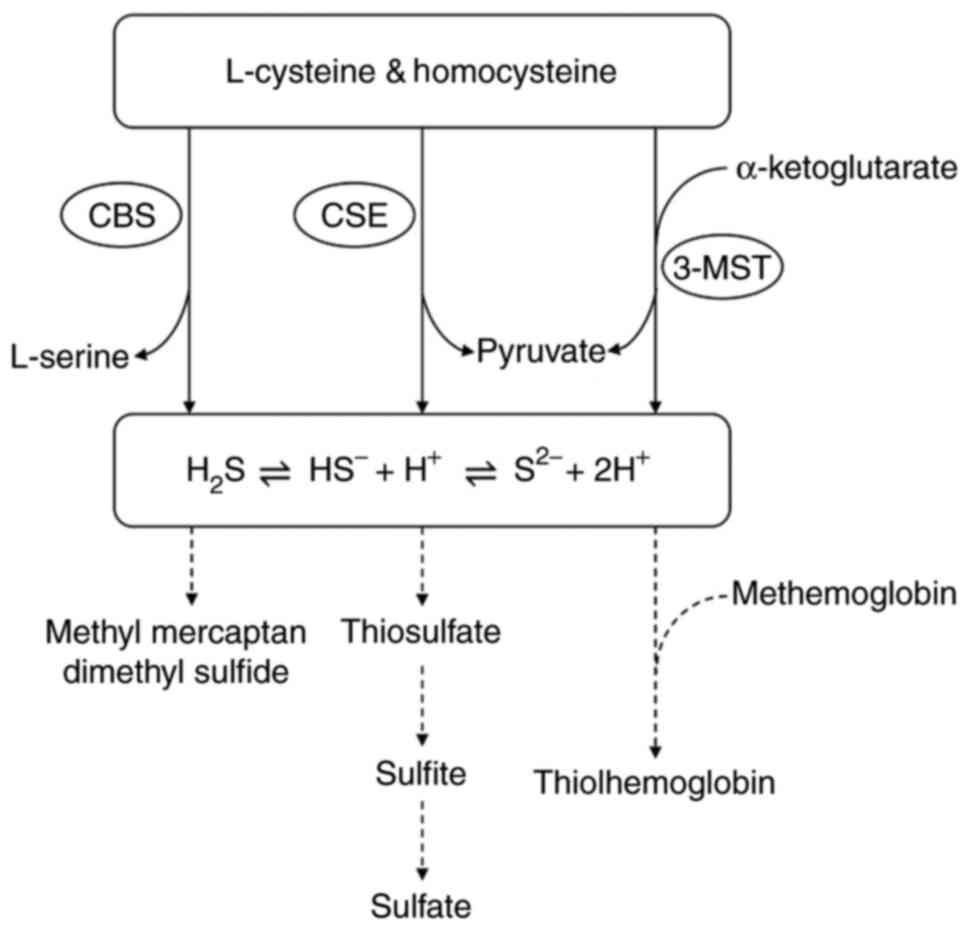
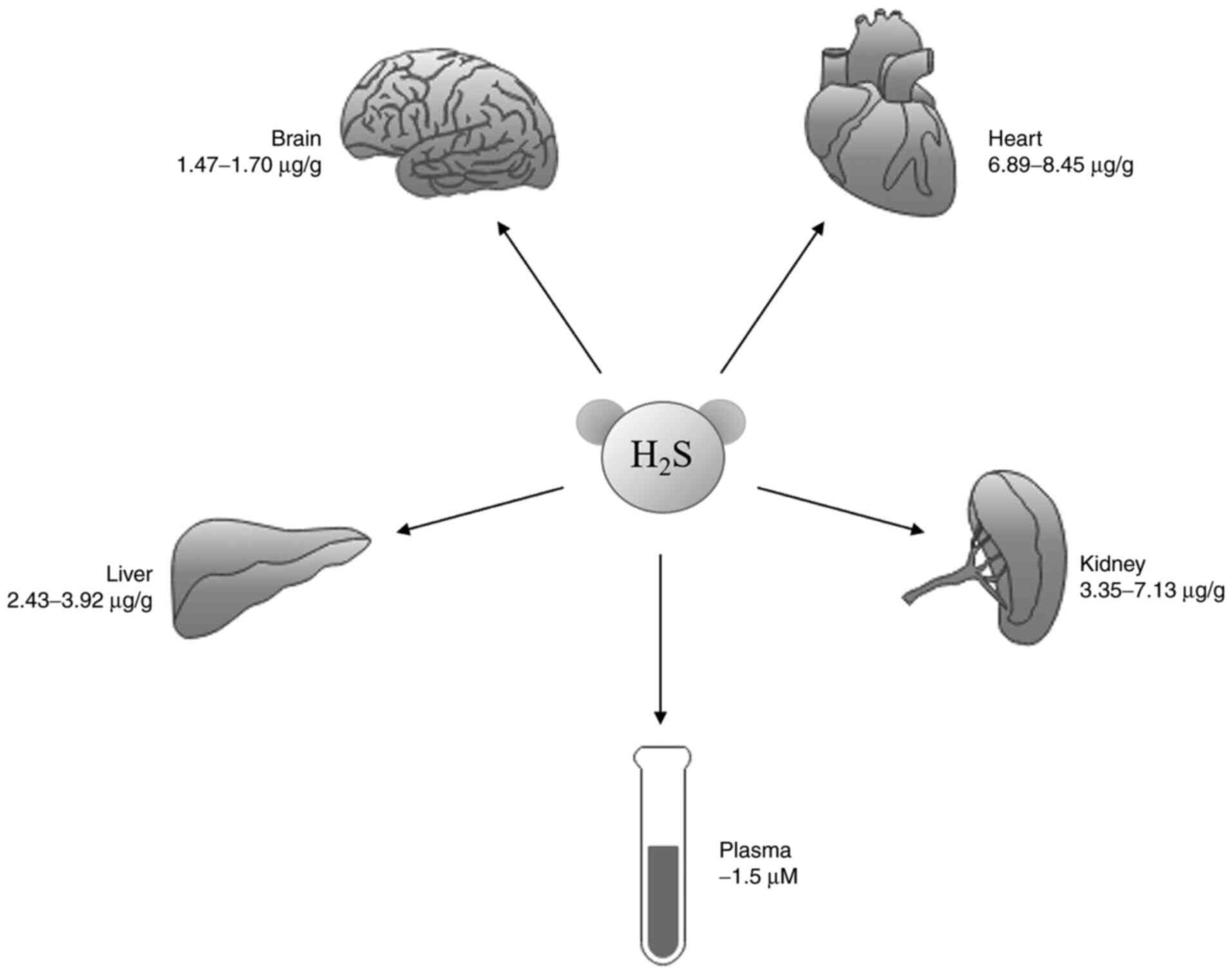
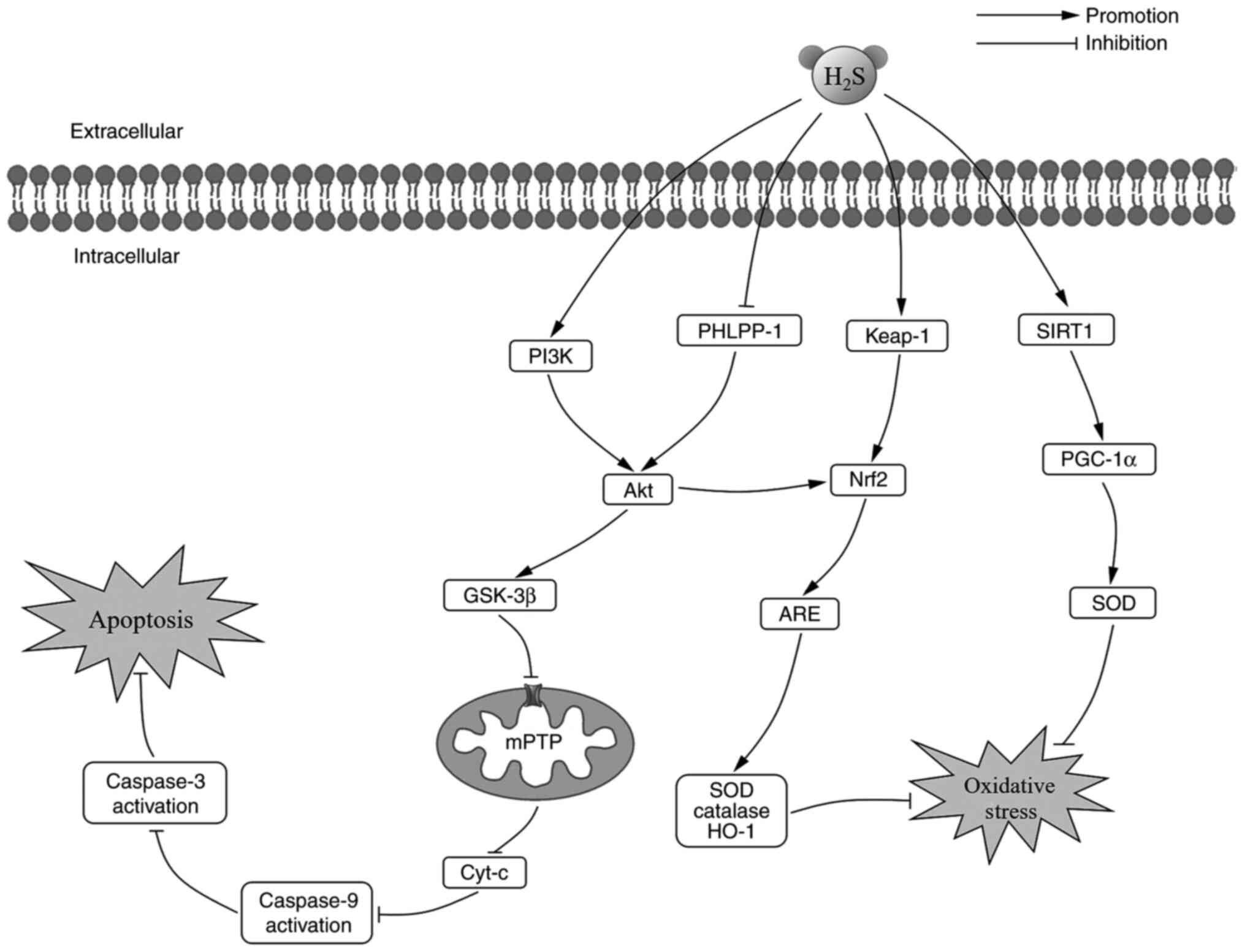
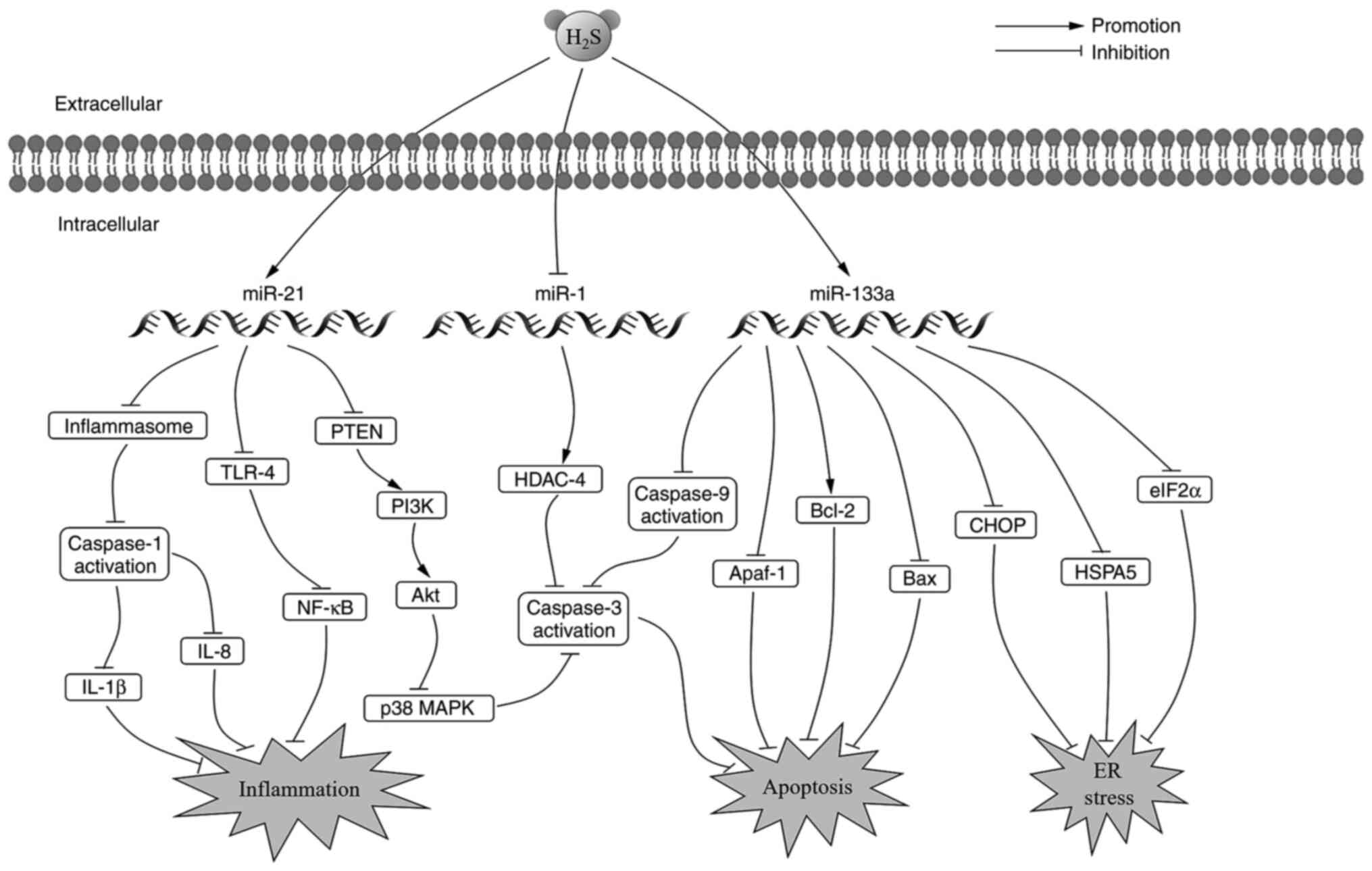
Cardiovascular diseases (CVD) contribute to a high
morbidity and mortality burden globally (1). In 2019, the number of patients with
CVD was ~523 and ~18.6 million cases succumbed to CVD (2). Myocardial ischemia is a common
clinical symptom resulting from atherosclerosis and myocardial
infarction (3). Reperfusion is
often used to repair myocardial structure damage and improve
cardiac function following ischemia. However, reperfusion may also
result in myocardial ischemia-reperfusion injury (MIRI), which
aggravates cardiac dysfunction. Therapeutic strategies, such as
preconditioning, postconditioning and administration of
antiplatelet or antithrombotic agents, have been utilized to
alleviate MIRI (4).
