Introduction
The human spalt-like transcription factor 4
(SALL4; OMIM no. 607343) gene was cloned and named by
Kohlhase et al (1). It is
located on chromosome 20q13.2 and consists of 3,162 bp and four
exons (2). The protein consists
of 1,053 amino acids. SALL4 is a transcription factor that
is widely expressed in the early stage of embryo development
(3). It is a master regulator
that contributes to the growth and differentiation of cancer stem
cells (4). Homozygous
loss-of-function SALL4 mutations are embryo lethal. The
SALL4 protein has multiple zinc finger motifs (ZFMs) of the C2H2
type. The fragments near the amino and carboxyl terminals each
encode one ZFM, while the middle fragment encodes two ZFMs
(5). These ZFMs serve an
important role in maintaining protein function (6). Variants of SALL4 cause
Okihiro syndrome [also known as Duane radial-ray syndrome (DRRS);
OMIM no. 607323], which is an autosomal dominant genetic disease
(7). Okihiro syndrome was first
discovered by Okihiro et al (8) in 1977. This syndrome is
characterized by radial malformation associated with the Duane
anomaly, which is defined as a limitation of abduction and
narrowing of the palpebral fissure with retraction of the globe on
adduction (7). Patients with
Okihiro syndrome exhibit different degrees of radial line
hypoplasia and other types of deformity, which include hypoplasia
of the thenar, radius and forearm, spinal and external ear
deformity, hearing impairment and heart and kidney abnormality
(9–11). SALL4 is highly expressed in
the developing midbrain, branchial arch and limbs in healthy
individuals, but also serves an important role in the development
of skeletal and ocular structures associated with Okihiro syndrome
(1,12). The present study investigated the
clinical characteristics of a case of Okihiro syndrome. Gene
sequencing and bioinformatics analysis were performed to determine
whether a SALL4 variant was the molecular cause of the
syndrome.
Materials and methods
Case data
The proband was from Yunnan, China, had profound
congenital sensorineural deafness and went to the hospital in July
2020. He received a cochlear implant from Kunming Children's
Hospital (KCH). Clinical data from the proband (male, 23 months
old) and his father (28 years old) and mother (29 years old) were
collected via questionnaires. The patient underwent examination of
audiology, ophtalmology, hair, skin, limb joints, blood and
digestive system. Otoacoustic emission, acoustic immittance,
auditory brain stem response (ABR), multiple steady-state response
and verbal hearing aid tests were performed to assess audition of
the proband. In addition, temporal CT and cranial MRI scans were
also performed prior to cochlear implant. Other routine imaging
examinations were performed on the patient: These included color
duplex ultrasonography of the liver, bile, pancreas, spleen and
kidney, in addition to cardiac ultrasonography and brain MRI.
Genetic diagnoses were performed with consent from the proband and
his parents. The present study was approved by the Medical Ethics
Committee of KCH.
DNA library preparation
Genomic DNA was extracted from proband and his
parents whole blood (3 ml) using QIAamp DNA Mini kit (Qiagen GmbH),
in accordance with the manufacturer's instructions. DNA was
quantified using Nanodrop 2000 (Thermo Fisher Scientific, Inc.). A
total of ≥3 µg DNA was used for the indexed Illumina libraries, in
accordance with the manufacturer's instructions (Generic DNA
Library preparation Kit v5.0; cat. no. MG0126; MyGenostics, Inc.).
DNA fragments 350–450 bp in length and included adapter sequences
were selected for DNA libraries. The final libraries were qualified
using an Agilent 2100 Bioanalyzer system using the Agilent High
Sensitivity DNA kit (cat. no. 5067-4626).
Whole-exome sequencing
Before sequencing, the molar mass of the library was
calculated according to its concentration and fragment size from
the 2100 Bioanalyzer system, using the formula as following:
Library molar concentration (nM)=Library concentration (ng/µl) ×
106/656.6 × Length. The loading concentration of the
final library was 2 pmol. The enriched libraries were sequenced on
NovaSeq6000 sequencer (Illumina, Inc.) using NovaSeq6000 S4 Reagent
Kit v1.5 (300 cycles; cat. no. 20028312) to obtain paired-end reads
with a length of 150 bp.
Data analysis and interpretation
The output data from Illumina NovaSeq were converted
from bcl to fastq files by Bcl2Fastq software (Bcl2Fastq 2.18.0.12;
Illumina, Inc.). Both Illumina sequencing adapters and low-quality
reads (<80 bp) were filtered using fastp (13). The clean reads were mapped to the
UCSC hg19 human reference genome using BWA software (v0.7.12-r1044;
http://bio-bwa.sourceforge.net/)
(14). The duplicated reads were
removed using Picard (v2.2.3; http://broadinstitute.github.io/picard/) and mapped
reads were used for the detection of variants. Single nucleotide
polymorphism (SNP) and insertion/deletion variants were detected
using HaplotypeCaller from the GATK software (v4.1.4.0; http://software.broadinstitute.org/gatk/). The data
were converted to VCF format. Variants were annotated using ANNOVAR
software (16 Apr 2018; http://annovar.openbioinformatics.org/en/latest/).
