Introduction
Cardiovascular disease may lead to death and affects
approximately 17.9 million individuals annually (1–3).
Atherosclerosis (AS) is a prevalent cardiovascular disease and a
leading cause of vascular-related death (4,5).
The critical function of vascular smooth muscle cells (VSMCs)
phenotypic transformation during AS progression has been previously
identified (6,7). VSMC phenotypic transformation from a
contractile to a synthetic status in vessel walls, stimulated by
adverse microenvironmental provocations, accompanied by VSMC
proliferation and migration, is crucial to AS development (8,9).
Phenotypic regulation of VSMCs from the contractile and quiescent
phenotype to the synthetic type is a critical step for the vascular
remodeling of AS (10). In
addition, it has been identified that VSMCs also experience notable
phenotypic transformation stimulated by some signals, such as
platelet-derived growth factor-BB (PDGF-BB) and angiotensin II
(AngII) (11). Accordingly, the
exploration of the potential candidate that is able to regulate
VSMCs phenotypic transformation may benefit the treatment of
AS.
Atorvastatin is a 3-hydroxy-3-methyl-glutaryl
coenzyme A (HMG-CoA) reductase inhibitor and is commonly applied
for the treatment of AS patients (12). Atorvastatin can enhance plaque
instability and relieve the coronary artery inflammation response,
decreasing the morbidity and mortality incidences of cardiovascular
disorder (13). Besides,
atorvastatin has several other functions, such as vascular
endothelial function improvement, anti-oxidation, and
anti-inflammation (14).
Moreover, it has been reported that histone deacetylases (HDACs)
are involved in the modulation of VSMCs (15). In addition, forkhead box O4
(FOXO4) and Akt signaling play a critical role in VSMCs phenotypic
transformation during AS (16,17). In addition, FOXO4 activity is
regulated by the PI3K/Akt signaling-mediated phosphorylation
(18). Moreover, it has been
reported that serum response factor (SRF)/myocardin signaling
participates in the regulation of VSMCs shiftiness (19). However, the effect of atorvastatin
on these factors in the development of VSMCs phenotypic
transformation remains obscure.
In this study, the role and underlying mechanism of
atorvastatin in the modulation of VSMCs phenotypic transformation
was investigated. A novel function of atorvastatin in regulating
VSMCs phenotypic transformation by epigenetically modulating
contractile proteins and mediating Akt/FOXO4 axis.
Materials and methods
Cell culture and treatment
Primary VSMCs were isolated from normal rat aortas
(N=5, male, 8 weeks) by applying the explant method as previously
reported (20). Briefly, the
aortas were isolated and the endothelial cells (ECs) were removed,
the smooth muscle layer was stripped and chopped into small
fragments (~1 mm3) in 0.5 ml of fetal bovine serum. The
fragments, together with the fetal bovine serum, were transferred
to a 25 cm2 flask and maintained upside down in an
incubator at 37°C for 4 h. The flask was turned over gently and
incubated for 4–7 days after the addition of 2 ml of DMEM. VSMCs
migrated out of the explants 4–7 days later, and passage was
performed 10–14 days after isolation. The VSMCs were cultured in
the medium of DMEM (Gibco; Thermo Fisher Scientific, Inc.)
containing 10% fetal bovine serum (Gibco; Thermo Fisher Scientific,
Inc.), 0.1 mg/ml streptomycin (Beijing Solarbio Science &
Technology Co., Ltd.) and 100 U/ml penicillin (Beijing Solarbio
Science & Technology Co., Ltd.) at a condition of 37°C with 5%
CO2. The primary VSMCs were identified using α-SMA. For
all the analyses, VSMCs (2–5 generations) were applied followed by
quiescence for 12 h. The VSMCs were treated with AngII (10 nM;
MilliporeSigma), trichostatin A (TSA, 0.01 ng/ml; MilliporeSigma),
atorvastatin (30 µmol/l; MilliporeSigma), mevalonic acid (MVA, 500
µM; MilliporeSigma), PDGF-BB (20 ng/ml; MilliporeSigma), and
LY294002 (10 µM, Sigma, USA) as indicated. The morphology of VSMCs
was analyzed using a Nikon microscope (Tokyo).
The average weight of the rats at the start of the
experiment was 187 g. The rats were fed in a condition of 25°C and
50% relative humidity with a 12-h light/dark cycle and free access
to standard chow and tap water. The rats were euthanized with an
overdose of pentobarbital sodium (150 mg/kg, iv). Animal care and
method procedure were authorized by the Animal Ethics Committee of
Tianjin Fifth Central Hospital (approval no. 2019-0619-37). The
procedures were conformed to the Guide for the Care and Use of
Laboratory Animal published by the US National Institutes of Health
(NIH publication, 8th edition, 2011).
Reverse transcription-quantitative PCR
(RT-qPCR)
The total RNAs from the mice and cells were
extracted by TRIzol® (Invitrogen; Thermo Fisher
Scientific, Inc.) from the VSMCs. The first-strand cDNA was
manufactured as per the manufacturer's instructions (Thermo Fisher
Scientific, Inc.). The RT-qPCR was carried out by applying SYBR
Real-time PCR I kit (Takara Bio, Inc.). The standard control for
mRNA was GAPDH. Quantitative determination of the RNA levels was
conducted in triplicate independent experiments (n=3). The
following thermocycling conditions were used for qPCR: Initial
denaturation at 95°C for 30 sec; followed by 39 cycles of 95°C for
5 sec and 60°C for 30 sec; and a final extension at 72°C for 5 min.