Oxygen homeostasis plays a vital role in the
maintenance of physiological functions. Reactive oxygen species
(ROS) are generated during the normal metabolism of oxygen and
participate in signal transduction. ROS are then scavenged by
various endogenous free radical scavenging enzymes, such as
superoxide dismutase (SOD), catalase, glutathione peroxidase and
thioredoxin (9). However,
overproduction of ROS or insufficient enzyme activity may impair
the equilibrium between ROS and antioxidants, resulting in damage
to proteins, DNA and lipids (10). SOD1 knockout mice were shown to
have excessive oxidative stress and aggravated myocardial injuries
following acute myocardial ischemia (11). Moreover, excessive ROS impairs
heart contraction by modifying excitation-contraction coupling
proteins. Excessive ROS also activates various signaling kinases
and transcription factors associated with myocardial hypertrophy.
In addition, the proliferation of cardiac fibroblast and the
activity of MMP are promoted by ROS (12,13).
The mitochondria are the main source of ROS
production. ROS are generated in the electron transport chain (ETC)
located on the mitochondrial membrane during the process of ATP
production, namely oxidative phosphorylation. Electrons are then
transported by a train of proteins known as the mitochondrial
complex via oxidation-reduction reactions and combine with oxygen
molecules to produce water. During this process, some oxygen
molecules are reduced to form ROS (14).
Mitochondria may also act as a target of ROS damage.
During the early process of reperfusion, the excessive ROS
generated may induce oxidative stress, leading to the abnormal
opening of the mitochondrial permeability transition pore (mPTP).
Opening of the mPTP leads to mitochondrial Ca2+
overload, usually accompanied by oxidative or nitrosative stress
and ATP depletion. Abnormal opening of mPTP also causes loss of
mitochondrial membrane potential (15), respiratory chain uncoupling and
impaired ATP synthesis. The impaired mitochondrial function results
in mitochondrial swelling, rupture and cell apoptosis or necrosis
(16,17). Mitochondria morphological changes
observed during a MIRI in rat myocardial tissues mainly manifest as
mitochondrial cristae and membrane damage, disordered fiber
arrangement and larger perinuclear space (18). Furthermore, inhibition of mPTP
opening using pharmaceutical agents, such as cyclosporine A, has
been shown to reduce myocardial infarct size in acute
ischemia-reperfusion injury (IRI) animals (19).
Autophagy plays a key role in cell survival by
transferring damaged proteins and organelles to lysosomes for
degradation. However, the autophagy process is controversial in
MIRI. Autophagy is activated via the AMP-activated protein kinase
pathway during ischemia to promote cell survival. However, during
reperfusion, autophagy exerts a harmful role via Beclin activation
(20). Loos et al
(21) observed the activation of
autophagy in mild ischemia. However, severe ischemia did not
activate autophagy. This demonstrates that autophagy induction is
closely associated with the degree of MIRI.
Researchers have also synthesized derivatives of
naturally occurring sulfur-containing organic compounds, such as
S-propargyl-cysteine, S-allycysteine and diallyl sulfide, to
improve the effectiveness of the H2S donors. In contrast
to conventional H2S donors that release H2S
directly, Allium sativum extract derivatives increase the
levels of H2S by increasing the expression and activity
of CSE and CBS. This is advantageous as the levels of
H2S are controlled and, thus, have a lower risk of
toxicity.
NaHS (10 µmol/l) postconditioning was revealed to
decrease the myocardial infarct size of isolated rat hearts and
inhibit oxidative stress by stimulating SOD activity and reducing
malondialdehyde levels via the activation of the
sirtuin1/peroxisome proliferator-activated receptor-γ
coactivator-1α pathway in an ex vivo study (73).
On the contrary, AP39 exhibited antioxidative
effects via ROS generation rather than scavenging. The alleviation
of myocardial infarction induced by AP39 during MIRI partly arose
from reduced production of ROS in interfibrillar and subsarcolemmal
mitochondria of cardiomyocytes, which were dose-dependent (Fig. 3) (74).
The cardioprotective effects mediated by exogenous
NaHS depend on mitochondrial ETC enzymes. Hemodynamic parameters
and mitochondrial ETC functional assessment revealed that the
cardioprotective effects of H2S require active
mitochondria (75). Following
MIRI, mouse hearts showed mitochondrial swelling, disorganized
cristae and lower matrix density. However, treatment with
H2S during reperfusion resulted in significantly
improved mitochondrial structure, stimulated mitochondrial
respiration and oxygen consumption (76). Karwi et al (74) reported that AP39 inhibited ROS
generation and mPTP opening during MIRI. However, inhibition of the
PI3K/Akt pathway, endothelial nitric oxide (NO) synthase (eNOS) or
soluble guanylyl cyclase did not reverse the protective effects of
AP39. Further research is required to investigate the association
of these effects to post-translation modifications mediated by
H2S and the interaction with NO in mitochondria.
