected to confirm the successful
ALI model establishment. miR-351-5p agomir (20 nmol), antagomir (50
nmol) and their corresponding NCs were injected into the tail vein
before LPS stimulation, as previously described (30). To inhibit AMPK, the mice were
intraperitoneally injected with CC (20 mg/kg) every other day for a
total of three treatments prior to miR-351-5p antagomir
administration (34). The mice also
received a single intraperitoneal injection of DDA (0.1 mg/kg) or
H89 (2 mg/kg) at 6 h prior to miR-351-5p antagomir administration.
For survival analysis, a lethal dose of LPS (25 mg/kg) was used as
previously described (7). The mice
were euthanized by a single intraperitoneal injection of sodium
pentobarbital (200 mg/kg), and the lung, liver and brain were
collected for further analyses. All experimental procedures
strictly complied with the Animal Research: Reporting of In
Vivo Experiments (ARRIVE) guidelines and were approved by the
Animal Ethics Committee of Renmin Hospital of Wuhan University.
BALF collection and analysis
BALFs were collected through 3-round intratracheal
injections with 1 ml pre-cooled sterile saline and then centrifuged
at 4°C for 10 min at 200 × g. The cell-free supernatants were used
to measure total protein concentrations using the Pierce BCA
protein assay kit, and TNF-α levels in BALFs were determined by
ELISA kits. To determine leukocyte extravasation, the pelleted
cells in BALFs were resuspended in sterile saline and counted with
a hemocytometer and Wright-Giemsa staining.
Lung wet/dry (W/D) ratio
The fresh lungs were dissected and weighed
immediately to obtain the lung wet weight after the blood was
removed. Next, the lungs were placed in an oven at 80°C for 96 h to
obtain the constant lung dry weight (7). The lung W/D ratio was calculated as a
measure of the degree of pulmonary edema.
Respiratory function measurement
A Buxco Resistance and Compliance system (Buxco
Electronics, Inc.) was used to monitor the respiratory data as
previously described (4). The
analysis focused on the tidal volume, dynamic lung compliance and
respiratory rate.
Arterial blood gas analysis
To further determine respiratory function, arterial
blood gas analysis was performed to evaluate pulmonary gas exchange
as previously described (35).
Briefly, 100 µl arterial blood samples were collected from the
right common carotid artery of mice prior to euthanasia with a
heparinized PE10 polyethylene catheter and then analyzed with an
automatic blood gas analyzer.
Hematoxylin and eosin (H&E)
staining
H&E staining was performed using the standard
protocols. Briefly, the lungs were excised and fixed in 4%
formaldehyde solution for 48 h, which were then embedded in
paraffin and sectioned at 5 µm. The sections were dewaxed, hydrated
and then incubated with hematoxylin for 10 min and eosin for 1 min
at room temperature. The images were captured by light microscopy
and appraised in a blinded manner.
Detection of oxidative stress
ROS levels in the lungs were detected by the DCFH-DA
method (36,37). In brief, the lung homogenates were
incubated with DCFH-DA (20 µmol/l) at 37°C for 1 h in the dark and
then analyzed using a microplate reader at excitation/emission
wavelengths of 485/535 nm (7,38,39).
The levels of MDA, protein carbonyls and GSH, and the activities of
TAOC, SOD, GPx and CAT were assessed by the respective kits,
following the manufacturers' instructions.
Measurements of the activities of LDH,
NRF2, MPO and PKA and the levels of cAMP in the lungs
Lung homogenates were prepared in the assay buffer
by a high-speed homogenizer (Wuhan Servicebio Technology Co., Ltd.)
according to the respective manufacturer's instructions to
determine the activities of LDH, MPO and PKA and the levels of cAMP
using commercial kits. NRF2 transcription activity was detected
using the TransAM® NRF2 kit, according to the
manufacturer's instructions. Briefly, nuclear extracts were
prepared from fresh lung tissues and incubated in plates coated
with oligonucleotides containing an antioxidant responsive element.
Subsequently, a primary antibody against NRF2 and a horseradish
peroxidase (HRP)-conjugated secondary antibody were added. Then,
the absorbance was read at 450 nm on a spectrophotometer with a
reference wavelength of 655 nm.
Western blot analysis
Frozen lung tissues were homogenized by a high-speed
homogenizer (Wuhan Servicebio Technology Co., Ltd.) and lysed in
RIPA lysis buffer (cat. no. G2002; Wuhan Servicebio Technology Co.,
Ltd.), and total protein concentrations were measured using a
Pierce BCA protein assay kit (40–42).
Next, 20 µg total proteins were separated on a 10% SDS-PAGE gel
according to standard protocols and electrotransferred to PVDF
membranes. To block the non-specific binding of the primary
antibodies, 5% BSA was used for 1 h at room temperature. Then, the
membranes were incubated with the indicated primary antibodies at
4°C overnight, followed by HRP-conjugated secondary antibodies at
1:5,000 dilution (cat. no. sc-2004 and sc-2005; Santa Cruz
Biotechnology, Inc.) at room temperature for an additional 1 h. The
protein bands were scanned with an electrochemiluminescence reagent
and the relative band intensity was quantified using Image Lab
Analyzer software (version 6.0; Bio-Rad Laboratories, Inc.).
Reverse transcription-quantitative
polymerase chain reaction (RT-qPCR)
Total RNA was extracted from the lungs using TRIzol™
reagent (Thermo Fisher Scientific, Inc.) and used for reverse
transcription and cDNA synthesis (36,43–46).
Next, qPCR was performed using the SYBR Premix Ex Taq kit (Takara
Biotechnology Co., Ltd.) on a CFX96 real-time PCR detection system
(Bio-Rad Laboratories, Inc.). The thermocycling conditions were as
follows: 95°C for 10 min, 40 cycles of 95°C for 15 sec, 60°C for 30
sec and 70°C for 30 sec. Gene expression was measured by the
2−ΔΔCq method and normalized to GAPDH (47). The primer sequences were: IL-1β,
forward, 5′-CCGTGGACCTTCCAGGATGA-3′ and reverse,
5′-GGGAACGTCACACACCAGCA-3′; IL-6, forward,
5′-AGTTGCCTTCTTGGGACTGA-3′ and reverse, 5′-TCCACGATTTCCCAGAGAAC-3′;
TNF-α, forward, 5′-AGCCCCCAGTCTGTATCCTT-3′ and reverse,
5′-CTCCCTTTGCAGAACTCAGG-3′; NLRP3, forward,
5′-TACGGCCGTCTACGTCTTCT-3′ and reverse, 5′-CGCAGATCACACTCCTCAAA-3′;
ASC, forward, 5′-GACAGTACCAGGCAGTTCGT-3′ and reverse,
5′-AGTCCTTGCAGGTCAGGTTC-3′; Pro-caspase-1, forward,
5′-CACAGCTCTGGAGATGGTGA-3′ and reverse, 5′-CTTTCAAGCTTGGGCACTTC-3′;
GAPDH, forward, 5′-ACTCCACTCACGGCAAATTC-3′ and reverse,
5′-TCTCCATGGTGGTGAAGACA-3′.
Dual-luciferase reporter assay
The wild-type (WT) or mutant (MUT) 3′-UTR of
adenylate cyclase type 6 (Adcy6) was cloned into the pGL3
luciferase reporter plasmid (Promega Corporation), which was then
co-transfected with miR-351-5p agomir or agomir NC into
H&EK293T cells using Lipofectamine™ 3000 (Thermo Fisher
Scientific, Inc.). H&EK293T cells were purchased from the
American Type Culture Collection. After 48 h, the cells were lysed
and subjected to a dual-luciferase reporter assay using the
Dual-Luciferase Reporter Assay System (Promega Corporation) and the
data were normalized to Renilla luciferase activity
according to the manufacturer's instructions.
Statistical analysis
All data were expressed as the mean ± SD and
statistical analysis was performed using SPSS 23.0 (SPSS, Inc.).
Comparisons between two groups were analyzed by two-tailed unpaired
Student's t-test, and comparisons among multiple groups were
analyzed by one-way ANOVA followed by Tukey's post hoc test where
appropriate. Survival analysis was performed by the Kaplan-Meier
method followed by a Mantel-Cox log rank test. P<0.05 was
considered to indicate a statistically significant difference.
Results
miR-351-5p antagomir alleviates
LPS-induced ALI
First, the expression levels of miR-351-5p were
detected in the lungs of mice challenged with LPS for 0, 1, 2, 6
and 12 h. As presented in Fig. 1A,
the miR-351-5p levels were significantly increased in response to
LPS injection. To determine the role of miR-351-5p upregulation in
the process of ALI, mice were treated with miR-351-5p antagomir.
Tail vein injection of miR-351-5p antagomir significantly reversed
the LPS-mediated upregulation of miR-351-5p in the mouse lungs
(Fig. 1B). Consistent with previous
reports, LPS challenge caused severe lung injury and pulmonary
edema, as evidenced by H&E staining, the increased LDH
activity, lung W/D ratio and total protein concentrations in BALFs,
which were remarkably alleviated by the miR-351-5p antagomir
(Fig. 1C-F). In addition, the mice
treated with the miR-351-5p antagomir had increased tidal volume,
lung compliance and respiratory rate (Fig. 1G-I). Consistently, the partial
pressure of oxygen (PaO2) was restored, while the
partial pressure of carbon dioxide (PaCO2) was decreased
following miR-351-5p antagomir treatment (Fig. 1J and K). Notably, pretreatment with
the miR-351-5p antagomir significantly improved the survival rate
of LPS-challenged mice (Fig. 1L).
