Introduction
Hepatitis B virus (HBV) infection is a serious
global public health problem especially in Africa and Asia, and is
the cause of nearly 1 million deaths from liver disease each year
(1). HBV is a small, enveloped,
dsDNA virus with a full length of 3,200 bp. HBV covalently closed
circular DNA (cccDNA) acts as the key intracellular replication
template of the virus. Following infection of hepatocytes, HBV
relaxed cDNA (rcDNA) enters the nucleus to form cccDNA pool using
host proteins. During this period, HBV ds linear DNA (dslDNA) can
integrate into the host genome when DNA strand breaks (DSBs) occur
(2). HBV integration is one of the
major pathogenic causes of HBV-associated hepatocellular carcinoma
(HCC) and the second leading cause of cancer-associated death
worldwide (3). The mechanisms may
include chromosomal instability, dysfunction of cell cycle
progression, host genes and apoptosis and increasing cell
proliferation (4). However, due to
the lack of in vitro infection models with detectable
integration events, the molecular mechanism of the formation of HBV
integration and its carcinogenic effects are not well-understood
(5).
Traditional methods, such as Southern blotting and
Alu-PCR, which are either time-consuming or laborious, have
demonstrated that HBV DNA can randomly integrate into the host
genome (6). Next generation
sequencing (NGS) assay of HBV integration detection identified a
large group of hotspots among the inserted host genes, for example,
myeloid/lymphoid or mixed-lineage leukemia 4, telomerase reverse
tranase (TERT), cyclin E1 (CCNE1) and lysine-specific
methyltransferase 2B (KMT2B) (7–9). To
the best of our knowledge, however, there remains a paucity of
in vitro studies of these integration sites. P21-activated
kinase 3 (PAK3) is an important oncogene in HCC (10); our previous study reported the HBV
integration site chrX: 111009033, which inserted into PAK3 gene in
HepG2.2.15 cells (11). The
present study aimed to detect the full sequence of HBV DNA
fragments of this site using an improved inverse nested PCR
(invPCR), its genetic regulation and the expression of PAK3 protein
in HepG2 and HepG2.2.15 cells with and without
H2O2 treatment, which exacerbates HBV
integration (12). The present
study may help to elucidate the pathogenesis and carcinogenesis of
chrX: 111009033 integration site.
Materials and methods
Cell culture
HepG2.2.15 and HepG2 liver cancer cells were
purchased from China Center for Type Culture Collection (Wuhan,
China). Cell lines were authenticated by STR identification and
cultured with DMEM with 10% FBS (Sangon Biotech Co., Ltd.) and 200
µg/ml G418 (Sangon Biotech Co., Ltd.) at 37°C with 5%
CO2. Subclones (termed C1-4) were transferred from
parental cells to 4-well plates and expanded in culture to
5.0×106−1.0×107 cells before collecting.
Nuclear RNA and DNA from cells were extracted using the Genomic RNA
and DNA Mini kits according to the manufacturer's instructions,
respectively (both Sangon Biotech Co., Ltd.). For quantitative
analysis of HBV integration and PAK3 gene, HepG2 and HepG2.2.15
cells (control group) were collected in 24-well plates at a density
of 3.5×103 cells/well. Additionally, as the treatment
group, 24-well culture plates with HepG2.2.15 cells and
H2O2 at 20 µmol/l were cultured, as
previously described (12).
PCR and sequencing validation
The chrX: 111009033 integration site was detected by
conventional PCR in HepG2.2.15 cells, with the primers forward (F)1
(5′-AGAGCCCCTGAGGGTTTT-3′) and reverse (R)1
(5′-CCCGTCTGTGCCTTCTCA-3′). PCR mix was prepared with 10 µM F and R
primers (1 µl each), 20 ng DNA (1 µl), 22 µl H2O and 25
µl 2X Taq Buffer (Sangon Biotech Co., Ltd.), and thermocycling
conditions were conducted as follows: Initial denaturation for 5
min at 95°C followed by 40 cycles of denaturation for 10 sec at
95°C, annealing for 10 sec at 54°C and extension for 3 min at 72°C
and final extension for 10 min at 72°C (11). The PCR products were
electrophoresed (10 µg/lane) using a 1.3% agarose gel, extracted
and sequenced by Sanger sequencing (Sangon Biotech Co., Ltd.).
Finally, HBV-cell DNA junction was confirmed using Basic Local
Alignment Search Tool (BLAST; http://blast.ncbi.nlm.nih.gov/Blast.cgi#alnHdr).
