Introduction
Premature ovarian insufficiency (POI) refers to the
presence of ovarian atrophy and permanent amenorrhea in women under
the age of 40, characterized by hypergonadotropic hypogonadism, and
presenting with either primary or secondary amenorrhea (1). In women of reproductive age, POI is
one of the most commonly diagnosed endocrine diseases, with a
worldwide prevalence of ~1% (2).
As well as menstrual disturbance, the main symptoms of POI are
decreased estradiol levels and increased plasma
follicle-stimulating hormone (FSH) levels (>25 mIU/ml on two
occasions, >4 weeks apart) (3,4).
The etiology of POI is highly heterogeneous and
complex, including genetic, autoimmune, infectious and iatrogenic
factors, among which genetic causes explain the presentation in
20–25% of patients worldwide (5).
Over the past few years, novel methods using next-generation
sequencing (NGS), particularly whole-exome sequencing (WES), have
led to the identification of numerous candidate genes that cause
POI. These genes are mainly involved in meiosis, DNA damage repair
and homologous recombination, including X-linked genes (e.g.,
FMR1, BMP15 and PGRMC1) and autosomal genes (e.g.,
FSHR, NOBOX, FIGLA, GDF9, FOXL2 and STAG3) (5–7).
In 2011, WES revealed PSMC3IP (MIM 608665) as a novel
candidate gene associated with autosomal recessive ovarian
dysgenesis (8). PSMC3IP is
important for homologous pairing and homologous recombination in
meiosis (9,10), which is indicated by its yeast
ortholog HOP2. In a previous study, female
PSMC31P-deficient mice displayed a significantly reduction
in ovarian volume and a lack of follicles (11,12), suggesting that loss of this
DNA-repair protein may be associated with infertility phenotypes.
To date, rare variants of PSMC3IP have been reported in POI
(8,13,14).
The current study presented a case of an adopted
Chinese woman suffering from POI. WES was performed on DNA obtained
from the patient to identify potential causative genes or
PSMC3IP mutations, which were associated with POI.
Identified sequences were subjected to extensive bioinformatics
analyses and screening against several databases to predict the