Collectively, these data indicated that the miR-351-5p antagomir
alleviated LPS-induced ALI.
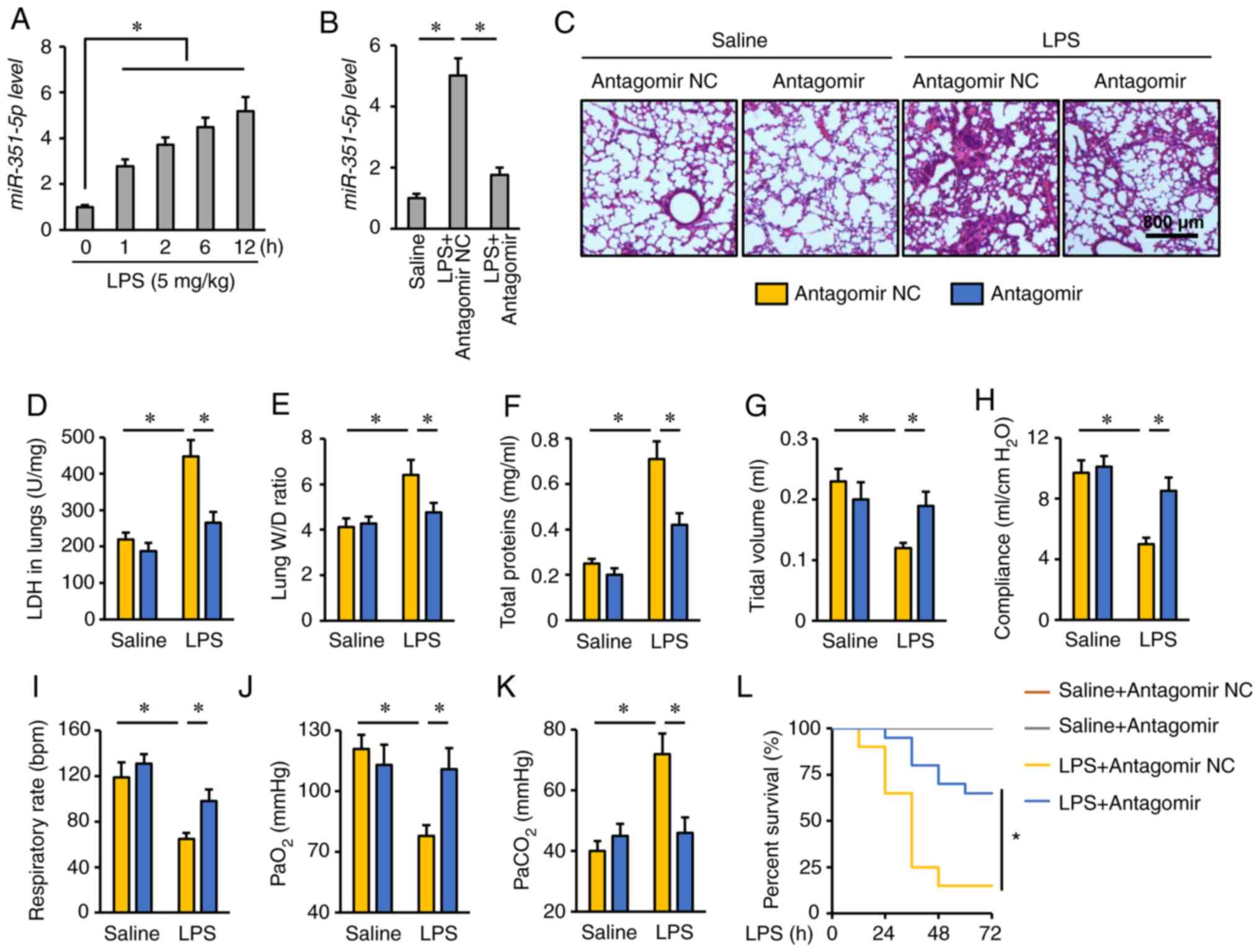 | Figure 1.miR-351-5p antagomir alleviates
LPS-induced ALI. (A and B) Relative miR-351-5p levels in the lungs
(n=6). (C) Lung histopathology determined by hematoxylin and eosin
staining (n=6). (D) LDH activity in the lungs with or without
miR-351-5p antagomir treatment upon LPS challenge (n=6). (E) Lung
W/D ratio (n=8). (F) Total protein concentrations in
bronchoalveolar lavage fluids (n=6). (G-I) Quantification of
respiratory functional parameters, including tidal volume, lung
compliance and respiratory rate (n=6). (J and K) Quantification of
PaO2 and PaCO2 during arterial blood gas
analysis (n=6). (L) Survival analysis of mice with or without
miR-351-5p antagomir treatment upon LPS challenge (n=20). All data
are expressed as mean ± SD. *P<0.05 with comparisons shown by
lines. miR, microRNA; LPS, lipopolysaccharide; ALI, acute lung
injury; LDH, lactate dehydrogenase; W/D, wet/dry weight;
PaO2, partial pressure of oxygen; PaCO2,
partial pressure of carbon dioxide; NC, negative control. |
miR-351-5p antagomir reduces
LPS-induced oxidative stress in the lungs
Oxidative stress is a key feature in LPS-induced ALI
and accelerates the progression of pulmonary injury and dysfunction
(4,7). As presented in Fig. 2A, LPS induced excessive ROS
generation in the lungs, and this was suppressed by the miR-351-5p
antagomir. Accordingly, the levels of MDA and protein carbonyls
were also decreased in mice treated with the miR-351-5p antagomir
(Fig. 2B). NRF2 functions as a
redox-sensitive transcription factor and is required for the
synthesis of various antioxidant enzymes to scavenge excessive free
radicals (20). As presented in
Fig. 2C and D, NRF2 protein
expression was inhibited in mice following LPS challenge but was
preserved when these mice were co-treated with miR-351-5p
antagomir. In addition, LPS-mediated suppression of NRF2
transcription activity in LPS-challenged lungs was reversed by the
miR-351-5p antagomir (Fig. 2E).
Consistently, endogenous antioxidant capacity in the lungs of mice
treated with miR-351-5p antagomir was significantly increased, as
evidenced by the preserved TAOC, total SOD activity, CAT activity,
GPx activity and GSH levels (Fig.
2F-J). These results suggested that the miR-351-5p antagomir
reduced LPS-induced oxidative stress in the lungs.
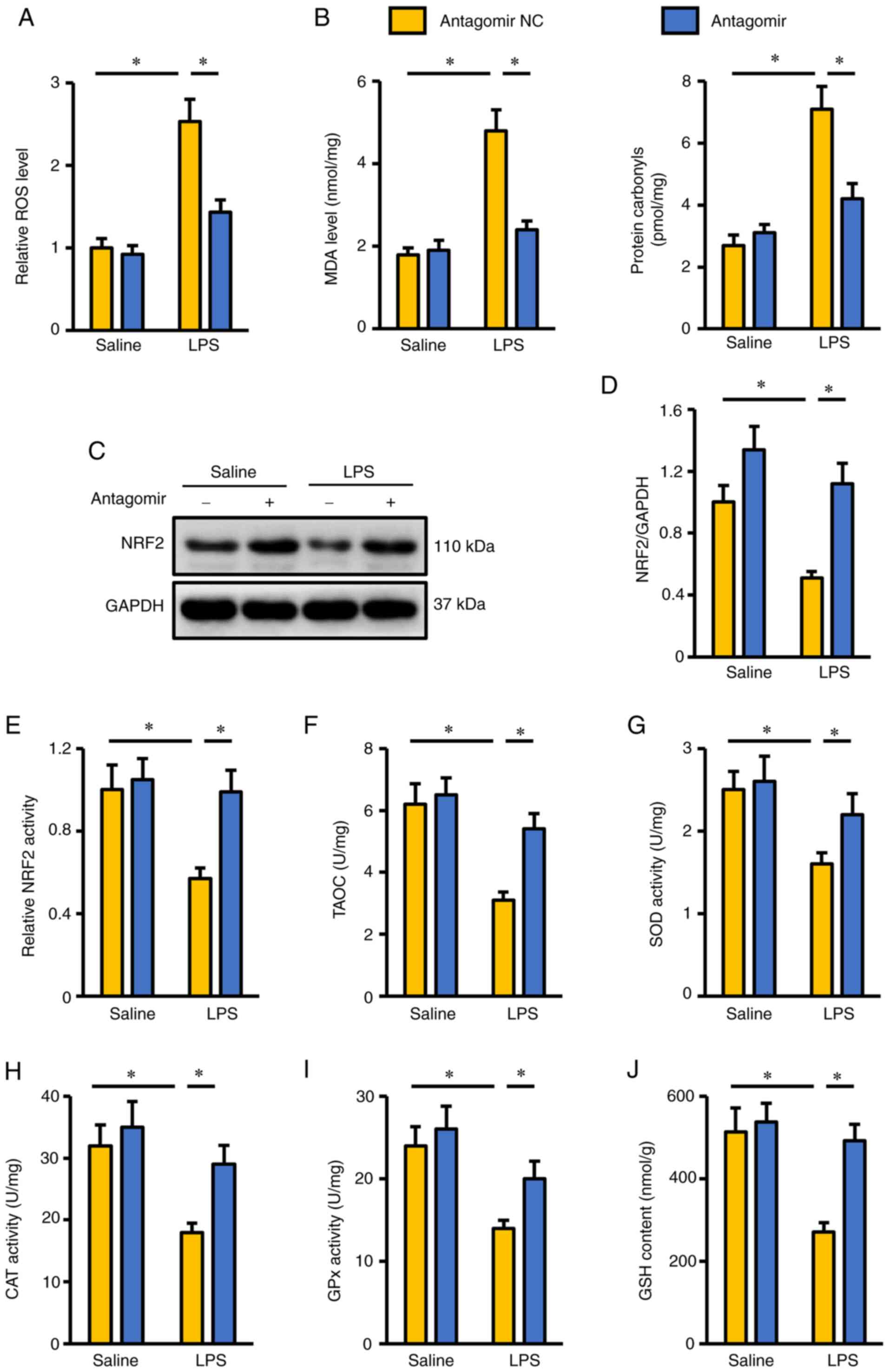 | Figure 2.miR-351-5p antagomir reduces
LPS-induced oxidative stress in the lungs. (A) Relative ROS levels
in the lungs with or without miR-351-5p antagomir treatment upon
LPS challenge (n=6). (B) MDA levels and protein carbonyls in the
lungs (n=6). (C) Representative blots and (D) quantification of
NRF2 protein expression levels in the lungs determined by western
blot analysis (n=6). (E) Quantification of NRF2 transcription
activity in the lungs (n=6). (F-J) Intracellular antioxidant
capacity of the lungs determined by TAOC, total SOD activity, CAT
activity, GPx activity and GSH content (n=6). All data are
expressed as mean ± SD. *P<0.05 with comparisons shown by lines.