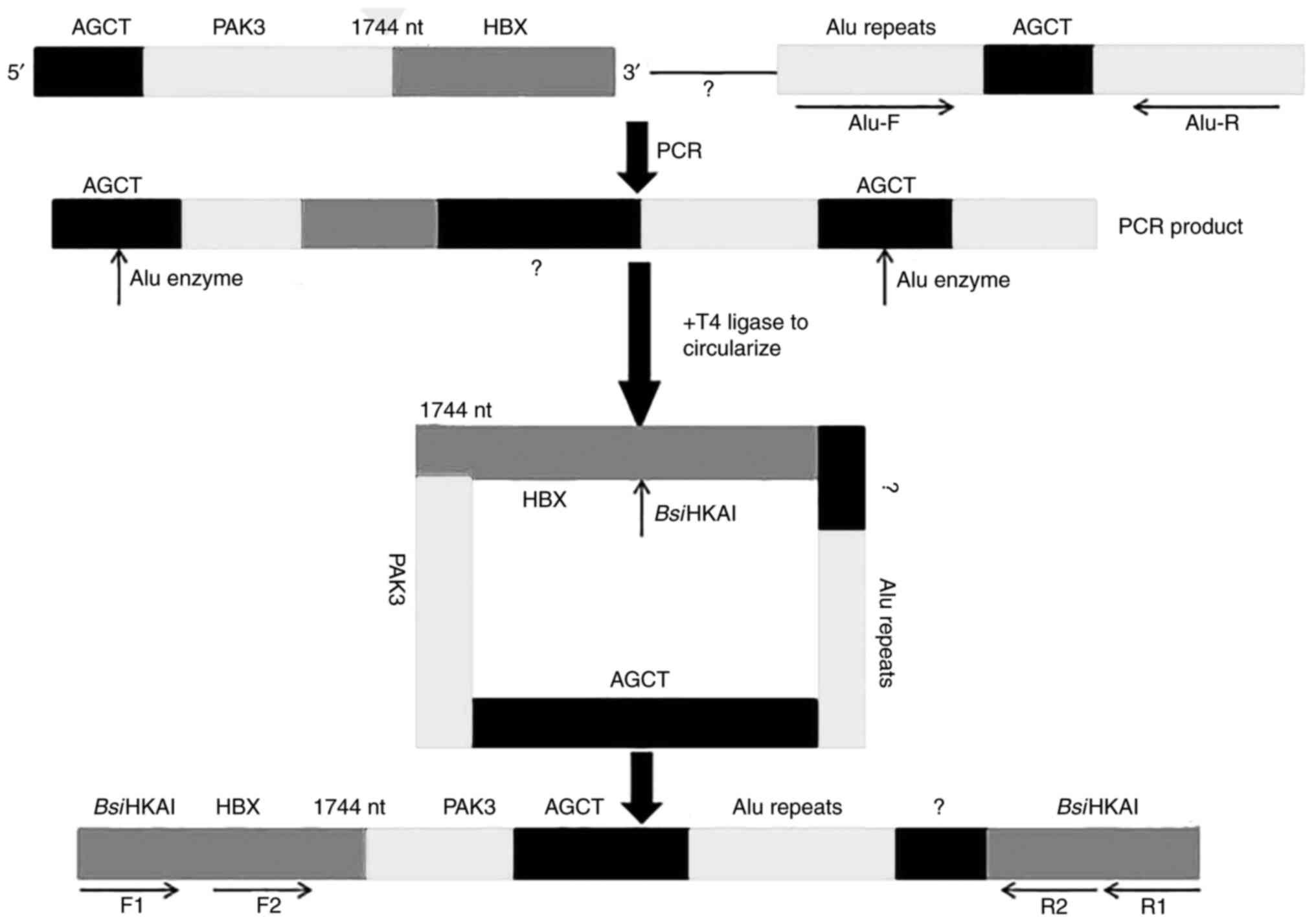
InvPCR
For verification of the full inserted HBV fragment
of the chrX: 111009033 integration site, an improved invPCR
(Fig. 1) was designed as described
previously (13). Firstly, to
amplify a chimeric fragment which partially aligned to host gene
including Alu repeats and unknown integrated HBV gene, primer
chimeric R (5′-AGCTTTTAATACCCAACTCCTCCC-3′), which included
integrated HBV (1,732–1,744 nt) and PAK3 sequence
(chrX111009033-111009027) and an Alu site (AGCT) at its 5′ end, was
designed. Because the orientation of the nearest Alu sequence is
unknown, primers Alu F1 (5′-CGGATCACCTGAGGTCAG-3′) and Alu R1
(5′-ACGGAGTCTCGCTCTGTC-3′) with opposite orientations were designed
and added to two reaction mixes with primer chimeric R. The primer
mix was prepared as aforementioned. Thermocycling conditions were
as follows: Initial denaturation for 5 min at 95°C followed by 40
cycles of denaturation for 10 sec at 95°C, annealing for 10 sec at
56°C and extension for 3 min at 72°C and final extension for 10 min
at 72°C.
Secondly, the specific amplification product (1 µg)
was digested with 1 µl Alu restriction enzyme [(New England
BioLabs, Inc. (NEB)] for 1 h at 37°C then inactivated for 20 min at
80°C, followed by addition of 500 U T4 DNA ligase (NEB). The
mixture was incubated at room temperature for 2 h, after which
ligase was inactivated for 20 min at 80°C, followed by purification
using PCR Purification kit according to the manufacturer's protocol
(Sangon Biotech Co., Ltd.). The DNA was divided into two parts for
either double digestion with 5 U BsiHKAI (1 h, 65°C) and 5 U
SphI (1 h, 37°C) or single digestion with 5 U BsiHKAI
(both NEB).
The digested DNA was serially subjected to nested
PCR. The first round of PCR was performed with the primers F1
(5′-TTCGCTTCACCTCTGCACG-3′) and R1 (5′-AAAGGACGTCCCGCGCAG-3′) for
25 cycles, the products of which were diluted with double distilled
water to 1:10, 1:100 and 1:1,000 and used as the template for the
second round of PCR with the primers F2 (5′-CGCATGGAGACCACCGTGA-3′)
and R2 (5′-CACAGCCTAGCAGCCATGG-3′). The nested PCR conditions were
as described previously (13) and
the products were extracted and sequenced as aforementioned.
According to the sequencing outcome of invPCR
conducted above, PCR primers IN F2 (5′-AGGCTGCCTTCCTGTCTG-3′) and
Alu R2 (5′-CCACGCCCGGCTAATTTT-3′) were designed to obtain the left
end of the viral-host junction. The primer mix and reaction
conditions in PCR were as aforementioned, with extension for 14
sec. The products were extracted and sequenced as aforementioned.
As a control, PCR was conducted in both HepG2 and HepG2.2.15
cells.
Reverse transcription-quantitative
(RT-q)PCR
RT-qPCR was performed with an OneStep RT-PCR system
(Sangon Biotech Co., Ltd.) in HepG2.2.15 cells. The cDNA was
synthesized using Sangon cDNA Synthesis kit according to the
manufacturer's protocol (Sangon Biotech, Co., Ltd.) using the
following reaction conditions: 42°C for 15 min and 85°C for 5 min.
The expression of HBV-PAK3 fusion transcript was detected using
primers IN F1 and IN R1. PCR and sequencing were performed as
previously described (11).
Total RNA was extracted from HepG2.2.15 cells with
and without H2O2 treatment and HepG2 cells
and then reverse-transcribed into cDNA as aforementioned. To detect
the copy numbers of the chX: 11009033 integration site in
HepG2.2.15 cells with and without H2O2
treatment, RT-qPCR with the primers IN F1 and IN R1 was performed
as described previously (11).