The 2−ΔΔCq method was used (21). The sequences were designed
according to the references (Genscript, Nanjing, China). Primer
sequences are listed in Table
I.
 | Table I.Primer sequences. |
Table I.
Primer sequences.
| Gene name | Primer | Genebank accession
no. | Product sizes
(bp) |
|---|
| α-SMA |
5′-GAAGAGCTACGAACTGCCTGATG-3′ | NM_007392.3 | 417 |
|
|
5′-TAGAAGCATTTGCGGTGGAC-3′ |
|
|
| SM-MHC |
5′-TTTGCCATTGAGGCCTTAGG-3′ | NG_009299 | 427 |
|
|
5′-GTTCACACGGCTGAGAATCCA-3′ |
|
|
| SM22α |
5′-TTGTAATGCAGTGTGGCCCT-3′ | NM_003186 | 369 |
|
|
5′-CAGGCTGTTCACCAACTTG-3′ |
|
|
| GAPDH |
5′-AAGAAGGTGGTGAAGCAGGC-3′ | XM_036165840.1 | 203 |
|
|
5′-TCCACCACCCAGTTGCTGTA-3′ |
|
|
| Calponin |
5′-GTCTGGGCATGGAACACTGT-3′ | NM_031747.2 | 477 |
|
|
5′-GAGGTACTTACTTGTGAGGGAAT-3′ |
|
|
Western blot analysis
Total proteins were extracted from the cells or mice
tissues with RIPA buffer (Cell Signaling Technology, Inc.). Protein
concentrations were measured by using the BCA Protein
Quantification Kit. The same concentration of protein (25 µg) was
divided by SDS-PAGE (12% polyacrylamide gels), and transferred to
PVDF membranes (MilliporeSigma) in the subsequent step. The
membranes were blocked with 5% milk and incubated overnight at 4°C
with the primary antibodies for α-SMA (1:1,000; cat. no. ab7817;
Abcam), SM-MHC (1:1,000; cat. no. ab133567; Abcam), SM22α (1:1,000;
cat. no. ab14106; Abcam), FOXO4 (1:1,000; cat. no. ab128908;
Abcam), p-FOXO4 (1:1,000; cat. no. ab126594; Abcam), Akt (1:1,000;
cat. no. ab8805; Abcam), p-Akt (1:1,000; cat. no. ab38449; Abcam),
SRF (1:1,000; cat. no. ab252868; Abcam), myocardin (1:1,000; cat.
no. ab107301; Abcam), and β-actin (1:1,000; cat. no. ab7817;
Abcam), in which β-actin served as the control. Then, the
corresponding secondary antibodies (1:1,000; cat. no.
ab96899/ab96879; Abcam) were used for incubating the membranes 1 h
at room temperature, followed by the visualization by using an
Odyssey CLx Infrared Imaging System. The experiments were conducted
in triplicate independent experiments (n=3). The densitometry
analysis was performed using ImageJ software (NIH, v1.8.0).
Immunofluorescence analysis
Cells were solidified at 4% paraformaldehyde for 30
min, treated with Triton X-100 (0.2%) for 10 min and treated with
BSA (2%) for 30 min. The slides were hatched with the primary
antibody overnight at 4°C, then hatched with secondary antibodies
(Proteintech) for 1 h at 37°C. The slides were stained with DAPI
(Beyotime Institute of Biotechnology) for 10 min at 25°C. A Nikon
microscope (Tokyo) was utilized to analyze the immunofluorescence.
The experiments were conducted in triplicate independent
experiments (n=3).
ChIP analysis
Chromatin immunoprecipitation (ChIP) was performed
using a SimpleChIP Enzymatic Chromatin IP Kit (Cell Signaling
Technology, Inc.) according to the manufacturer's instructions.
Chromatin prepared from the cells in a 15-cm dish was used to
determine total DNA input and was incubated overnight with specific
antibodies, H3K9ac (1:500; cat. no. ab32129; Abcam), H4Ac (1:500,
39243, Thermo Fisher, Cambridge, MA, USA), HDAC1 (1:500; cat. no.
ab109411; Abcam), HDAC3 (1:500; cat. no. ab32369; Abcam), P300
(1:500; cat. no. ab275378; Abcam), or normal rabbit IgG. Then, the
binding DNA was analyzed by qPCR assays and the primer sequences as
indicated in Table I. The
experiments were conducted as triplicate independent experiments
(n=3).
HDAC activity analysis
The activities of HDAC were analyzed by applying an
HDAC Activity Colorimetric Assay Kit (Biovision, Inc.) in the VSMCs
according to the manufacturer's protocol. The experiments were
conducted as triplicate independent experiments (n=3).
Immunoprecipitation assays
The interaction of the proteins was analyzed by
immunoprecipitation in the VSMCs. IP was conducted by Pierce
Co-Immunoprecipitation kit (Thermo Fisher Scientific, Inc.,
Germany) according to the manufacturer's instructions. The
experiments were conducted in triplicate independent experiments
(n=3).