miR, microRNA; LPS, lipopolysaccharide; ROS, reactive oxygen
species; MDA, malondialdehyde; NRF2, nuclear factor erythroid-2
related factor 2; TAOC, total antioxidant capacity; SOD, superoxide
dismutase; CAT, catalase; GPx, glutathione peroxidase; GSH,
glutathione; NC, negative control. |
miR-351-5p antagomir inhibits
LPS-induced inflammation in the lungs
Next, the present study investigated the effect of
the miR-351-5p antagomir on LPS-induced intrapulmonary inflammatory
responses in mice. The data indicated that miR-351-5p antagomir
treatment effectively inhibited the mRNA expression levels of
IL-1β, IL-6 and TNF-α in the lungs (Fig. 3A). Accordingly, TNF-α protein
expression levels in the lungs and BALFs were both reduced by the
miR-351-5p antagomir (Fig. 3B). The
NLRP3 inflammasome is a multiprotein complex responsible for the
cleavage and activation of caspase-1, which subsequently promotes
the maturation and release of proinflammatory cytokines, such as
IL-1β and IL-18 (7,48). As presented in Fig. 3C, the miR-351-5p antagomir
significantly decreased the mRNA expression levels of NLRP3,
apoptosis-associated speck-like protein containing a CARD (ASC) and
pro-caspase-1 in LPS-challenged lungs. The protein expression
levels of NLRP3 and active caspase-1 p10 were also suppressed by
the miR-351-5p antagomir in response to LPS injection (Fig. 3D and E). In addition, the miR-351-5p
antagomir significantly inhibited the LPS-associated induction of
caspase-1 activity (Fig. 3F). Of
note, the levels of the downstream IL-1β and IL-18 were both
reduced in the lungs following miR-351-5p antagomir treatment
(Fig. 3G). Furthermore,
pretreatment with the miR-351-5p antagomir significantly reduced
the number of total cells, macrophages and neutrophils in BALFs and
MPO activity in the lungs of ALI mice (Fig. 3H and I). Taken together, the present
results demonstrated that the miR-351-5p antagomir inhibited
LPS-induced inflammation in the lungs.
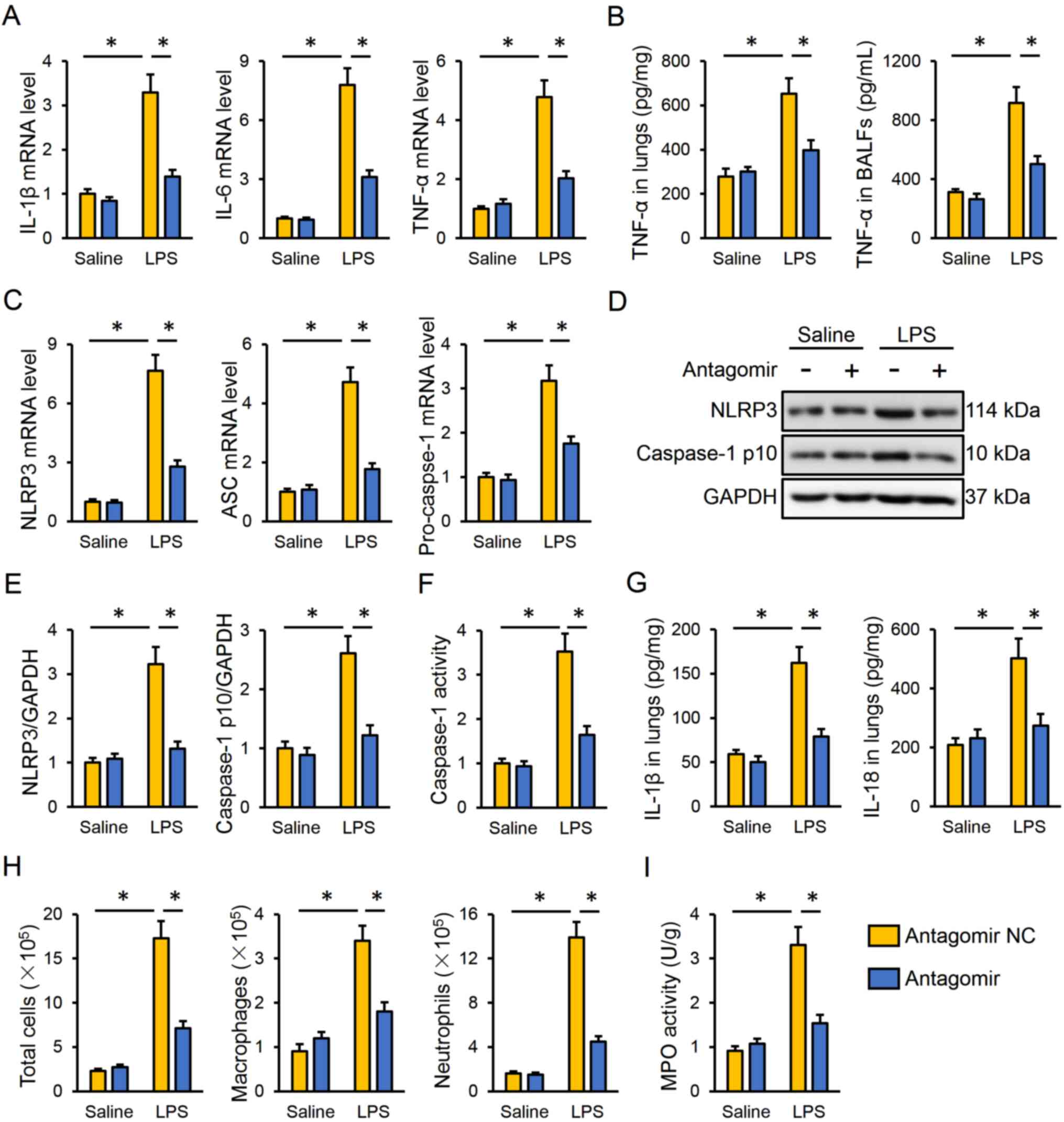 | Figure 3.miR-351-5p antagomir inhibits
LPS-induced inflammation and NLRP3 inflammasome in the lungs. (A)
Relative mRNA expression levels of proinflammatory cytokines IL-1β,
IL-6 and TNF-α in the lungs (n=6). (B) TNF-α levels in the lungs
and BALFs measured by ELISA (n=6). (C) Relative mRNA expression
levels of NLRP3, ASC and pro-caspase-1 in the lungs (n=6). (D)
Representative blots and (E) quantification of NLRP3 and caspase-1
p10 protein expression levels determined by western blot analysis
(n=6). (F) Relative caspase-1 activity (n=6). (G) IL-1β and IL-18
levels in the lungs measured by ELISA (n=6). (H) Total cells,
macrophages and neutrophils in BLAFs were counted (n=6). (I) MPO
activity in the lungs with or without miR-351-5p antagomir
treatment upon LPS challenge (n=6). All data are expressed as mean
± SD. *P<0.05 with comparisons shown by lines. miR, microRNA;
LPS, lipopolysaccharide; NLRP3, NACHT, LRR and PYD
domain-containing protein 3; BALF, bronchoalveolar lavage fluid;
ASC, apoptosis-associated speck-like protein containing a CARD;
MPO, myeloperoxidase; NC, negative control. |
miR-351-5p agomir aggravates
LPS-induced ALI
The present study also used a miR-351-5p agomir to
clarify whether overexpression of miR-351-5p in the lungs could
aggravate LPS-induced ALI (Fig.