potential effect of the mutations on protein function. Mutations
were confirmed with Sanger sequencing and screened against negative
control DNA from healthy female individuals.
Materials and methods
Case presentation
The proband, a 29-year-old woman from Fujian origin,
was admitted to Women's and Children's Hospital affiliated to
Xiamen University. She had primary amenorrhea, had been married for
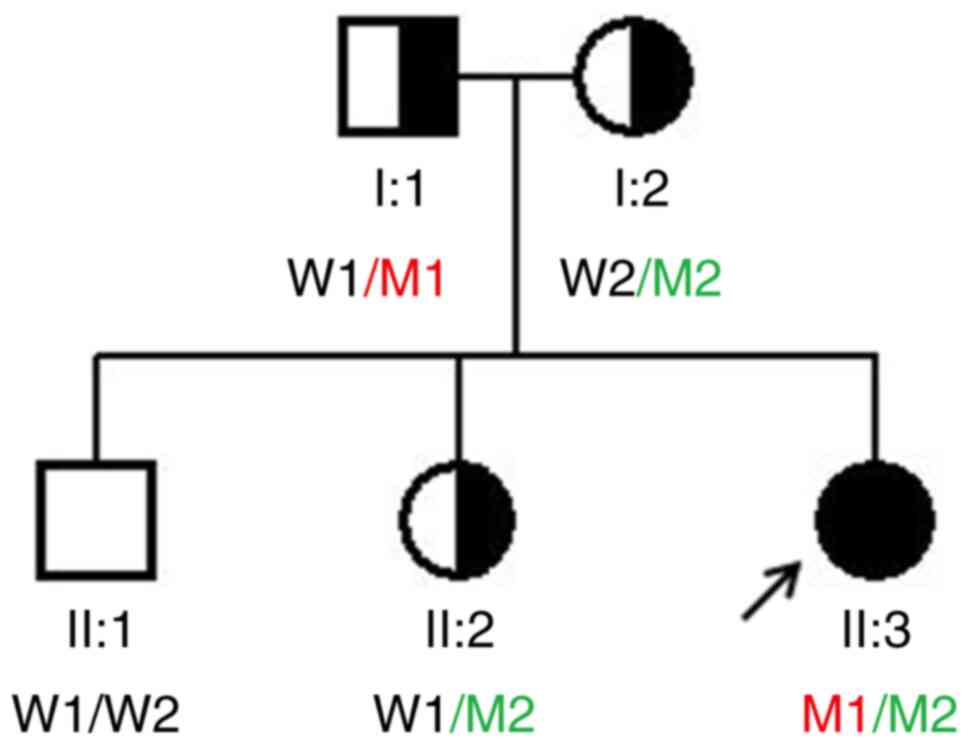
5 years without conceiving and was diagnosed with POI (Fig. 1). She had a normal target height
(160 cm) and normal weight (55 kg). Physical examination showed no
dysmorphic features or breast development, and normal intellectual
development. From a gynecological examination, it was clear that
the patient had a sparse amount of pubic and armpit hair. A
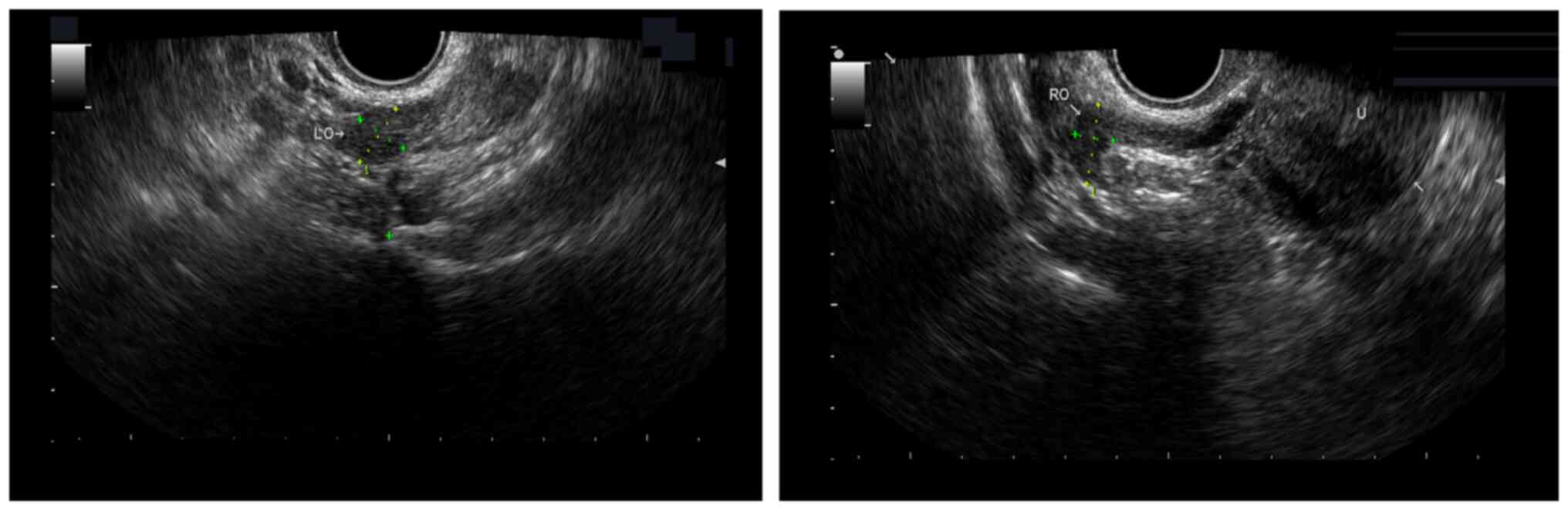
transvaginal ultrasound examination revealed that the bilateral
ovaries were abnormally small (the left ovary was 1.23×1.00 cm, and
the right ovary was 1.55×0.74 cm), and no obvious antral follicles
were observed (Fig. 2). Her basic
hormone levels were as follows: FSH, 62.5–78.6 mIU/l (reference
range: 1.5–10 mIU/ml before ovulation; 8–20 mIU/ml ovulation
period; 2–10 mIU/ml after ovulation); luteinizing hormone,
20.4–25.4 mIU/l (reference range: 5–25 mIU/ml non-ovulation period;
30–100 mIU/ml ovulation period; 4–10 mIU/ml after ovulation);
estradiol, 13.0–42.5 pmol/l (reference range: 48–521 pmol/l before
ovulation; 70–1835 pmol/l ovulation period; 272–793 pmol/l after
ovulation). The patient had a normal 46,XX karyotype and
FMR1 repeat lengths, and a negative test for the adrenal
cortical antibody. The biological parents of the proband were
healthy and non-consanguineous. The proband's biological mother and
sister had normal menstrual histories, and the family did not
report any history of systemic diseases or solid tumors. The
present study fully complied with the tenets of the Declaration of
Helsinki and was approved by the Ethics Board of the Women's and
Children's Hospital affiliated to Xiamen University (approval
number XY-2019-059). Written informed consent was obtained from all
participants prior to testing.