Multiple databases, including 1000 Genomes (201508 collection v5b),
ESP6500 (esp6500siv2), dbSNP (dbSNP150), EXAC (http://exac.broadinstitute.org/) and Human Gene
Mutation Database (HGMD, http://www.hgmd.cf.ac.uk/ac/index.php) were used to
annotate variants to estimate their frequency. Variants were
predicted using SIFT (http://provean.jcvi.org/index.php), PolyPhen-2
(http://genetics.bwh.harvard.edu/pph2/), MutationTaster
(http://www.mutationtaster.org/) and
GERP++ (http://mendel.stanford.edu/SidowLab/downloads/gerp/).
Both the CNVKit (CNVkit 0.9.8 documentation) and the delly software
(v0.9.1; www.korbel.embl.de/software.html) were used to call
copy number and structural variation across all regions. Candidate
copy number variations (CNVs) were annotated by analyzing the genes
contained in the CNVs and the CNV intervals themselves with the
databases Decipher (v11.9, http://decipher.sanger.ac.uk/), dbVar (http://www.ncbi.nlm.nih.gov/dbvar), ClinGen
(https://www.clinicalgenome.org/), and
OMIM (http://www.ncbi.nlm.nih.gov/omim). The candidate CNVs
were then filtered using the normal frequency databases, such as
DGV (http://dgv.tcag.ca/dgv/app/home) and
gnomAD (https://macarthurlab.org/2017/02/27/the-genome-aggregation-database-gnomad/).
CNVs carried by the normal population that are not pathogenic were
filtered out to look for rare CNVs which may being pathogenic. Each
database had the size of the pathogenic CNVs and is automatically
updated. The variants were interpreted in accordance with the
American College of Medical Genetics and Genomics guidelines
(15) and patient phenotype.
Sanger sequencing
Venous blood (3.5 ml) was collected form the proband
and his parents. Genomic DNA was extracted using QIAamp DNA Mini
kit (Qiagen GmbH) and used for Sanger sequencing. The data from
high-throughput sequencing were used to design primers for
suspected pathological variants using PRIMER3 (version 0.4.0,
http://bioinfo.ut.ee/primer3-0.4.0/),
(forward, 5′-CGTGATTGTAGCACTTGCCTG-3′ and reverse,
5′-GAATGTGGACCCTGTTGTGTG-3′). PCR was performed using KAPA 2G Fast
PCR kit (Roche, cat. no. KK5009), as follows: Initial denaturation
at 95°C for 5 min, followed by 32 cycles of denaturation at 94°C
for 30 sec, annealing at 60°C for 30 sec and extension at 72°C for
45 sec and final extension at 72°C for 5 min. PCR products (length,
412 bp) were analyzed by gel electrophoresis and purified.
Capillary electrophoretic sequencing was performed using an ABI
PRISM 3730 sequencer (Thermo Fisher Scientific, Inc.) and variants
were analyzed. The Sanger sequencing results were aligned with
SALL4 reference sequence (NM_020436, http://www.ncbi.nlm.nih.gov/nuccore/NM_020436)
using SeqMan 7.1 software (16).
Protein structure prediction
The amino acid sequence of SALL4 was obtained from
National Center for Biotechnology Information. The structure of
SALL4 protein and mutant was predicted using
zhanglab.dcmb.med.umich.edu/I-TASSER website. Spbdv (https://spdbv.unil.ch/) was used to visualize and
label the results of the I-TASSER predictions.