4A). As expected, lung injury and pulmonary edema in mice with
LPS challenge were more severe in the presence of miR-351-5p
agomir, as evidenced by H&E staining, the increased LDH
activity, lung W/D ratio and total protein concentrations in BALFs
(Fig. 4B-E). In addition,
miR-351-5p agomir treatment further reduced tidal volume, lung
compliance and respiratory rate in LPS-challenged mice (Fig. 4F-H). Accordingly, LPS-associated
impairment of blood gas exchange was further aggravated by the
miR-351-5p agomir, as evidenced by the decreased PaO2
and increased PaCO2 (Fig.
4I). Furthermore, the survival time of miR-351-5p
agomir-treated mice upon LPS challenge was no more than 24 h (data
not shown). Overall, these results revealed that miR-351-5p
overexpression aggravated LPS-induced ALI.
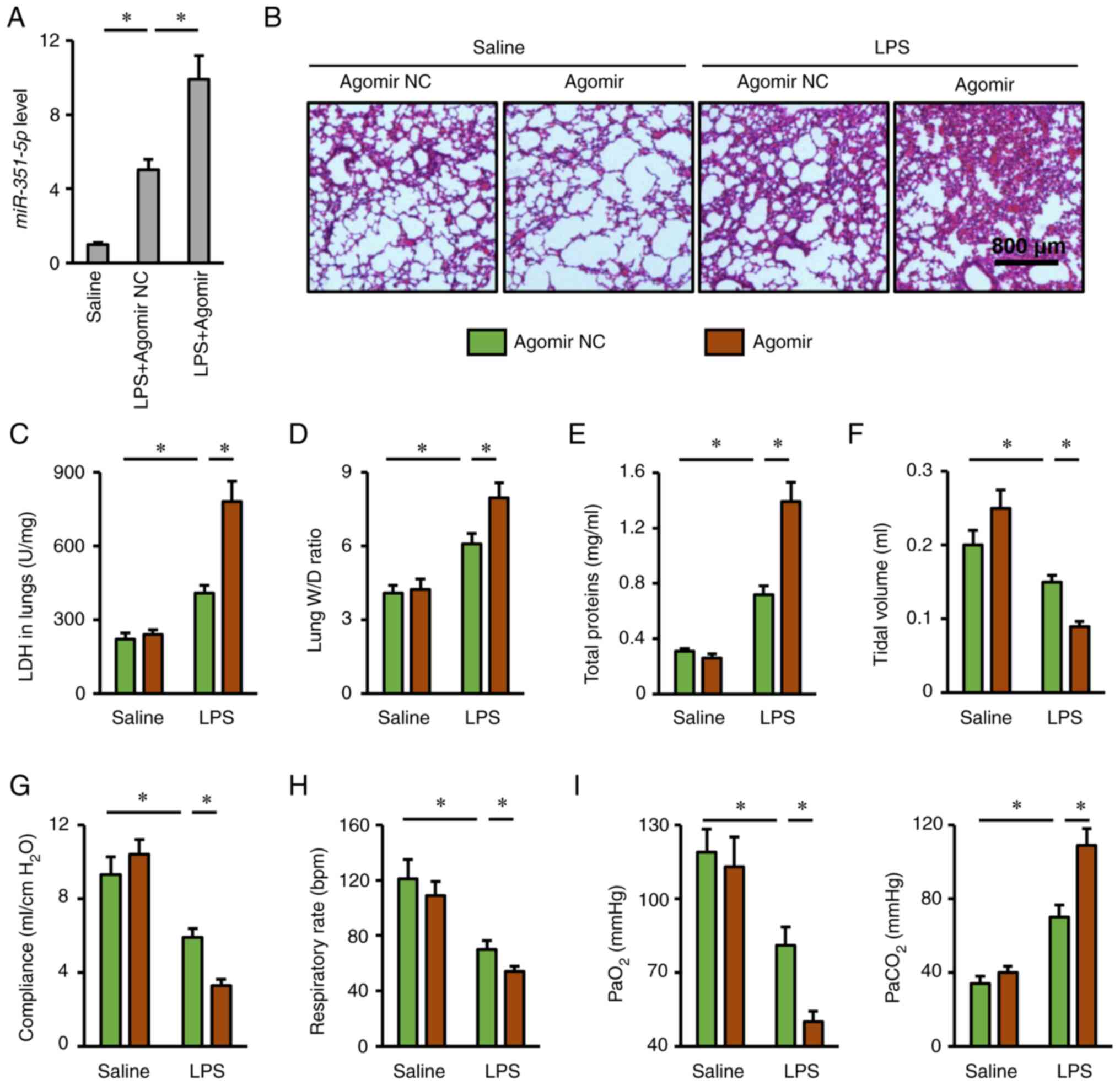 | Figure 4.miR-351-5p agomir aggravates
LPS-induced ALI. (A) Relative miR-351-5p expression levels in the
lungs (n=6). (B) Lung histopathology determined by hematoxylin and
eosin staining (n=6). (C) LDH activity in the lungs with or without
miR-351-5p agomir treatment upon LPS challenge (n=6). (D) Lung W/D
ratio (n=8). (E) Total prtein concentrations in bronchoalveolar
lavage fluids (n=6). (F-H) Respiratory functional parameters,
including tidal volume, lung compliance and respiratory rate (n=6).
(I) Quantification of PaO2 and PaCO2 during
arterial blood gas analysis (n=6). All data are expressed as mean ±
SD. *P<0.05 with comparisons shown by lines. miR, microRNA; LPS,
lipopolysaccharide; ALI, acute lung injury; LDH, lactate
dehydrogenase; W/D, wet/dry weight; PaO2, partial
pressure of oxygen; PaCO2, partial pressure of carbon
dioxide; NC, negative control. |
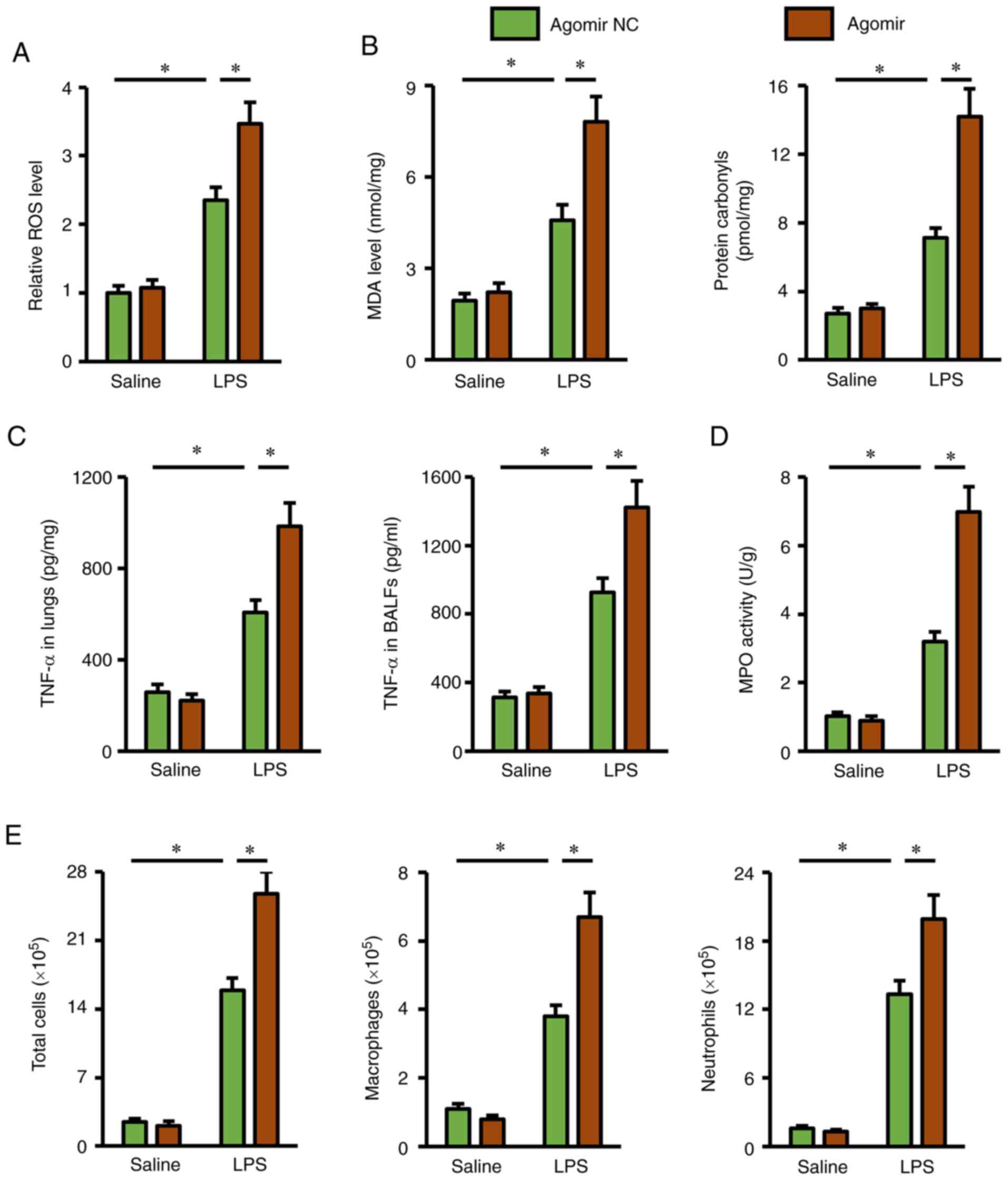
miR-351-5p agomir exacerbates
LPS-induced oxidative stress and inflammation in the lungs
Next, several important biomarkers related to
oxidative stress and inflammation were investigated, in order to
further explore the role of miR-351-5p agomir in mice. Consistent
with the ALI phenotype, the miR-351-5p agomir remarkably promoted
ROS generation in LPS-challenged lungs, and the production of MDA
and protein carbonyls was also increased (Fig. 5A and B). In addition, pretreatment
with the miR-351-5p agomir significantly elevated TNF-α levels in
the lungs and BALFs, and MPO activity in lung tissues (Fig. 5C and D). Furthermore, the numbers of
total cells, macrophages and neutrophils in BALFs from
LPS-challenged mice were significantly increased by the miR-351-5p
agomir (Fig. 5E). Thus, the present
data demonstrated that the miR-351-5p agomir exacerbated
LPS-induced oxidative stress and inflammation in the lungs.