Kelch-like ECH-associated protein-1
(Keap-1)/Nrf2/antioxidant response elements (ARE) pathway is a
primary pathway involved in the cellular defense against oxidative
stress. In response to oxidative stress, H2S dissociates
Nrf2 from Keap1 (81). During
early preconditioning, H2S promotes the nuclear
translocalization of Nrf2 and increases the phosphorylation of
protein kinase C epsilon and STAT-3. Moreover, H2S
increases the expression of heme oxygenase-1 and thioredoxin 1
during late preconditioning (82). As a result of Nrf2 nuclear
translocation, ARE is activated and enhances the transcription of
SOD, catalase and heme oxygenase-1 (83). PH domain leucine-rich repeat
protein phosphatase-1 (PHLPP-1) has recently been shown to
dephosphorylate Akt at Ser473, which increases infarct size and
aggravates MIRI (84,85). During MIRI, the levels of cardiac
malondialdehyde are increased, while the expression levels of SOD
and heme oxygenase-1 are downregulated. Pretreatment with GYY4137
was shown to reverse the oxidative stress induced by MIRI. GYY4137
also increased the protein expression levels of Akt and Nrf2 by
downregulating the level of PHLPP-1. Thus, the antioxidant effect
of H2S in MIRI partly depended on the PHLPP-1/Akt/Nrf2
pathway (41). PI3K, an upstream
factor of Akt, is considered an important molecule in the
underlying mechanism of H2S protection against
ischemia-reperfusion. The PI3K/Akt/Nrf2 pathway has been reported
to play a major role in alleviating cerebral ischemia-reperfusion
injury (86). However, to the
best of our knowledge, this mechanism has not been reported in the
cardiovascular system.
Endoplasmic reticulum (ER) stress is activated to
protect cells when they are exposed to hypoxia. However, sustained
activation of ER stress causes apoptosis (93). Ren et al (94) reported that the expression levels
of ER stress biomarkers, heat shock protein family A (Hsp70) member
5, CHOP and eukaryotic initiation factor-2α, were significantly
increased during ischemia/reperfusion. However, in vitro and
in vivo results revealed that pretreatment with
H2S alleviated ER stress and subsequent apoptosis via
the miR-133a signaling pathway by reversing the cardiomyocyte
trauma induced by MIRI. The combination of H2S
intervention and miR-133a overexpression notably increased the
proliferation, migration and invasion of cardiomyocytes. miR-133a
was also observed to promote anti-apoptotic protein Bcl-2
expression and inhibit pro-apoptotic protein Bax, caspase-3,
caspase-9 and apoptotic peptidase activating factor-1 expression.
Consequently, decreasing apoptosis in the cardiomyocytes (95,96).
In recent years, increased attention has been paid
to S-sulfhydration, a post-translational modification between
H2S and cysteine residues of proteins that modifies the
structure and biological activities of protein targets (99). Pharmacological postconditioning
performed at the onset of reperfusion with NaHS significantly
increased S-nitrosylation of cardioprotective proteins, as well as
reduced post-ischemic contractile dysfunction and infarct size
(100). However, the
S-sulfhydration of proteins in MIRI has not been fully studied.
H2S was reported to S-sulfhydrate Keap1 in response to
oxidative stress, thereby mediating the dissociation of Nrf2 from
Keap1, and as a result, promoting Nrf2 translocation in sulfur
mustard-induced lung injury (81). A similar mechanism was confirmed
in diabetic mice, wherein, H2S attenuated
diabetes-accelerated atherosclerosis by S-sulfhydrating Keap1 at
Cys151, resulting in activation of Nrf2 signaling (51). These mechanisms may contribute to
the potential role of H2S in MIRI.
Not applicable.
Sponsored by Shanghai Pujiang Program (grant no.
2020PJD055) and Shanghai Key Specialty Construction Project of
Clinical Pharmacy (2018).
Data sharing not applicable to this article as no
datasets were generated or analyzed during the current study.
YG wrote the original manuscript and the prepared
figures and tables. DW modified the manuscript according to the
reviewers and editors' comments. DZ contributed to the revision of
the article. All authors have read and approved the final
manuscript. Data sharing not applicable.
Not applicable.
Not applicable.
The authors declare that they have no competing
interests.
|
1
|
Townsend N, Wilson L, Bhatnagar P,
Wickramasinghe K, Rayner M and Nichols M: Cardiovascular disease in
Europe: Epidemiological update 2016. Eur Heart J. 37:3232–3245.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Roth GA, Mensah GA, Johnson CO, Addolorato
G, Ammirati E, Baddour LM, Barengo NC, Beaton AZ, Benjamin EJ,
Benziger CP, et al: Global burden of cardiovascular diseases and
risk factors, 1990–2019: Update from the GBD 2019 Study. J Am Coll
Cardiol. 76:2982–3021. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Wu MY, Yiang GT, Liao WT, Tsai AP, Cheng
YL, Cheng PW, Li CY and Li CJ: Current mechanistic concepts in
ischemia and reperfusion injury. Cell Physiol Biochem.
46:1650–1667. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Ibáñez B, Heusch G, Ovize M and Van de
Werf F: Evolving therapies for myocardial ischemia/reperfusion
injury. J Am Coll Cardiol. 65:1454–1471. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Gorini F, Bustaffa E, Chatzianagnostou K,
Bianchi F and Vassalle C: Hydrogen sulfide and cardiovascular
disease: Doubts, clues, and interpretation difficulties from
studies in geothermal areas. Sci Total Environ. 743:1408182020.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Wu D, Hu Q, Tan B, Rose P, Zhu D and Zhu
YZ: Amelioration of mitochondrial dysfunction in heart failure
through S-sulfhydration of Ca2+/calmodulin-dependent
protein kinase II. Redox Biol. 19:250–262. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Wang ZJ, Wu J, Guo W and Zhu YZ:
Atherosclerosis and the hydrogen sulfide signaling
pathway-therapeutic approaches to disease prevention. Cell Physiol
Biochem. 42:859–875. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Donnarumma E, Trivedi RK and Lefer DJ:
Protective actions of H2S in acute myocardial infarction and heart
failure. Compr Physiol. 7:583–602. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Xu T, Ding W, Ji X, Ao X, Liu Y, Yu W and
Wang J: Oxidative stress in cell death and cardiovascular diseases.