Results
Clinical features of proband with
Okihiro syndrome
The proband (Table
I) was a 23-month-old boy of Han nationality (height, 86.5 cm;
weight, 12.0 kg) who presented to KCH because he was born deaf. The
patient was severely affected (Table
II): Both hands flexed to the radial side and he had missing
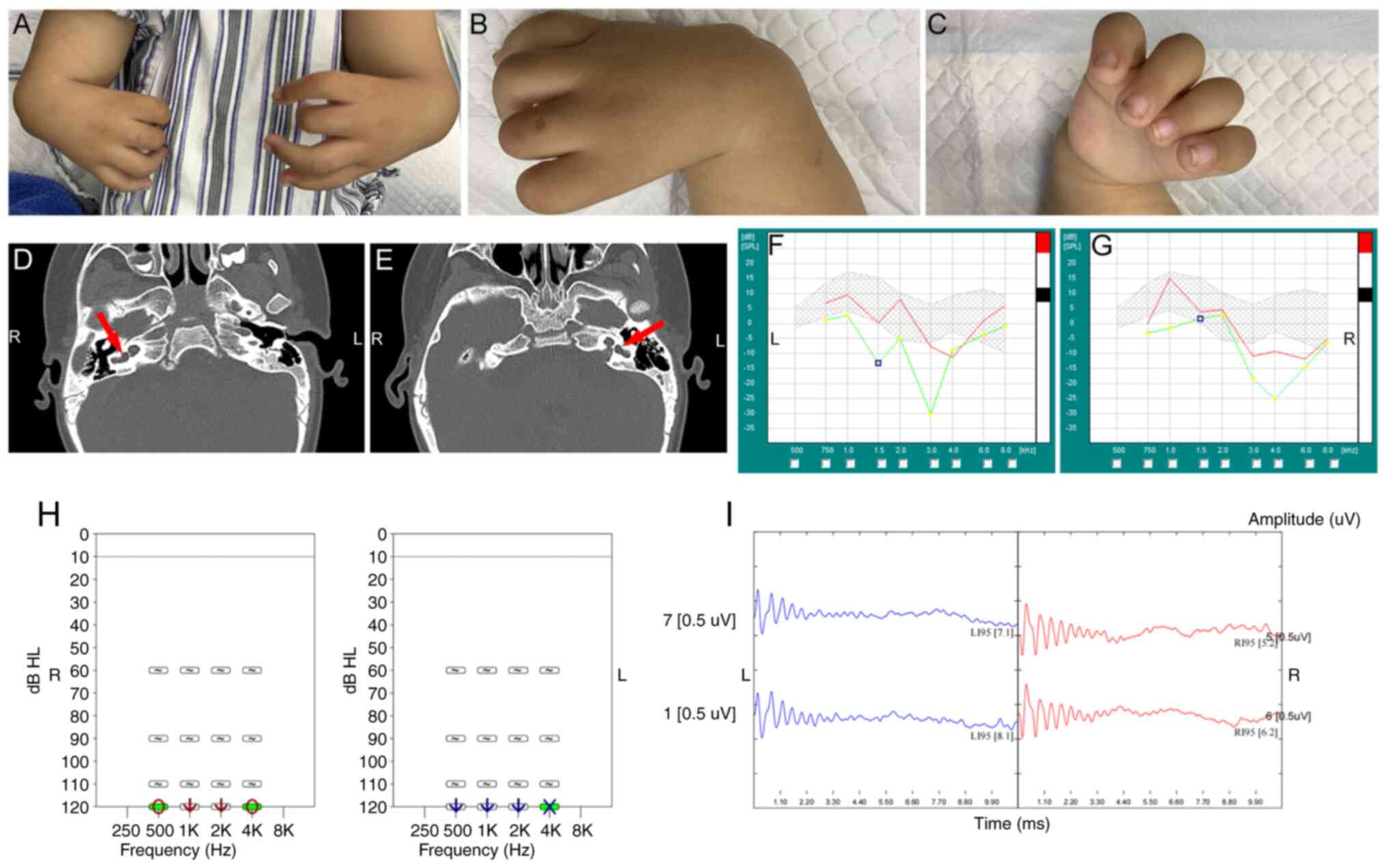
radii and thumbs (Fig. 1A-C). His
forearms were short and there was dysplasia of the left humerus.
Nasal obstruction without respiratory distress was described.
Radiographs of the upper limbs showed that the bilateral radius,
first palm and phalanx were absent. The bilateral humerus and ulna
were curved, with a slightly short and thick ulna. The junction of
the ulna and the humerus did not join correctly (data not shown).
The bilateral cochlea and vestibule had not developed and showed
cystic changes (Fig. 1D and E).
The proband presented with severe bilateral sensorineural hearing
loss. Clinical audiological examination showed failed bilateral
otoacoustic emission (Fig. 1F and
G), auditory steady-state response threshold >110 dB hearing
level (HL; Fig. 1H) under
500-4,000 Hz and with bilateral ABR threshold >95 dB HL
(Fig. 1I). There was no evidence
of Duane abnormality on ophthalmologic evaluation. Abdomen B-scan
ultrasounds did not identify any abnormalities. The results of the
intelligence developmental assessment were normal except for
delayed speech development. Red and white blood cell and platelet
counts were normal (data not shown). The proband with the
aforementioned clinical manifestations was diagnosed as a probable
Okihiro syndrome patient (1,9).
Both parents of the patient were healthy and non-consanguineous
marriage. There was no genetic history of Okihiro syndrome in the
family. The proband received a cochlear implant in his right ear at
the KCH on 17 August 2020.
 | Table I.Sociodemographic information of the
proband. |
Table I.
Sociodemographic information of the
proband.
| Characteristic | Description |
|---|
| Sex | Male |
| Age | 23.0 months |
| Height | 86.5 cm |
| Weight | 12.0 kg |
| Cochlear
implant | Right ear |
| Table II.Clinical characteristics of the
proband. |
Table II.