To further confirm the therapeutic potential of
miR-315-5p in mice, its effect on LPS-induced inflammation and
oxidative stress in livers and brains was also evaluated. As shown
in Fig. S1A, miR-315-5p
antagomir-treated mice had lower intracellular ROS levels in the
livers, accompanied by reduced lipid and protein peroxidation. In
addition, the miR-315-5p antagomir decreased the hepatic TNF-α
levels and MPO activity upon LPS stimulation (Fig. S1B and C). By contrast, the
miR-315-5p agomir further aggravated LPS-induced oxidative stress
and inflammation in the livers (Fig.
S1D-F). Consistent with the findings in the lungs and livers,
it was observed that the miR-315-5p antagomir decreased, while the
miR-315-5p agomir further increased cerebral oxidative stress and
inflammation upon LPS injection in mice (Fig. S2). These data further validate the
therapeutic value of miR-315-5p against LPS-induced ALI.
miR-351-5p antagomir attenuates
LPS-induced ALI via activating AMPK
The potential role of AMPK in the protective effects
of the miR-351-5p antagomir was evaluated next. As presented in
Fig. 6A and B, the miR-351-5p
antagomir restored, while the miR-351-5p agomir further reduced
AMPK phosphorylation in LPS-treated lungs. To inhibit AMPK, the
specific inhibitor CC was used as previously described (34). CC treatment completely abrogated the
inhibitory effects of the miR-351-5p antagomir on oxidative stress
and inflammation, as evidenced by the increased ROS, MDA, protein
carbonyls, TNF-α levels and MPO activity in the lungs and BALFs
(Fig. 6C-F). Accordingly, the
reductions of the lung LDH activity and W/D ratio in miR-351-5p
antagomir-treated mice upon LPS challenge were blocked by CC
(Fig. 6G and H). Of note, the
miR-351-5p antagomir significantly restored tidal volume and
PaO2 in LPS-treated mice, but not in those pretreated
with CC (Fig. 6I-J). Collectively,
it can be deduced that the miR-351-5p antagomir attenuated
LPS-induced ALI via activating AMPK.
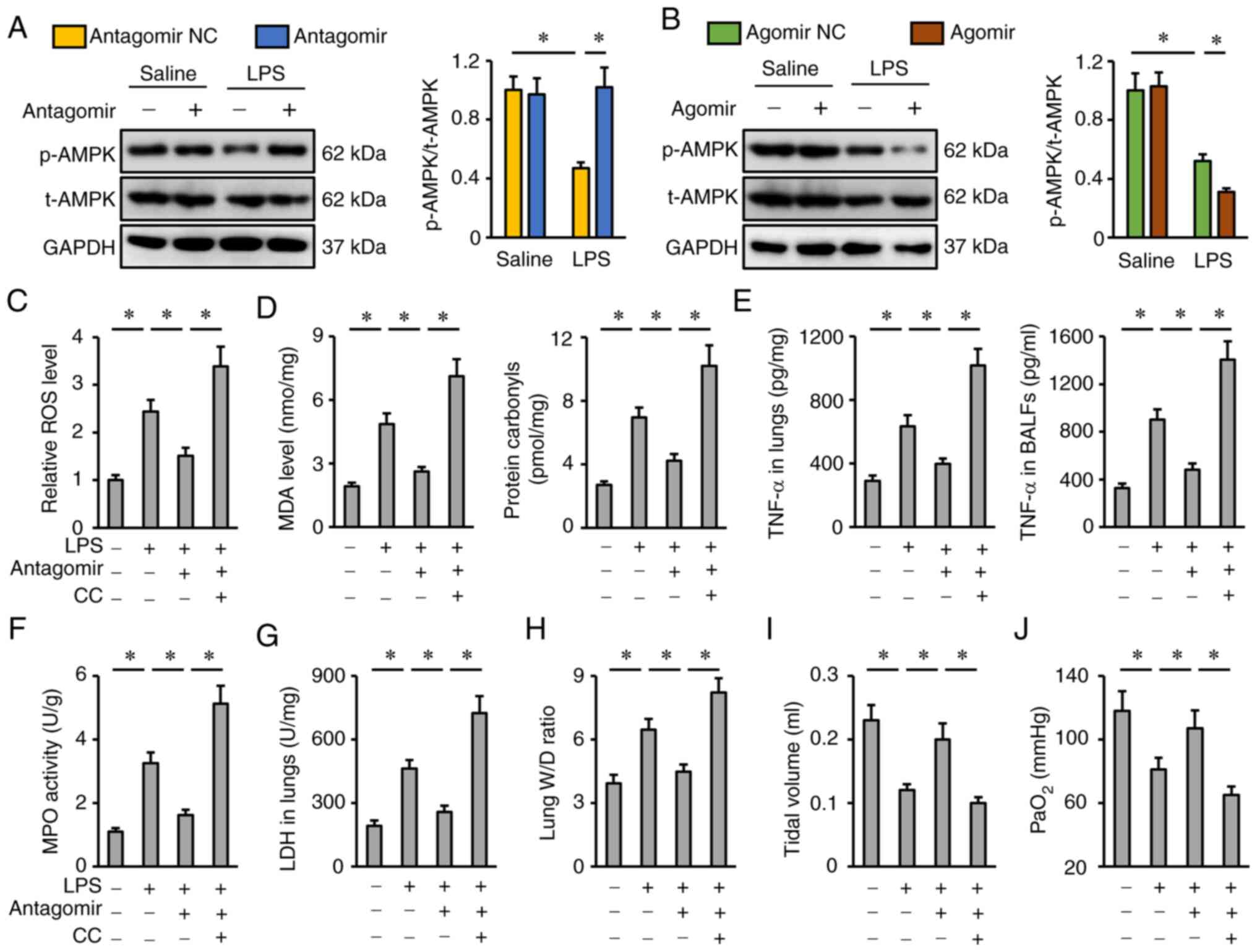 | Figure 6.miR-351-5p antagomir attenuates
LPS-induced ALI via activating AMPK. (A and B) Representative blots
and quantification of t-AMPK and p-AMPK protein expression levels
(n=6). (C) Relative ROS levels in miR-351-5p antagomir-treated
lungs with or without CC administration upon LPS challenge (n=6).
(D) MDA level and protein carbonyls in the lungs (n=6). (E) TNF-α
levels in the lungs and BALFs measured by ELISA (n=6). (F) MPO
activity in miR-351-5p antagomir-treated lungs with or without CC
administration upon LPS challenge (n=6). (G) LDH activity in the
lungs (n=6). (H) Lung W/D ratio (n=8). (I) Tidal volume in
miR-351-5p antagomir-treated mice with or without CC administration
upon LPS challenge (n=6). (J) Quantification of PaO2
during arterial blood gas analysis (n=6). All data are expressed as
mean ± SD. *P<0.05 with comparisons shown by lines. miR,
microRNA; LPS, lipopolysaccharide; ALI, acute lung injury; AMPK,
AMP-activated protein kinase; t-, total; p-, phosphorylated; ROS,
reactive oxygen species; CC, compound C; MDA, malondialdehyde;
BALF, bronchoalveolar lavage fluid; MPO, myeloperoxidase; LDH,
lactate dehydrogenase; W/D, wet/dry weight; PaO2,
partial pressure of oxygen; NC, negative control. |
miR-351-5p antagomir activates AMPK
via the cAMP/PKA axis
Finally, the present study investigated the
potential molecular basis by which the miR-351-5p antagomir may
activate AMPK. Using the TargetScan database, it was found that
miR-351-5p might directly bind to the 3′-UTR of three isoforms of
AC (AC1, AC5 and AC6), which catalyze the conversion of ATP to
cAMP, and induce a rapid elevation of the levels of intracellular
cAMP and the activities of PKA, the upstream activator of the AMPK
pathway (Fig. 7A) (49). To validate the putative interaction
between miR-351-5p and AC, a dual-luciferase reporter assay was
performed. AC6 (encoded by the Adcy6 gene) functions as an
important AC in the development of multiple lung disease;
therefore, the WT and MUT 3′-UTR of Adcy6 were cloned to perform
dual-luciferase reporter assays (50,51).