PAK3 PCR F (5′-CAACCGGGATTCTTCAGCACT-3′) and R primer
(5′-CACATGAATCGTATGCTCAAAGTCTG-3′) were designed and SYBR Green I
(Sangon Biotech Co., Ltd.) was used for RT-qPCR as reported
previously (10).
Western blot analysis
Proteins were extracted from HepG2.2.15 cells with
and without H2O2 treatment and HepG2 cells
using Cell Protein Extraction kit according to the manufacturer's
instruction (Sangon Biotech Co., Ltd.). GAPDH, (cat. No. AF7021;
1:1,000, Affinity Biosciences) was used as the loading control.
Following determination using a Bicinchoninic Acid (BCA) kit
according to the manufacturer's instruction (Beyotime Institute of
Biotechnology), the cellular proteins (20 µg/lane) were loaded onto
8% SDS-PAGE gels and transferred to PVDF membranes (both Sangon
Biotech Co., Ltd.). The membranes were blocked with 5% non-fat milk
at 25°C for 1 h, then incubated with primary antibodies against
PAK3 (cat. no. AF7659; 1:1,000, Affinity Biosciences) overnight at
4°C. Following primary antibody incubation, the membranes were
incubated with secondary antibodies (cat. no. ZB-2305; 1:20,000;
OriGene Technologies, Inc.) for 1 h at 25°C. Finally, protein bands
were visualized using ECL Western Blotting Substrate kit according
to the manufacturer's protocol (Thermo Fisher Scientific, Inc.),
and images were captured for densitometry using ImageJ software
(version 1.46; National Institutes of Health).
Statistical analysis
Continuous variables are expressed as the mean ±
standard deviation and analyzed by SPSS 20 (IBM Corp.). Student's t
test (unpaired) was used to assess differences between two groups;
one-way ANOVA followed by Tukey's post hoc test was used for
comparison of multiple groups. Correlations were analyzed using
Pearson's correlation coefficient. Two-sided P<0.05 was
considered to indicate a statistically significant difference.
Results
HBV integration site chrX: 111009033
is detected in HepG2.2.15 cells
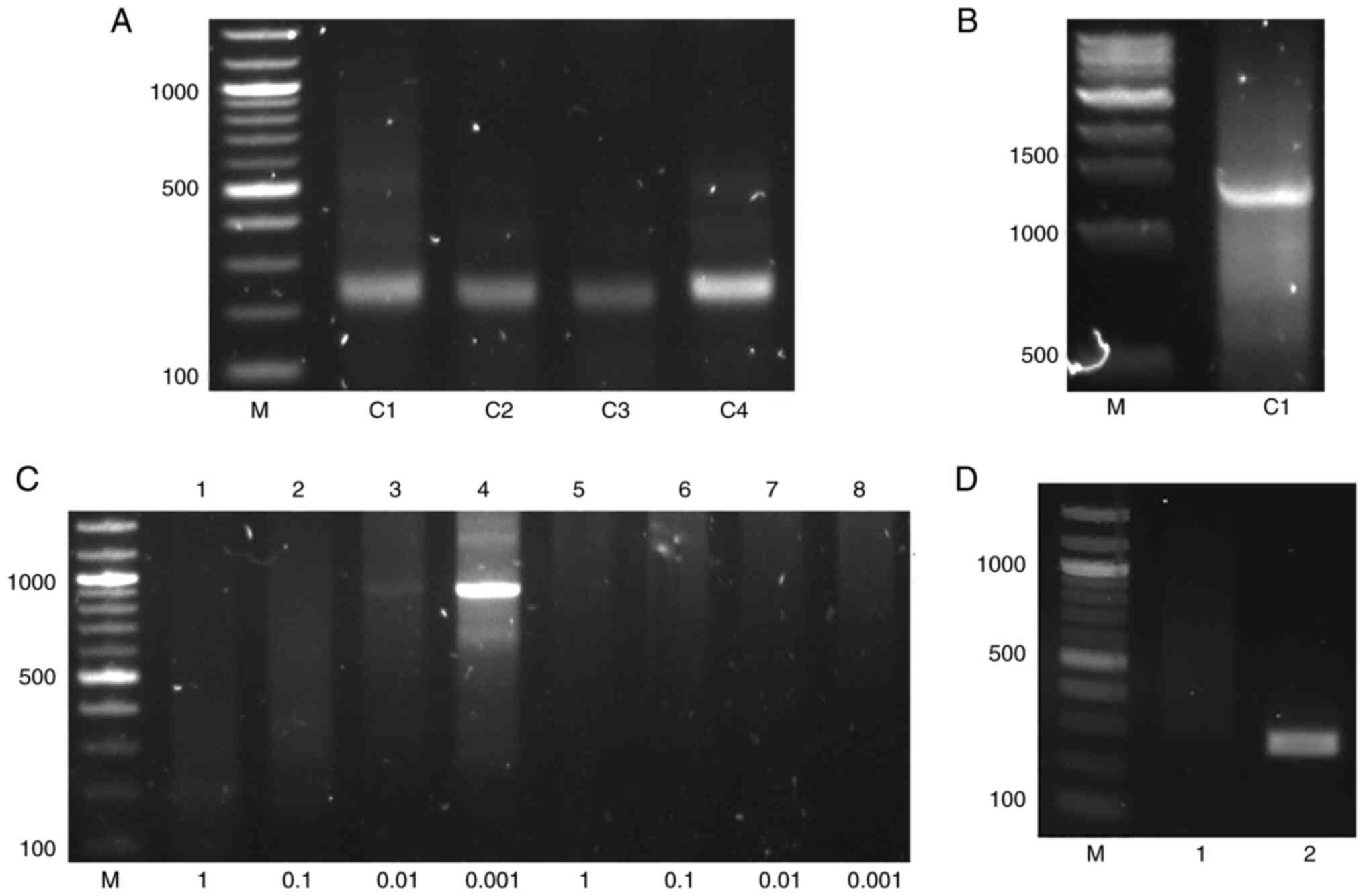
A fragment of 227 bp was acquired by conventional
PCR in DNA extracted from HepG2.2.15 cells (Fig. 2A). BLAST analysis indicated that
this was the right end of the junction of the chrX: 111009033
integration site (11).