Oxid Med Cell Longev. 2019:90305632019. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Zorov DB, Juhaszova M and Sollott SJ:
Mitochondrial reactive oxygen species (ROS) and ROS-induced ROS
release. Physiol Rev. 94:909–950. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Bai YD, Yang YR, Mu XP, Lin G, Wang YP,
Jin S, Chen Y, Wang MJ and Zhu YC: Hydrogen sulfide alleviates
acute myocardial ischemia injury by modulating autophagy and
inflammation response under oxidative stress. Oxid Med Cell Longev.
2018:34028092018. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Tsutsui H, Kinugawa S and Matsushima S:
Oxidative stress and heart failure. Am J Physiol Heart Circ
Physiol. 301:H2181–H2190. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
van der Pol A, van Gilst WH, Voors AA and
van der Meer P: Treating oxidative stress in heart failure: Past,
present and future. Eur J Heart Fail. 21:425–435. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Li X, Fang P, Mai J, Choi ET, Wang H and
Yang XF: Targeting mitochondrial reactive oxygen species as novel
therapy for inflammatory diseases and cancers. J Hematol Oncol.
6:192013. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Briston T, Selwood DL, Szabadkai G and
Duchen MR: Mitochondrial permeability transition: A molecular
lesion with multiple drug targets. Trends Pharmacol Sci. 40:50–70.
2019. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Bauer TM and Murphy E: Role of
mitochondrial calcium and the permeability transition pore in
regulating cell death. Circ Res. 126:280–293. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Kwong JQ and Molkentin JD: Physiological
and pathological roles of the mitochondrial permeability transition
pore in the heart. Cell Metab. 21:206–214. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Li HW and Xiao FY: Effect of hydrogen
sulfide on cardiomyocyte apoptosis in rats with myocardial
ischemia-reperfusion injury via the JNK signaling pathway. Eur Rev
Med Pharmacol Sci. 24:2054–2061. 2020.PubMed/NCBI
|
|
19
|
Ong S, Samangouei P, Kalkhoran SB and
Hausenloy DJ: The mitochondrial permeability transition pore and
its role in myocardial ischemia reperfusion injury. J Mol Cell
Cardiol. 78:23–34. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Matsui Y, Takagi H, Qu X, Abdellatif M,
Sakoda H, Asano T, Levine B and Sadoshima J: Distinct roles of
autophagy in the heart during ischemia and reperfusion: Roles of
AMP-activated protein kinase and Beclin 1 in mediating autophagy.
Circ Res. 100:914–922. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Loos B, Genade S, Ellis B, Lochner A and
Engelbrecht AM: At the core of survival: Autophagy delays the onset
of both apoptotic and necrotic cell death in a model of ischemic
cell injury. Exp Cell Res. 317:1437–1453. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Hausenloy DJ and Yellon DM: New directions
for protecting the heart against ischaemia-reperfusion injury:
Targeting the Reperfusion Injury Salvage Kinase (RISK)-pathway.
Cardiovasc Res. 61:448–460. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Hausenloy DJ and Yellon DM: Reperfusion
injury salvage kinase signalling: Taking a RISK for
cardioprotection. Heart Fail Rev. 12:217–234. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Peake BF, Nicholson CK, Lambert JP, Hood
RL, Amin H, Amin S and Calvert JW: Hydrogen sulfide preconditions
the db/db diabetic mouse heart against ischemia-reperfusion injury
by activating Nrf2 signaling in an Erk-dependent manner. Am J
Physiol Heart Circ Physiol. 304:H1215–H1224. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Cao X, Ding L, Xie ZZ, Yang Y, Whiteman M,
Moore PK and Bian JS: A review of hydrogen sulfide synthesis,
metabolism, and measurement: Is modulation of hydrogen sulfide a
novel therapeutic for cancer? Antioxid Redox Signal. 31:1–38. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Bełtowski J and Jamroz-Wiśniewska A:
Hydrogen sulfide and endothelium-dependent vasorelaxation.
Molecules. 19:21183–21199. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Panthi S, Chung HJ, Jung J and Jeong NY:
Physiological importance of hydrogen sulfide: Emerging potent
neuroprotector and neuromodulator. Oxid Med Cell Longev.
2016:90497822016. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Wilinski B, Wilinski J, Somogyi E,
Goralska M and Piotrowska J: Paracetamol (acetaminophen) decreases
hydrogen sulfide tissue concentration in brain but increases it in
the heart, liver and kidney in mice. Folia Biol (Krakow). 59:41–44.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Wilinski B, Wilinski J, Somogyi E,
Goralska M and Piotrowska J: Ramipril affects hydrogen sulfide
generation in mouse liver and kidney. Folia Biol (Krakow).
58:177–180. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Wilinski J, Wilinski B, Somogyi E,
Piotrowska J, Kameczura T and Zygmunt M: Nicotine affects hydrogen
sulfide concentrations in mouse kidney and heart but not in brain
and liver tissues. Folia Med Cracov. 57:55–64. 2017.PubMed/NCBI
|
|
31
|
Tan B, Jin S, Sun J, Gu Z, Sun X, Zhu Y,
Huo K, Cao Z, Yang P, Xin X, et al: New method for quantification
of gasotransmitter hydrogen sulfide in biological matrices by
LC-MS/MS. Sci Rep. 7:462782017. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Wu D, Hu Q and Zhu YZ; Therapeutic
application of hydrogen sulfide donors, : The potential and
challenges. Front Med. 10:18–27. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Pei J, Wang F, Pei S, Bai R, Cong X, Nie Y
and Chen X: Hydrogen sulfide promotes cardiomyocyte proliferation
and heart regeneration via ROS scavenging. Oxid Med Cell Longev.
2020:14126962020. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Feng A, Ling C, Xin-duo L, Bing W, San-Wu
W, Yu Z, Yu-Lan H and You-En Z: Hydrogen sulfide protects human
cardiac fibroblasts against H2O2-induced
injury through regulating autophagy-related proteins. Cell
Transplant. 27:1222–1234. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Huang Z, Dong X, Zhuang X, Hu X, Wang L
and Liao X: Exogenous hydrogen sulfide protects against high
glucose-induced inflammation and cytotoxicity in H9c2 cardiac
cells. Mol Med Rep. 14:4911–4917. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Yuan C, Hou HT, Chen HX, Wang J, Wang ZQ,
Chen TN, Novakovic A, Marinko M, Yang Q, Liu ZG, et al: Hydrogen
sulfide-mediated endothelial function and the interaction with eNOS
and PDE5A activity in human internal mammary arteries. J Int Med
Res. 47:3778–3791. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Wang GG and Li W: Hydrogen sulfide
improves vessel formation of the ischemic adductor muscle and wound
healing in diabetic db/db mice. Iran J Basic Med Sci. 22:1192–1197.