Statistical analysis
Data are presented as mean ± SD, and the statistical
analysis was performed by GraphPad prism 7 (GraphPad Software,
Inc.). The unpaired Student's t-test was applied for comparing two
groups, and the one-way analysis of variance (ANOVA) followed by a
post hoc Tukey's test was applied for comparing among multiple
groups. P<0.05 was considered statistically significant.
Results
The isolation and identification of
rat primary VSMCs
During AS, VSMCs present a significant phenotypic
transformation from the quiescent contractile status to the
stimulated synthetic status and are featured by induced migration
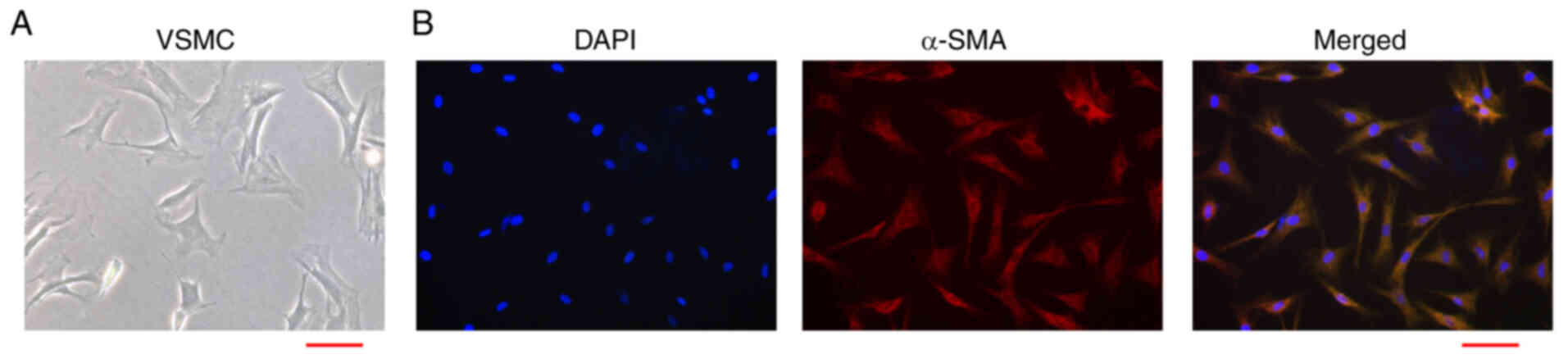
and proliferation ability. Firstly, we isolated the primary rat
VSMCs and the morphology was shown (Fig. 1A). In addition, the purity of
VSMCs was validated by the phenotypic transformation markers termed
α-SMA in the VSMCs (Fig. 1B).
Atorvastatin regulates
AngII-associated VSMCs phenotypic transformation by epigenetically
regulating contractile proteins
Next, we explored the effect of atorvastatin on the
AngII-associated VSMC phenotypic transformation. To this end, the
VSMCs were treated with AngII, atorvastatin, or co-treated with
AngII and atorvastatin, AngII and HDAC inhibitor termed
trichostatin A (2).
Significantly, the protein expression of contractile proteins,
including α-SMA, SM-MHC, and SM22α, as the VSMCs-specific genes,
was reduced by AngII and enhanced by atorvastatin, in which
atorvastatin could reverse the effect of AngII in the VSMCs
(Fig. 2A and B). In addition, the
treatment of TSA was able to enhance the AngII-inhibited expression
of α-SMA and SM-MHC (Fig. 2A and
B). Furthermore, ChIP assays showed that the interaction of
HDAC, P300, SRF, histone H3 lysine 9 acetylation (H3K9ac), histone
H4 acetylation (H4ac) on the promoters of α-SMA and SM-MHC, but not
SM22α, in which AngII could regulate the interaction while the
co-treatment of atorvastatin or TSA could reverse the effect of
AngII in the system (Fig. 2C).
Consistently, the treatment of AngII reduced the HDAC activities
and atorvastatin presented a reversed effect, in which atorvastatin
could attenuate the effect of AngII in the VSMCs (Fig. 2D). Together these suggest that
atorvastatin regulates VSMCs phenotypic transformation by
modulating HDAC3.