As shown in Fig. 7B, the miR-315-5p
agomir significantly inhibited luciferase activity in cells
transfected with the WT 3′-UTR of Adcy6 but failed to inhibit
luciferase activity in cells transfected with the MUT 3′-UTR. As
expected, it was found that the miR-351-5p antagomir increased,
while the miR-351-5p agomir decreased cAMP levels and PKA
activities in LPS-treated lungs (Fig.
7C and D). Accordingly, AMPK activation by the miR-351-5p
antagomir was blocked after the inhibition of AC by DDA or the
inhibition of PKA by H89 (Fig. 7E).
The miR-351-5p antagomir also lost its inhibitory effects on
LPS-induced pulmonary oxidative damage and inflammation in the
presence of DDA or H89 treatment (Fig.
7F-H). In addition, the reductions of lung LDH activity and W/D
ratio in miR-351-5p antagomir-treated mice upon LPS challenge were
blocked by DDA or H89 (Fig. 7I and
J). Accordingly, DDA or H89 also blocked the restoration of
tidal volume and PaO2 by the miR-351-5p antagomir in
LPS-treated mice (Fig. 7K and L).
Taken together, it can be concluded that the miR-351-5p antagomir
activated AMPK via the cAMP/PKA axis.
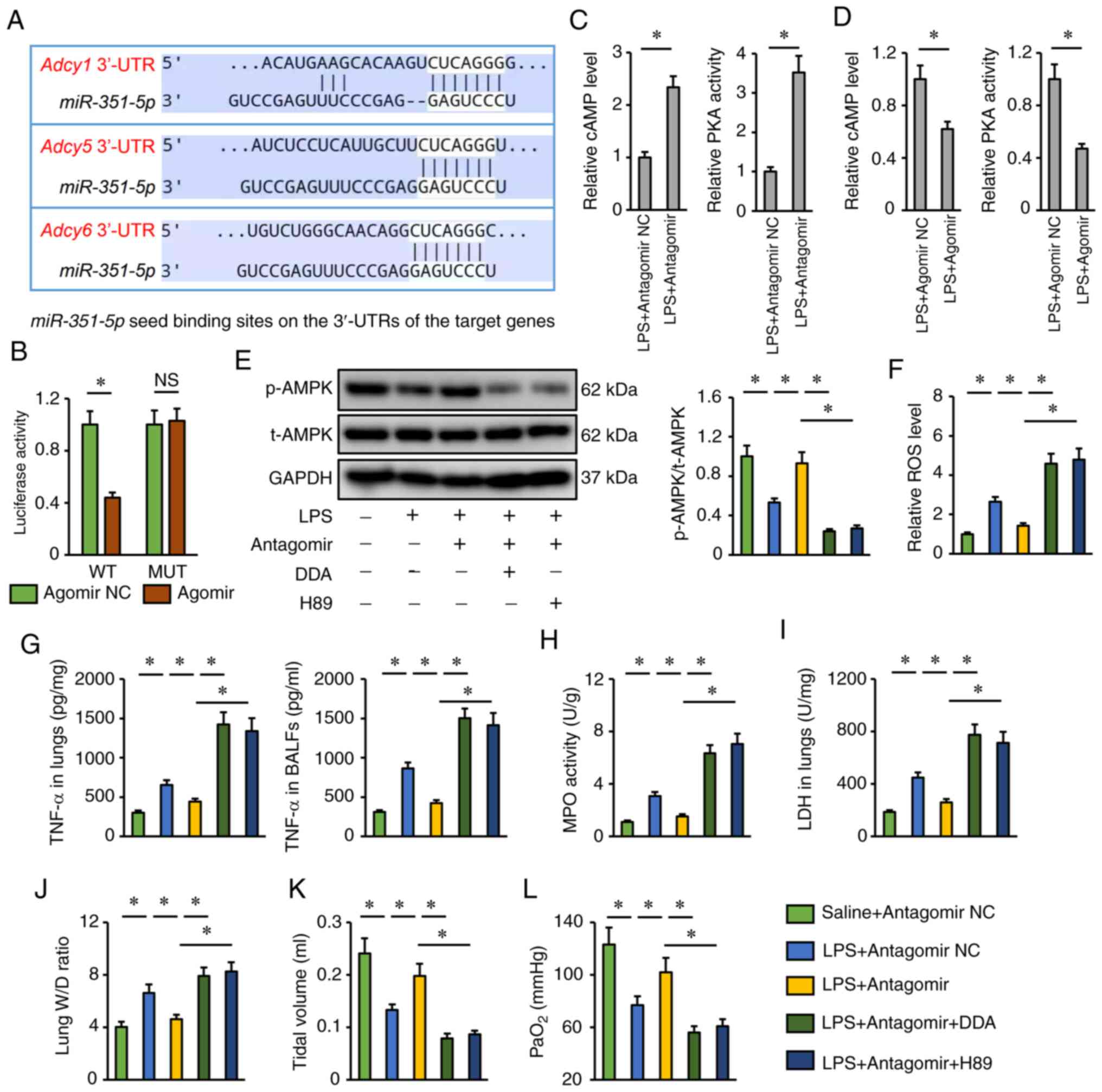 | Figure 7.miR-351-5p antagomir activates AMPK
via the cAMP/PKA axis. (A) Putative miR-351-5p seed binding sites
on the 3′-UTRs of the target genes. (B) Relative luciferase
activity in miR-351-5p antagomir-treated cells following
transfection with a WT or MUT Adcy6 3′-UTR reporter (n=6). (C and
D) Relative cAMP levels and PKA activities in LPS-challenged lungs
with miR-351-5p antagomir or agomir treatment (n=6). (E) Western
blot analysis of t-AMPK and p-AMPK protein expression levels (n=6).
(F) Relative ROS levels in miR-351-5p antagomir-treated lungs with
DDA or H89 administration upon LPS challenge (n=6). (G) TNF-α
levels in the lungs and BALFs measured by ELISA (n=6). (H) MPO
activity in miR-351-5p antagomir-treated lungs with DDA or H89
administration upon LPS challenge (n=6). (I) LDH activity in the
lungs (n=6). (J) Lung W/D ratio (n=8). (K) Tidal volume in
miR-351-5p antagomir-treated mice with DDA or H89 administration
upon LPS challenge (n=6). (L) Quantification of PaO2
during arterial blood gas analysis (n=6). All data are expressed as
mean ± SD. *P<0.05 with comparisons shown by lines. miR,
microRNA; AMPK, AMP-activated protein kinase; PKA, protein kinase
A; UTR, untranslated region; WT, wild-type; MUT, mutant; LPS,
lipopolysaccharide; t-, total; p-, phosphorylated; ROS, reactive
oxygen species; DDA, 2′,5′-dideoxyadenosine; BALF, bronchoalveolar
lavage fluid; MPO, myeloperoxidase; LDH, lactate dehydrogenase;
W/D, wet/dry weight; PaO2, partial pressure of oxygen;
NC, negative control; NS, not significant. |
Discussion
In the present study, LPS challenge was found to
upregulate miR-351-5p expression in the lungs, which then directly
targeted AC to decrease the intracellular cAMP levels and PKA
activities, ultimately resulting in AMPK suppression and ALI
progression. By contrast, miR-351-5p antagomir treatment restored
AMPK phosphorylation, and significantly reduced oxidative stress
and inflammation in LPS-treated mice. Overall, the present study
for the first time demonstrated the involvement of miR-351-5p in
the development of ALI and identified it as a promising therapeutic
candidate to treat ALI.
Oxidative stress is a major pathogenic factor during
ALI and directly induces oxidative damage to proteins, lipids and
nucleic acids, which ultimately causes cellular dysfunction and
even cell death (4,7). NRF2 functions as a signaling hub in
the regulation of redox homeostasis and is essential for the
transcription of multiple antioxidant enzymes (20). Upon LPS stimulation, the NRF2
protein levels are reduced, and the expression of downstream
antioxidant enzymes is suppressed, resulting in a deficiency in
scavenging excessive free radicals. Consistently, the present study
found that the intracellular antioxidant capacity was inhibited in
the lungs of mice injected with LPS, but partially preserved in
those pretreated with the miR-351-5p antagomir. Extensive
inflammation also contributes to the initiation and progression of
LPS-induced ALI. LPS is an exogenous ligand of Toll-like receptors
and can promote NF-κB nuclear translocation to induce the
transcription of various proinflammatory cytokines (35,52).