Clinical characteristics of the
proband.
| Characteristic | Description |
|---|
| Upper limb
deformity | Both hands flexed
to the radial side; absent radii and thumbs; short forearms;
dysplasis of the left humerus |
| Upper limb
X-ray | Absent bilateral
radius, first palm, and phalanx were absent; curved humerus and
ulna; short, thick ulna; poor match between ulna and ulnohumeral
joint |
| Temporal CT | Undeveloped
bilateral cochlea and vestibule |
| Ophthalmic
assessment | Normal |
| Otoacoustic
emission assessment | Failed |
| Auditory
steady-state response | >110 dB HL |
| Auditory brain stem
response | >95 dB HL |
| Abdomen B-scan
ultrasound | Normal |
Identification of a de novo SALL4
frameshift mutation
A heterozygous variant, c.3060delG, in exon 4 of
SALL4 (NM_020436) was detected in the proband and resulted
in a Gln(Q) to His(H) (p. Q1020Hfs*57) change at amino acid 1,020,
which is a frameshift mutation. Sanger sequencing showed that this
site was wild-type in the parents (Fig. 2A and B), which indicated that the
variant was a de novo variant of the proband. From the point
of the variation (amino acid 1,020), the gene encoded the following
amino acids: R V S V Q M W K N Q V L T A F P N T S F L T S W K K T
R L R S A K G E L A W K E Q C R H S E I S R I C F V L. Translation
was then terminated and the resulting mutant protein was 1,076
amino acids in length (Fig. 2C).
This variant was predicted to exhibit PVS1_PM4 + PS2 + PM2
pathology, based on ACMG guidelines (15). PVS1_PM4 is a frameshift mutation,
which may cause loss of function (15). There were no variants at PS2 locus
in the parents of the proband. PS2 variant is a spontaneous
variant. The mean of PM2 was variant frequency is extremely low,
even was not included in the normal population databases 1,000
Genomes and gnomAD. To the best of our knowledge, this variant site
has not previously been reported in the literature or ClinVar
database (15). The amino acid
sequence at position (p. Q1020) was conserved between multiple
homologous species (Fig. 2D). The
amino acid variant was located in the final 10% of the protein (red
box; Fig. 2F). The configuration
of the mutant SALL4 protein showed multiple changes compared with
the wild-type (Fig. 2E and F).
Due to the change of the amino acid sequence, the α-helix primary
structure and β-sheet, irregular coil and other secondary
structures of the mutant protein have been changed, the protein
cannot be folded correctly, and the spatial configuration was
changed.
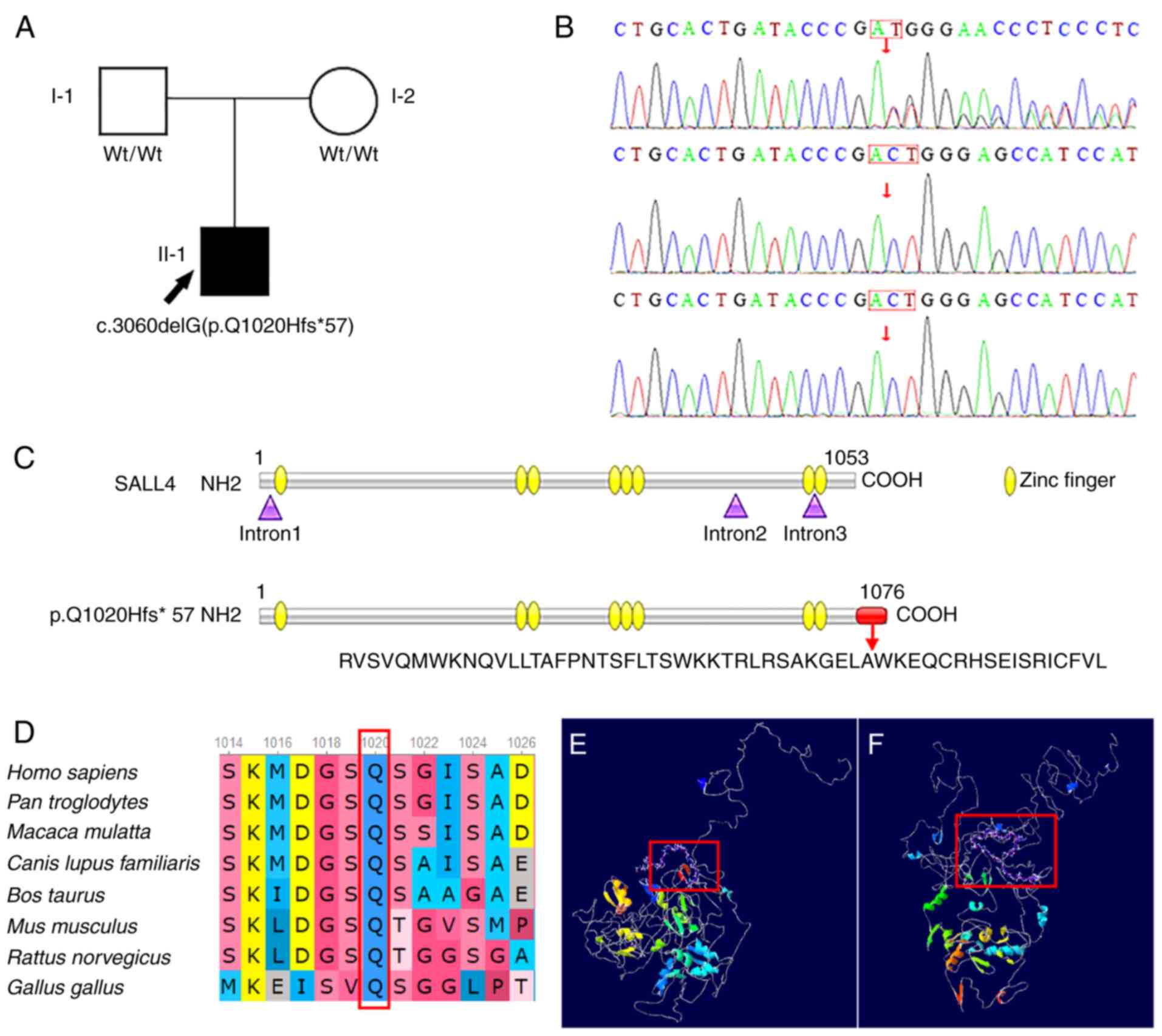 | Figure 2.Family pedigree, results of gene
sequencing, and bioinformatics analysis. (A) Family pedigree. WT,
wild-type; I-1, father of the proband; I-2, mother of the proband;
II-1, proband; Q, Gln, glutamine; H, His, histidine; black,
patient; white, normal; box, male; circle, female. (B) De
novo variant in SALL4 (NM_033326; variant c.3060delG).