Atorvastatin regulates PDGF-BB-induced
VSMCs phenotypic transformation by modulating PI3K signaling
PDGF-BB is able to induce the phenotypic
transformation of VSMCs and plays a critical role in the regulation
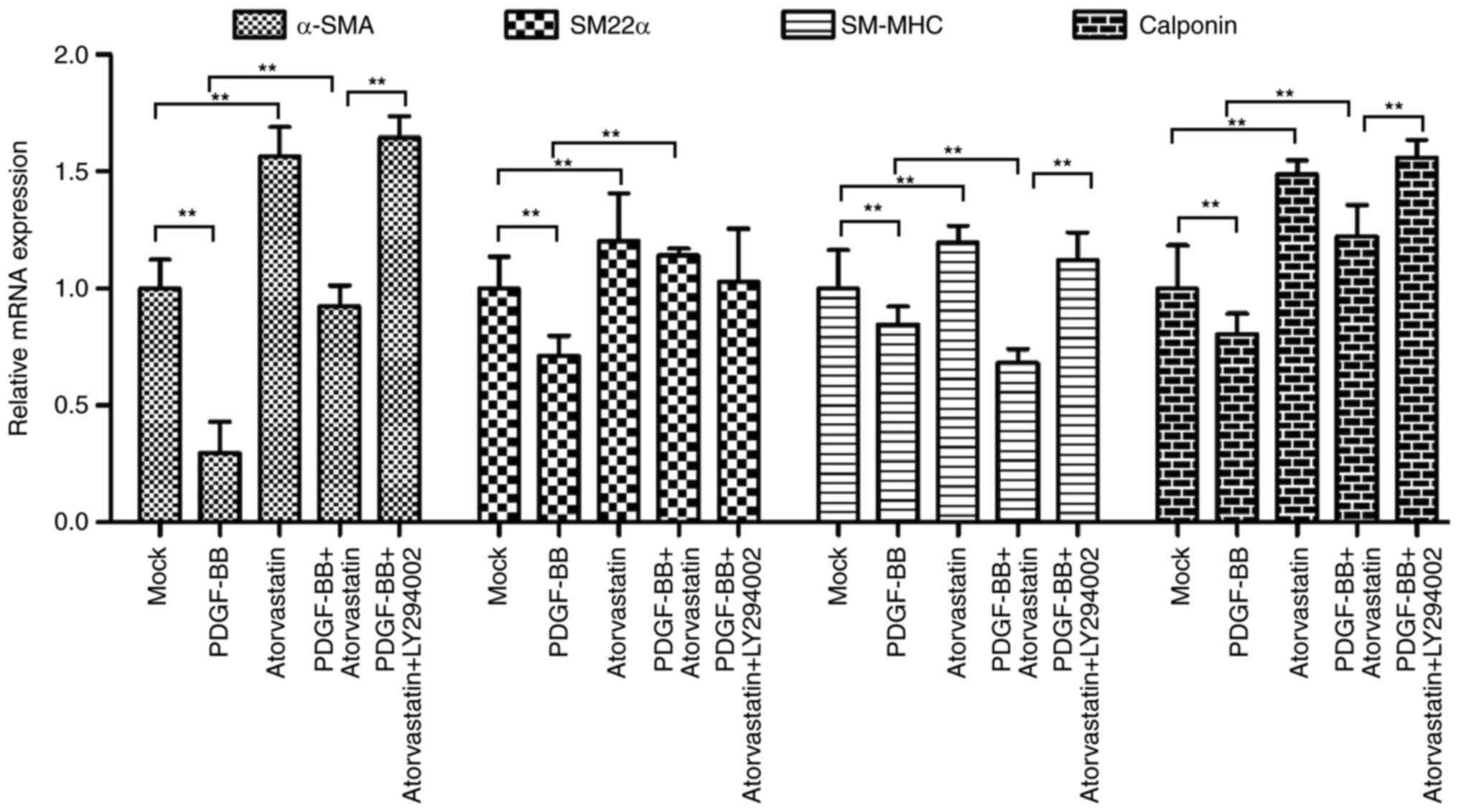
of VSMCs phenotypes. Accordingly, we further evaluated the effect
of atorvastatin on the PDGF-BB-induced VSMCs phenotypic
transformation. For this purpose, the VSMCs were treated with
PDGF-BB, atorvastatin, or co-treated with PDGF-BB and atorvastatin,
PDGF-BB, atorvastatin, and PI3K inhibitor LY294002. The results
showed that the mRNA expression of contractile proteins, such as
α-SMA, SM-MHC, SM22α, and calponin, was inhibited by PDGF-BB but
enhanced by atorvastatin, in which co-treatment of atorvastatin
with PDGF-BB could rescue the PDGF-BB-reduced phenotypes in the
VSMCs (Fig. 3). Meanwhile,
LY294002 was able to enhance α-SMA, SM-MHC, SM22α, and calponin
expression in the PDGF-BB and atorvastatin co-treated VSMCs
(Fig. 3).