A total of 100 unrelated ethnically matched healthy
female individuals (age, 22–40 years; mean age, 28 years) were
recruited as controls. The healthy controls menstruated regularly,
had normal FSH levels (range, 2.5–10.1 IU/l; mean, 3.6±1.9 IU/l)
and normal pelvic ultrasound imaging.
Targeted exon capturing and NGS
Total genomic DNA was extracted from peripheral
blood leukocytes using the magnetic bead method with the Blood
Genomic DNA Mini kit (cat no: 51106; Qiagen, Inc.). The
concentration of DNA (ng/µl) in each sample was analyzed using a
NanoDrop 2000 spectrophotometer (Thermo Fisher Scientific, Inc.).
The genomic DNA (3 µg) was fragmented into ~150 bp. In-solution
hybridization-based enrichment was performed using the SureSelect
Human All Exon V6 (Agilent Technologies, Inc.) according to the
manufacturer's protocol. The concentrations of the library was
measured by a Qubit 4.0 Fluorometer (Invitrogen; Thermo Fisher
Scientific, Inc.) and the loading concentration of the final
library was 16pM. The library pool was sequenced on the Illumina
HiSeq 2500 sequencing platform(cat nos: PE-401-3001 and
FC-401-3001,Illumina, Inc.) in high output run mode with 150 bp
paired-end reads.
Bioinformatics analyses
After Illumina HiSeq sequencing, raw NGS data were
imported into FastQC for assessing the quality, and high-quality
reads were aligned to the human reference genome (GRCh37/hg19)
using Burrows-Wheeler Aligner software (BWA 0.7.17-r1188;
http://bio-bwa.sourceforge.net).
Subsequently, variant calling and annotation were performed using
GATK 4.0 software (https://software.broadinstitute.org/gatk). Several
databases, such as the Single Nucleotide Polymorphism Database
(dbSNP)138 (https://www.ncbi.nlm.nih.gov/snp), the 1000 Genome
Project (http://www.internationalgenome.org), the Exome
Aggregation Consortium (ExAc; http://exac.broadinstitute.org), ClinVar (https://www.ncbi.nlm.nih.gov/clinvar)
and the Genome Aggregation Database (gnomAD; http://gnomad-sg.org/) were employed to select all
variants with frequencies >5%. In addition, online tools such as
Human Splicing Finder (http://www.umd.be/HSF3/HSF.shtml), PolyPhen-2
(http://genetics.bwh.harvard.edu/pph2), SIFT
(http://sift.jcvi.org), Clustal W 2.0 (http://www.clustal.org) and Ensembl (https://asia.ensembl.org/index.html) were applied
to predict the potential effect on protein function.
Confirmation by Sanger sequencing
Mutations of PSMC3IP were further confirmed
by Sanger sequencing; two pair primers were designed to amplify the
exon 4 and exon 7 of PSMC3IP (NM_016556). Exon4-F:
5′-GCCCAGCAAAGGGGTCTTAG-3′; Exon4-R: 5′-GCTGGTTCCTGAGCATATCCA-3′.
Exon7-F: 5′-GCCAGTGCAAGACATCTCAC-3′; Exon7-R:
5′-CCAGATCAGCCGCTACACAAT-3′. The PCR amplifications were performed
as per the following procedure: Initial denaturation of 95°C for 5
min, 33 cycles of denaturation at 95°C for 30 sec, annealing at
64°C/62°C for 30 sec, extension at 72°C for 30 sec and a final
extension of 72°C for 10 min. The polymerase chain reaction
products were sequenced on an ABI 3730×l DNA Analyzer (Applied
Biosystems; Thermo Fisher Scientific, Inc.). Sequencing results
were analyzed using Lasergene software version 7.0 (DNASTAR,
Inc.).
Results
Mutation identification by NGS and
Sanger sequencing
Overall, the coverage of the target region was 99.3%
with an average sequencing depth of >130X and with a variant
accuracy of >99.97%. After filtering out all existing mutations
with a minor allele frequency >0.05 as determined with dbSNP138,
1000 Genomes, ExAc, ClinVar and gnomAD, a total of 18 variants
remained.