2019.PubMed/NCBI
|
|
38
|
Wang CN, Liu YJ, Duan GL, Zhao W, Li XH,
Zhu XY and Ni X: CBS and CSE are critical for maintenance of
mitochondrial function and glucocorticoid production in adrenal
cortex. Antioxid Redox Signal. 21:2192–2207. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Li L, Whiteman M, Guan YY, Neo KL, Cheng
Y, Lee SW, Zhao Y, Baskar R, Tan CH and Moore PK: Characterization
of a novel, water-soluble hydrogen sulfide-releasing molecule
(GYY4137): New insights into the biology of hydrogen sulfide.
Circulation. 117:2351–2360. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Castelblanco M, Lugrin J, Ehirchiou D,
Nasi S, Ishii I, So A, Martinon F and Busso N: Hydrogen sulfide
inhibits NLRP3 inflammasome activation and reduces cytokine
production both in vitro and in a mouse model of inflammation. J
Biol Chem. 293:2546–2557. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Qiu Y, Wu Y, Meng M, Luo M, Zhao H, Sun H
and Gao S: GYY4137 protects against myocardial ischemia/reperfusion
injury via activation of the PHLPP-1/Akt/Nrf2 signaling pathway in
diabetic mice. J Surg Res. 225:29–39. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Yang H, Mao Y, Tan B, Luo S and Zhu Y: The
protective effects of endogenous hydrogen sulfide modulator,
S-propargyl-cysteine, on high glucose-induced apoptosis in
cardiomyocytes: A novel mechanism mediated by the activation of
Nrf2. Eur J Pharmacol. 761:135–143. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Qian X, Li X, Ma F, Luo S, Ge R and Zhu Y:
Novel hydrogen sulfide-releasing compound, S-propargyl-cysteine,
prevents STZ-induced diabetic nephropathy. Biochem Biophys Res
Commun. 473:931–938. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Kan J, Guo W, Huang C, Bao G, Zhu Y and
Zhu YZ: S-propargyl-cysteine, a novel water-soluble modulator of
endogenous hydrogen sulfide, promotes angiogenesis through
activation of signal transducer and activator of transcription 3.
Antioxid Redox Signal. 20:2303–2316. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Zhao FL, Fang F, Qiao PF, Yan N, Gao D and
Yan Y: AP39, a mitochondria-targeted hydrogen sulfide donor,
supports cellular bioenergetics and protects against Alzheimer's
disease by preserving mitochondrial function in APP/PS1 mice and
neurons. Oxid Med Cell Longev. 2016:83607382016. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Szczesny B, Módis K, Yanagi K, Coletta C,
Le Trionnaire S, Perry A, Wood ME, Whiteman M and Szabo C: AP39, a
novel mitochondria-targeted hydrogen sulfide donor, stimulates
cellular bioenergetics, exerts cytoprotective effects and protects
against the loss of mitochondrial DNA integrity in oxidatively
stressed endothelial cells in vitro. Nitric Oxide. 41:120–130.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Chao C, Zatarain JR, Ding Y, Coletta C,
Mrazek AA, Druzhyna N, Johnson P, Chen H, Hellmich JL,
Asimakopoulou A, et al: Cystathionine-beta-synthase inhibition for
colon cancer: Enhancement of the efficacy of aminooxyacetic acid
via the prodrug approach. Mol Med. 22:361–379. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Lilyanna S, Peh MT, Liew OW, Wang P, Moore
PK, Richards AM and Martinez EC: GYY4137 attenuates remodeling,
preserves cardiac function and modulates the natriuretic peptide
response to ischemia. J Mol Cell Cardiol. 87:27–37. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Zhou X, Tang S, Hu K, Zhang Z, Liu P, Luo
Y, Kang J and Xu L: DL-Propargylglycine protects against myocardial
injury induced by chronic intermittent hypoxia through inhibition
of endoplasmic reticulum stress. Sleep Breath. 22:853–863. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Szabo C, Ransy C, Modis K, Andriamihaja M,
Murghes B, Coletta C, Olah G, Yanagi K and Bouillaud F: Regulation
of mitochondrial bioenergetic function by hydrogen sulfide. Part I.
Biochemical and physiological mechanisms. Br J Pharmacol.
171:2099–2122. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Xie L, Gu Y, Wen M, Zhao S, Wang W, Ma Y,
Meng G, Han Y, Wang Y, Liu G, et al: Hydrogen sulfide induces Keap1
S-sulfhydration and suppresses diabetes-accelerated atherosclerosis
via Nrf2 activation. Diabetes. 65:3171–3184. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Meng G, Liu J, Liu S, Song Q, Liu L, Xie
L, Han Y and Ji Y: Hydrogen sulfide pretreatment improves
mitochondrial function in myocardial hypertrophy via a
SIRT3-dependent manner. Br J Pharmacol. 175:1126–1145. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Sun Y, Lu F, Yu X, Wang B, Chen J, Lu F,
Peng S, Sun X, Yu M, Chen H, et al: Exogenous H2S
promoted USP8 sulfhydration to regulate mitophagy in the hearts of
db/db mice. Aging Dis. 11:269–285. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Yu M, Du H, Wang B, Chen J, Lu F, Peng S,
Sun Y, Liu N, Sun X, Shiyun D, et al: Exogenous H2S
induces Hrd1 S-sulfhydration and prevents CD36 translocation via
VAMP3 ubiquitylation in diabetic hearts. Aging Dis. 11:286–300.
2020. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Kar S, Shahshahan HR, Hackfort BT, Yadav
SK, Yadav R, Kambis TN, Lefer DJ and Mishra PK: Exercise training
promotes cardiac hydrogen sulfide biosynthesis and mitigates
pyroptosis to prevent high-fat diet-induced diabetic
cardiomyopathy. Antioxidants. 8:6382019. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Shimizu Y, Polavarapu R, Eskla KL,
Nicholson CK, Koczor CA, Wang R, Lewis W, Shiva S, Lefer DJ and
Calvert JW: Hydrogen sulfide regulates cardiac mitochondrial
biogenesis via the activation of AMPK. J Mol Cell Cardiol.