Atorvastatin modulates PDGF-BB-induced
VSMC phenotypic transformation by modulating PI3K/FOXO4 axis
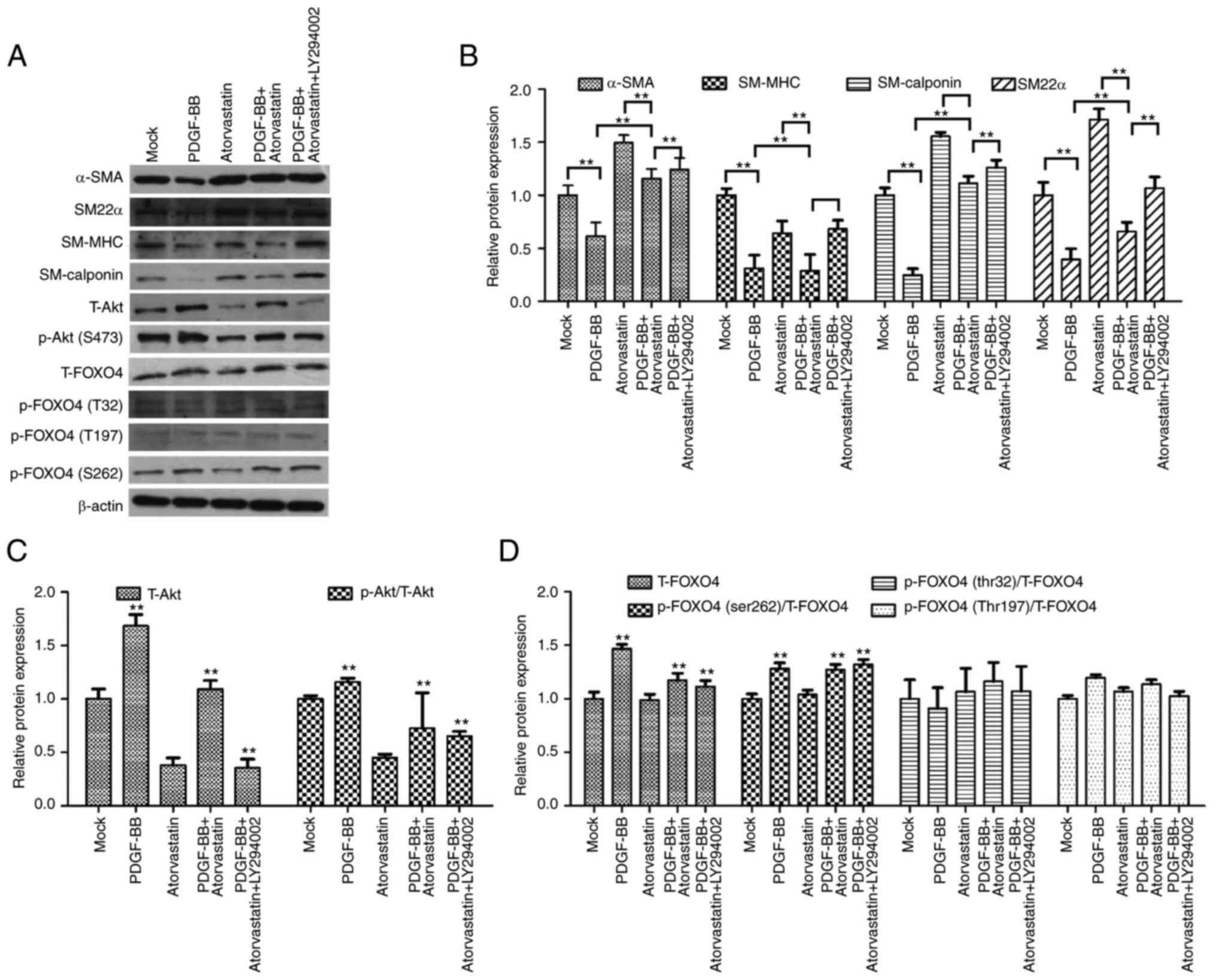
Moreover, the protein levels of α-SMA, SM-MHC,
SM22α, and calponin, were reduced by PDGF-BB but upregulated by
atorvastatin, in which the co-treatment of atorvastatin with
PDGF-BB could rescue the PDGF-BB-inhibited phenotypes in the VSMCs
(Fig. 4A and B). Furthermore,
LY294002 induced α-SMA, SM-MHC, SM22α, and calponin expression in
the PDGF-BB and atorvastatin co-treated VSMCs (Fig. 4A and B). In addition, the
expression and phosphorylation of Akt were increased by PDGF-BB, in
which atorvastatin could attenuate the effect of atorvastatin and
LY294002 was able to further inhibit the phenotype (Fig. 4A and C). Significantly, the
expression and phosphorylation of FOXO4, especially the serine 262
phosphorylation, was induced by PDGF-BB, while atorvastatin reduced
the effect in the system and LY294002 could further reinforce the
inhibitor phenotype in the VSMCs (Fig. 4A and D), suggesting that
atorvastatin modulates PDGF-BB-induced VSMC phenotypic
transformation by modulating the PI3K/FOXO4 axis.