Full sequence of HBV fragments are
detected in the integration site chrX: 111009033
To detect the full HBV integration sequence,
improved invPCR was used. Using the primers Chimeric R and Alu R1,
a chimeric fragment of 1,255 bp was amplified by conventional PCR
(Fig. 2B), which partially
contained the Alu repeats. The second round of invPCR specifically
amplified a fragment of 900 bp using the product of double
digestion diluted 1,000 times (Fig.
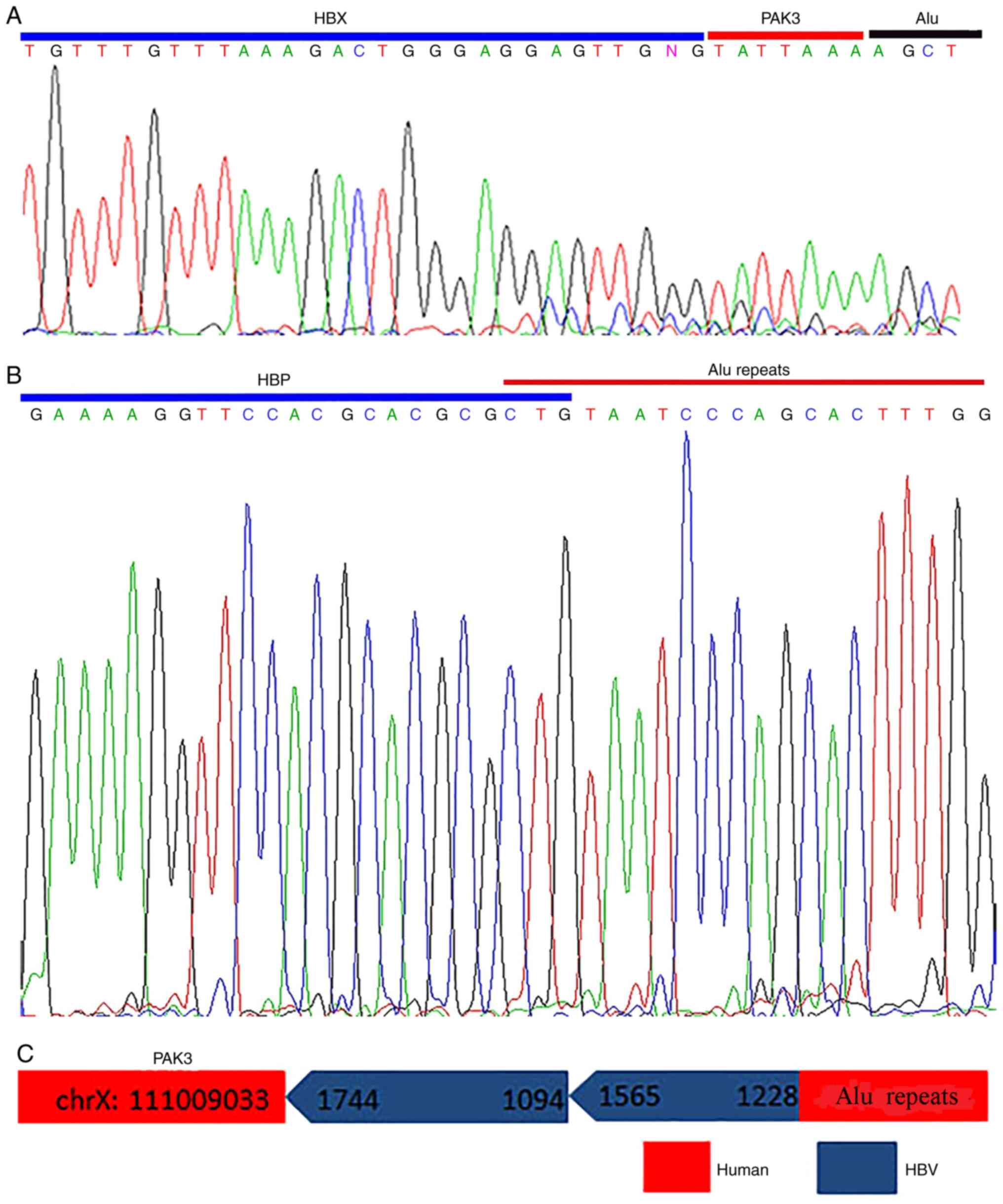
2C), as shown by Sanger sequencing (Fig. 3A); the product of the first round
of invPCR yielded no product following single digestion (Fig. 2C). To obtain the left end of the
HBV-human junction, a pair of PCR primers were designed (IN F2 and
Alu R2), by which a fragment of 223 bp was found in HepG2.2.15 but
not in HepG2 cells (Fig. 2D).
Sanger sequencing indicated that the left HBV-human junction
located in 1,228 nt, belonging to HBV P region, and inserted into
the Alu repeats with 3 bp (CTG) of shared sequences (Fig. 3B). As a result, it was concluded
that the full length of inserted HBV fragment of the chrX: 11009033
integration site was 987 bp, which contained two fragments of
dslDNA with the same orientation (1,744–1,094 and 1,565–1,228 nt;
Fig. 3C).
