Introduction
Cognitive dysfunction as a result of
sepsis-associated encephalopathy (SAE) is a serious complication of
sepsis (1). Lipopolysaccharide
(LPS)-activated macrophages can directly permeate into the central
nervous system (CNS) to produce an inflammatory response. In
addition, LPS can indirectly produce neuroinflammation by
peripheral inflammatory factors diffusing into the CNS through the
blood-brain barrier, thus leading to CNS inflammatory injury
(1). Symptoms of SAE-related
cognitive dysfunction ranges from mild delirium to coma, which can
persist for months to years (2).
They are frequently associated with reduced quality of life and
poor prognosis, in addition to increased morbidity and mortality
(3). Several studies previously
revealed that cognitive dysfunction is associated with microglial
activation and hypoxic-ischemic injury, which then activates the
inflammatory response in the hippocampus (4,5).
This leads to pathological changes that are comparable to those
observed during neurodegenerative diseases (6,7).
However, the specific mechanism of this remains elusive.
The pathogenesis of SAE involves a number of
factors, including neuroinflammation, collapse of the blood brain
barrier (BBB), ischemic injury, alterations in the neurotransmitter
profile and mitochondrial dysfunction. Aberrant neuroinflammation
has been reported to at least in part mediate the pathogenesis of
SAE and multiple organ dysfunction syndrome (MODS) (8,9).
This is especially the case for the CNS injury that occurs during
MODS, which is vulnerable to inflammatory insults (8,9).
Additionally, neuroinflammation has been previously found to be
responsible for the extensive apoptosis of various cell types in
the brain, including microglial cells, neurons and vascular
endothelial cells (10).
Microglia-mediated neuroinflammation reportedly mediates a
substantial portion of the inflammatory response in the CNS, which
results in worse outcomes due to septic complications (11–13).
Forkhead box C1 (Foxc1) is a transcription factor
that is involved in various pathophysiological processes, such as
myocardial ischemia (14), facial
paralysis (15) and colorectal
cancer (16). Recent studies
revealed that Foxc1 can promote defense against oxidative stress,
inflammation and apoptosis (17,18). In addition, Foxc1 has been found
to serve key roles in a number of biological processes, including
cell proliferation, differentiation, migration and survival
(19,20). A previous study demonstrated that
upregulation of Foxc1 expression promoted Schwann cell migration
(15). By contrast, knockdown of
Foxc1 expression has been shown to significantly suppress the
migration of cervical and breast cancer cells (21,22). In terms of myeloid tissue
regeneration, Foxc1 was demonstrated to promote regenerative
functions by promoting mveloid tissue bone marrow mesenchymal stem
cell migration (23). However,
the biological function of Foxc1 in microglial cells remains poorly
understood.
NF-κB consists of a family of transcription factors
that are involved in the regulation of inflammation, cell
proliferation, migration, differentiation and survival (24,25). NF-κB can regulate the expression
of genes involved in a wide range of biological processes,
including cancer cell proliferation, migration and apoptosis
(25). A previous study has
revealed that suppression of NF-κB signaling can attenuate
inflammation and cell migration in LPS-treated BV-2 microglial
cells (26). However, the
relationship between Foxc1 and the NF-κB pathway in microglial
physiology remains unclear.
In the present study, the potential role of Foxc1 in
SAE-associated cognitive dysfunction was investigated using an
LPS-induced microglial cell model and a cecal ligation and
perforation (CLP) mouse model. In addition, the effects of Foxc1 on
microglial migration in the hippocampus and NF-κB signaling were
also focused upon. These results may prove beneficial for the
development of therapeutic strategies for SAE.
Materials and methods
Cell culture and treatment
BV-2 is a murine-derived immortalized microglial
cell line and were purchased from ScienCell Research Laboratories,
Inc., which were routinely cultured in DMEM (Gibco; Thermo Fisher
Scientific, Inc.) supplemented with 10% FBS (Gibco; Thermo Fisher
Scientific, Inc.) and 2 mM L-glutamine, penicillin (100 U/ml) and
streptomycin (100 g/ml; Invitrogen; Thermo Fisher Scientific, Inc.)
at 37°C with 5% CO2 and 95% humidity. For treatment,
BV-2 cells were incubated in a six-well plate at a density of
1×105 cells/ml at 37°C with 5% CO2 for 24 h,
and were then cultured in the absence or presence of 100 ng/ml LPS
(Sigma-Aldrich; Merck KGaA) for 6 h (27).
Foxc1 adenovirus transfection
Foxc1 adenovirus (Ad-Foxc; adenoviral plasmid name:
GV345; target gene ID: 17300; sequence: GGTATAAGAGGCGCGACCAG;
accession no. NC_000079; 4×108 TU/ml; Shanghai GeneChem
Co., Ltd.) or the adenovirus control (Ad-Ctrl), which is the
adenovirus containing the empty vector without the Foxc1 gene
(1.5×109 TU/ml; Shanghai GeneChem Co., Ltd.), was added
into the media for transfection into BV-2 cells at a 60–80%
confluence. Transfection was for 8 h at 37°C with 5% CO2
(Multiplicity of infection, 100). The medium was then removed and
replaced with fresh medium. Cells stably overexpressing Foxc1 were
then selected using puromycin incubation (2.5 µg/ml; cat. no.
ST551; Beyotime Institute of Biotechnology) at 37°C with 5%
CO2 for 7 days. Reverse transcription-quantitative PCR
(RT-qPCR), western blotting and immunofluorescence analysis were
used to detect transfection efficiency.
Small interfering RNA (siRNA)
transfection
BV-2 cells were seeded in 6-well plates at a density
of 5×104 cells/ml for culturing at 37°C with 5%
CO2 for 12 h before siRNA transfections (50 nM) were
performed at ~80% confluency. IκBα expression in BV-2 cells was
knocked down using siRNA (siRNA-IκBα sense,
5′-ACUCAUUGGUUCCUUUAAGGG-3′ and antisense,
5′-CUUAAAGGAACCAAUGAGUCC-3′). A non-targeting siRNA (siRNA-NT) was
used as a negative control (Santa Cruz Biotechnology, Inc.).
Transfection of siRNAs into BV-2 cells was performed using the
Lipofectamine® 3000 reagent (Invitrogen; Thermo Fisher
Scientific, Inc.) at 37°C with 5% CO2 for 10 h.
Transfection efficiency was measured using RT-qPCR and western blot
analysis.
Animals
A total of 48 C57BL/6J mice, aged 8–10 weeks
(weight, 25±5 g), were obtained from the Department of Experimental
Animal Science, School of Medicine, Shanghai Jiao Tong University
(license no. SYXK 2018-0027; Shanghai, China). All study procedures
were approved by the Institutional Animal Care and Use Committee of
Shanghai Jiao Tong University School of Medicine. All experimental
procedures were conducted in accordance with the National
Institutes of Health Guide for the Care and Use of Laboratory
Animals (National Institutes of Health, revised in 1996) (28). Mice were housed in a temperature
of 23±2°C and humidity of 40–60% on a 12-h light-dark cycle with
free access to food and water. Only male mice were used in the
present study to minimize the risk of heterogeneity due to sex
differences in the pathology of encephalopathy.
For the generation of Foxc1-overexpressing (Foxc1
OE) mice, embryonic stem (ES) cells (B6/BLU; cat. no. SCSP-226; The
Cell Bank of Type Culture Collection of the Chinese Academy of
Sciences) were cultured with the ES complete medium [600 ml,
comprising DMEM (cat. no. 12430; Gibco; Thermo Fisher Scientific,
Inc.) 497 ml; fetal bovine serum (Gibco; Thermo Fisher Scientific,
Inc.), 90 ml; Glutamax (cat. no. 35050-061; Gibco; Thermo Fisher
Scientific, Inc.) 6 ml; MEM Non-Essential Amino Acids Solution
(cat. no. 11140-050; Gibco; Thermo Fisher Scientific, Inc.), 6 ml;
LIF (cat. no. ESG1107; MilliporeSigma), 60 µl (concentration, 1,000
U/ml), 2-Mercaptoethanol (cat. no. 21985023; Gibco; Thermo Fisher
Scientific, Inc.) 1.5 ml] at 37°C with 5% CO2 and
obtained from School of Medicine, Shanghai Jiao Tong University and
transfected with the Foxc1 overexpression adenovirus (MOI, 100) for
72 h in vitro. The Foxc1-overexpressing ES cells were then
transplanted into the uterus of female mice (10 male mice and 20
female mice were used in this study). In total, two litters of
first-generation male and female mice were bred to obtain the
Foxc1-overexpressing mice, where the Foxc1-overexpressing mice were
reproduced indefinitely.
Experimental design
The sepsis mouse model was established using CLP
surgery as described previously (29). Mice were randomly divided into the
following four groups (n=12 per group): i) Naive; ii) sham
operated; iii) CLP; and iv) CLP + Foxc1 overexpression (OE). A
total of 6 days before and 10 days after CLP surgery, mice were
subjected to the Morris water maze (MWM) trial for 6 days. The mice
were then sacrificed after MWM trial, and the hippocampus tissues
were removed for RT-qPCR and western blot analysis.
Mice with the following symptoms (humane endpoints)
were be euthanized during the experiment: i) Inactivity or do not
respond to gentle stimulation; ii) dyspnea, including signs of
salivation and/or cyanosis from the nose and mouth; iii) diarrhea
or urinary incontinence; iv) 20% weight loss; v) Inability to eat
or drink; vi) clear signs of anxiety and irritability in animals;
vii) paralysis, persistent epilepsy or rigid behavior and viii)
infection and pus at the site of operation.
CLP surgery
For the CLP surgery itself (29), the mice were anesthetized using
intraperitoneal injections of ketamine (80 mg/kg) and xylazine (5
mg/kg). After disinfection an incision was made below the xiphoid
to expose the abdominal cavity. The cecum was then isolated,
ligated and punctured twice using a 22-gauge needle. Intestinal
contents were gently extruded into the peritoneal cavity. The
abdomen was then sutured after the cecum was reinserted into the
peritoneum. For the sham operated mice, the cecum was only exposed
without ligation or perforation, before 1 ml sterile saline
pre-warmed to 37°C was put into the abdominal cavity. After
surgery, mice were returned to their cages with a warm cotton pad
and free access to food and water. Naive mice did not undergo
laparotomy.