(C) Schematic representation of SALL4 protein and mutant. (D)
Conservation analysis of mutation site. Predicted structure of (E)
SALL4 protein and (F) mutant. SALL4, spalt-like transcription
factor 4; Wt, wild-type. |
Discussion
Okihiro syndrome is rare, with previous reports
indicating that most cases are sporadic (17,18). Poznanski indicated that the
familial incidence is 5% in Okihiro syndrome patients (19). The phenotype of patients with
variants in SALL4 is highly variable: Certain patients show
the full range of clinical phenotypes, while others exhibit only
partial clinical manifestations (11). The phenotypes may be different
between patients within a family, which means family members may be
mistakenly thought to have two different diseases when phenotypes
are considered in isolation (7).
Duane anomaly is considered to be one of the primary symptoms of
Okihiro syndrome but approximately one third of patients with
variant SALL4 do not exhibit this abnormality; in affected
families, at least one family member may not have this defect
(20). Alves et al
(9) showed that five out of 28
patients with variant SALL4 did not exhibit Duane
abnormality. Similarly, the proband in the present study did not
exhibit Duane abnormality. Patients with Okihiro syndrome present
with upper limb deformity, such as hypoplasia of the thumb,
three-phalanged thumb, inward flexion of the radii or absence of
the forearm (7,12). In a simple, speculative model, it
has been predicted that loss of ZFM domains results in more severe
upper limb deformity (5).
However, it has also been predicted that truncated protein retains
more ZFMs, which leads to a more severe phenotype (21). Therefore, the size of the
truncated protein or number of ZFMs does not correspond to the
phenotypical severity of upper limb abnormality in the upper limb.
Okihiro syndrome causes face, ear, heart, kidney, vertebral body
and lower limb abnormality (11).
In addition, Parentin et al (22) reported a case of Okihiro syndrome
with developmental delay in 2003. The SALL4 missense variant
is predicted to result in increased DNA binding affinity and causes
Okihiro syndrome with holoprosencephalic features (5). Dental abnormality has also been
reported (5). The association of
autosomal recessive missense/nonsense variants with mild mental
retardation has been demonstrated (23,24). However, mental retardation has not
been described in SALL4 genetic syndromes. The phenotype
caused by Okihiro syndrome involves multiple systems and certain
clinical phenotypes overlap with Townes-Brocks, Holt-Oram,
acro-reno-ocular, oculo-oto-radial and cervico-oculo-acoustic
syndrome (7,25). Diagnosis of Okihiro syndrome is
therefore difficult. As it is rare and certain phenotypical
features of SALL4 variant may be due to chance, the syndrome
needs to be investigated in further pedigree studies. All affected
members of a family should undergo clinical examination to obtain
greater understanding of the phenotype of the SALL4
variant.