116:29–40. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Wu T, Li H, Wu B, Zhang L, Wu SW, Wang JN
and Zhang YE: Hydrogen sulfide reduces recruitment of
CD11b+Gr-1+ cells in mice with myocardial
infarction. Cell Transplant. 26:753–764. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Ye P, Gu Y, Zhu YR, Chao YL, Kong XQ, Luo
J, Ren XM, Zuo GF, Zhang DM and Chen SL: Exogenous hydrogen sulfide
attenuates the development of diabetic cardiomyopathy via the FoxO1
pathway. J Cell Physiol. 233:9786–9798. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Ellmers LJ, Templeton EM, Pilbrow AP,
Frampton C, Ishii I, Moore PK, Bhatia M, Richards AM and Cameron
VA: Hydrogen sulfide treatment improves post-infarct remodeling and
long-term cardiac function in CSE knockout and wild-type mice. Int
J Mol Sci. 21:42842020. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Sun X, Zhao D, Lu F, Peng S, Yu M, Liu N,
Sun Y, Du H, Wang B, Chen J, et al: Hydrogen sulfide regulates
muscle RING finger-1 protein S-sulfhydration at Cys44 to
prevent cardiac structural damage in diabetic cardiomyopathy. Br J
Pharmacol. 177:836–856. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Meng G, Xiao Y, Ma Y, Tang X, Xie L, Liu
J, Gu Y, Yu Y, Park CM, Xian M, et al: Hydrogen sulfide regulates
krüppel-like factor 5 transcription activity via specificity
protein 1 s-sulfhydration at Cys664 to prevent myocardial
hypertrophy. J Am Heart Assoc. 5:e0041602016. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Yu W, Liao Y, Huang Y, Chen SY, Sun Y, Sun
C, Wu Y, Tang C, Du J and Jin H: Endogenous hydrogen sulfide
enhances carotid sinus baroreceptor sensitivity by activating the
transient receptor potential cation channel subfamily V Member 1
(TRPV1) Channel. J Am Heart Assoc. 6:e0049712017. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Jin S, Teng X, Xiao L, Xue H, Guo Q, Duan
X, Chen Y and Wu Y: Hydrogen sulfide ameliorated L-NAME-induced
hypertensive heart disease by the Akt/eNOS/NO pathway. Exp Biol Med
(Maywood). 242:1831–1841. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Meng G, Zhu J, Xiao Y, Huang Z, Zhang Y,
Tang X, Xie L, Chen Y, Shao Y, Ferro A, et al: Hydrogen sulfide
donor GYY4137 protects against myocardial fibrosis. Oxid Med Cell
Longev. 2015:6910702015. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Huang C, Kan J, Liu X, Ma F, Tran BH, Zou
Y, Wang S and Zhu YZ: Cardioprotective effects of a novel hydrogen
sulfide agent-controlled release formulation of
S-propargyl-cysteine on heart failure rats and molecular
mechanisms. PLoS One. 8:e692052013. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Zhong X, Wang L, Wang Y, Dong S, Leng X,
Jia J, Zhao Y, Li H, Zhang X, Xu C, et al: Exogenous hydrogen
sulfide attenuates diabetic myocardial injury through cardiac
mitochondrial protection. Mol Cell Biochem. 371:187–198. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Sodha NR, Clements RT, Feng J, Liu Y,
Bianchi C, Horvath EM, Szabo C, Stahl GL and Sellke FW: Hydrogen
sulfide therapy attenuates the inflammatory response in a porcine
model of myocardial ischemia/reperfusion injury. J Thorac
Cardiovasc Surg. 138:977–984. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Meng G, Wang J, Xiao Y, Bai W, Xie L, Shan
L, Moore PK and Ji Y: GYY4137 protects against myocardial ischemia
and reperfusion injury by attenuating oxidative stress and
apoptosis in rats. J Biomed Res. 29:203–213. 2015.PubMed/NCBI
|
|
69
|
King AL, Polhemus DJ, Bhushan S, Otsuka H,
Kondo K, Nicholson CK, Bradley JM, Islam KN, Calvert JW, Tao YX, et
al: Hydrogen sulfide cytoprotective signaling is endothelial nitric
oxide synthase-nitric oxide dependent. Proc Natl Acad Sci USA.
111:3182–3187. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Karwi QG, Bice JS and Baxter GF: Pre- and
postconditioning the heart with hydrogen sulfide (H2S)
against ischemia/reperfusion injury in vivo: A systematic review
and meta-analysis. Basic Res Cardiol. 113:62018. View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Xiong Q, Wang Z, Yu Y, Wen Y, Suguro R,
Mao Y and Zhu YZ: Hydrogen sulfide stabilizes atherosclerotic
plaques in apolipoprotein E knockout mice. Pharmacol Res.
144:90–98. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
72
|
Sun X, Wang W, Dai J, Jin S, Huang J, Guo
C, Wang C, Pang L and Wang Y: A Long-term and slow-releasing
hydrogen sulfide donor protects against myocardial
ischemia/reperfusion injury. Sci Rep. 7:35412017. View Article : Google Scholar : PubMed/NCBI
|
|
73
|
Hu MZ, Zhou B, Mao HY, Sheng Q, Du B, Chen
JL, Pang QF and Ji Y: Exogenous hydrogen sulfide postconditioning
protects isolated rat hearts from ischemia/reperfusion injury
through Sirt1/PGC-1α signaling pathway. Int Heart J. 57:477–482.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
74
|
Karwi QG, Bornbaum J, Boengler K,
Torregrossa R, Whiteman M, Wood ME, Schulz R and Baxter GF: AP39, a
mitochondria-targeting hydrogen sulfide (H2 S) donor, protects
against myocardial reperfusion injury independently of salvage
kinase signalling. Br J Pharmacol. 174:287–301. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
75
|
Nandi S, Ravindran S and Kurian GA: Role
of endogenous hydrogen sulfide in cardiac mitochondrial
preservation during ischemia reperfusion injury. Biomed
Pharmacother. 97:271–279. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
76
|
Elrod JW, Calvert JW, Morrison J, Doeller
JE, Kraus DW, Tao L, Jiao X, Scalia R, Kiss L, Szabo C, et al:
Hydrogen sulfide attenuates myocardial ischemia-reperfusion injury
by preservation of mitochondrial function. Proc Natl Acad Sci USA.
104:15560–15565. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
77
|
Ji Y, Pang Q, Xu G, Wang L, Wang J and
Zeng Y: Exogenous hydrogen sulfide postconditioning protects
isolated rat hearts against ischemia-reperfusion injury. Eur J
Pharmacol. 587:1–7. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
78
|
Testai L, Marino A, Piano I, et al: The
novel H2S-donor 4-carboxyphenyl isothiocyanate promotes
cardioprotective effects against ischemia/reperfusion injury
through activation of mitoKATP channels and reduction of oxidative
stress. Pharmacol Res. 113:290–299. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
79
|
Yao LL, Huang XW, Wang YG, Cao YX, Zhang
CC and Zhu YC: Hydrogen sulfide protects cardiomyocytes from
hypoxia/reoxygenation-induced apoptosis by preventing
GSK-3beta-dependent opening of mPTP. Am J Physiol Heart Circ
Physiol. 298:H1310–H1319. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
80
|
Lambert JP, Nicholson CK, Amin H, Amin S
and Calvert JW: Hydrogen sulfide provides cardioprotection against
myocardial/ischemia reperfusion injury in the diabetic state
through the activation of the RISK pathway. Med Gas Res. 4:202014.