MWM trial
Spatial learning and memory were assessed using the
MWM trial as described previously (30). Briefly, the water temperature was
kept at 22–24°C. Acquisition test was performed four times per day
for 5 consecutive days; mice were put into the pool from four
different directions and allowed to swim freely until they climbed
up to platform under the water surface. If they did not find the
platform within 60 sec, they were be guided to the platform and
stayed on the platform for 10 sec. Four tests were conducted 20 min
apart and their swimming track was recorded. The time of climbing
to the hidden platform was defined as the escape latency. The
platform was then removed on day 6 before retention tests were
performed, mice were put into the pool from the same orientation
and allowed to swim freely in the pool for 60 sec and the swimming
track was recorded. The time of climbing to the hidden platform,
the number of crossings over the platform area and the swimming
speed were recorded. Experimental parameters were recorded using a
SuperMaze Morris Water Maze video analysis system (model, XR-XM101;
Shanghai XinRuan Information Technology Co., Ltd.).
Animal sample collection
From March to June 2021, mice were anesthetized
using an intraperitoneal injection of 1% pentobarbital sodium (50
mg/kg). Mice were sacrificed by CO2 inhalation using
30%/min volume displacement rate after cardiac saline perfusion,
and the hippocampus tissues were then collected for subsequent
experimentation.
RT-qPCR
RT-qPCR was performed as described previously
(31). In brief, total RNA was
extracted from BV-2 cells and hippocampal tissues using RNAiso Plus
reagent (Takara Bio, Inc.), and then subjected to reverse
transcription using a PrimeScript™ RT Master Mix kit (cat. no.
RR036A; Takara Bio, Inc.) according to manufacturer's protocol.
cDNA was used for qPCR with a TB Green® Premix Ex Taq™
(Tli RNaseH Plus) kit (cat. no. RR420A; Takara Bio, Inc.) according
to the manufacturer's protocol. All procedures were performed in
triplicate. The mRNA expression levels were calculated relative to
the GAPDH using the the 2−ΔΔCq method. The sequences of
the primers used were as follows: Foxc1 forward,
5′-AAGACGGAGAACGGTACGTG-3′ and reverse, 5′-TCACCGGGGAGTTGTTCAAG-3′;
IκBα forward, 5′-TGTGCTTCGAGTGACTGACC-3′ and reverse,
5′-TCACCCCACATCACTGAACG-3′ and GAPDH forward,
5′-AGGTCGGTGTGAACGGATTTG-3′ and reverse,
5′-TGTAGACCATGTAGTTGAGGTCA-3′. The full thermocycling conditions
used for qPCR were as follows: Initial denaturation at 95°C for 30
sec; followed by 40 cycles at 95°C for 5 sec, 60°C for 34 sec, 95°C
for 15 sec, 60°C for 60 sec and 95°C for 15 sec.
Western blot analysis
Western blot analysis was performed as described
previously (32). The total
protein content of cells and hippocampal samples was extracted
using RIPA solution (cat. no. P0013B; Beyotime Institute of
Biotechnology) for 30 min, and a BCA assay kit (cat. no. P0012S;
Beyotime Institute of Biotechnology) was used to detect the total
protein concentration. The extracted proteins (30 µg/lane) were
separated by sodium dodecyl sulfate-polyacrylamide gel
electrophoresis on 10% gels (Invitrogen; Thermo Fisher Scientific,
Inc.) and were then transferred onto nitrocellulose membranes
(MilliporeSigma), which were blocked with 5% non-fat milk at room
temperature for 2 h, incubated with the corresponding primary at
4°C overnight and secondary antibodies at room temperature for 2 h.
The mouse antibodies used in the present study are listed: Foxc1
(1:1,000; cat. no. ab227977; Abcam), p65 (1:1,000; cat. no.
ab32536; Abcam), IκBα (1:1,000; cat. no. ab32518; Abcam), allograft
inflammatory factor 1 (Iba-1; 1:1,000; cat. no. ab178846; Abcam),
IL-1β (1:1,000; cat. no. ab254360; Abcam), TNF-α (1:1,000; cat. no.
ab215188; Abcam), and β-actin (1:1,000; cat. no. ab179467; Abcam),
HRP-conjugated goat anti-rabbit IgG H&L secondary antibody
(1:2,000; cat. no. ab7090; Abcam). Protein bands were detected
using an enhanced chemiluminescence reagent (cat. no. WBKLS0100;
MilliporeSigma), visualized using a Bio-Rad ChemiDoc XRS imaging
system (Bio-Rad Laboratories, Inc.) and analyzed by Image J
software (version no: 1.51, National Institute of Health) and
normalized to that of β-actin.
Cell proliferation assay
The viability of BV-2 cells was evaluated using the
Cell Counting Kit-8 (CCK-8; MedChemExpress) assay in accordance
with the manufacturer's protocol. Briefly, the BV-2 cells with or
without Foxc1 overexpression were seeded into 96-well plates
(2×105 cells/well) and cultured at 37°C for 12 h. After
treatment with or without LPS at 37°C for 6 h, 10 ml CCK-8 solution
was added into each well and cultured at 37°C for 2 h, after which
the optical density of each well was determined at 450 nm using a
multiplate reader.
Transwell migration assay
The effect of Foxc1 on the migration of BV-2 cells
was examined using Transwell chambers (pore size, 8-µm). Cells in
different groups suspended in serum-free DMEM were seeded into the
top chamber at a density of 3×105 cells/well, whereas
500 µl complete DMEM containing 10% FBS was added to the lower
chamber. The cells were allowed to migrate for 24 h at 37°C in 5%
CO2. Cells on the upper surfaces of each chamber were
removed using a cotton swab, before the chambers were fixed with 4%
paraformaldehyde at room temperature for 30 min followed by 0.4%
crystal violet staining at room temperature for 15 min. In total,
five random fields of view were counted per chamber using an
inverted microscope under a light field (×40 magnification; Olympus
BX51; Olympus Corporation).
H&E staining
The hippocampal tissues were fixed with 4%
paraformaldehyde at room temperature overnight, embedded in
paraffin, sliced into 40-µm sections and dewaxed at 65°C for 30
min. Sections were then incubated twice with xylene for 8 min and
once for 5 min, followed by incubated three times with 100% alcohol
for 5 min and once with 75% alcohol for 3 min, and rinsed with
water for 5 min. Finally, slices were stained at room temperature
with hematoxylin for 3 min and eosin for 25 sec, and were observed
using an inverted microscope under a light field (×40
magnification; Olympus BX51; Olympus Corporation).
Immunofluorescence analysis
After tissue fixation, embedding, slicing and
dewaxing as aforementioned, the antigen retrieval was performed
using Improved Citrate Antigen Retrieval Solution (cat. no. P0083;
Beyotime Institute of Biotechnology) at 95–100°C for 20 min. Slices
were then blocked with Immunol Staining Blocking Buffer (cat. no.
P0102; Beyotime Institute of Biotechnology) at room temperature for
60 min, and were incubated with primary antibodies as follows:
Iba-1 (1:100; cat. no. ab178846; Abcam) at 4°C overnight.
Cells were fixed with 4% paraformaldehyde at room
temperature for 15 min followed by permeabilization with 0.03%
Triton X-100 and blocked with 5% BSA (cat. no. ST025; Beyotime
Institute of Biotechnology) at room temperature for 30 min. They
were then incubated with the Foxc1 antibody (1:100; cat. no.
ab227977; Abcam) at 4°C overnight in a humidified box. Antibodies
were diluted using Immunol Staining Primary and secondary Antibody
Dilution Buffer (cat. nos. P0103 and P0108; Beyotime Institute of
Biotechnology).
On the following morning, the cells or tissue
sections were washed with 1× TBST solution (cat. no. XY51287;
Shanghai Xinyu Biotechnology Co., Ltd.) and incubated with Alexa
Fluor® 647-conjugated goat anti-mouse IgG (1:100, cat.
no. ab150115, Abcam) at room temperature for 1 h and labeled with
DAPI (1 mg/ml; cat. no. 62247; Thermo Fisher Scientific, Inc.) at
room temperature for 5 min. The fluorescence intensity was observed
in three fields of view using fluorescence microscopy (×40
magnification; Olympus BX51; Olympus Corporation) and analyzed by
ImageJ software.
TUNEL assay
Apoptosis was detected using the TUNEL Apoptosis
Assay kit (cat. no. C1088; Beyotime Institute of Biotechnology).
Briefly, after tissue fixation, embedding, slicing, dewaxing and
antigen retrieval as aforementioned, 40-µm tissue sections were
treated with 0.1% Triton X-100 for 10 min, and then incubated with
TUNEL reagent at 37°C in the dark for 60 min. The nuclei were
stained with 5 µg/ml DAPI at room temperature for 5 min. The
morphological changes of apoptotic cells were observed under a
fluorescence microscope in three fields of view (Olympus BX51;
Olympus Corporation). Green fluorescence was considered to indicate
apoptotic cells.
ELISA
BV-2 cells with or with Foxc1 overexpression were
seeded into 12-well plates (5×105 cells/well) and
cultured at 37°C for 12 hBV-2 cells were treated with or without
LPS at 37°C for 6 h. Finally, IL-1β and TNF-α in supernatants were
measured by ELISA kits (cat. nos. 432601 and 430907; BioLegend,
Inc.) following the manufacturers' protocols.
Flow cytometry analysis
HT-22 neuronal cells were purchased from The Cell
Bank of Type Culture Collection of the Chinese Academy of Sciences
to explore the effects of microglia-mediated inflammatory response
on neuronal cells. HT-22 cells were routinely cultured in DMEM
(Gibco; Thermo Fisher Scientific, Inc.) supplemented with 10% FBS
(Gibco; Thermo Fisher Scientific, Inc.) and 2 mM L-glutamine,
penicillin (100 U/ml) and streptomycin (100 g/ml; Gibco; Thermo
Fisher Scientific, Inc.) at 37°C with 5% CO2. HT-22
cells (1×105 cells/ml) were cultured with the BV-2
cell-conditioned medium under different conditions at 37°C with 5%
CO2 for 12 h. Suspended HT-22 cells were washed twice
with PBS containing 2% FBS and stained with Annexin V FITC Apop
Dtec Kit (cat. no. 556547; Becton-Dickinson and Company). In brief,
100 µl binding buffer before 5 µl FITC-labelled Annexin V (20
µg/ml) and 5 µl PI (50 µg/ml) were added and incubated in the dark
at room temperature for 15 min. A tube without Annexin V FITC and
PI was used as Blank, whereas Annexin V or PI was used the
single-standard control. In total, 400 µl binding buffer was added
at the end of the experiment and then assessed by flow cytometry
(CytoFlex; Beckman Coulter, Inc.). The percentage of early- and
late-stage apoptotic cells was calculated using FlowJo v10 software
(FlowJo, LLC).
Statistical analysis
The data are presented as the mean ± standard
deviation from three independent experiments. Differences
between/among groups were determined using an unpaired Student's
t-test or one-way ANOVA followed by Tukey's post hoc test. A
two-way ANOVA was performed for cell proliferation and a mixed
ANOVA was used to analyze swimming speed and latency data from the
MWM followed by Bonferroni correction for multiple testing.