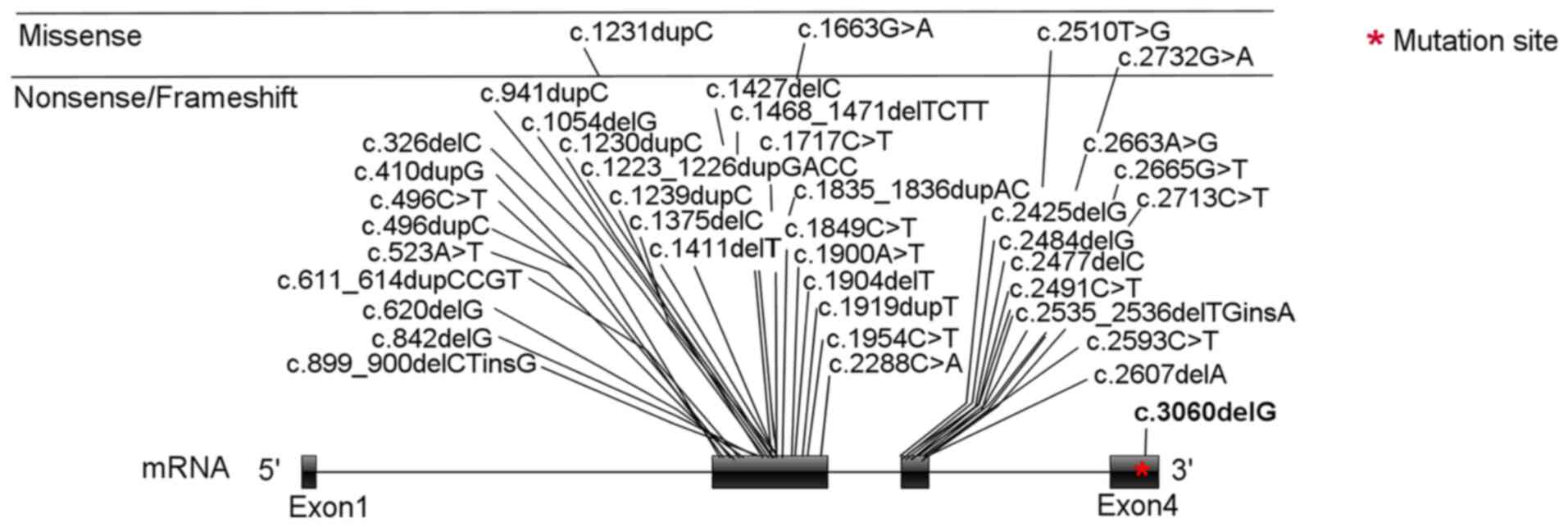
As of December 2021, HGMD
(hgmd.cf.ac.uk/ac/index.php) comprises 53 SALL4 variants
associated with Okihiro syndrome. Of these, 40 are point variants
(Fig. 3) in exons 2 and 3. The
variant site of the present proband was located in exon 4 of
SALL4 (Fig. 3); to the
best of our knowledge, this is the first variant to be reported in
this exon. Only four of the 40 point variants are missense
variants; the rest are nonsense/frameshift variants. There are also
13 types of copy number variation of SALL4 that have been
included in HGMD (hgmd.cf.ac.uk/ac/index.php). The phenotype of
patients with Okihiro syndrome with SALL4 exon deletion is
no more severe than the phenotype of patients with point variants
(20). To the best of our
knowledge, previous case studies have not found a clear association
between genotype and phenotype (1,10,21).
Nonsense and frameshift mutations or exon deletions
of SALL4 are the primary types of variant that lead to
production of truncated proteins (19). Studies have shown that truncated
proteins may inhibit the expression of wild-type SALL4 protein in a
dominant negative manner (7,12,26,27). The lack of SALL4 protein or
production of non-functional ZFMs causes haploinsufficiency, which
results in Okihiro syndrome (27). Nonsense-mediated decay (NMD)
degrades mRNA of nonsense mutations and decreases accumulation of
truncated proteins in the body (28). However, NMD may not effectively
eliminate all SALL4 variants that contain premature stop
codons (29,30). SALL4 is only expressed in
germ cells in adult tissue, which hinders the study of the
molecular mechanism underlying SALL4-associated limb defect
(31). Here, the SALL4
c.3060delG (p. Q1020Hfs*57) variant did not cause the protein to be
truncated. By contrast, the mutant protein was longer and the
variant was located in the final exon, which did not stimulate NMD.
It was hypothesized that the variant sequence at the carboxyl
terminal affected configuration of the protein, which caused the
Okihiro syndrome.
The current understanding of Okihiro syndrome is not
comprehensive and there is no clinical diagnostic standard; further
case studies are required to improve clinical understanding and
establish a diagnostic standard. However, more attention should be
given to the role of genetic testing for rare genetic diseases,
which can be accurately diagnosed via genetic testing combined with
clinical phenotypes. Okihiro syndrome leads to acral deformities,
with upper limb deformity being the most common (21). These deformities may be detected
during prenatal check-ups, allowing for early intervention
(32).
Sanger sequencing can identify patients with
clinical features suspected to be associated with Okihiro/DRRS. To
the best of our knowledge, there are few reports on Okihiro
syndrome and there may be novel genes that lead to the occurrence
of the disease. Whole-exome sequencing may be used to quickly
obtain genetic information to diagnose and rule out other potential
disease-causing genes (33).
Here, a de novo variant of SALL4, c.3060delG (p.
Q1020Hfs *57), was the molecular cause of Okihiro syndrome in the
proband. Gene sequencing should be used to help clinicians in the
diagnosis of rare disease.
Acknowledgements
Not applicable.
Funding
The present study was supported by Yunnan Applied Basic Research
Joint Special Project of Kunming Medical University (grant no.