Atorvastatin is able to regulate
FOXO4/SRF/myocardin axis
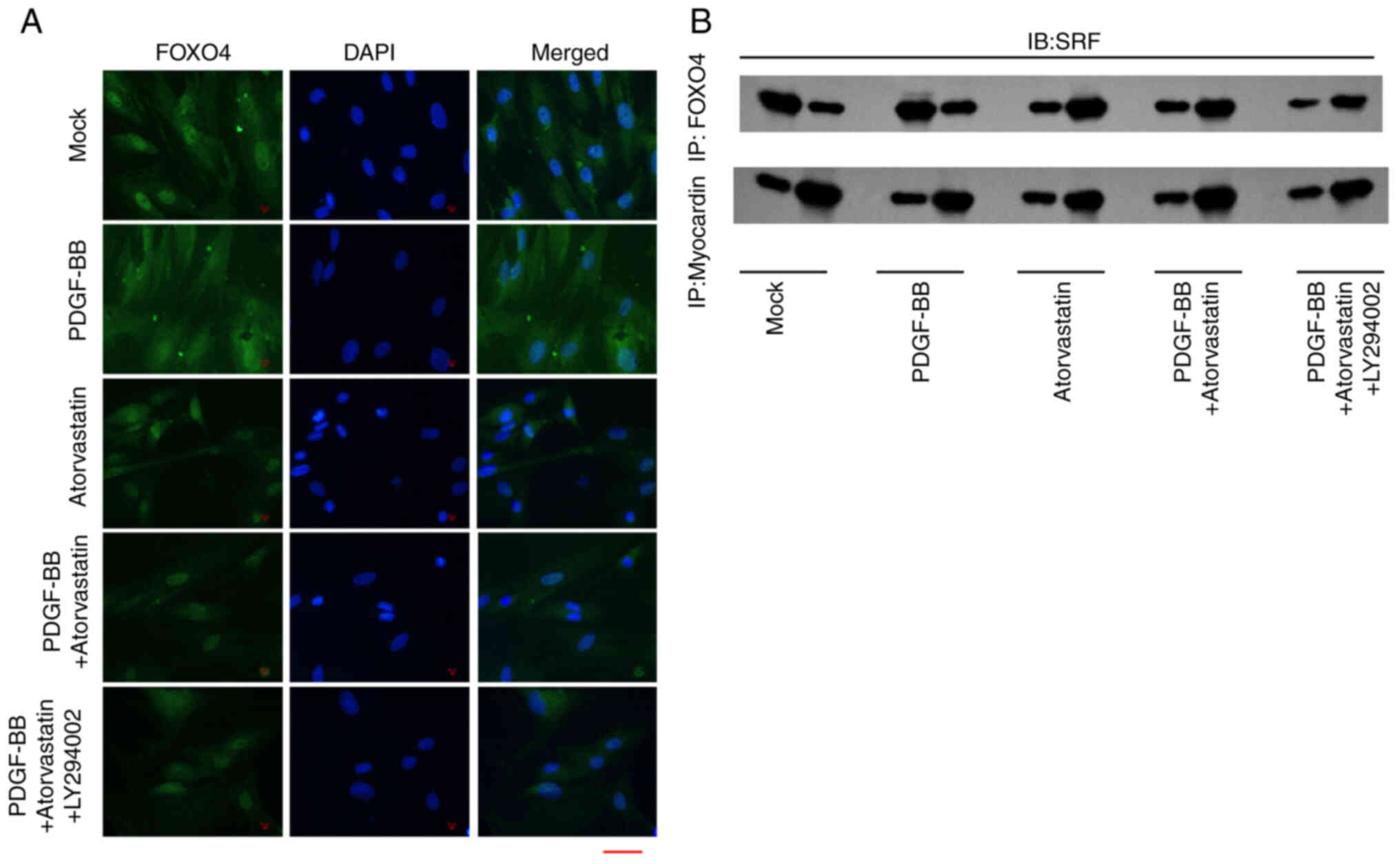
The immunofluorescence analysis revealed that
PDGF-BB enhanced the accumulation of FOXO4 in the VSMCs, while the
treatment of atorvastatin was able to attenuate this effect and the
co-treatment of LY294002 could further inhibit the phenotype
(Fig. 5A). Subsequently, the
treatment of PDGF-BB enhanced the interaction of SRF with FOXO4 and
myocardin in the VSMCs, in which the co-treatment of atorvastatin
and LY294002 could reverse the effect of PDGF-BB in the system
(Fig. 5B), suggesting that
atorvastatin is able to regulate FOXO4/SRF/myocardin axis.
Discussion
AS is a predominant type of cardiovascular disease
with severe morbidity and high mortality (4). Atorvastatin has presented
anti-inflammatory effects and improvements in vascular endothelial
function. Nevertheless, the role and the potential mechanism of
atorvastatin in the regulation of VSMCs phenotypic transformation
is still unclear. In this study, we discovered a novel function of
atorvastatin in modulating VSMCs phenotypic transformation by
epigenetically modulating contractile proteins and mediating
Akt/FOXO4 axis.
Previous findings identified the function of
atorvastatin in AS and VSMCs regulation. It has been reported that
atorvastatin upregulates ACE2 expression by epigenetic histone
modifications in rabbit AS mode (22). Atorvastatin reduces pyroptosis via
NEXN-AS1/NEXN signaling in vascular endothelial cells during AS
(23). Atorvastatin represses the
PDGF-ββ-stimulated migration and proliferation of VSMCs by G0/G1
cell cycle suppression and the arrest of PDGFRβ/PI3K/Akt signaling
(24). Atorvastatin enhances
apoptosis of VSMCs by downregulating Rho A prenylation and Bcl-2
expression (25). Results of the
present study showed that, atorvastatin was able to regulate
PDGF-BB/AngII-induced VSMCs phenotypic transformation. This is a
novel function of atorvastatin in VSMCs regulation, providing
valuable evidence for the fundamental role of atorvastatin in the
development of AS.
Moreover, HDAC is known to participate in VSMCs and
AS development (26,27). Protein kinase B/AKT regulates
insulin-like growth factor 1-accociated phosphorylation and nuclear
export of HDAC5 by activating NADPH oxidase 4 in VSMCs (26). TSA suppresses VSMCs proliferation
by enhancing WAF1 (27).