Transcription of the chrX: 11009033
integration site and quantitative detection with that of PAK3
HBV-PAK3 chimeric transcript of the chrX: 11009033
integration site was successfully observed in HepG2.2.15 cells by
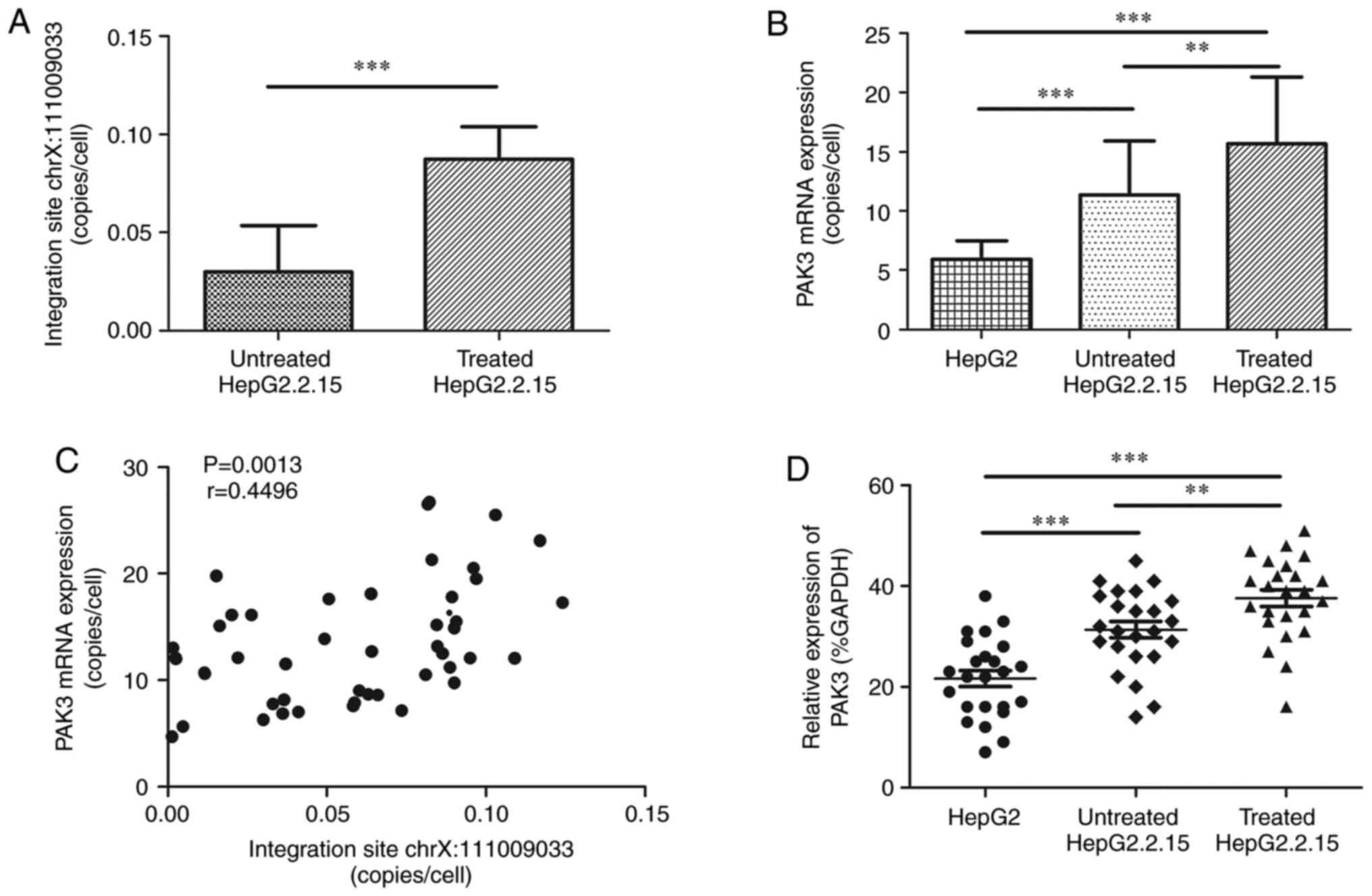
RT-qPCR (Fig. 2A). As
H2O2 can exacerbate HBV integration (12), copy number of this integration site
in cDNA was detected in HepG2.2.15 cells with and without
H2O2 treatment. Its average level was
3.02×10−2±2.33×10−2 copies/cell (range,
1.36×10−3−8.30×10−2 copies/cell) in cells
without H2O2 treatment and
8.73×10−2±1.65×10−2 copies/cell
(5.87×10−2−1.24×10−1 copies/cell) in the
cells with H2O2 treatment with significant
difference between them (P<0.0001; Fig. 4A). The level of PAK3 was 15.67±5.65
copies/cell (7.88-26.70 copies/cell) in HepG2.2.15 cells with
H2O2 treatment, significantly higher than
that in the cells without H2O2 treatment,
11.34±4.58 copies/cell (4.68-21.30 copies/cell, P=0.0076), and that
in HepG2 cells, 5.92±1.54 copies/cell (3.90-9.50 copies/cell,
P<0.0001). Significant difference was also found between
HepG2.2.15 cells without H2O2 treatment and
HepG2 cells (P<0.0001, Fig.
4B). The copy numbers of chrX: 11009033 integration site were
positively correlated with those of PAK3 (P=0.0013, Fig. 4C).
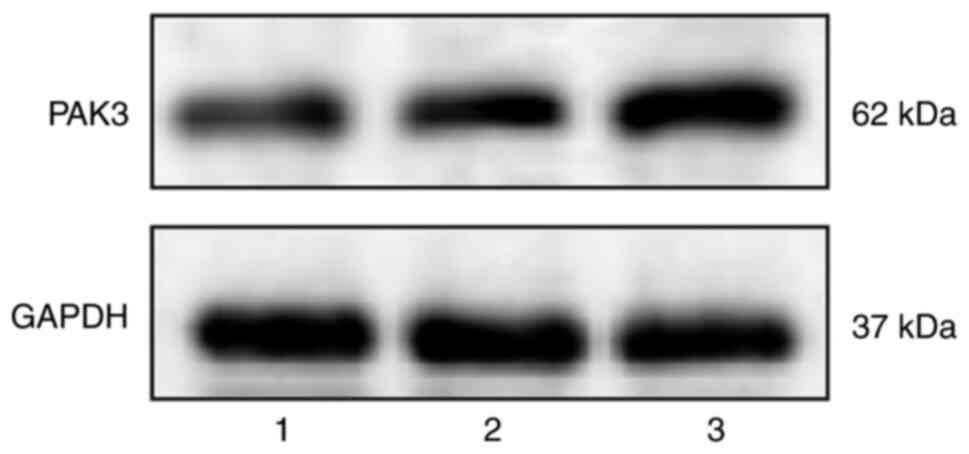
PAK3 expression is high in HepG2.2.15
cells
To detect protein expression of PAK3, western
blotting was performed on HepG2 and HepG2.2.15 cells with and
without H2O2 treatment (Fig. 5). The overall PAK3 expression was
significantly increased in HepG2.2.15 cells with
H2O2 treatment compared with that in
HepG2.2.15 cells without H2O2 treatment
(37.63±8.16 and 31.38±7.94, P=0.008) and HepG2 cells (21.67±7.88,
P<0.0001), while a significant difference was also found between
HepG2.2.15 cells without H2O2 treatment and
HepG2 cells (31.38±7.94 and 21.67±7.88, P=0.0002, Fig. 4D).