202001AY070001-170), Haibo Wang Expert Workstation in Yunnan
Province (grant no. 202105AF150056) and Reserve Talents for Young
and Middle-aged Academic and Technical Leaders in Yunnan Province
(grant no. 2019HB102).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
XM and RH designed the study, collected data and
wrote the manuscript. GL and JM designed the study, analyzed data
and wrote the manuscript. JM performed the literature review and
revised the manuscript. TZ conceived and designed the study and
revised the manuscript. XM and JM confirm the authenticity of all
the raw data. All authors have read and approved the final version
of the manuscript.
Ethics approval and consent to
participate
The presented study was performed in accordance with
the Declaration of Helsinki and approved by The Ethics Committee of
Kunming Children's Hospital (Kunming, China, grant no.
2020-01-009-H01). Written informed consent was obtained from all
participants.
Patient consent for publication
The mother of the proband provided consent for
publication of images of the proband.
Competing interests
The authors declare that they have no competing
interests.
Glossary
Abbreviations
Abbreviations:
|
DRRS
|
Duane-radial ray syndrome
|
|
ZFM
|
zinc finger motif
|
|
KCH
|
Kunming Children's Hospital
|
|
ABR
|
auditory brain stem response
|
|
CNV
|
copy number variation
|
|
HGMD
|
Human Gene Mutation Database
|
|
NMD
|
nonsense-mediated decay
|
References
|
1
|
Kohlhase J, Heinrich M, Liebers M,
Fröhlich Archangelo L, Reardon W and Kispert A: Cloning and
expression analysis of SALL4, the murine homologue of the gene
mutated in Okihiro syndrome. Cytogenet Genome Res. 98:274–277.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Kohlhase J, Heinrich M, Schubert L,
Liebers M, Kispert A, Laccone F, Turnpenny P, Winter RM and Reardon
W: Okihiro syndrome is caused by SALL4 mutations. Hum Mol Genet.
11:2979–2987. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Lim CY, Tam WL, Zhang J, Ang HS, Jia H,
Lipovich L, Ng HH, Wei CL, Sung WK, Robson P, et al: Sall4
regulates distinct transcription circuitries in different
blastocyst-derived stem cell lineages. Cell Stem Cell. 3:543–554.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Yang J: SALL4 as a transcriptional and
epigenetic regulator in normal and leukemic hematopoiesis. Biomark
Res. 6:12018. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Miertus J, Borozdin W, Frecer V, Tonini G,
Bertok S, Amoroso A, Miertus S and Kohlhase J: A SALL4 zinc finger
missense mutation predicted to result in increased DNA binding
affinity is associated with cranial midline defects and mild
features of Okihiro syndrome. Hum Genet. 119:154–161. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
de Celis JF and Barrio R: Regulation and
function of Spalt proteins during animal development. Int J Dev
Biol. 53:1385–1398. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Hayes A, Costa T and Polomeno RC: The
Okihiro syndrome of Duane anomaly, radial ray abnormalities, and
deafness. Am J Med Genet. 22:273–280. 1985. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Okihiro MM, Tasaki T, Nakano KK and
Bennett BK: Duane syndrome and congenital upper-limb anomalies. A
familial occurrence. Arch Neurol. 34:174–179. 1977. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Alves LU, Perez AB, Alonso LG, Otto PA and
Mingroni-Netto RC: Novel frameshift variant in gene SALL4 causing
Okihiro syndrome. Eur J Med Genet. 59:80–85. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Kohlhase J, Schubert L, Liebers M, Rauch
A, Becker K, Mohammed SN, Newbury-Ecob R and Reardon W: Mutations
at the SALL4 locus on chromosome 20 result in a range of clinically
overlapping phenotypes, including Okihiro syndrome, Holt-Oram
syndrome, acro-renal-ocular syndrome, and patients previously
reported to represent thalidomide embryopathy. J Med Genet.
40:473–478. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Terhal P, Rösler B and Kohlhase J: A
family with features overlapping Okihiro syndrome, hemifacial
microsomia and isolated Duane anomaly caused by a novel SALL4
mutation. Am J Med Genet A. 140:222–226. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Sakaki-Yumoto M, Kobayashi C, Sato A,
Fujimura S, Matsumoto Y, Takasato M, Kodama T, Aburatani H,
Asashima M, Yoshida N and Nishinakamura R: The murine homolog of
SALL4, a causative gene in Okihiro syndrome, is essential for
embryonic stem cell proliferation, and cooperates with Sall1 in
anorectal, heart, brain and kidney development. Development.
133:3005–3013. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Chen S, Zhou Y, Chen Y and Gu J: Fastp: An
ultra-fast all-in-one FASTQ preprocessor. Bioinformatics.
34:i884–i890. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Li H and Durbin R: Fast and accurate short
read alignment with Burrows-Wheeler transform. Bioinformatics.
25:1754–1760. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Richards S, Aziz N, Bale S, Bick D, Das S,
Gastier-Foster J, Grody WW, Hegde M, Lyon E, Spector E, et al:
Standards and guidelines for the interpretation of sequence
variants: A joint consensus recommendation of the American college
of medical genetics and genomics and the association for molecular
pathology. Genet Med. 17:405–424. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Swindell SR and Plasterer TN: SEQMAN.