Statistical analyses were performed using the SPSS software
(version 26.0; IBM Corp.). P<0.05 was considered to indicate a
statistically significant difference.
Results
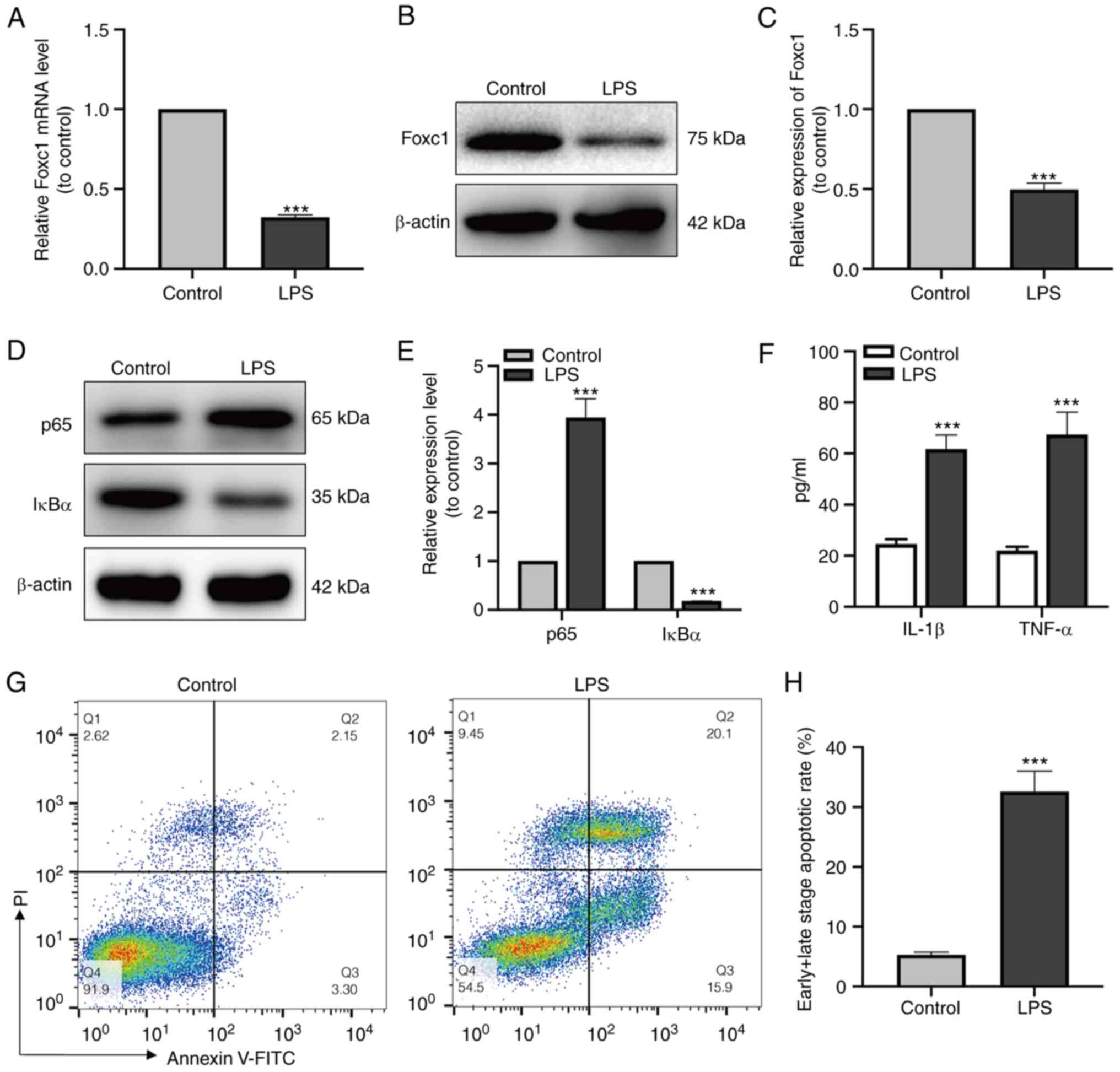
LPS triggers the inflammatory response
in microglia and promotes neuronal apoptosis
After LPS treatment, the expression levels of Foxc1,
p65 and IκBα of the NF-κB signaling pathway in microglial cells was
measured by both RT-qPCR and western blot analysis. Foxc1 and IκBα
expression was found to be significantly decreased, whereas that of
p65 was significantly increased compared with that in the control
group (Fig. 1A-E). In addition,
the secretion of IL-1β and TNF-α (Fig. 1F) by BV-2 cells was next examined
using ELISA, where the results showed that LPS significantly
increased the secretion of IL-1β and TNF-α compared with that in
the control group. Subsequently, HT-22 neurons were cultured with
the media conditioned by BV-2 cells in the control or LPS-treated
groups, respectively. The percentage of neuronal apoptosis was then
measured by flow cytometry, which showed that the levels of HT-22
cell apoptosis in the LPS-treated BV-2 media group were
significantly higher compared with those in the control group
(Fig. 1G and H). These results
suggest that microglia-mediated inflammatory response induced by
LPS resulted in neuronal apoptosis.
Overexpression of Foxc1 inhibits
microglia-mediated inflammatory response and neuronal apoptosis
induced by LPS
After adenoviral transfection, the expression of
Foxc1 in BV-2 cells with or without LPS treatment was significantly
increased compared with that in the Ad-Ctrl group, according to
western blotting (Fig. 2A, B, D and
E), RT-qPCR (Fig. 2C) and
immunofluorescence analysis observations (Fig. 2F and G). Subsequently, the
expression of p65 and IκBα was measured using western blot
analysis, whereas the secretion of IL-1β and TNF-α by BV-2 cells
was examined by ELISA. Foxc1 overexpression significantly reduced
the expression of p65 whilst upregulating the expression of IκBα
compared with those in the Ad-Ctrl group in the presence of LPS
(Fig. 2H and I). The secretion of
IL-1β and TNF-α by BV-2 cells also exhibited a similar trend as
p65, in that significant reversals of the LPS-induced upregulation
in secretion were observed (Fig.
2J). HT-22 neurons were next cultured with the media
conditioned by BV-2 cells in the control, Ad-Ctrl and Ad-Foxc1
groups with LPS treatment. The extent of neuronal apoptosis in the
Ad-Foxc1 group was significantly lower compared with that in the
Ad-Ctrl group (Fig. 2K and L).
These results suggest that overexpression of Foxc1 inhibited
LPS-induced inflammatory responses and neuronal apoptosis by
deactivating of NF-κB signaling.
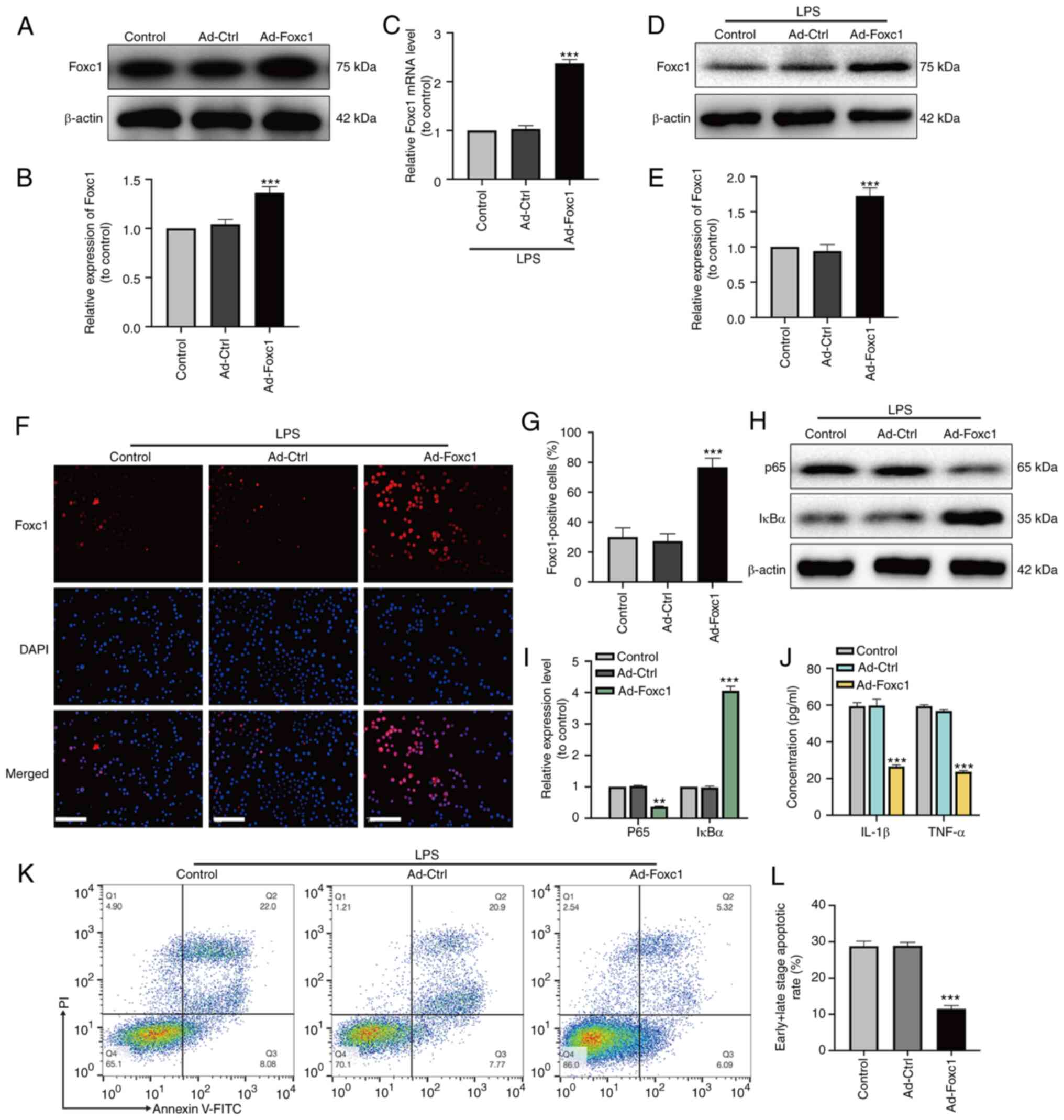 | Figure 2.Overexpression of Foxc1 inhibits
microglia-mediated inflammatory response and neuronal apoptosis
induced by LPS. After adenoviral transfection, transfection
efficiency was analyzed by (A) western blot analysis and (B) was
semi-quantified. After LPS treatment, the expression of Foxc1 was
analyzed by (C) reverse transcription-quantitative PCR and (D)
western blot analysis, (E) the latter of which was quantified. (F)
Immunofluorescence assay was performed to detect Foxc1
overexpression in BV-2 cells following LPS-treatment. Untreated
BV-2 cells were used as the control group. Scale bar, 20 µm. (G)
Percentage of Foxc1-positive BV-2 cells. (H) In the presence of
LPS, the expression levels of p65 and IκBα in BV-2 cells in the
three groups were detected by western blot analysis and (I)
quantified. (J) In the presence of LPS, the secretion of IL-1β and
TNF-α into the culture supernatant by BV-2 cells in the three
groups were detected by ELISA. (K) Flow cytometry analysis of HT-22
cell apoptosis after incubation in the media conditioned by
microglia. (L) The percentage of early and late apoptotic HT-22
cells was examined by flow cytometry analysis. Data represents the
mean ± SD of three independent experiments. **P<0.01 and
***P<0.001 vs. Ad-Ctrl group. LPS, lipopolysaccharide; Ad-,
adenovirus; Ctrl, control; Foxc1, Forkhead box C1; IκBα, NF-κB
inhibitor α. |
Overexpression of Foxc1 inhibits
LPS-induced microglial migration, inflammation and neuronal
apoptosis through the inhibition of IκBα/NF-κB signaling
To investigate the underlying regulatory mechanism
of Foxc1 on the inflammatory response, microglial migration and
neuronal apoptosis induced by LPS, the effect of Foxc1
overexpression on microglial migration, NF-κB signaling and
neuronal apoptosis was next investigated. Transwell assay revealed
that LPS markedly increased microglial migration compared with that
in the blank group (BV-2 cells without any treatment). Foxc1
overexpression significantly inhibited microglial migration
compared with that in the Ad-Ctrl group in the presence of LPS
(Fig. 3A and B). Subsequently,
the effect of Foxc1 overexpression and LPS treatment on the
viability of BV-2 cells was also evaluated using CCK-8 assay, which
showed that neither Foxc1 overexpression nor LPS treatment could
influence the viability of BV-2 cells (Fig. 3C and D). These results suggest
that Foxc1 overexpression inhibited microglial migration induced by
LPS without affecting viability. Subsequently, to determine the
effect of Foxc1 overexpression on NF-κB signaling during the
LPS-induced inflammatory response, the expression of IκBα in
Foxc1-overexpressing BV-2 cells was knocked down using siRNA-IκBα
in the absence or in the presence of LPS. Both western blot
analysis and RT-qPCR results showed that the expression of IκBα in
the siRNA-IκBα group was significantly decreased compared with that
in the siRNA-NT (the negative silenced) group, though no
significant difference could be found between the siRNA-NT and
control groups (Fig. 3E-I). In
addition, the expression of p65 in the siRNA-IκBα group was
significantly increased compared with that in siRNA-NT group, but
no significant difference could be found between the siRNA-NT and
the control groups (Fig. 3H and
I). Transwell assay results demonstrated that knocking down
IκBα expression significantly promoted microglial migration
compared with that in the siRNA-NT group (Fig. 3J and K). ELISA results revealed
that the secretion of IL-1β and TNF-α by BV-2 cells were
significantly increased in the siRNA-IκBα group compared with that
in the siRNA-NT group (Fig. 3L).