Discussion
HepG2.2.15 cells are derived from HepG2 with stable
HBV expression (14). Although the
HepG2.2.15 cell line may not be used to study the process of HBV
infection, it is an ideal tool to study the role of ubiquitination
modification on HBV replication, HCC progression and immune
tolerance (15). Using HepG2 and
HepG2.2.15 cells, molecular mechanisms in hepatocarcinogenesis have
been found to cause HBV integration, such as increased apoptosis,
DNA hypermethylation and ubiquitylome and proteome modification in
host cells (4,15,16).
We previously reported the chrX: 11009033 integration site located
in the intron of PAK3 gene in HepG2.2.15 cells cultivated in INSERM
U1052 (10). Studies have
indicated that HBV integration sites are preferentially enriched in
the intron of human gene, which may result in host gene dysfunction
(17,18). PAKs are serine/threonine protein
kinases that are overactivated or overexpressed in many types of
tumor including breast cancer, pancreatic melanoma, thyroid cancer
(19). PAK3 is a human member of
the PAK gene family mapping on chromosome X, which is closely
associated with tumor invasion, migration and proliferation
(20). The aberrant activation and
expression of PAK3 occurs during tumor pathogenesis in gastric and
pancreatic cancer, causing it to be considered a tumor enhancer
protein (21,22). Epithelial-mesenchymal transition
(EMT), a cellular reprogramming process, is key for tumor
metastasis and serves a key role in hepatocarcinogenesis by which
epithelial cells alter their shape (23). PAK3 serves as an oncogene in HCC by
enhancing EMT (10).
In the present study the chrX: 11009033 integration
site was detected in HepG2.2.15 cells, indicating this site
originated from primary human hepatocyte (PHH) but not from clone
hepatocyte and may be an ideal integration event for in
vitro study (12). Because
PAK3 may contribute to HCC, this finding also provides an
opportunity to evaluate molecular mechanisms of tumorigenesis of
HBV integration. Detection of the full sequence of inserted HBV DNA
fragments of this site is difficult by conventional methods.
Firstly, the integrated HBV fragments, ranging in size from 28 to
3,215 bp, exist different kinds of rearrangements, such as forward
simple junction, reverse simple junction, forward and reverse
complicated junctions. Secondly, according to Illumina long-reads
sequencing, the presence of structural abnormity of the host genome
containing HBV integration results in host genome instability
(24–27).
The present study used an improved invPCR technology
using AluI instead of NcoI restriction enzyme. The
invPCR technology can amplify single copies of DNA template with
high specificity and sensitivity. An Alu-repeated sequence is
interspersed at the average interval of ~4 kb in the human genome,
which is often used to amplify unknown flanking sequences (6). To amplify the chimeric fragments, the
sequence of which partially aligned to Alu repeats and the unknown
integrated HBV genome, two Alu primers with opposite orientation
and one chimeric primer were designed, which may be beneficial for
Alu digestion and T4 ligation from the two sides of the amplified
fragment. Furthermore, double digestion (BsiHKAI and
SphI-HF) was performed to avoid non-specific PCR
amplification. Dilution step may be key for successful specific
amplification in the second round of invPCR. While HBV X region had
more integration opportunities than other parts of HBV, the left
junction of inserted viral fragments of this site was located in
1,228 nt, providing a novel viral inserting region (HBV P region).
On the other hand, 3 bp (CTG) of microhomology homologous (MH) was
found, indicating the host chromosomal DNA DSB in this junction was
repaired by microhomology-mediated end joining (MMEJ) (28,29).
MMEJ is an alternative DNA DSB repair system in mitochondrial DNA
lesions (30). As there is
enriched MH between HBV and human genome sequences, MMEJ may also
be an important mechanism mediating virus integration besides
non-homologous end joining (31).
Additionally, the presence of HBV-PAK3 chimeric transcripts
suggested this integration site may regulate PAK3 gene
expression.
E-cadherin is an epithelial marker gene. During HCC
progression, hepatocytes lose cell-to-cell contact and acquire
migration abilities to spread to distant or surrounding tissue due
to the loss of E-cadherin, thus promoting the invasion and
migration of cancer cells (32).
The occurrence and promotion of EMT decreases expression of
E-cadherin, chiefly via cellular transcription processes involving
three transcription factor families: Twist, ZEB and Snail (33). Once activated, these transcription
factors can inhibit the expression of epithelial marker genes such
as E-cadherin and promote the expression of stromal genes
simultaneously (34).
During HCC progression and migration, increased
TGF-β signaling is critical for promotion of EMT (35). Smad proteins are downstream targets
of TGF-β signaling, which may serve critical roles in proliferation
and regulation of transcription and cell differentiation (36). Recent research indicated that in
hepatoma cells, PAK3 gene acts through the Smad-dependent TGF-β
pathway to enhance EMT (10). In
the present study, RT-qPCR and western blot analysis demonstrated
that PAK3 was significantly upregulated in HepG2.2.15 cells
compared with HepG2 cells, suggesting that the occurrence of HBV
integration in HepG2.2.15 cells may affect gene functions such as
that of PAK3 (4).