In addition, LPS-associated ROS generation increases the binding of
thioredoxin-interacting protein to NLRP3, which subsequently
activates the NLRP3 inflammasome to accelerate the maturation and
release of proinflammatory cytokines (7). The present findings demonstrated that
the miR-351-5p antagomir decreased, while the miR-351-5p agomir
increased NLRP3 inflammasome activation. Based on these data, it
can be speculated that miR-351-5p may serve as a therapeutic target
to treat ALI. However, the exact cell populations mediating the
effects of miR-351-5p/AMPK during LPS-induced ALI remain unclear
and need further investigation.
AMPK is a multifunctional kinase with
anti-inflammatory and antioxidant capacities that has already been
identified as a strategic cellular target to treat ALI (7,17,19).
AC works downstream of G protein-coupled receptors and is essential
for the rapid induction of intracellular cAMP synthesis, which
ultimately causes PKA activation (53). PKA then phosphorylates and activates
AMPK (54). miRNAs are a group of
intracellular genetic regulators that also participate in
regulating ALI progression. The present study found that miR-351-5p
directly targeted AC and functioned as an endogenous inhibitor of
the AMPK pathway. miR-351-5p antagomir treatment significantly
restored intracellular cAMP levels and PKA activities, and
subsequently activated AMPK to inhibit LPS-induced oxidative
stress, inflammation and pulmonary dysfunction in ALI mice.
Taken together, the present results demonstrated
that miR-351-5p aggravated LPS-induced ALI via inhibiting AMPK and
that targeting miR-351-5p may facilitate the development of
efficient therapeutic approaches for treating ALI.
Supplementary Material
Supporting Data
Acknowledgements
Not applicable.
Funding
No funding was received.
Availability of data and materials
All data generated or analyzed during this study are
included in this published article.
Authors' contributions
FH, XFD, WXL and RYL designed the experiments; FH,
XFD, WXL, JFL and FL performed the experiments; JFL, FL, GS and GQH
analyzed the experimental results and interpreted the data; FH, XFD
and RYL wrote and revised the manuscript. All authors read and
approved the final manuscript.
Ethics approval and consent to
participate
All experimental procedures involving animals were
approved by the Animal Ethics Committee of Renmin Hospital of Wuhan
University.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Rubenfeld GD, Caldwell E, Peabody E,
Weaver J, Martin DP, Neff M, Stern EJ and Hudson LD: Incidence and
outcomes of acute lung injury. N Engl J Med. 353:1685–1693. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Liu X, Zheng X, Wang J, Zhang N, Leung KS,
Ye X and Cheng L: A long non-coding RNA signature for diagnostic
prediction of sepsis upon ICU admission. Clin Transl Med.
10:e1232020. View
Article : Google Scholar : PubMed/NCBI
|
|
3
|
Chen H, Li Y, Wu J, Li G, Tao X, Lai K,
Yuan Y, Zhang X, Zou Z and Xu Y: RIPK3 collaborates with GSDMD to
drive tissue injury in lethal polymicrobial sepsis. Cell Death
Differ. 27:2568–2585. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Yang HH, Duan JX, Liu SK, Xiong JB, Guan
XX, Zhong WJ, Sun CC, Zhang CY, Luo XQ, Zhang YF, et al: A
COX-2/sEH dual inhibitor PTUPB alleviates
lipopolysaccharide-induced acute lung injury in mice by inhibiting
NLRP3 inflammasome activation. Theranostics. 10:4749–4761. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Shah TG, Predescu D and Predescu S:
Mesenchymal stem cells-derived extracellular vesicles in acute
respiratory distress syndrome: A review of current literature and
potential future treatment options. Clin Transl Med. 8:252019.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Jiang J, Huang K, Xu S, Garcia J, Wang C
and Cai H: Targeting NOX4 alleviates sepsis-induced acute lung
injury via attenuation of redox-sensitive activation of
CaMKII/ERK1/2/MLCK and endothelial cell barrier dysfunction. Redox.
36:1016382020. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Huang XT, Liu W, Zhou Y, Sun M, Yang HH,
Zhang CY and Tang SY: Galectin-1 ameliorates
lipopolysaccharide-induced acute lung injury via AMPK-Nrf2 pathway
in mice. Free Radic Biol Med. 146:222–233. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Aggarwal S, Lazrak A, Ahmad I, Yu Z,
Bryant A, Mobley JA, Ford DA and Matalon S: Reactive species
generated by heme impair alveolar epithelial sodium channel
function in acute respiratory distress syndrome. Redox Biol.
36:1015922020. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Li Y, Cao Y, Xiao J, Shang J, Tan Q, Ping
F, Huang W, Wu F, Zhang H and Zhang X: Inhibitor of
apoptosis-stimulating protein of p53 inhibits ferroptosis and
alleviates intestinal ischemia/reperfusion-induced acute lung
injury. Cell Death Differ. 27:2635–2650. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Jia Y, Cui R, Wang C, Feng Y, Li Z, Tong
Y, Qu K, Liu C and Zhang J: Metformin protects against intestinal
ischemia-reperfusion injury and cell pyroptosis via
TXNIP-NLRP3-GSDMD pathway. Redox Biol. 32:1015342020. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Dai Y, Zhang J, Xiang J, Li Y, Wu D and Xu
J: Calcitriol inhibits ROS-NLRP3-IL-1beta signaling axis via
activation of Nrf2-antioxidant signaling in hyperosmotic stress
stimulated human corneal epithelial cells. Redox Biol.
21:1010932019. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Wang Z, Xu G, Gao Y, Zhan X, Qin N, Fu S,
Li R, Niu M, Wang J, Liu Y, et al: Cardamonin from a medicinal herb
protects against LPS-induced septic shock by suppressing NLRP3
inflammasome. Acta Pharm Sin B. 9:734–744. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Zhou X, Wu Y, Ye L, Wang Y, Zhang K, Wang
L, Huang Y, Wang L, Xian S, Zhang Y and Chen Y: Aspirin alleviates
endothelial gap junction dysfunction through inhibition of NLRP3
inflammasome activation in LPS-induced vascular injury. Acta Pharm
Sin B. 9:711–723. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Zhang X, Ma ZG, Yuan YP, Xu SC, Wei WY,
Song P, Kong CY, Deng W and Tang QZ: Rosmarinic acid attenuates
cardiac fibrosis following long-term pressure overload via
AMPKα/smad3 signaling. Cell Death Dis. 9:1022018. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Qi G, Zhou Y, Zhang X, Yu J, Li X, Cao X,
Wu C and Guo P: Cordycepin promotes browning of white adipose
tissue through an AMP-activated protein kinase (AMPK)-dependent
pathway. Acta Pharm Sin B. 9:135–143. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Rodríguez C, Contreras C, Sáenz-Medina J,
Muñoz M, Corbacho C, Carballido J, Garcia-Sacristán A, Hernandez M,
Lopez M, Rivera L and Prieto D: Activation of the AMP-related
kinase (AMPK) induces renal vasodilatation and downregulates
nox-derived reactive oxygen species (ROS) generation. Redox Biol.
34:1015752020. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Hu C, Zhang X, Wei W, Zhang N, Wu H, Ma Z,
Li L, Deng W and Tang Q: Matrine attenuates oxidative stress and
cardiomyocyte apoptosis in doxorubicin-induced cardiotoxicity via
maintaining AMPK α/UCP2 pathway. Acta Pharm Sin B. 9:690–701. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Zhao P, Wong KI, Sun X, Reilly SM, Uhm M,
Liao Z, Skorobogatko Y and Saltiel AR: TBK1 at the crossroads of
inflammation and energy homeostasis in adipose tissue. Cell.
172:731–743. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Jiang WL, Zhao KC, Yuan W, Zhou F, Song
HY, Liu GL, Huang J, Zou JJ, Zhao B and Xie SP: MicroRNA-31-5p
exacerbates lipopolysaccharide-induced acute lung injury via
inactivating cab39/AMP α pathway. Oxid Med Cell Longev.
2020:88223612020. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Warpsinski G, Smith MJ, Srivastava S,
Keeley TP, Siow R, Fraser PA and Mann GE: Nrf2-regulated redox
signaling in brain endothelial cells adapted to physiological
oxygen levels: Consequences for sulforaphane mediated protection
against hypoxia-reoxygenation. Redox Biol. 37:1017082020.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Deng S, Essandoh K, Wang X, Li Y, Huang W,
Chen J, Peng J, Jiang DS, Mu X, Wang C, et al: Tsg101 positively
regulates P62-Keap1-Nrf2 pathway to protect hearts against
oxidative damage. Redox Biol. 32:1014532020. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Zhang X, Hu C, Kong CY, Song P, Wu HM, Xu
SC, Yuan YP, Deng W, Ma ZG and Tang QZ: FNDC5 alleviates oxidative
stress and cardiomyocyte apoptosis in doxorubicin-induced
cardiotoxicity via activating AKT. Cell Death Differ. 27:540–555.