Subsequently, HT-22 neurons were cultured with the media
conditioned by BV-2 cells in the control, siRNA-NT and siRNA-IκBα
groups in Foxc1-overexpressing BV-2 cells treated with LPS. HT-22
cell apoptosis in siRNA-IκBα group was found to be significantly
increased compared with that in siRNA-NT group, but no statistical
significance was found between the siRNA-NT and control groups
(Fig. 3M and N). These data
suggest that Foxc1 overexpression attenuated LPS-induced
inflammatory response, microglial migration and neuronal apoptosis
by inhibiting IκBα/NF-κB pathway.
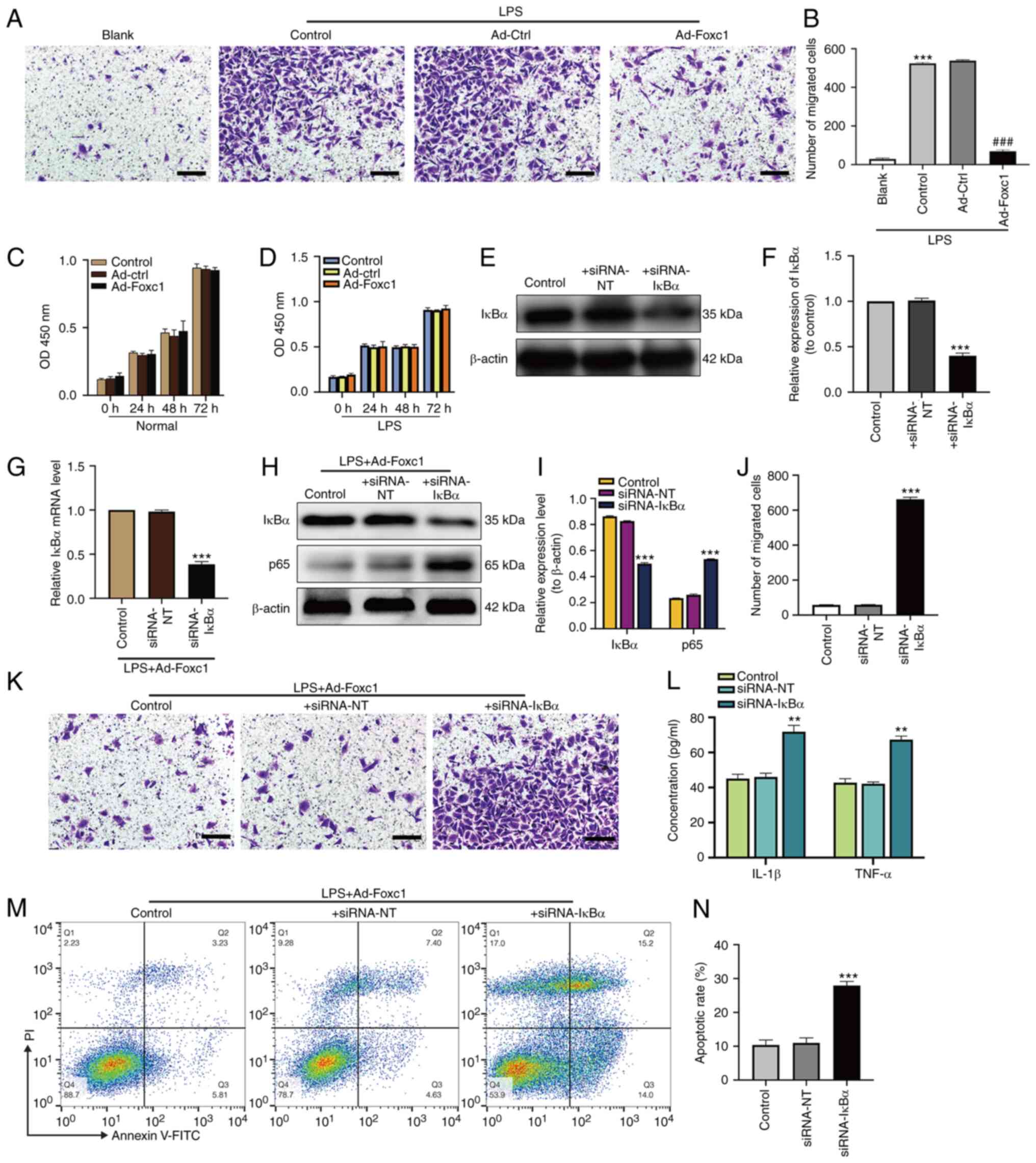 | Figure 3.Overexpression of Foxc1 inhibits
LPS-induced microglial migration, inflammatory response and
neuronal apoptosis through the NF-κB pathway. (A) Representative
images showing migrated BV-2 microglial cells with or without Foxc1
overexpression under normal or LPS conditions using Transwell
assays, (B) which were quantified. Scale bar, 50 µm. ***P<0.001
vs. blank group; ###P<0.001 vs. Ad-Ctrl group.
Viability of BV-2 cells under (C) normal or (D) LPS conditions with
or without Foxc1 overexpression were determined by the CCK-8 assay.
After siRNA transfection, transfection efficiency was analyzed by
(E) western blot analysis and (F) was semi-quantified. In the
presence of LPS, BV-2 cells with Foxc1 overexpression were
transfected with siRNA-IκBα or with siRNA-NT. Foxc1-overexpressing
BV-2 cells without siRNA transfection were used as the control
group. siRNA-mediated transfection efficiency on IκBα and p65
expression was determined by (G) reverse transcription-quantitative
PCR and (H) western blot analysis, (I) the latter of which was
quantified. (H) Quantification of BV-2 cell migration after Foxc1
overexpression in the presence of LPS using Transwell assays. (J)
Representative Transwell assay images, (K) which were quantified.
Scale bar, 50 µm. (L) After siRNA transfection, the secretion of
IL-1β and TNF-α into the cell culture supernatant by BV-2 cells
with Foxc1 overexpression in the presence of LPS was measured by
ELISA. (M) Flow cytometry analysis of HT-22 cell apoptosis after
incubation in the media conditioned by microglia. (N) The
percentage of early and late apoptotic HT-22 cells was measured by
flow cytometry analysis. Data represents the mean ± SD of three
independent experiments. **P<0.01 and ***P<0.001 vs. +
siRNA-NT group. LPS, lipopolysacharide; Ad-, adenovirus; Ctrl,
control; siRNA, small interfering RNA; Foxc1, Forkhead box C1;
IκBα, NF-κB inhibitor α. |
Cognitive impairments and
downregulation of Foxc1/IκBα in sepsis-associated
encephalopathy
Establishment of the mouse septic encephalopathy
model by CLP, animal behavior and molecular experiments were
performed as shown in Fig. 4A.
Before CLP surgery, mouse cognitive ability, including learning,
memory, and spatial orientation, were evaluated using the MWM test.
Results showed that the learning and memory abilities of spatial
orientation were not significantly different among the four groups
(Fig. S1), suggesting that Foxc1
overexpression did not alter the cognitive ability of the mice.
After CLP surgery, MWM test results showed that the swimming speed
exhibited no statistical difference throughout the 6 consecutive
days (Fig. 4B), suggesting that
neither Foxc1 overexpression nor CLP surgery affected the motor
ability of the mice. However, mice in the CLP surgery group
exhibited significantly longer escape latency compared with that in
the sham-operated group (Fig.
4C). Dwell time in the target quadrant and the frequency of
passing through the target platform area were also significantly
reduced in the CLP surgery group compared with those in the
sham-operated group (Fig. 4D and
E). The escape latency of mice in the Foxc1 overexpression
group was significantly shorter compared with that in CLP group,
whereas the dwell time in the target quadrant and the frequency of
passing through the target platform area were significantly
increased in the Foxc1 overexpression group compared with those in
the CLP group (Fig. 4C and E).