Contig assembly. Methods Mol Biol. 70:75–89. 1997.PubMed/NCBI
|
|
17
|
Jourdain AS, Petit F, Odou MF, Balduyck M,
Brunelle P, Dufour W, Boussion S, Brischoux-Boucher E, Colson C,
Dieux A, et al: Multiplex targeted high-throughput sequencing in a
series of 352 patients with congenital limb malformations. Hum
Mutat. 41:222–239. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
van de Putte R, Dworschak GC, Brosens E,
Reutter HM, Marcelis CLM, Acuna-Hidalgo R, Kurtas NE, Steehouwer M,
Dunwoodie SL, Schmiedeke E, et al: A genetics-first approach
revealed monogenic disorders in patients with ARM and VACTERL
Anomalies. Front Pediatr. 8:3102020. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Poznanski AK, Garn SM and Holt JF: The
thumb in the congenital malformation syndromes. Radiology.
100:115–129. 1971. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Borozdin W, Graham JM Jr, Bohm D, Bamshad
MJ, Spranger S, Burke L, Leipoldt M and Kohlhase J: Multigene
deletions on chromosome 20q13.13-q13.2 including SALL4 result in an
expanded phenotype of Okihiro syndrome plus developmental delay.
Hum Mutat. 28:8302007. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Borozdin W, Wright MJ, Hennekam RC,
Hannibal MC, Crow YJ, Neumann TE and Kohlhase J: Novel mutations in
the gene SALL4 provide further evidence for acro-renal-ocular and
Okihiro syndromes being allelic entities, and extend the phenotypic
spectrum. J Med Genet. 41:e1022004. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Parentin F and Perissutti P: Solitary
median maxillary central incisor, Duane retraction syndrome, growth
hormone deficiency and duplicated thumb phalanx: A case report.
Clin Dysmorphol. 12:141–142. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Perez Y, Wormser O, Sadaka Y, Birk R,
Narkis G and Birk OS: A Rare Variant in PGAP2 causes autosomal
recessive hyperphosphatasia with mental retardation syndrome, with
a mild phenotype in heterozygous carriers. Biomed Res Int.
2017:34702342017. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Scala M, Mojarrad M, Riazuddin S, Brigatti
KW, Ammous Z, Cohen JS, Hosny H, Usmani MA, Shahzad M, Riazuddin S,
et al: RSRC1 loss-of-function variants cause mild to moderate
autosomal recessive intellectual disability. Brain. 143:e312020.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Paradisi I and Arias S: IVIC syndrome is
caused by a c.2607delA mutation in the SALL4 locus. Am J Med Genet
A. 143:326–332. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Kiefer SM, McDill BW, Yang J and Rauchman
M: Murine Sall1 represses transcription by recruiting a histone
deacetylase complex. J Biol Chem. 277:14869–14876. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Kiefer SM, Ohlemiller KK, Yang J, McDill
BW, Kohlhase J and Rauchman M: Expression of a truncated Sall1
transcriptional repressor is responsible for Townes-Brocks syndrome
birth defects. Hum Mol Genet. 12:2221–2227. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Al-Baradie R, Yamada K, St Hilaire C, Chan
WM, Andrews C, McIntosh N, Nakano M, Martonyi EJ, Raymond WR,
Okumura S, et al: Duane radial ray syndrome (Okihiro syndrome) maps
to 20q13 and results from mutations in SALL4, a new member of the
SAL family. Am J Hum Genet. 71:1195–1199. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Kiefer SM, Robbins L, Barina A, Zhang Z
and Rauchman M: SALL1 truncated protein expression in Townes-brocks
syndrome leads to ectopic expression of downstream genes. Hum
Mutat. 29:1133–1140. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Liberalesso PBN, Cordeiro ML, Karuta SCV,
Koladicz KRJ, Nitsche A, Zeigelboim BS, Raskin S and Rauchman M:
Phenotypic and genotypic aspects of Townes-Brock syndrome: Case
report of patient in southern Brazil with a new SALL1 hotspot
region nonsense mutation. BMC Med Genet. 18:1252017. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Miettinen M, Wang Z, McCue PA,
Sarlomo-Rikala M, Rys J, Biernat W, Lasota J and Lee YS: SALL4
expression in germ cell and non-germ cell tumors: A systematic
immunohistochemical study of 3215 cases. Am J Surg Pathol.
38:410–420. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Becker R, Horn D, Knoll U, Stumm M, Wegner
RD, Peters H and Sarioglu N: First-trimester prenatal diagnosis of
Okihiro syndrome. Fetal Diagn Ther. 27:222–226. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Li B, Chen S, Sun K, Xu R and Wu Y:
Genetic analyses identified a SALL4 gene mutation associated with
holt-oram syndrome. DNA Cell Biol. 37:398–404. 2018. View Article : Google Scholar : PubMed/NCBI
|