2020. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Yang L, Li X, Jiang A, Li X, Chang W, Chen
J and Ye F: Metformin alleviates lead-induced mitochondrial
fragmentation via AMPK/Nrf2 activation in SH-SY5Y cells. Redox
Biol. 36:1016262020. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Matzinger M, Fischhuber K, Pölöske D,
Mechtler K and Heiss EH: AMPK leads to phosphorylation of the
transcription factor Nrf2, tuning transactivation of selected
target genes. Redox Biol. 29:1013932020. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Carbonell T and Gomes AV: MicroRNAs in the
regulation of cellular redox status and its implications in
myocardial ischemia-reperfusion injury. Redox Biol. 36:1016072020.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Wang G, Yuan J, Cai X, Xu Z, Wang J,
Ocansey DK, Yan Y, Qian H, Zhang X, Xu W and Mao F: HucMSC-Exosomes
carrying miR-326 inhibit neddylation to relieve inflammatory bowel
disease in mice. Clin Transl Med. 10:e1132020. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Wang YM, Zheng YF, Yang SY, Yang ZM, Zhang
LN, He YQ, Gong XH, Liu D, Finnell RH, Qiu ZL, et al: MicroRNA-197
controls ADAM10 expression to mediate MeCP2′s role in the
differentiation of neuronal progenitors. Cell Death Differ.
26:1863–1879. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Wei X, Yi X, Lv H, Sui X, Lu P, Li L, An
Y, Yang Y, Yi H and Chen G: MicroRNA-377-3p released by mesenchymal
stem cell exosomes ameliorates lipopolysaccharide-induced acute
lung injury by targeting RPTOR to induce autophagy. Cell Death Dis.
11:6572020. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Xie SJ, Li JH, Chen HF, Tan YY, Liu SR,
Zhang Y, Xu H, Yang JH, Liu S, Zheng LL, et al: Inhibition of the
JNK/MAPK signaling pathway by myogenesis-associated miRNAs is
required for skeletal muscle development. Cell Death Differ.
25:1581–1597. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Hu Y, Tao X, Han X, Xu L, Yin L, Sun H, Qi
Y, Xu Y and Peng J: MicroRNA-351-5p aggravates intestinal
ischaemia/reperfusion injury through the targeting of MAPK13 and
sirtuin-6. Br J Pharmacol. 175:3594–3609. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Hu Y, Mao Z, Xu L, Yin L, Tao X, Tang Z,
Qi Y, Sun P and Peng J: Protective effect of dioscin against
intestinal ischemia/reperfusion injury via adjusting
miR-351-5p-mediated oxidative stress. Pharmacol Res. 137:56–63.
2018. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Zheng L, Han X, Hu Y, Zhao X, Yin L, Xu L,
Qi Y, Xu Y, Han X, Liu K and Peng J: Dioscin ameliorates intestinal
ischemia/reperfusion injury via adjusting
miR-351-5p/MAPK13-mediated inflammation and apoptosis. Pharmacol
Res. 139:431–439. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
da Silva W, dos Santos RA and Moraes KC:
Mir-351-5p contributes to the establishment of a pro-inflammatory
environment in the H9c2 cell line by repressing PTEN expression.
Mol Cell Biochem. 411:363–371. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Ma ZG, Dai J, Zhang WB, Yuan Y, Liao HH,
Zhang N, Bian ZY and Tang QZ: Protection against cardiac
hypertrophy by geniposide involves the GLP-1 receptor/AMPKα
signalling pathway. Br J Pharmacol. 173:1502–1516. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Wang Z, Yan J, Yang F, Wang D, Lu Y and
Liu L: MicroRNA-326 prevents sepsis-induced acute lung injury via
targeting TLR4. Free Radic Res. 54:408–418. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Hu C, Zhang X, Song P, Yuan YP, Kong CY,
Wu HM, Xu SC, Ma ZG and Tang QZ: Meteorin-Like protein attenuates
doxorubicin-induced cardiotoxicity via activating cAMP/PKA/SIRT1
pathway. Redox Biol. 37:1017472020. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Hu C, Zhang X, Zhang N, Wei WY, Li LL, Ma
ZG and Tang QZ: Osteocrin attenuates inflammation, oxidative
stress, apoptosis, and cardiac dysfunction in doxorubicin-induced
cardiotoxicity. Clin Transl Med. 10:e1242020. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Qi S, Guo L, Yan S, Lee RJ, Yu S and Chen
S: Hypocrellin A-based photodynamic action induces apoptosis in
A549 cells through ROS-mediated mitochondrial signaling pathway.
Acta Pharm Sin B. 9:279–293. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Pintado-Berninches L, Fernandez-Varas B,
Benitez-Buelga C, Manguan-Garcia C, Serrano-Benitez A, Iarriccio L,
Carrillo J, Guenechea G, Egusquiaguirre SP, Pedraz JL, et al: GSE4
peptide suppresses oxidative and telomere deficiencies in ataxia
telangiectasia patient cells. Cell Death Differ. 26:1998–2014.
2019. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Zhang X, Zhu JX, Ma ZG, Wu HM, Xu SC, Song
P, Kong CY, Yuan YP, Deng W and Tang QZ: Rosmarinic acid alleviates
cardiomyocyte apoptosis via cardiac fibroblast in
doxorubicin-induced cardiotoxicity. Int J Biol Sci. 15:556–567.
2019. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Zhou F, Mei J, Han X, Li H, Yang S, Wang
M, Chu L, Qiao H and Tang T: Kinsenoside attenuates osteoarthritis
by repolarizing macrophages through inactivating NF-κB/MAPK
signaling and protecting chondrocytes. Acta Pharm Sin B. 9:973–985.
2019. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Ou T, Yang W, Li W, Lu Y, Dong Z, Zhu H,
Sun X, Dong Z, Weng X, Chang S, et al: SIRT5 deficiency enhances
the proliferative and therapeutic capacities of adipose-derived
mesenchymal stem cells via metabolic switching. Clin Transl Med.
10:e1722020. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Zhang X, Hu C, Zhang N, Wei WY, Li LL, Wu
HM, Ma ZG and Tang QZ: Matrine attenuates pathological cardiac
fibrosis via RPS5/p38 in mice. Acta Pharmacol Sin. 42:573–584.
2020. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Yi W, Tu MJ, Liu Z, Zhang C, Batra N, Yu
AX and Yu AM: Bioengineered miR-328-3p modulates GLUT1-mediated
glucose uptake and metabolism to exert synergistic
antiproliferative effects with chemotherapeutics. Acta Pharm Sin B.
10:159–170. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Wu L, Xiang S, Hu X, Mo M, Zhao C, Cai Y,
Tong S, Jiang H, Chen L, Wang Z, et al: Prostate-specific antigen
modulates the osteogenic differentiation of MSCs via the cadherin
11-akt axis. Clin Transl Med. 10:363–373. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Zhang X, Hu C, Yuan YP, Song P, Kong CY,
Wu HM, Xu SC, Ma ZG and Tang QZ: Endothelial ERG alleviates cardiac
fibrosis via blocking endothelin-1-dependent paracrine mechanism.
Cell Biol Toxicol. 20:doi:10.1007. 2021.PubMed/NCBI
|
|
47
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Wang Y, Liu X, Shi H, Yu Y, Yu Y, Li M and
Chen R: NLRP3 inflammasome, an immune-inflammatory target in
pathogenesis and treatment of cardiovascular diseases. Clin Transl
Med. 10:91–106. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Agarwal V, Bell GW, Nam JW and Bartel DP:
Predicting effective microRNA target sites in mammalian mRNAs.
Elife. 4:e050052015. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Birrell MA, Bonvini SJ, Wortley MA,
Buckley J, Yew-Booth L, Maher SA, Dale N, Dubuis ED and Belvisi MG:
The role of adenylyl cyclase isoform 6 in beta-adrenoceptor
signalling in murine airways. Br J Pharmacol. 172:131–141. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Sayner SL, Balczon R, Frank DW, Cooper DM
and Stevens T: Filamin A is a phosphorylation target of membrane
but not cytosolic adenylyl cyclase activity. Am J Physiol Lung Cell
Mol Physiol. 301:L117–L124. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Schappe MS, Szteyn K, Stremska ME, Mendu
SK, Downs TK, Seegren PV, Mahoney MA, Dixit S, Krupa JK, Stipes EJ,
et al: Chanzyme TRPM7 mediates the Ca2+ influx essential
for lipopolysaccharide-induced toll-like receptor 4 endocytosis and
macrophage activation. Immunity. 48:59–74. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Tang T, Hammond HK, Firth A, Yang Y, Gao
MH, Yuan JX and Lai NC: Adenylyl cyclase 6 improves calcium uptake
and left ventricular function in aged hearts. J Am Coll Cardiol.
57:1846–1855. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Han F, Hou N, Liu Y, Huang N, Pan R, Zhang
X, Mao E and Sun X: Liraglutide improves vascular dysfunction by
regulating a cAMP-independent PKA-AMPK pathway in perivascular
adipose tissue in obese mice. Biomed Pharmacother. 120:1095372019.
View Article : Google Scholar : PubMed/NCBI
|