This suggests that CLP surgery impaired the cognitive ability of
the mice, which was partially prevented by Foxc1 overexpression.
Subsequently, the mRNA expression levels of Foxc1 and IκBα in the
hippocampus tissues of the mice on days 3, 7 and 14 after CLP
surgery were measured by RT-qPCR, which showed that the expression
levels of Foxc1 (Fig. 4F) and
IκBα (Fig. 4G) were significantly
reduced compared with those in the sham-operated group on days 3, 7
and 14 after CLP surgery. These in vivo results suggest that
CLP resulted in cognitive impairment in addition to the
downregulation of Foxc1 and IκBα expression in the hippocampus of
the mice. Consequently, Foxc1 and IκBα may regulate the cognitive
function of mice during inflammation.
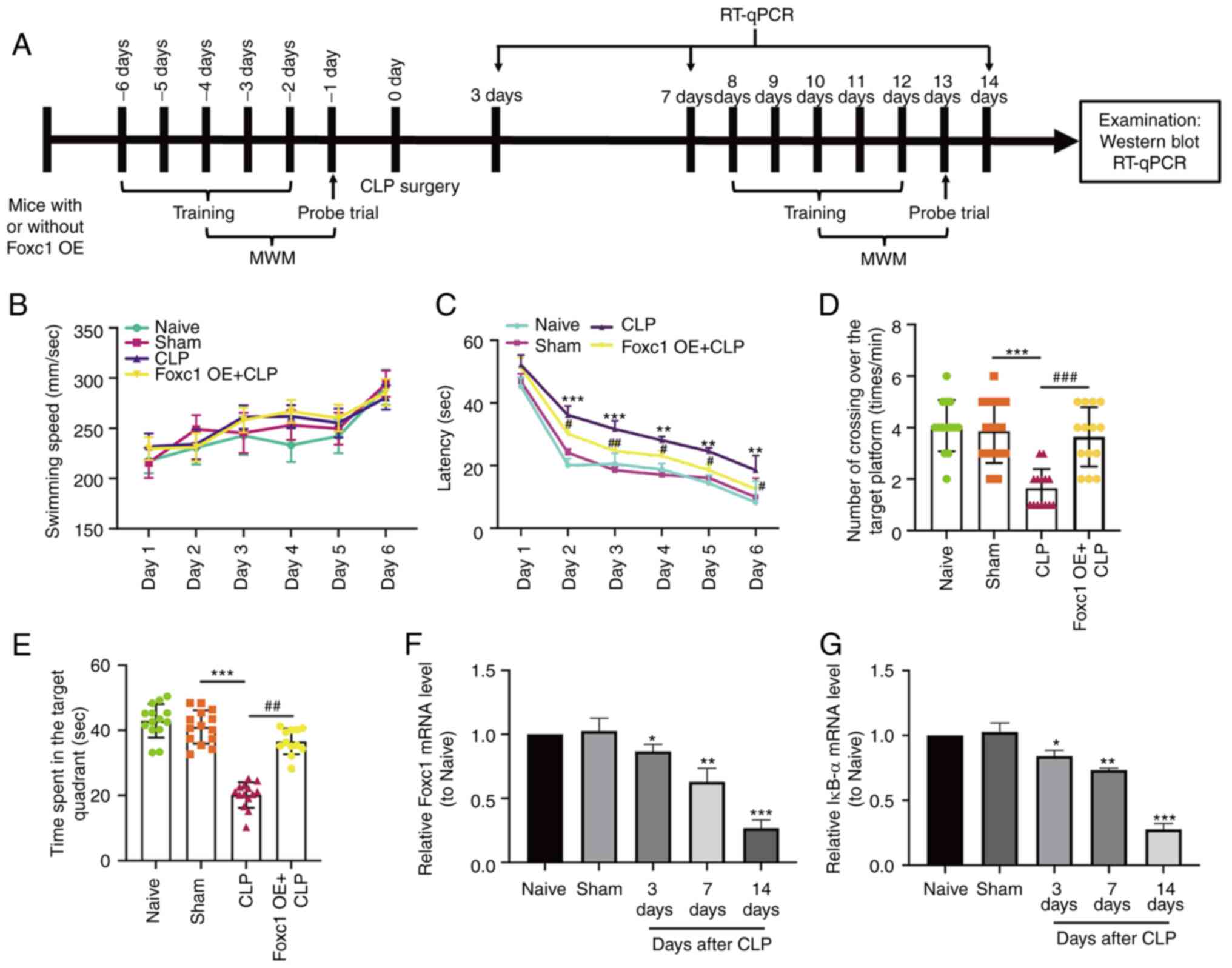 | Figure 4.Cognitive impairment and
downregulation of Foxc1 and IκBα expression in sepsis-associated
encephalopathy. (A) Timeline of the Morris Water Maze (MWM)
protocol, RT-qPCR and western blotting in naive, sham-operated,
CLP-operated and CLP + Foxc1 OE mice. (B-E) Cognitive function of
mice in each treatment group was analyzed by MWM test. (B) Average
swimming speed throughout the 6 consecutive days among the four
experimental groups. (C) Latency, defined as the time the mouse
took to find the hidden platform or first passed through the area
where the platform was located. (D) The frequency of passing
through the area where the platform was located was recorded. (E)
Dwell time in the quadrant where the platform was located. n=14.
The mRNA expression of (F) Foxc1 and (G) IκBα in the hippocampus
tissues of mice with or without CLP surgery were detected by
RT-qPCR. Data represents the mean ± SD of three independent
experiments. *P<0.05, **P<0.01 and ***P<0.001 vs. Sham.
#P<0.05, ##P<0.01 and
###P<0.001 vs. CLP group. CLP, cecal ligation and
perforation; OE, overexpression; MWM, Morris Water Maze; Foxc1,
Forkhead box C1; IκBα, NF-κB inhibitor α. |
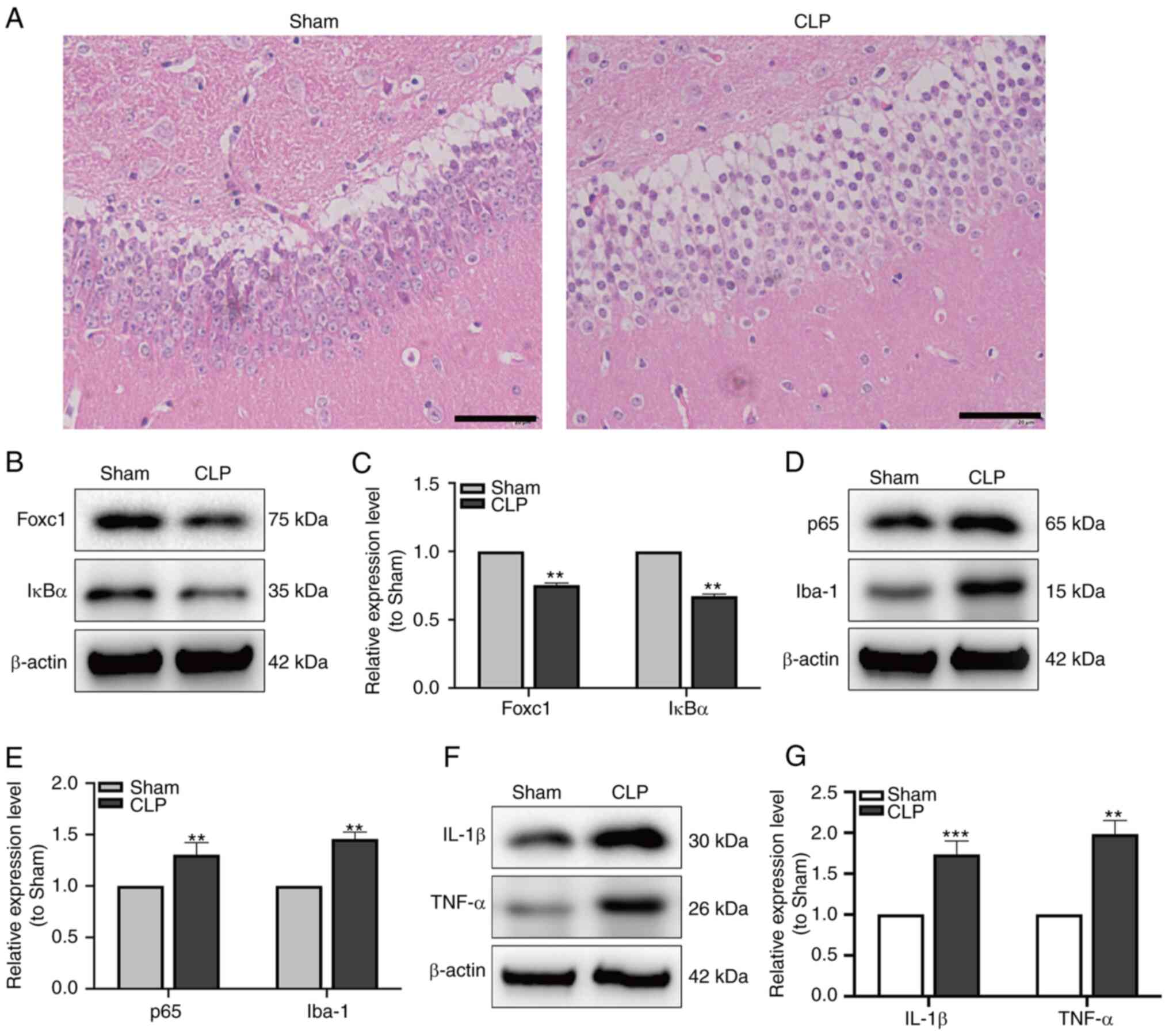
NF-κB signaling is involved in
mediating the hippocampal inflammatory response and microglial
migration in mice with SAE
After CLP surgery, to verify the establishment of
SAE, MWM trial and H&E staining were performed. Mice in the
group showed significantly longer latency and shorter dwell times
in the goal quadrant compared with those in the sham-operated group
(Fig. 4C-E). H&E staining
demonstrated that in the CLP group, the hippocampal structure was
disordered, where the nuclei of neurons were condensed and the
cytoplasm were swollen (Fig. 5A).
The expression of Foxc1 and IκBα in hippocampal tissue of mice were
measured by western blot analysis. The expression of Foxc1 and IκBα
were significantly reduced in the CLP group compared with that in
the Sham group (Fig. 5B and C).
Additionally, the expression of p65, a subunit in NF-κB family and
Iba-1, a microglia marker, were detected by western blot analysis.