Persistent HBV infection can increase the production
of reactive oxygen species in hepatocytes, leading to oxidative
damage, host DNA mutation and exacerbating HBV infection (37). This provides opportunity for the
selected clonal expansion of HBV-integrated hepatocytes, which may
contribute to HCC progression. While H2O2 may
induce DSB in hepatoma cells (38,39),
treating HepG2.2.15 cells with H2O2 may mimic
ROS production and promote HBV integration (40). Here, the average levels of the
chrX: 11009033 integration site and PAK3 expression, which were
positively correlated, were significantly higher in HepG2.2.15
cells with H2O2 treatment compared with the
cells without treatment. Multiple factors may contribute to
upregulation of PAK3 expression in HepG2.2.15 cells, including
occurrence and clonal expansion of chrX: 11009033 integration site,
as shown by the significant difference in levels of PAK3 between
HepG2 and HepG2.2.15 cells and its positive correlation with chrX:
11009033 integration site in the latter. The mechanism at the
molecular level underlying the functions of EMT and TGF-β/Smad
signaling need to be further investigated.
In summary, the chrX: 11009033 integration site,
originated from PHH and may be an ideal integration event for in
vitro study. The full length of inserted HBV fragment of this
site was 987 bp, containing two fragments of dslDNA with the same
orientation. The occurrence of the chrX: 11009033 integration site
and its clonal expansion may upregulate PAK3 expression and serve a
role in hepatocarcinogenesis. Further investigation is required to
determine its molecular mechanisms.
Acknowledgements
Not applicable.
Funding
The present study was supported by the National Natural Science
Foundation of Hubei Province (grant no. 2020CFB608).
Availability of data and materials
The datasets used and/or analyzed during the present
study are available from the corresponding author on reasonable
request.
Authors' contributions
PR and JS confirm the authenticity of all the raw
data, designed and conceived the study. PR, RZ and HSY conducted
cell culture and molecular biological experiments. MJL and CH
performed western blot experiments. CPH and XFD conducted the
statistical analysis. PR wrote the manuscript. All authors have
read and approved the final manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
GBD 2016 Causes of Death Collaborators, .
Global, regional, and national age-sex specific mortality for 264
causes of death, 1980–2016: A systematic analysis for the global
burden of disease study 2016. Lancet. 390:1151–1210. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Ruan P, Zhou B, Dai X, Sun Z, Guo X, Huang
J and Gong Z: Predictive value of intrahepatic hepatitis B virus
covalently closed circular DNA and total DNA in patients with acute
hepatitis B and patients with chronic hepatitis B receiving
anti-viral treatment. Mol Med Rep. 9:1135–1141. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Lin SY, Zhang A, Lian J, Wang J, Chang TT,
Lin YJ, Song W and Su YH: Recurrent HBV integration targets as
potential drivers in hepatocellular carcinoma. Cells. 10:12942021.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Hu X, Jiang J, Ni C, Xu Q, Ye S, Wu J, Ge
F, Han Y, Mo Y, Huang D and Yang L: HBV integration-mediated cell
apoptosis in HepG2.2.15. J Cancer. 10:4142–4150. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Tu T, Budzinska MA, Vondran FWR, Shackel
NA and Urban S: Hepatitis B virus DNA integration occurs early in
the viral life cycle in an in vitro infection model via sodium
taurocholate cotransporting polypeptide-dependent uptake of
enveloped virus particles. J Virol. 92:e020007–17. 2018. View Article : Google Scholar
|
|
6
|
Minami M, Poussin K, Bréchot C and
Paterlini P: A novel PCR technique using Alu-specific primers to
identify unknown flanking sequences from the human genome.
Genomics. 29:403–408. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Ding D, Lou X, Hua D, Yu W, Li L, Wang J,
Gao F, Zhao N, Ren G, Li L and Lin B: Recurrent targeted genes of
hepatitis B virus in the liver cancer genomes identified by a
next-generation sequencing-based approach. PLoS Genet.
8:e10030652012. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Lau CC, Sun T, Ching AK, He M, Li JW, Wong
AM, Co NN, Chan AW, Li PS, Lung RW, et al: Viral-human chimeric
transcript predisposes risk to liver cancer development and
progression. Cancer Cell. 25:335–349. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Svicher V, Salpini R, Piermatteo L,
Carioti L, Battisti A, Colagrossi L, Scutari R, Surdo M,
Cacciafesta V, Nuccitelli A, et al: Whole exome HBV DNA integration
is independent of the intrahepatic HBV reservoir in HBeAg-negative
chronic hepatitis B. Gut. 70:2337–2348. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Gao Z, Zhong M, Ye Z, Wu Z, Xiong Y, Ma J,
Chen H, Zhu Y, Yang Y, Zhao Y and Zhang Z: PAK3 promotes the
metastasis of hepatocellular carcinoma by regulating EMT process. J
Cancer. 13:153–161. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Ruan P, Dai X, Sun J, He C, Huang C, Zhou
R and Chemin I: Integration of hepatitis B virus DNA into
p21-activated kinase 3 (PAK3) gene in HepG2.2.15 cells. Virus
Genes. 56:168–173. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Dandri M, Burda MR, Bürkle A, Zuckerman
DM, Will H, Rogler CE, Greten H and Petersen J: Increase in de novo
HBV DNA integrations in response to oxidative DNA damage or
inhibition of poly(ADP-ribosyl)ation. Hepatology. 35:217–223. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Tu T and Jilbert AR: Detection of
hepatocyte clones containing integrated hepatitis B virus DNA using
inverse nested PCR. Methods Mol Biol. 1540:97–118. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Sells MA, Chen ML and Acs G: Production of
hepatitis B virus particles in Hep G2 cells transfected with cloned
hepatitis B virus DNA. Proc Natl Acad Sci USA. 84:1005–1009. 1987.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Yuan S, Tanzeel Y, Tian X, Zheng D,