View Article : Google Scholar : PubMed/NCBI
|
|
81
|
Meng W, Pei Z, Feng Y, Zhao J, Chen Y, Shi
W, Xu Q, Lin F, Sun M and Xiao K: Neglected role of hydrogen
sulfide in sulfur mustard poisoning: Keap1 S-sulfhydration and
subsequent Nrf2 pathway activation. Sci Rep. 7:94332017. View Article : Google Scholar : PubMed/NCBI
|
|
82
|
Calvert JW, Jha S, Gundewar S, Elrod JW,
Ramachandran A, Pattillo CB, Kevil CG and Lefer DJ: hydrogen
sulfide mediates cardioprotection through Nrf2 signaling. Circ Res.
105:365–374. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
83
|
Tu W, Wang H, Li S, Liu Q and Sha H: The
anti-inflammatory and anti-oxidant mechanisms of the Keap1/Nrf2/ARE
signaling pathway in chronic diseases. Aging Dis. 10:637–651. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
84
|
Gao T, Furnari F and Newton AC: PHLPP: A
phosphatase that directly dephosphorylates Akt, promotes apoptosis,
and suppresses tumor growth. Mol Cell. 18:13–24. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
85
|
Miyamoto S, Purcell NH, Smith JM, Gao T,
Whittaker R, Huang K, Castillo R, Glembotski CC, Sussman MA, Newton
AC and Brown JH: PHLPP-1 negatively regulates Akt activity and
survival in the heart. Circ Res. 107:476–484. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
86
|
Ji K, Xue L, Cheng J and Bai Y:
Preconditioning of H2S inhalation protects against cerebral
ischemia/reperfusion injury by induction of HSP70 through
PI3K/Akt/Nrf2 pathway. Brain Res Bull. 121:68–74. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
87
|
Kang B, Li W, Xi W, Yi Y, Ciren Y, Shen H,
Zhang Y, Jiang H, Xiao J and Wang Z: Hydrogen sulfide protects
cardiomyocytes against apoptosis in ischemia/reperfusion through
MiR-1-regulated histone deacetylase 4 pathway. Cell Physiol
Biochem. 41:10–21. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
88
|
Muñoz-Planillo R, Nuñez G, Franchi L and
Eigenbrod T: The inflammasome: A caspase-1-activation platform that
regulates immune responses and disease pathogenesis. Nat Immunol.
10:241–247. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
89
|
Toldo S, Das A, Mezzaroma E, Chau VQ,
Marchetti C, Durrant D, Samidurai A, Van Tassell BW, Yin C, Ockaili
RA, et al: Induction of microRNA-21 with exogenous hydrogen sulfide
attenuates myocardial ischemic and inflammatory injury in mice.
Circ Cardiovasc Genet. 7:311–320. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
90
|
Zhu WD, Xu J, Zhang M, Zhu TM, Zhang YH
and Sun K: MicroRNA-21 inhibits lipopolysaccharide-induced acute
lung injury by targeting nuclear factor-κB. Exp Ther Med.
16:4616–4622. 2018.PubMed/NCBI
|
|
91
|
Yan X, Liu Y, Kong X, Ji J, Zhu H, Zhang
Z, Fu T, Yang J, Zhang Z, Liu F and Gu Z: MicroRNA-21-5p are
involved in apoptosis and invasion of fibroblast-like synoviocytes
through PTEN/PI3K/AKT signal. Cytotechnology. 71:317–328. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
92
|
Huang W, Tian SS, Hang PZ, Sun C, Guo J
and Du ZM: Combination of microRNA-21 and microRNA-146a attenuates
cardiac dysfunction and apoptosis during acute myocardial
infarction in mice. Mol Ther Nucleic Acids. 5:e2962016. View Article : Google Scholar : PubMed/NCBI
|
|
93
|
Karaskov E, Scott C, Zhang L, Teodoro T,
Ravazzola M and Volchuk A: Chronic palmitate but not oleate
exposure induces endoplasmic reticulum stress, which may contribute
to INS-1 pancreatic beta-Cell apoptosis. Endocrinology.
147:3398–3407. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
94
|
Ren L, Wang Q, Chen Y, Ma Y and Wang D:
Involvement of MicroRNA-133a in the protective effect of hydrogen
sulfide against ischemia/reperfusion-induced endoplasmic reticulum
stress and cardiomyocyte apoptosis. Pharmacology. 103:1–9. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
95
|
He B, Xiao J, Ren AJ, Zhang YF, Zhang H,
Chen M, Xie B, Gao XG and Wang YW: Role of miR-1 and miR-133a in
myocardial ischemic postconditioning. J Biomed Sci. 18:222011.
View Article : Google Scholar : PubMed/NCBI
|
|
96
|
Dakhlallah D, Zhang J, Yu L, Marsh CB,
Angelos MG and Khan M: MicroRNA-133a engineered mesenchymal stem
cells augment cardiac function and cell survival in the infarct
heart. J Cardiovasc Pharmacol. 65:241–251. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
97
|
Predmore BL, Kondo K, Bhushan S,
Zlatopolsky MA, King AL, Aragon JP, Grinsfelder DB, Condit ME and
Lefer DJ: The polysulfide diallyl trisulfide protects the ischemic
myocardium by preservation of endogenous hydrogen sulfide and
increasing nitric oxide bioavailability. Am J Physiol Heart Circ
Physiol. 302:H2410–H2418. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
98
|
Minamishima S, Bougaki M, Sips PY, Yu JD,
Minamishima YA, Elrod JW, Lefer DJ, Bloch KD and Ichinose F:
Hydrogen sulfide improves survival after cardiac arrest and
cardiopulmonary resuscitation via a nitric oxide synthase
3-dependent mechanism in mice. Circulation. 120:888–896. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
99
|
Bibli SI, Hu J, Looso M, Weigert A, Ratiu
C, Wittig J, Drekolia MK, Tombor L, Randriamboavonjy V, Leisegang
MS, et al: Mapping the endothelial Cell S-sulfhydrome highlights
the crucial role of integrin sulfhydration in vascular function.
Circulation. 143:935–948. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
100
|
Sun J, Aponte AM, Menazza S, Gucek M,
Steenbergen C and Murphy E: Additive cardioprotection by
pharmacological postconditioning with hydrogen sulfide and nitric
oxide donors in mouse heart: S-sulfhydration vs. S-nitrosylation.
Cardiovasc Res. 110:96–106. 2016. View Article : Google Scholar : PubMed/NCBI
|