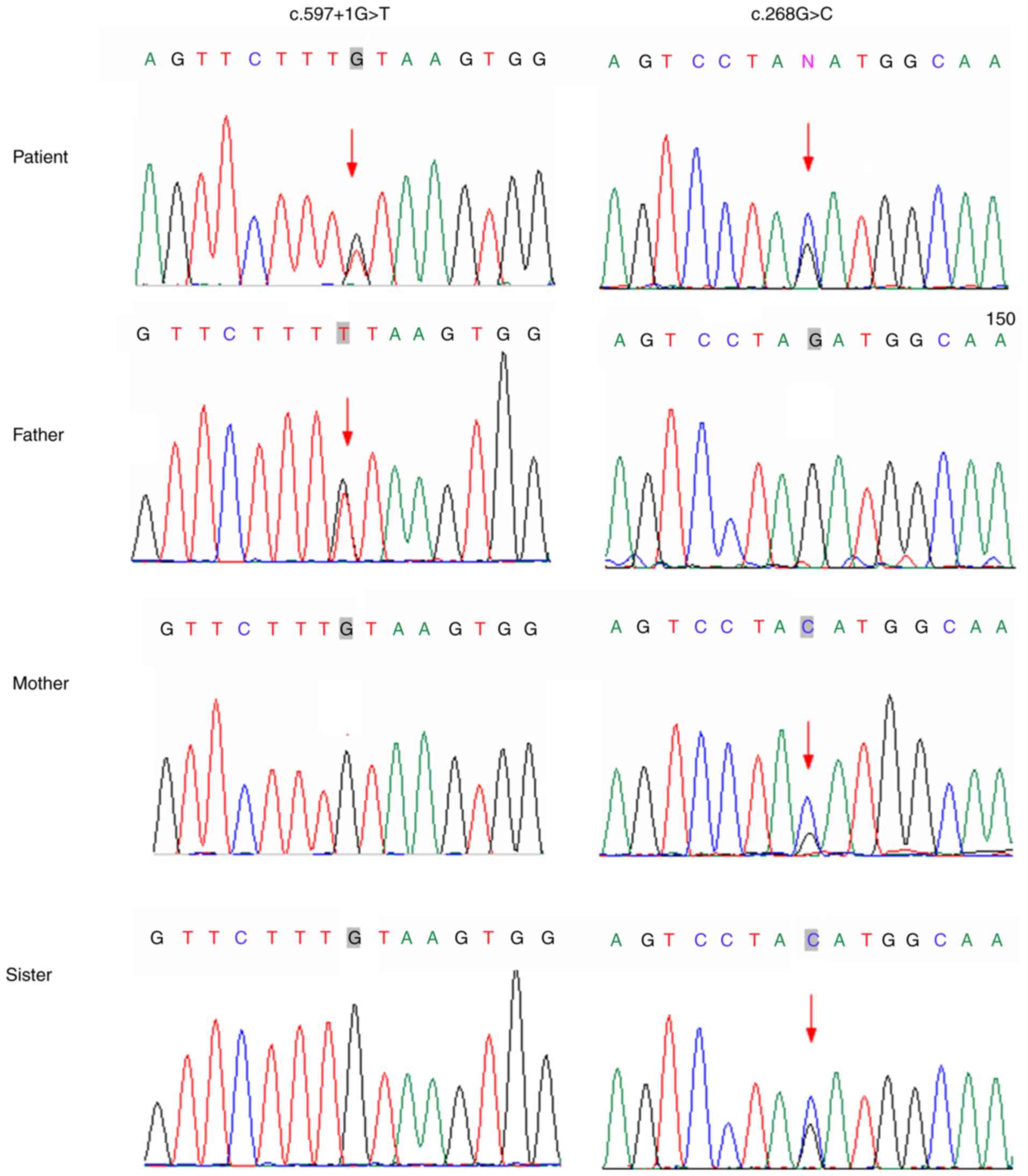
In combination with the clinical phenotype and
database analyses, two compound heterozygous mutations of
PSMC3IP, c.597+1G>T and c.268G>C, were considered as
pathogenic. Furthermore, Sanger sequencing on the DNA obtained from
the family members confirmed that the c.597+1G>T mutation was
inherited from the father, whereas the missense mutation
(c.268G>C) was observ 3 ed in the mother and younger sister,
showing complete co-segregation of the mutations with the disease
phenotype (Fig. 3).