The expression of both p65 and Iba-1 in the hippocampal tissues of
mice were found to be significantly increased in the CLP group
compared with that in the Sham group (Fig. 5D and E). Subsequent western blot
analysis of IL-1β and TNF-α expression in the hippocampal tissues
also showed that the expression both of these cytokines were
significantly increased in the CLP group compared with that in the
Sham group (Fig. 5F and G). These
data suggest that NF-κB pathway may serve important roles in the
hippocampal inflammatory response and microglial migration during
SAE.
Overexpression of Foxc1 attenuates
cognitive dysfunction by inhibiting microglial migration,
inflammation and neuronal apoptosis in the hippocampus of mice with
SAE through the IκBα/NF-κB pathway
The expression of Foxc1 in the hippocampus of mice
with or without Foxc1 overexpression before CLP surgery was
assessed by western blot analysis, which revealed the success of
Foxc1 overexpression in the hippocampus of mice (Fig. 6A and B). After CLP surgery,
H&E staining demonstrated that the hippocampal structure
appeared to be more ordered with cell volume smaller, where the
extent of cytosolic edema was decreased in tissues from the Foxc1
overexpression group compared with those in the CLP group (Fig. 6C). To investigate the role of the
NF-κB pathway in the hippocampal inflammatory response, microglial
migration and neuronal apoptosis in mice with SAE,
Foxc1-overexpressing mice were used. After CLP surgery, the
expression of Foxc1 and IκBα in the hippocampus of mice were
examined using western blot analysis. Results showed that the
expression levels of Foxc1 and IκBα were significantly higher in
the CLP + Foxc1 OE group compared with that in the CLP group
(Fig. 6D and E). The effect of
Foxc1 overexpression on the expression of p65, Iba-1, IL-1β and
TNF-α in the hippocampus tissues of mice were next detected by
western blot analysis, which showed that Foxc1 overexpression
significantly inhibited the expression of p65, Iba-1, IL-1β and
TNF-α in the compared with that in the CLP group (Fig. 6F-I). Subsequently, microglial
migration and neuronal apoptosis in the hippocampus of mice were
measured using immunofluorescence and TUNEL staining. The number of
Iba-1-positive and TUNEL-positive cells were both marked increased
in the CLP group compared with those in the Sham group, but the
number of Iba-1-positive and TUNEL-positive cells in the CLP +
Foxc1 OE group were both markedly decreased compared with those in
the CLP group (Fig. 6J-L). These
results suggest that Foxc1 overexpression can inhibit microglial
migration, inflammation and neuronal apoptosis in the hippocampus
of mice with SAE through the NF-κB pathway.
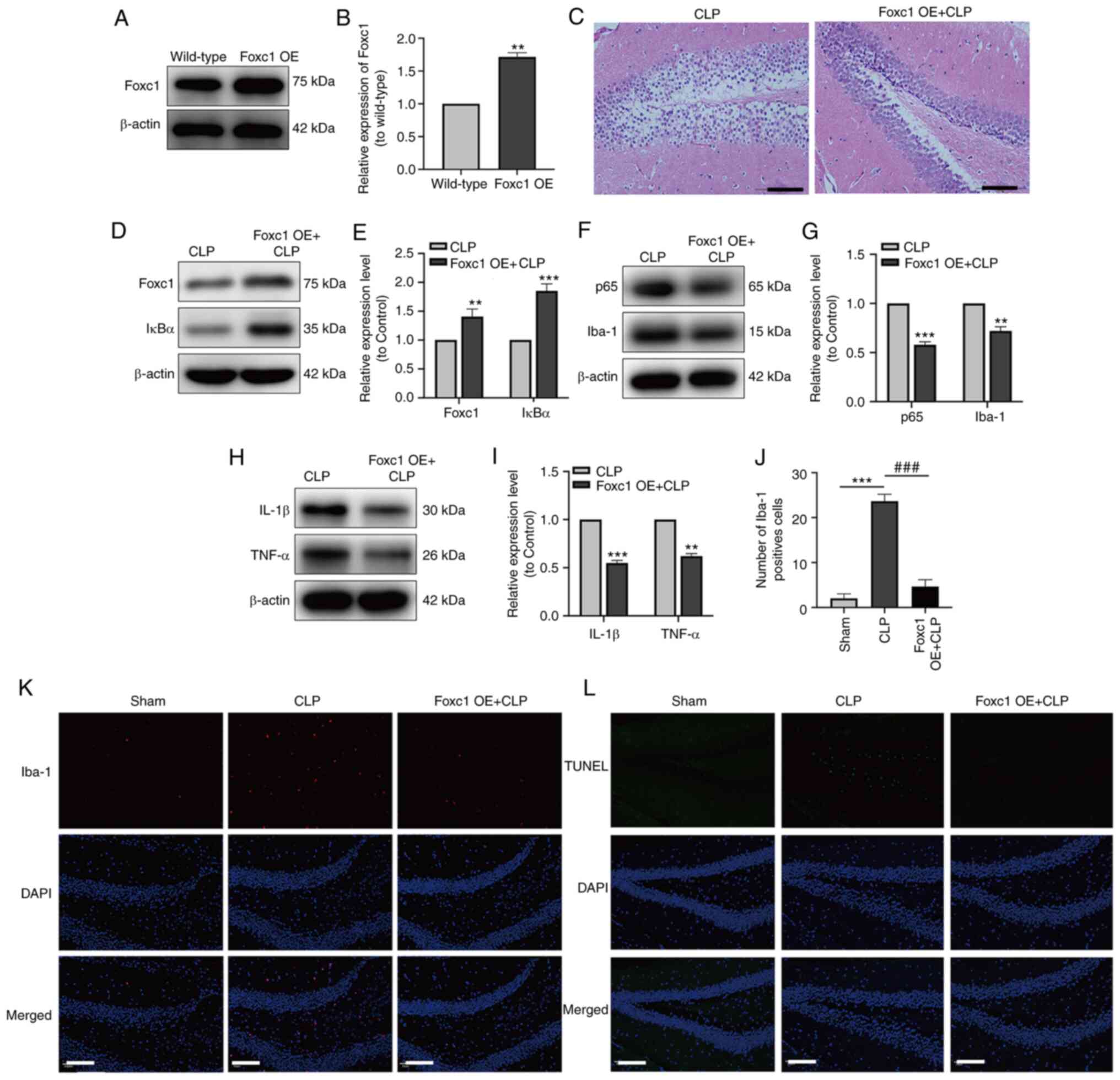 | Figure 6.Overexpression of Foxc1 inhibits
microglial migration, suppresses the inflammatory response and
neuronal apoptosis in the hippocampus of mice with
sepsis-associated encephalopathy through the NF-κB pathway. The
expression of Foxc1 in the hippocampus of mice with or without
Foxc1 overexpression was analyzed by (A) western blot analysis and
(B) was semi-quantified, **P<0.01 vs. wild-type group. (C)
H&E staining of the mouse hippocampal tissue. Scale bar, 100
µm. (D) The expression of Foxc1 and IκBα in hippocampal tissue of
mice was measured by western blotting (E) and quantified. (F) The
expression of p65 and Iba-1 in the hippocampal tissues of the mice
was measured by western blotting (G) and quantified. (H) The
expression of IL-1β and TNF-α in the hippocampal tissues of mice
was measured by western blot analysis (I) and quantified. (K)
Immunofluorescence staining quantification of Iba-1 in the mouse
hippocampal tissue. (J) Representative immunofluorescence images of
Iba-1 staining, where red dots represent microglia. Scale bar, 20
µm; n=3/group. (L) TUNEL staining, where green dots represent
apoptotic neurons. Scale bar, 20 µm. **P<0.01 and ***P<0.001
vs. CLP or as indicated; ###P<0.001 as indicated.
Foxc1, Forkhead box C1; OE, overexpression; IκBα, NF-κB inhibitor
α; CLP, cecal ligation and perforation; Iba-1, allograft
inflammatory factor 1. |
Discussion
The present study provided information on the
potential mechanism underlying cognitive dysfunction in SAE. LPS
induced inflammatory responses in the microglia, increased
microglial migration and neuronal apoptosis in addition to
downregulating Foxc1 and IκBα expression in vitro.
Furthermore, sepsis induced by CLP surgery led to cognitive
dysfunctions accompanied with increased longer escape latency,
decreased dwell time in the target quadrant and decreased frequency
of passing through the target platform area in the MWM test.
Decreased expression of Foxc1 and IκBα, increased expression of
p65, Iba-1, IL-1β and TNF-α and increased neuronal apoptosis were
also observed in the hippocampal tissues of mice following CLP. It
was found that Foxc1 overexpression promoted IκBα expression,
inhibited microglial migration, inflammation and neuronal apoptosis
both in vitro and in vivo, which prevented cognitive
dysfunction in mice induced by CLP. In addition, IκBα knockdown
using siRNA in microglial cells reversed the inhibitory effects of
Foxc1 overexpression on the inflammatory response, microglial
migration and neuronal apoptosis. Collectively, these data suggest
that Foxc1 overexpression can inhibit microglial migration,
hippocampal inflammatory response and neuronal apoptosis to
alleviate sepsis-induced cognitive dysfunction.
In the present study, LPS reduced the expression of
Foxc1 in microglial BV-2 cells, whilst CLP surgery also
downregulated the expression of Foxc1 in the hippocampus of mice
compared with their corresponding control groups. Foxc1 contains a
common 100-amino acid winged-helix DNA-binding domain that serves
key roles in cell proliferation, differentiation migration and
survival (19,33). A previous study showed that Foxc1
promoted the migration of hepatocellular carcinoma (HCC) cells, as
knockdown of Foxc1 inhibited the migration of HCC cells (34). Additionally, overexpression of
Foxc1 in breast cancer cells was found to increase proliferation,
migration and invasion, whilst Foxc1 knockdown exerted opposite
effects (22). The present study
revealed that the overexpression of Foxc1 inhibited the LPS-induced
microglial migration both in vitro and in vivo. This
finding is not consistent with the previously reported effects of
Foxc1 on cancer cellular migration.
Microglia is a highly specialized resident
population of macrophage-like immune cells in the CNS that serves
ambiguous functions, since they can mediate both neuroprotective
and neurotoxic effects (35).