Additionally, FOXO4 plays a critical role in the progression of
VSMCs and AS. MiR-23b downregulation enhances phenotypic switching
of VSMCs by targeting FOXO4 (28). MiR-128-3p decreases VSMCs
migration and proliferation by inhibiting FOXO4 signaling (29). In addition, Akt signaling is
involved in the modulation of VSMCs and AS (18,30). In the present study, atorvastatin
modulated VSMCs phenotypic transformation potentially by
epigenetically regulating contractile proteins and regulating
PI3K/FOXO4 signaling. These data identify the unreported
correlation of atorvastatin with these critical factors in the
modulation of VSMCs during AS development.
In conclusion, findings of the present study showed
that atorvastatin regulated VSMCs phenotypic transformation by
epigenetically modulating contractile proteins and mediating
Akt/FOXO4 axis. This finding provides new insights into the
mechanism by which atorvastatin modulates VSMCs, providing valuable
evidence for the application of atorvastatin in the treatment of
AS.
Acknowledgements
Not applicable.
Funding
This study was supported by the Sichuan Provincial Science and
Technology Department Project (2018JY0403), Youth innovative
Scientific Research Project of Sichuan Medical Association
(Q16076), and Technology and Human resources' Bureau of Luzhou
(17256).
Availability of data and materials
All data generated or analyzed during this study are
included in this published article.
Authors' contributions
XD, XL and GZ designed the study; XD performed the
experiments in figures 1,2,3,4.
ZS, ZC, XZ, DW, and KW performed the experiments in figures 4 and 5. XD, XL and GZ wrote the manuscript.
XD, XL and GZ confirm the authenticity of all the raw data. All
authors reviewed and approved the final manuscript.
Ethics approval and consent to
participate
Animal care and method procedure were authorized by
the Animal Ethics Committee of Tianjin Fifth Central Hospital
(approval no.: 2019-0619-37). The procedures were conformed to the
Guide for the Care and Use of Laboratory Animal published by the US
National Institutes of Health (NIH publication, 8th edition,
2011).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Peng J, Xiao X, Hu M and Zhang X:
Interaction between gut microbiome and cardiovascular disease. Life
Sci. 214:153–157. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Nitsa A, Toutouza M, Machairas N, Mariolis
A, Philippou A and Koutsilieris M: Vitamin D in cardiovascular
disease. In Vivo. 32:977–981. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Leong DP, Joseph PG, McKee M, Anand SS,
Teo KK, Schwalm JD and Yusuf S: Reducing the global burden of
cardiovascular disease, part 2: Prevention and treatment of
cardiovascular disease. Circ Res. 121:695–710. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Schaftenaar F, Frodermann V, Kuiper J and
Lutgens E: Atherosclerosis: The interplay between lipids and immune
cells. Curr Opin Lipidol. 27:209–215. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Barrington WT and Lusis AJ:
Atherosclerosis: Association between the gut microbiome and
atherosclerosis. Nat Rev Cardiol. 14:699–700. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
He X, Lian Z, Yang Y, Wang Z, Fu X, Liu Y,
Li M, Tian J, Yu T and Xin H: Long Non-coding RNA PEBP1P2
suppresses proliferative VSMCs phenotypic switching and
proliferation in atherosclerosis. Mol Ther Nucleic Acids. 22:84–98.
2020. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Xu J, Zhang Y, You S, Guo Y, Chen S, Chang
Y, Zhang N and Sun Y: Paired box 9 regulates VSMC phenotypic
transformation, proliferation, and migration via sonic hedgehog.
Life Sci. 257:1180532020. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Fu Y, Chang Y, Chen S, Li Y, Chen Y, Sun
G, Yu S, Ye N, Li C and Sun Y: BAG3 promotes the phenotypic
transformation of primary rat vascular smooth muscle cells via
TRAIL. Int J Mol Med. 41:2917–2926. 2018.PubMed/NCBI
|
|
9
|
Lu QB, Wan MY, Wang PY, Zhang CX, Xu DY,
Liao X and Sun HJ: Chicoric acid prevents PDGF-BB-induced VSMC
dedifferentiation, proliferation and migration by suppressing
ROS/NFĸB/mTOR/P70S6K signaling cascade. Redox Biol. 14:656–668.
2018. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Zhang M, Li F, Wang X, Gong J, Xian Y,
Wang G, Zheng Z, Shang C, Wang B, He Y, et al: MiR-145 alleviates
Hcy-induced VSMC proliferation, migration, and phenotypic switch
through repression of the PI3K/Akt/mTOR pathway. Histochem Cell
Biol. 153:357–366. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Song W, Gao K, Huang P, Tang Z, Nie F, Jia
S and Guo R: Bazedoxifene inhibits PDGF-BB induced VSMC phenotypic
switch via regulating the autophagy level. Life Sci.