Prediction of the pathogenic
significance of the mutations
According to the classification standards of
American College of Medical Genetics and Genomics (15), c.597+1G>T and c.268G>C are
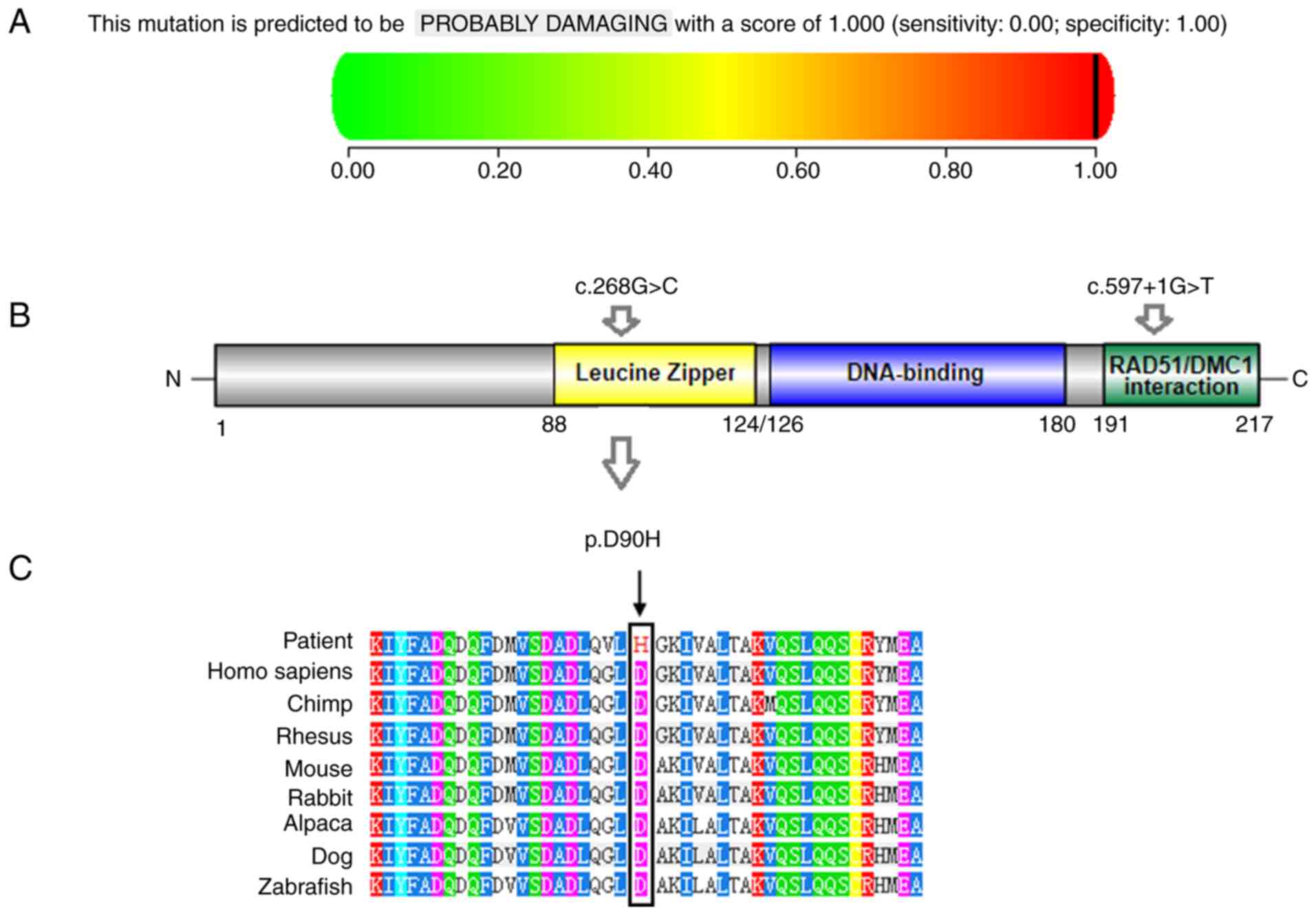
classified as suspected pathogenic mutations. The splicing
mutation, c.4106+2T>C, was predicted to alter the splice donor
site, most likely influenced by splicing, according to Human
Splicing Finder the c.268G>C mutation is a missense mutation and
results in a substitution of aspartate with histidine at amino acid
position 90 (p.D90H). According to Clustal W/Ensembl online
software UCD Conway Institute), the species conservation analysis
confirmed that the ninetieth aspartic acid residues were highly
conserved among different species (Fig. 4C). The mutation was described as
‘probably damaging’ by the online disease prediction software,
PolyPhen-2 (Fig. 4A), and it was
suggested to ‘affect protein function’ by SIFT. Neither of the two
mutations has been reported in the Human Gene Mutation Database,
dbSNP138, the ExAC database, the 1000 Genomes database or in any
other single-nucleotide polymorphism database. In addition, to the
best of our knowledge, no relevant literature has reported on this
mutation. Furthermore, neither of the heterozygous mutations were
found in 100 unrelated control individuals from the same ethnic
origin (data not shown). Taken together, these results powerfully
support that PSMC3IP mutations are disease-causing mutations
in this family.