Microglial cells serve essential roles in brain development and are
responsible for maintaining homeostasis in the CNS (36). In addition, microglia-mediated
neuroinflammation serves important roles in neuronal repair and the
regulation of inflammation in the CNS (37,38). However, microglia have also been
shown to promote neuroinflammation-associated cognitive impairment
(39). Activated microglia can
polarize towards the M1 and M2 phenotypes
(40). The M1
phenotype mainly migrates towards the site of neuroinflammation,
where it secretes a number of proinflammatory cytokines, including
IL-1β, TNF-α and adhesion molecules (such as CD86) (40). These cytokines then disrupt the
BBB, which potentiates further microglia activation, leading to a
vicious cycle and eventual neuronal damage (41–43). By contrast, the M2
phenotype is generally associated with tissue regeneration by
secreting anti-inflammatory cytokines to inhibit microglia
polarization into the M1 phenotype (44,45). In the present study, LPS induced
the secretion of IL-1β and TNF-α by the microglial cells,
suggesting that LPS induced microglial polarization into the
M1 phenotype. In addition, increased IL-1β and TNF-α
expression was observed in the hippocampal tissues of mice
following CLP surgery. This is consistent with the findings from a
previous hypoxic-ischemic brain injury study, which found that
hypoxia-ischemia facilitated M1 phenotype polarization
whilst attenuating M2 phenotype activation (46). It is noteworthy that the
overexpression of Foxc1 decreased the expression of IL-1β and TNF-α
in the present study, suggesting that Foxc1 overexpression promoted
microglial polarization into the M2 phenotype whilst
suppressing the M1 phenotype. Under physiological
conditions, microglial cells are sparsely distributed in the CNS.
After the CNS becomes infected or damaged, microglia will rapidly
proliferate and migrate to the damaged site (47). In the present study, LPS was found
to induce microglial migration in vitro, whereas the
expression of Iba-1, a microglia marker protein, was also
significantly increased in the hippocampal tissues of mice
following CLP surgery. In addition, increased neuroinflammation was
accompanied with increased microglial migration, suggesting that
microglial migration promoted the inflammatory response. Given its
key role in mediating neuroinflammation, the regulation of
migration and phenotype polarization in microglia is likely to be
an important factor in SAE. However, the regulatory mechanism
remains unclear at present.
The NF-κB signaling pathway serves an important role
in the regulation of inflammation; NF-κB normally exists in the
cytosol as an inactive form, where it forms a complex with
inhibitory IκBα proteins (48,49). NF-κB pathway has been previously
reported to be part of the mechanism underlying SAE-related CNS
damage (50). Members of the
NF-κB family include p50, p52, p65, RelB and c-Rel subunits. In
total, two separate activation pathways for NF-κB have been
reported, namely the canonical pathway and the alternative pathway
(51). In the canonical pathway,
phosphorylated IκBα is degraded by the proteasome, causing it to
separate from the IκBα/p65 complex (51). The p65 subunit is then released,
where it translocates into the nucleus to promote NF-κB-dependent
gene transcription (51). This
results in the accumulation of NF-κB homodimers and heterodimers in
the nucleus (51). Therefore,
decreased expression of IκBα can be applied to indirectly measure
the status of NF-κB activation (48). The alternative pathway can be
activated by a small subset of tumor necrosis factors, such as CD40
ligand and lymphotoxin B. By contrast, the canonical pathway can
also be activated by proinflammatory cytokines, such as TNF-α and
IL-1, usually leading to the activation of NF-κB pathway; briefly,
mitogen kinase and phosphorylated-IκBα protein kinase are
activated, leading to the degradation of NF-κB p100 via
phosphorylation, and the formation of the P52/RelB heterodimer and
NF-κB P50/RelB heterodimer, which enter the nucleus and regulate
the transcription of target genes (48). A previous study has reported that
inflammatory encephalopathy enhanced NF-κB activity primarily
through the canonical pathway (52). The present study showed that LPS
treatment downregulated the expression of IκBα but upregulated the
expression of p65 in microglia, suggesting that the activation of
NF-κB was likely through the canonical pathway. This notion was
supported further by observations of increased IL-1β and TNF-α
secretion/expression. However, the underlying mechanism of this
LPS-induced activation of NF-κB pathway in microglia require
further study.
To examine the association between Foxc1 and IκBα in
SAE-related hippocampal inflammatory response, the expression of
Foxc1 was overexpressed in microglia. Overexpression of Foxc1 was
found to upregulate the expression of IκBα in microglia, in
addition to inhibiting microglial migration and the inflammatory
response, suggesting that overexpression of Foxc1 stabilized the
IκBα/p65 complex and inhibited p65 activation to suppress the
transcription of proinflammatory cytokines in microglia.
Subsequently, the expression of IκBα was then knocked down in
microglia, which reversed the inhibitory effects of Foxc1
overexpression on microglial migration and inflammation. This
suggests that Foxc1 overexpression inhibits the NF-κB pathway by
promoting the expression of IκBα, which then inhibits microglial
migration and inflammation during SAE. Microglia-mediated
neuroinflammatory response has been reported to at least partially
mediate the pathology of various neurodegenerative diseases,
including Alzheimer's disease, Parkinson's disease (PD) and
multiple sclerosis (53–55). A number of studies have documented
that microglia-mediated inflammation can aggravate neuronal axon
and synaptic damage, increase demyelination, destroy the integrity
of the white matter and induce neuronal apoptosis, all of which
eventually leading to progressive neurodegeneration or even death
(40,56,57). The present study also showed that
mice following CLP surgery manifested cognitive impairments
accompanied with increased neuroinflammation and neuronal apoptosis
in the hippocampus, in accordance with the hypothesis that
neuroinflammation is deleterious to CNS. In addition,
overexpression of Foxc1 was found to inhibit neuroinflammation,
neuronal apoptosis whilst alleviating cognitive dysfunction in the
present study, this suggests that the anti-inflammatory function of
Foxc1 may contribute to protective effects against sepsis-induced
neuronal impairments and cognitive dysfunction.
In conclusion, in vitro and in vivo
approaches were both applied to reveal a novel function of Foxc1 on
microglia-mediated inflammatory response, migration and neuronal
apoptosis during sepsis-induced cognitive dysfunction. The present
study also provided evidence that Foxc1 can negatively regulate the
microglia-mediated inflammatory response, microglial migration and
neuronal apoptosis through the NF-κB pathway. The adds support for
the future therapeutic targeting of Foxc1 for the treatment of
sepsis-related cognitive dysfunction in SAE.
In summary, CLP surgery induced-sepsis caused
inflammatory responses in the hippocampus mediated by microglia,
resulting in deficits of cognitive function. Foxc1 overexpression
prevented sepsis-induced neuroinflammation, microglial migration,
neuronal apoptosis and cognitive dysfunction, which was likely
mediated through its effects on IκBα and NF-κB signaling.
Therefore, targeting Foxc1/IκBα signaling may serve as a viable
therapeutic strategy for treating or preventing cognitive
dysfunction in SAE.
Supplementary Material
Supporting Data
Acknowledgements
Not applicable.
Funding
The present study was supported by Chinese International Medical
Exchange Foundation 2021 (grant no. Z-2016-23-2101-37),
Cardiovascular Multidisciplinary Integrated Thinking Research Fund
(grant no. z-2016-23-2101-37) and Key Scientific Research Project
Plan of Henan University (grant no. 20A320007).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
Hongyu W conducted most of the experiments and
drafted the manuscript. YS, CL and XQ made substantial
contributions to data analysis and data interpretation. DZ and XJ
were involved in performing some of the experiments and data
analysis. Hongwei W and SZ made substantial contributions to
conception and design, were involved in revising the manuscript
critically for important intellectual content, and gave their final
approval of the published version. All authors read and approved
the final version of the manuscript. Hongyu W and Hongwei W
confirmed the authenticity of all the raw data.
Ethics approval and consent to
participate
All study procedures were approved by the
Institutional Animal Care and Use Committee of Shanghai Jiao Tong
University School of Medicine (approval no. XHEC-F-2019-030;
Shanghai, China).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no conflict of
interests.
References
|
1
|
Gofton TE and Young GB: Sepsis-associated
encephalopathy. Nat Rev Neurol. 8:557–566. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Czempik PF, Pluta MP and Krzych ŁJ:
Sepsis-associated brain dysfunction: A review of current
literature. Int J Environ Res Public Health. 17:58522020.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Sonneville R, de Montmollin E, Poujade J,
Garrouste-Orgeas M, Souweine B, Darmon M, Mariotte E, Argaud L,
Barbier F, Goldgran-Toledano D, et al: Potentially modifiable
factors contributing to sepsis-associated encephalopathy. Intensive
Care Med. 43:1075–1084. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Abe N, Nishihara T, Yorozuya T and Tanaka
J: Microglia and macrophages in the pathological central and
peripheral nervous systems. Cells. 9:21322020. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Michels M, Abatti M, Vieira A, Ávila P,
Goulart AI, Borges H, Córneo E, Dominguini D, Barichello T and
Dal-Pizzol F: Modulation of microglial phenotypes improves
sepsis-induced hippocampus-dependent cognitive impairments and
decreases brain inflammation in an animal model of sepsis. Clin Sci
(Lond). 134:765–776. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Wang LM, Wu Q, Kirk RA, Horn KP, Ebada
Salem AH, Hoffman JM, Yap JT, Sonnen JA, Towner RA, Bozza FA, et
al: Lipopolysaccharide endotoxemia induces amyloid-β and p-tau
formation in the rat brain. Am J Nucl Med Mol Imaging. 8:86–99.
2018.PubMed/NCBI
|
|
7
|
Gunther ML, Morandi A, Krauskopf E,
Pandharipande P, Girard TD, Jackson JC, Thompson J, Shintani AK,
Geevarghese S, Miller RR III, et al: The association between brain
volumes, delirium duration, and cognitive outcomes in intensive
care unit survivors: The VISIONS cohort magnetic resonance imaging
study*. Crit Care Med. 40:2022–2032. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Adam N, Kandelman S, Mantz J, Chrétien F
and Sharshar T: Sepsis-induced brain dysfunction. Expert Rev Anti
Infect Ther. 11:211–221. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Maddux AB, Hiller TD, Overdier KH, Pyle LL
and Douglas IS: Innate immune function and organ failure recovery
in adults with sepsis. J Intensive Care Med. 34:486–494. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Dal-Pizzol F, Tomasi CD and Ritter C:
Septic encephalopathy: Does inflammation drive the brain crazy?
Braz J Psychiatry. 36:251–258. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Michels M, Vieira AS, Vuolo F, Zapelini
HG, Mendonça B, Mina F, Dominguini D, Steckert A, Schuck PF,
Quevedo J, et al: The role of microglia activation in the
development of sepsis-induced long-term cognitive impairment. Brain
Behav Immun. 43:54–59. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Margotti W, Giustina AD, de Souza Goldim
MP, Hubner M, Cidreira T, Denicol TL, Joaquim L, De Carli RJ,
Danielski LG, Metzker KL, et al: Aging influences in the
blood-brain barrier permeability and cerebral oxidative stress in
sepsis. Exp Gerontol. 140:1110632020. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Shulyatnikova T and Verkhratsky A:
Astroglia in sepsis associated encephalopathy. Neurochem Res.