259:1183972020. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Taniguti EH, Ferreira YS, Stupp IJV,
Fraga-Junior EB, Doneda DL, Lopes L, Rios-Santos F, Lima E, Buss
ZS, Viola GG and Vandresen-Filho S: Atorvastatin prevents
lipopolysaccharide-induced depressive-like behaviour in mice. Brain
Res Bull. 146:279–286. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Li S, Yu Y, Jin Z, Dai Y, Lin H, Jiao Z,
Ma G, Cai W, Han B and Xiang X: Prediction of pharmacokinetic
drug-drug interactions causing atorvastatin-induced rhabdomyolysis
using physiologically based pharmacokinetic modelling. Biomed
Pharmacother. 119:1094162019. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Kogawa AC, Pires AEDT and Salgado HRN:
Atorvastatin: A review of analytical methods for pharmaceutical
quality control and monitoring. J AOAC Int. 102:801–809. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Ginnan R, Sun LY, Schwarz JJ and Singer
HA: MEF2 is regulated by CaMKIIδ2 and a HDAC4-HDAC5 heterodimer in
vascular smooth muscle cells. Biochem J. 444:105–114. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Zhao G, Fu Y, Cai Z, Yu F, Gong Z, Dai R,
Hu Y, Zeng L, Xu Q and Kong W: Unspliced XBP1 Confers VSMC
homeostasis and prevents aortic aneurysm formation via FoxO4
Interaction. Circ Res. 121:1331–1345. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Huang C, Huang W, Wang R and He Y:
Ulinastatin inhibits the proliferation, invasion and phenotypic
switching of PDGF-BB-Induced VSMCs via Akt/eNOS/NO/cGMP signaling
pathway. Drug Des Devel Ther. 14:5505–5514. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Lu W, Ni Z, Jiang S, Tong M, Zhang J, Zhao
J, Feng C, Jia Q, Wang J, Yao T, et al: Resveratrol inhibits bile
acid-induced gastric intestinal metaplasia via the PI3K/AKT/p-FoxO4
signalling pathway. Phytother Res. 35:1495–1507. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Zhou N, Lee JJ, Stoll S, Ma B, Wiener R,
Wang C, Costa KD and Qiu H: Inhibition of SRF/myocardin reduces
aortic stiffness by targeting vascular smooth muscle cell
stiffening in hypertension. Cardiovasc Res. 113:171–182. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Qin X, Hou X, Zhang M, Liang T, Zhi J, Han
L and Li Q: Relaxation of rat aorta by farrerol correlates with
potency to reduce intracellular calcium of VSMCs. Int J Mol Sci.
15:6641–6656. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(−Delta Delta C(T)) Method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Tikoo K, Patel G, Kumar S, Karpe PA,
Sanghavi M, Malek V and Srinivasan K: Tissue specific up regulation
of ACE2 in rabbit model of atherosclerosis by atorvastatin: Role of
epigenetic histone modifications. Biochem Pharmacol. 93:343–351.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Wu LM, Wu SG, Chen F, Wu Q, Wu CM, Kang
CM, He X, Zhang RY, Lu ZF, Li XH, et al: Atorvastatin inhibits
pyroptosis through the lncRNA NEXN-AS1/NEXN pathway in human
vascular endothelial cells. Atherosclerosis. 293:26–34. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Chen S, Dong S, Li Z, Guo X, Zhang N, Yu B
and Sun Y: Atorvastatin calcium inhibits PDGF-ββ-induced
proliferation and migration of VSMCs Through the G0/G1 cell cycle
arrest and suppression of activated PDGFRbeta-PI3K-Akt signaling
cascade. Cell Physiol Biochem. 44:215–228. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Blanco-Colio LM, Villa A, Ortego M,
Hernandez-Presa MA, Pascual A, Plaza JJ and Egido J:
3-Hydroxy-3-methyl-glutaryl coenzyme A reductase inhibitors,
atorvastatin and simvastatin, induce apoptosis of vascular smooth
muscle cells by downregulation of Bcl-2 expression and Rho A
prenylation. Atherosclerosis. 161:17–26. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Pietruczuk P, Jain A, Simo-Cheyou ER,
Anand-Srivastava MB and Srivastava AK: Protein kinase B/AKT
mediates insulin-like growth factor 1-induced phosphorylation and
nuclear export of histone deacetylase 5 via NADPH oxidase 4
activation in vascular smooth muscle cells. J Cell Physiol.
234:17337–17350. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Okamoto H, Fujioka Y, Takahashi A,
Takahashi T, Taniguchi T, Ishikawa Y and Yokoyama M: Trichostatin
A, an inhibitor of histone deacetylase, inhibits smooth muscle cell
proliferation via induction of p21(WAF1). J Atheroscler Thromb.
13:183–191. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Iaconetti C, De Rosa S, Polimeni A,
Sorrentino S, Gareri C, Carino A, Sabatino J, Colangelo M, Curcio A
and Indolfi C: Down-regulation of miR-23b induces phenotypic
switching of vascular smooth muscle cells in vitro and in vivo.
Cardiovasc Res. 107:522–533. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Qu C, Liu X, Guo Y, Fo Y, Chen X, Zhou J
and Yang B: MiR-128-3p inhibits vascular smooth muscle cell
proliferation and migration by repressing FOXO4/MMP9 signaling
pathway. Mol Med. 26:1162020. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Gong C, Ai J, Fan Y, Gao J, Liu W, Feng Q,
Liao W and Wu L: NCAPG promotes the proliferation of hepatocellular
carcinoma through PI3K/AKT signaling. Onco Targets Ther.
12:8537–8552. 2019. View Article : Google Scholar : PubMed/NCBI
|