Wajeeha N, Xu J, Ke Y, Zhang Z, Peng X, Lu L, et al: Global
analysis of HBV-mediated host proteome and ubiquitylome change in
HepG2.2.15 human hepatoblastoma cell line. Cell Biosci. 11:752021.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Oikawa R, Watanabe Y, Yotsuyanagi H,
Yamamoto H and Itoh F: DNA methylation at hepatitis B virus
integrants and flanking host mitochondrially encoded cytochrome C
oxidase III. Oncol Lett. 24:4242022. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Yan H, Yang Y, Zhang L, Tang G, Wang Y,
Xue G, Zhou W and Sun S: Characterization of the genotype and
integration patterns of hepatitis B virus in early- and late-onset
hepatocellular carcinoma. Hepatology. 61:1821–1831. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Li W, Cui X, Huo Q, Qi Y, Sun Y, Tan M and
Kong Q: Profile of HBV integration in the plasma DNA of
hepatocellular carcinoma patients. Curr Genomics. 20:61–68. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Ye DZ and Field J: PAK signaling in
cancer. Cell Logist. 2:105–116. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Zhou N, Ding B, Agler M, Cockett M and
McPhee F: Lethality of PAK3 and SGK2 shRNAs to human papillomavirus
positive cervical cancer cells is independent of PAK3 and SGK2
knockdown. PLoS One. 10:e01173572015. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Wang GJ, Yu TY, Li YR, Liu YJ and Deng BB:
Circ_0000190 suppresses gastric cancer progression potentially via
inhibiting miR-1252/PAK3 pathway. Cancer Cell Int. 20:3512020.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Wu HY, Yang MC, Ding LY, Chen CS and Chu
PC: p21-activated kinase 3 promotes cancer stem cell phenotypes
through activating the Akt-GSK3β-β-catenin signaling pathway in
pancreatic cancer cells. Cancer Lett. 456:13–22. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Santamaria PG, Moreno-Bueno G, Portillo F
and Cano A: EMT: Present and future in clinical oncology. Mol
Oncol. 11:718–738. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Hama N, Totoki Y, Miura F, Tatsuno K,
Saito-Adachi M, Nakamura H, Arai Y, Hosoda F, Urushidate T, Ohashi
S, et al: Epigenetic landscape influences the liver cancer genome
architecture. Nat Commun. 9:16432018. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Mason WS, Low HC, Xu C, Aldrich CE,
Scougall CA, Grosse A, Clouston A, Chavez D, Litwin S, Peri S, et
al: Detection of clonally expanded hepatocytes in chimpanzees with
chronic hepatitis B virus infection. J Virol. 83:8396–8408. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Yang L, Ye S, Zhao X, Ji L, Zhang Y, Zhou
P, Sun J, Guan Y, Han Y, Ni C, et al: Molecular characterization of
HBV DNA integration in patients with hepatitis and hepatocellular
carcinoma. J Cancer. 9:3225–3235. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Ruan P, Dai XF, Sun J, He C, Huang C, Zhou
R, Cao Z and Ye L: Different types of viral-host junction found in
HBV integration breakpoints in HBV-infected patients. Mol Med Rep.
19:1410–1416. 2019.PubMed/NCBI
|
|
28
|
Li W, Zeng X, Lee NP, Liu X, Chen S, Guo
B, Yi S, Zhuang X, Chen F, Wang G, et al: HIVID: An efficient
method to detect HBV integration using low coverage sequencing.
Genomics. 102:338–344. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Hu Z, Zhu D, Wang W, Li W, Jia W, Zeng X,
Ding W, Yu L, Wang X, Wang L, et al: Genome-wide profiling of HPV
integration in cervical cancer identifies clustered genomic hot
spots and a potential microhomology-mediated integration mechanism.
Nat Genet. 47:158–163. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Tadi SK, Sebastian R, Dahal S, Babu RK,
Choudhary B and Raghavan SC: Microhomology-mediated end joining is
the principal mediator of double-strand break repair during
mitochondrial DNA lesions. Mol Biol Cell. 27:223–235. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Lamouille S, Xu J and Derynck R: Molecular
mechanisms of epithelial-mesenchymal transition. Nat Rev Mol Cell
Biol. 15:178–196. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Mladenov E, Magin S, Soni A and Iliakis G:
DNA double-strand-break repair in higher eukaryotes and its role in
genomic instability and cancer: Cell cycle and
proliferation-dependent regulation. Semin Cancer Biol. 37–38.
51–64. 2016.PubMed/NCBI
|
|
33
|
Thiery JP: Epithelial-mesenchymal
transitions in development and pathologies. Curr Opin Cell Biol.
15:740–746. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Gui Y, Khan MGM, Bobbala D, Dubois C,
Ramanathan S, Saucier C and Ilangumaran S: Attenuation of
MET-mediated migration and invasion in hepatocellular carcinoma
cells by SOCS1. World J Gastroenterol. 23:6639–6649. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Zhang C, Zhang X, Xu R, Huang B, Chen AJ,
Li C, Wang J and Li XG: TGF-β2 initiates autophagy via Smad and
non-Smad pathway to promote glioma cells' invasion. J Exp Clin
Cancer Res. 36:1622017. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Ahmadi A, Najafi M, Farhood B and
Mortezaee K: Transforming growth factor-β signaling: Tumorigenesis
and targeting for cancer therapy. J Cell Physiol. 234:12173–12187.
2019. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Zhao LH, Liu X, Yan HX, Li WY, Zeng X,
Yang Y, Zhao J, Liu SP, Zhuang XH, Lin C, et al: Genomic and
oncogenic preference of HBV integration in hepatocellular
carcinoma. Nat Commun. 7:129922016. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Schraufstatter IU, Hyslop PA, Hinshaw DB,
Spragg RG, Sklar LA and Cochrane CG: Hydrogen peroxide-induced
injury of cells and its prevention by inhibitors of
poly(ADP-ribose) polymerase. Proc Natl Acad Sci USA. 83:4908–4912.
1986. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Schraufstatter IU, Hyslop PA, Jackson J
and Cochrane CC: Oxidant injury of cells. Int J Tissue React.
9:317–324. 1987.PubMed/NCBI
|
|
40
|
Mason WS, Jilbert AR and Summers J: Clonal
expansion of hepatocytes during chronic woodchuck hepatitis virus
infection. Proc Natl Acad Sci USA. 102:1139–1144. 2005. View Article : Google Scholar : PubMed/NCBI
|