45:83–99. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Zhang SP, Yang RH, Shang J, Gao T, Wang R,
Peng XD, Miao X, Pan L, Yuan WJ, Lin L and Hu QK: FOXC1
up-regulates the expression of toll-like receptors in myocardial
ischaemia. J Cell Mol Med. 23:7566–7580. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Xia W, Zhu J, Wang X, Tang Y, Zhou P, Wei
X, Chang B, Zheng X, Zhu W, Hou M and Li S: Overexpression of Foxc1
regenerates crushed rat facial nerves by promoting Schwann cells
migration via the Wnt/β-catenin signaling pathway. J Cell Physiol.
235:9609–9622. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Liu J, Zhang Z, Li X, Chen J, Wang G, Tian
Z, Qian M, Chen Z, Guo H, Tang G, et al: Forkhead box C1 promotes
colorectal cancer metastasis through transactivating ITGA7 and
FGFR4 expression. Oncogene. 37:5477–5491. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Xia S, Qu J, Jia H, He W, Li J, Zhao L,
Mao M and Zhao Y: Overexpression of Forkhead box C1 attenuates
oxidative stress, inflammation and apoptosis in chronic obstructive
pulmonary disease. Life Sci. 216:75–84. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Zhao L, Zhang R, Su F, Dai L, Wang J, Cui
J, Huang W and Zhang S: FoxC1-induced vascular niche improves
survival and myocardial repair of mesenchymal stem cells in
infarcted hearts. Oxid Med Cell Longev. 2020:78653952020.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Cao Q, Wang X, Shi Y, Zhang M, Yang J,
Dong M, Mi Y, Zhang Z, Liu K, Jiang L, et al: FOXC1 silencing
inhibits the epithelial-to-mesenchymal transition of glioma cells:
Involvement of β-catenin signaling. Mol Med Rep. 19:251–261.
2019.PubMed/NCBI
|
|
20
|
Nishimura DY, Swiderski RE, Alward WL,
Searby CC, Patil SR, Bennet SR, Kanis AB, Gastier JM, Stone EM and
Sheffield VC: The forkhead transcription factor gene FKHL7 is
responsible for glaucoma phenotypes which map to 6p25. Nat Genet.
19:140–147. 1998. View
Article : Google Scholar : PubMed/NCBI
|
|
21
|
Huang L, Huang Z, Fan Y, He L, Ye M, Shi
K, Ji B, Huang J, Wang Y and Li Q: FOXC1 promotes proliferation and
epithelial-mesenchymal transition in cervical carcinoma through the
PI3K-AKT signal pathway. Am J Transl Res. 9:1297–1306.
2017.PubMed/NCBI
|
|
22
|
Ray PS, Wang J, Qu Y, Sim MS, Shamonki J,
Bagaria SP, Ye X, Liu B, Elashoff D, Hoon DS, et al: FOXC1 is a
potential prognostic biomarker with functional significance in
basal-like breast cancer. Cancer Res. 70:3870–3876. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Omatsu Y, Seike M, Sugiyama T, Kume T and
Nagasawa T: Foxc1 is a critical regulator of haematopoietic
stem/progenitor cell niche formation. Nature. 508:536–540. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Hayden MS and Ghosh S: Shared principles
in NF-kappaB signaling. Cell. 132:344–362. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Dolcet X, Llobet D, Pallares J and
Matias-Guiu X: NF-kB in development and progression of human
cancer. Virchows Arch. 446:475–482. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Nguyen PL, Bui BP, Lee H and Cho J: A
Novel 1,8-Naphthyridine-2-carboxamide derivative attenuates
inflammatory responses and cell migration in LPS-Treated BV2 Cells
via the suppression of ROS generation and TLR4/Myd88/NF-κB
signaling pathway. Int J Mol Sci. 22:25272021. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Campesi I, Montella A and Franconi F:
Human monocytes respond to lipopolysaccharide (LPS) stimulation in
a sex-dependent manner. J Cell Physiol. Jul 12–2021.(Epub ahead of
print). doi: 10.1002/jcp.30503. PubMed/NCBI
|
|
28
|
National Research Council Institute for
Laboratory Animal R, . Guide for the Care and Use of Laboratory
Animals National Academies Press (US) Copyright 1996 by the
National Academy of Sciences. Washington, DC: 1996.
|
|
29
|
Yin J, Shen Y, Si Y, Zhang Y, Du J, Hu X,
Cai M, Bao H and Xing Y: Knockdown of long non-coding RNA SOX2OT
downregulates SOX2 to improve hippocampal neurogenesis and
cognitive function in a mouse model of sepsis-associated
encephalopathy. J Neuroinflammation. 17:3202020. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Gao H, Han Z, Huang S, Bai R, Ge X, Chen F
and Lei P: Intermittent hypoxia caused cognitive dysfunction relate
to miRNAs dysregulation in hippocampus. Behav Brain Res. 335:80–87.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Shan G, Tang T, Xia Y and Qian HJ: Long
non-coding RNA NEAT1 promotes bladder progression through
regulating miR-410 mediated HMGB1. Biomed Pharmacother.
121:1092482020. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) Method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Wang H, Yang T, Sun J, Zhang S and Liu S:
SENP1 modulates microglia-mediated neuroinflammation toward
intermittent hypoxia-induced cognitive decline through the
de-SUMOylation of NEMO. J Cell Mol Med. 25:6841–6854. 2021.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Wang J, Ray PS, Sim MS, Zhou XZ, Lu KP,
Lee AV, Lin X, Bagaria SP, Giuliano AE and Cui X: FOXC1 regulates
the functions of human basal-like breast cancer cells by activating
NF-κB signaling. Oncogene. 31:4798–4802. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Huang W, Chen Z, Zhang L, Tian D, Wang D,
Fan D, Wu K and Xia L: Interleukin-8 induces expression of FOXC1 to
promote transactivation of CXCR1 and CCL2 in hepatocellular
carcinoma cell lines and formation of metastases in mice.
Gastroenterology. 149:1053–1067.e14. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Chen Z and Trapp BD: Microglia and
neuroprotection. J Neurochem. 136 (Suppl 1):S10–S17. 2016.
View Article : Google Scholar
|
|
37
|
Kettenmann H, Hanisch UK, Noda M and
Verkhratsky A: Physiology of microglia. Physiol Rev. 91:461–553.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Colonna M and Butovsky O: Microglia
function in the central nervous system during health and
neurodegeneration. Annu Rev Immunol. 35:441–468. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Vaessen TJ, Overeem S and Sitskoorn MM:
Cognitive complaints in obstructive sleep apnea. Sleep Med Rev.
19:51–58. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Kiernan EA, Smith SMC, Mitchell GS and
Watters JJ: Mechanisms of microglial activation in models of
inflammation and hypoxia: Implications for chronic intermittent
hypoxia. J Physiol. 594:1563–1577. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
de Lima FFF, Mazzotti DR, Tufik S and
Bittencourt L: The role inflammatory response genes in obstructive
sleep apnea syndrome: A review. Sleep Breath. 20:331–338. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Calsolaro V and Edison P:
Neuroinflammation in Alzheimer's disease: Current evidence and
future directions. Alzheimers Dement. 12:719–732. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Yang Q, Wang Y, Feng J, Cao J and Chen B:
Intermittent hypoxia from obstructive sleep apnea may cause
neuronal impairment and dysfunction in central nervous system: The
potential roles played by microglia. Neuropsychiatr Dis Treat.
9:1077–1086. 2013.PubMed/NCBI
|
|
44
|
Zhai Q, Li F, Chen X, Jia J, Sun S, Zhou
D, Ma L, Jiang T, Bai F, Xiong L and Wang Q: Triggering receptor
expressed on myeloid cells 2, a novel regulator of immunocyte
phenotypes, confers neuroprotection by relieving neuroinflammation.
Anesthesiology. 127:98–110. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Xia CY, Zhang S, Gao Y, Wang ZZ and Chen
NH: Selective modulation of microglia polarization to M2 phenotype
for stroke treatment. Int Immunopharmacol. 25:377–382. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Hellström Erkenstam N, Smith PL, Fleiss B,
Nair S, Svedin P, Wang W, Boström M, Gressens P, Hagberg H, Brown
KL, et al: Temporal characterization of microglia/macrophage
phenotypes in a mouse model of neonatal hypoxic-ischemic brain
injury. Front Cell Neurosci. 10:2862016. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Dou Y, Wu HJ, Li HQ, Qin S, Wang YE, Li J,
Lou HF, Chen Z, Li XM, Luo QM and Duan S: Microglial migration
mediated by ATP-induced ATP release from lysosomes. Cell Res.
22:1022–1033. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Mitchell S, Vargas J and Hoffmann A:
Signaling via the NF-κB system. Wiley Interdiscip Rev Syst Biol
Med. 8:227–241. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Viatour P, Merville MP, Bours V and
Chariot A: Phosphorylation of NF-kappaB and IkappaB proteins:
Implications in cancer and inflammation. Trends Biochem Sci.
30:43–52. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Chen S, Tang C, Ding H, Wang Z, Liu X,
Chai Y, Jiang W, Han Y and Zeng H: Maf1 ameliorates
sepsis-associated encephalopathy by suppressing the NF-κB/NLRP3
inflammasome signaling pathway. Front Immunol. 11:5940712020.
View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Matthew SH and Sankar G: Shared principles
in NF-kappaB signaling. Cell. 132:344–362. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Oliver KM, Garvey JF, Ng CT, Veale DJ,
Fearon U, Cummins EP and Taylor CT: Hypoxia activates
NF-kappaB-dependent gene expression through the canonical signaling
pathway. Antioxid Redox Signal. 11:2057–2064. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Regen F, Hellmann-Regen J, Costantini E
and Reale M: Neuroinflammation and Alzheimer's Disease:
Implications for microglial activation. Curr Alzheimer Res.
14:1140–1148. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Ho MS: Microglia in Parkinson's Disease.
Adv Exp Med Biol. 1175:335–353. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Bjelobaba I, Savic D and Lavrnja I:
Multiple sclerosis and neuroinflammation: The overview of current
and prospective therapies. Curr Pharm Des. 23:693–730. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Hong S, Beja-Glasser VF, Nfonoyim BM,
Frouin A, Li S, Ramakrishnan S, Merry KM, Shi Q, Rosenthal A,
Barres BA, et al: Complement and microglia mediate early synapse
loss in Alzheimer mouse models. Science. 352:712–716. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Wang H, Xiong W, Hang S, Wang Y, Zhang S
and Liu S: Depletion of SENP1-mediated PPARγ SUMOylation
exaggerates intermittent hypoxia-induced cognitive decline by
aggravating microglia-mediated neuroinflammation. Aging (Albany
NY). 13:15240–15254. 2021. View Article : Google Scholar : PubMed/NCBI
|