Discussion
The present study analyzed samples from an adopted
29-year-old Chinese woman with POI and identified two biallelic
mutations, c.597+1G>T and c.268G>C in PSMC3IP. The two
mutations carried by the patient were inherited from her biological
mother (c.268G>C) and father (c.597+1G>T). PSMC3IP has
previously been linked to hereditary breast and ovarian cancer, and
has been reported to cause autosomal recessive POI (8,16,17). PSMC3IP defects can disrupt
estrogen-driven transcription activation of PSMC3IP. Impaired
estrogenic signaling can result in ovarian dysgenesis by
interfering with the follicular pool and failing to counteract
follicular atresia (8,18).
PSMC3IP is located at 17q21.2. The protein
product consists of 217 amino acids in its monomer form, encoding a
nuclear, tissue-specific protein with multiple functions, including
a role in meiotic recombination and acting as a coactivator of
ligand-dependent transcription mediated by nuclear hormone
receptors, which is conserved in evolution (19,20). Previous studies demonstrated that,
in PSMC3IP-knockout mice, the ovarian volume was reduced and
germ cells were missing (21,22). PSMC3IP is a DNA-binding protein
dimer, characterized by the presence of three domains including a
leucine zipper domain, a DNA-binding domain and a RAD51/DMC1
interaction domain (13). The
c.268G>C mutation occurs within the highly conserved leucine
zipper domain (Fig. 4B). In
vitro experiments previously revealed that a defect in the
leucine zipper eliminated the dimerization of PSMC3IP (19). In the present study, the
c.268G>C mutation was detected in the proband's mother and
sister with normal ovarian function. The splicing mutation,
c.597+1G>T, is predicted to alter the splice donor site thereby
interfering with splicing. However, the exact effects of splice
site mutations on mRNA cleavage are not clear and need to be
investigated further.
To date, four studies have described PSMC3IP
variants unique to patients with ovarian dysgenesis, including the
one reported in the present study (Table I). A total of six pathogenic
PSMC3IP mutations have been identified, comprising three
frameshift mutations, one nonsense mutation, one missense mutation
and a splicing mutation. In 2011, Zangen et al (8) first identified a homozygous 3 bp
in-frame deletion in exon 8 of PSMC3IP in a large
consanguineous Arab Palestinian pedigree with XX-female gonadal
dysgenesis, leading to the deletion of Glu201. Furthermore, in a
consanguineous Yemeni family of one brother with azoospermia and
four sisters with ovarian dysgenesis, Al-Agha et al
(13) identified a homozygous
C-terminal nonsense mutation (c.489C>G, p.Tyr163Ter) in
PSMC3IP, suggesting an important role of PSMC3IP in the
development of both male and female germ cells. In the present
study, the proband's father carried the heterozygous splice site
mutation, c.597+1G>T, but he did not show spermatogenesis
dysfunction. Previously, two compound heterozygous mutations of
PSMC3IP (c.430_431insGA, p.L144*; c.496_497delCT, p.R166Afs)
were found in a 28-year-old French woman who presented with POI
(14). By contrast, a cohort of
50 Swedish women with POI did not exhibit any pathogenic variants
of PSMC3IP (23). Our
study also screened 112 Chinese women suffering from POI using WES
and identified pathogenic mutations in known genes (ERCC6,
FIGLA and NOBOX), but no pathogenic mutation in
PSMC3IP gene was detected (unpublished data), highlighting
the genetic complexities that give rise to this syndrome. The
pathogenesis of POI caused by PSMC3IP is unknown. Because of the
limited number of cases of PSMC3IP mutations associated with
POI, we are not able to make a clear association between this
genotype and phenotype. Further functional studies are required to
evaluate the genotype and phenotype associations in large cohorts
containing patients of various ethnicities.
 | Table I.PSMC3IP mutations identified in
patients with primary ovarian insufficiency. |
Table I.
PSMC3IP mutations identified in
patients with primary ovarian insufficiency.
| First author,
year | Age at diagnosis,
years | Ethnic origin | CS | Karyotype | Nucleotide
change | Amino acid
change | Status | (Refs.) |
|---|
| Zangen et al,
2011 | 21 | Palestinian | Yes | 46,XX | c.[600_602del] | p.Glu201del | Ho | (8) |
| Yang et al,
2019 | 28 | French | No | 46,XX | c.[496_497delCT] +
[430_431insGA] | p.[R166Afs] +
[L144X] | He | (14) |
| Al-Agha et
al, 2018 | 27 | Yemeni | Yes | 46,XX | c.[489 C>G] | p.[Tyr163Ter] | Ho | (13) |
| Present study | 29 | Chinese | No | 46,XX | c.[597+1G>T] +
[268G>C] | P.[splicing] +
[p.D90H] | He | – |
In conclusion, the present study identified two
novel variants in PSMC3IP in a Chinese female patient with
POI. The present findings provide further evidence that the
PSMC3IP gene serves a role in the pathogenesis of POI and
support the application of NGS in the genetic diagnosis of female
infertility. Moreover, the findings extend the context of genotype
and phenotype in the POI patients and have important implications
for genetic counseling for the family.
Acknowledgements
Not applicable.
Funding
The present study was supported by the National Natural Science
Foundation of China (grant no. 31801044), the Medical and Health
Research Guidance Plan of Xiamen (grant no. 3502Z20209195) and the
Medical Science Research Foundation of Bethune (grant no.
QL002DS).
Availability of data and materials
All data generated or analyzed during this study are
included in this published article, except for next-generation
sequencing. The next-generation sequencing datasets generated
and/or analyzed during the current study are not publicly
available, as the patient did not consent to the public release of
these data.
Authors' contributions
PL and LM designed the research protocols. LM and LH
performed the experiments. XW and HH acquired the clinical data.
LM, YH, XH and ZS analyzed and interpreted the study data. LM and
LH wrote the manuscript. PL participated in the supervision and
critically reviewed the manuscript. LH and PL confirm the
authenticity of all the raw data. All authors have read and
approved the final manuscript.
Ethics approval and consent to
participate
The present study fully complied with the tenets of
the Declaration of Helsinki and was approved by the Ethics Board of
the Women's and Children's Hospital affiliated to Xiamen
University, China (approval number XY-2019-059). Written informed
consent was obtained from all participants before testing.
Patient consent for publication
Enrolled study participants and the patient provided
written informed consent for the publication of this manuscript;
however, the patients did not consent to having their data shared
out of concern for their privacy.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Nelson LM: Clinical practice. Primary
ovarian insufficiency. N Engl J Med. 360:606–614. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Tucker EJ, Grover SR, Bachelot A, Touraine
P and Sinclair AH: Premature ovarian insufficiency: New
perspectives on genetic cause and phenotypic spectrum. Endocr Rev.
37:609–635. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Gowri V, Al Shukri M, Al-Farsi FA,
Al-Busaidi NA, Dennison D, Al Kindi S, Daar S, Al Farsi K and
Pathare AV: Aetiological profile of women presenting with premature
ovarian failure to a single tertiary care center in Oman. Post
Reprod Health. 21:63–68. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
European Society for Human Reproduction
and Embryology (ESHRE) Guideline Group on POI, ; Webber L, Davies
M, Anderson R, Bartlett J, Braat D, Cartwright B, Cifkova R, de
Muinck Keizer-Schrama S, Hogervorst E, et al: ESHRE guideline:
management of women with premature ovarian insufficiency. Hum
Reprod. 31:926–937. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Jiao X, Ke H, Qin Y and Chen ZJ: Molecular
genetics of premature ovarian insufficiency. Trends Endocrinol
Metab. 29:795–807. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Rossetti R, Ferrari I, Bonomi M and
Persani L: Genetics of primary ovarian insufficiency. Clin Genet.
91:183–198. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Huhtaniemi I, Hovatta O, La Marca A,
Livera G, Monniaux D, Persani L, Heddar A, Jarzabek K, Laisk-Podar
T, Salumets A, et al: Advances in the molecular pathophysiology,
genetics, and treatment of primary ovarian insufficiency. Trends
Endocrinol Metab. 29:400–419. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Zangen D, Kaufman Y, Zeligson S, Perlberg
S, Fridman H, Kanaan M, Abdulhadi-Atwan M, Abu Libdeh A, Gussow A,
Kisslov I, et al: XX ovarian dysgenesis is caused by a PSMC3IP/HOP2
mutation that abolishes coactivation of estrogen-driven
transcriptio. Am J Hum Genet. 89:572–579. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Sansam CL and Pezza RJ: Connecting by
breaking and repairing: Mechanisms of DNA strand exchange in
meiotic recombination. FEBS J. 282:2444–2457. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Zhao W and Sung P: Significance of ligand
interactions involving Hop2-Mnd1 and the RAD51 and DMC1
recombinases in homologous DNA repair and XX ovarian dysgenesis.
Nucleic Acids Res. 43:4055–4066. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Petukhova GV, Romanienko PJ and
Camerini-Otero RD: The Hop2 protein has a direct role in promoting
interhomolog interactions during mouse meiosis. Dev Cell.
5:927–936. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Biswas L, Tyc K, El Yakoubi W, Morgan K,
Xing J and Schindler K: Meiosis interrupted: The genetics of female
infertility via meiotic failure. Reproduction. 161:R13–R35. 2021.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Al-Agha AE, Ahmed IA, Nuebel E, Moriwaki
M, Moore B, Peacock KA, Mosbruger T, Neklason DW, Jorde LB, Yandell
M and Welt CK: Primary ovarian insufficiency and azoospermia in
carriers of a homozygous PSMC3IP stop gain mutation. J Clin
Endocrinol Metab. 103:555–563. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Yang X, Touraine P, Desai S, Humphreys G,
Jiang H, Yatsenko A and Rajkovic A: Gene variants identified by
whole-exome sequencing in 33 French women with premature ovarian
insufficiency. J Assist Reprod Genet. 36:39–45. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Richards S, Aziz N, Bale S, Bick D, Das S,
Gastier-Foster J, Grody WW, Hegde M, Lyon E, Spector E, et al:
Standards and guidelines for the interpretation of sequence
variants: A joint consensus recommendation of the American college
of medical genetics and genomics and the association for molecular
pathology. Genet Med. 17:405–424. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Schubert S, Ripperger T, Rood M, Petkidis
A, Hofmann W, Frye-Boukhriss H, Tauscher M, Auber B, Hille-Betz U,
Illig T, et al: GT198 (PSMC3IP) germline variants in early-onset
breast cancer patients from hereditary breast and ovarian cancer
families. Genes Cancer. 8:472–483. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Achyut BR, Zhang H, Angara K, Mivechi NF,
Arbab AS and Ko L: Oncoprotein GT198 vaccination delays tumor
growth in MMTV-PyMT mice. Cancer Lett. 476:57–66. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Capdevila-Busquets E, Badiola N, Arroyo R,
Alcalde V, Soler-López M and Aloy P: Breast cancer genes PSMC3IP
and EPSTI1 play a role in apoptosis regulation. PLoS One.
10:e01153522015. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Nathansen J, Lukiyanchuk V, Hein L, Stolte
MI, Borgmann K, Löck S, Kurth I, Baumann M, Krause M, Linge A, et
al: Oct4 confers stemness and radioresistance to head and neck
squamous cell carcinoma by regulating the homologous recombination
factors PSMC3IP and RAD54L. Oncogene. 40:4214–4228. 2021.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Pang J, Gao J, Zhang L, Mivechi NF and Ko
L: GT198 is a target of oncology drugs and anticancer herbs. Front
Oral Health. 2:6794602020. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Lin T, Zhang Y, Zhang T, Steckler RA and
Yang X: Hop2 interacts with the transcription factor CEBPα and
suppresses adipocyte differentiation. J Biol Chem. 297:1012642021.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Zhao W, Saro D, Hammel M, Kwon Y, Xu Y,
Rambo RP, Williams GJ, Chi P, Lu L, Pezza RJ, et al: Mechanistic
insights into the role of Hop2-Mnd1 in meiotic homologous DNA
pairing. Nucleic Acids Res. 42:906–917. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Norling A, Hirschberg AL, Karlsson L,
Rodriguez-Wallberg KA, Iwarsson E, Wedell A and Barbaro M: No
mutations in the PSMC3IP gene identified in a Swedish cohort of
women with primary ovarian insufficiency. Sex Dev. 8:146–150. 2014.
View Article : Google Scholar : PubMed/NCBI
|