Introduction
Type-2 diabetes mellitus (T2DM) is one of the most
common metabolic diseases, characterized by chronic hyperlipidemia
and hyperglycemia. With the incidence of T2DM increasing, T2DM has
become a major public health concern. A study reported that ~425
million individuals worldwide were diagnosed with T2DM in 2017,
which is expected to be ~629 million by 2045 (1). However, the most concerning part of
T2DM is that a number of complications can arise over the prolonged
course of the disease which seriously threaten human health, such
as cardiovascular disease, retinopathy, neuropathy, and nephropathy
(2). Diabetic kidney disease
(DKD), the most frequent complication of diabetes mellitus (DM),
has been reported to be the main cause of poor prognoses in DM
patients (3,4). DKD is characterized by gradual renal
dysfunction and fibrosis caused by glycolipid metabolism disorder
and is the main cause of end-stage kidney disease (5). Of DM patients, ~30–40% have been
reported to develop DKD (6,7),
which has received increased attention due to the increase of T2DM
patients and its difficult treatment (8,9).
However, its pathogenesis is not fully clarified, therefore,
exploring the mechanisms of T2DM-induced DKD and seeking potential
therapeutic targets require further study.
Increasing evidence reveals that inflammation of the
kidney plays a pivotal role in T2DM-induced DKD (10). The persistent hyperglycemic and
hyperlipidemic environments in T2DM can cause renal inflammation
and damage, eventually resulting in renal fibrosis (11). Inflammasomes are a multi-protein
complex of intracytoplasmic pattern recognition receptors, which
are reported to recognize damage- and pathogen-associated molecular
patterns (12) and growing
evidence suggests that inflammasomes may play a critical role in
evoking inflammation by serving as a platform to recruit the
apoptosis-associated speck-like protein containing a CARD (ASC) and
procaspase-1, leading to the maturation of inflammatory cytokines
(13). Nod-like receptor protein 3
(NLRP3) inflammasomes are reported to be involved in the occurrence
and progression of several renal diseases, including DKD (14). NLRP3 inflammasomes promote the
release of the inflammatory cytokines, such as IL-1β, in the
pathogenesis of DKD, inducing sustained inflammation and renal
injury and promoting dysfunction and fibrosis of the kidney
(14). Inhibiting NLRP3
inflammasomes improves renal function, attenuates
glomerulosclerosis, interstitial fibrosis and inflammation and
decreases TGF-β and phosphorylated (p-)SMAD2/3 expression in the
kidney of mice with T2DM, thereby inhibiting renal fibrosis
(15). However, the regulatory
mechanisms of NLRP3 in T2DM-induced DKD remain to be
elucidated.
Intracellular calcium
([Ca2+]i) overload due to various reasons is
associated with the progression of DKD (16). The transient receptor potential
channel 6 (TRPC6), a canonical non-selective cation channel with
significant permeability to Ca2+, is widely expressed in
a number of tissues such as the brain and kidney (17,18).
According to data from the Human Protein Atlas (https://www.proteinatlas.org/ENSG00000137672-TRPC6/tissue),
TRPC6 mRNA and protein are widely expressed in human tissues,
including the digestive system, muscle tissues and the urinary
system, with Trpc6 being more abundantly expressed in the
kidney. In the kidney, TRPC6 is largely expressed in multiple
cells, such as podocytes, tubular epithelial cells and glomerular
mesangial cells (19–21). It has been reported that drastic
increases of TRPC6-mediated [Ca2+]i causes
podocyte hypertrophy and foot process effacement, resulting in
renal injury in DKD (19). TRPC6
inhibitors are reported to alleviate renal fibrosis in a unilateral
ureteral obstruction mouse model (22). Calcineurin (CN) is a type of
Ca2+-dependent phosphatase (23) and is reported to participate in
multiple cellular processes and signal pathways (24). As an important nuclear
transcription factor, nuclear factor of activated T cells (NFAT)
can be regulated by CN to regulate the expression of target genes
(25). NFAT2 is extensively
expressed in renal tissue and plays a pivotal role in promoting
podocyte damages in DKD, while the inhibitor of NFAT, 11R-VIVIT,
can reduce renal injury and renal fibrosis in DKD (26). However, the mechanism of
TRPC6-CN-NFAT2 regulation of NLRP3 inflammasomes, eventually
promoting renal injury and glomerular fibrosis in DKD, remains to
be elucidated.
The present study hypothesized that Trpc6
knockout might inhibit calcium overload and NFAT2 activation, thus
alleviating renal injury and fibrosis in T2DM-induced DKD. To test
this hypothesis, the roles of T2DM in renal fibrosis, renal damage
and changes in CN-NFAT2 signaling and NLRP3 inflammasomes were
observed in wild type mice (WT) mice. The present study also
explored the effects of Trpc6 knockout on the improvement of
T2DM-induced DKD and the regulation of CN-NFAT2 signaling and NLRP3
inflammasomes. The findings provided a more comprehensive
understanding of DKD and provided additional therapeutic targets
for treatment of T2DM-induced DKD.
Materials and methods
Animals and treatment
In the present study, the Trpc6 knockout
(Trpc6−/−) mice (C57BL/6J) were obtained by
Suzhou Cyagen Biosciences (Suzhou) Inc. The genotype of
Trpc6−/− homozygous mice was determined by PCR
amplification and agarose gel electrophoresis separation (Fig. S1). WT mice were obtained from the
same litter of Trpc6−/− mice. The mice were bred
in an environmentally controlled room (temperature, 22–24°C;
relative humidity, 50–60%) under a 12-h light/dark cycle with
unlimited access to food and water. After the acclimatization of 50
male mice (20–25 g) at 8 weeks of age, they were divided into five
groups: WT, Trpc6−/−, WT + high fat diet (HFD) +
streptozotocin (STZ), Trpc6−/− + HFD and
Trpc6−/− + HFD + STZ (n=10 per group). The mice
of the WT and Trpc6−/− groups were fed with a
growth-maintenance diet and the other mice were given a HFD of 20%
carbohydrates, 20% protein and 60% fat (cat. no. D12492, Jiangsu
Xietong Pharmaceutical Bio-engineering Co., Ltd.) for eight
weeks.
After eight weeks, the mice of the WT + HFD + STZ
and Trpc6−/− + HFD + STZ groups (fasting without
water restriction for 8 h) were treated intraperitoneally with STZ
(110 mg/kg, Shanghai Yuanye Bio-Technology Co., Ltd.), which was
dissolved in a 0.1 M sodium citrate buffer to induce the T2DM model
(27). The fasting blood glucose
(FBG) level was measured after 72 h. Mice with a FBG concentration
≥16.7 mM were considered optimal for further experiments. Next,
each group was given the same diet as described previously for
eight weeks. The health and behavior of the animals were monitored
daily. Throughout the experiment, one mouse in the
Trpc6−/− + HFD + STZ group succumbed to infection
at week 13 and one mouse in the WT + HFD + STZ group succumbed to
hyperglycemia at weeks 13 and 14, respectively. The remaining mice
were sacrificed. The mice were anesthetized using an
intraperitoneal (IP) injection of tribromoethanol (300 mg/kg) and
sacrificed by cervical dislocation after orbital blood extraction
(once; ~0.2 ml) when they did not respond to gentle stimulation.
All experiments were conducted under the approval of the Anhui
Medical University Ethics Committee (approval no. LLSC20190302).
When a click in the neck of the mouse was clearly heard, i.e.,
cervical spine detachment, spinal cord rupture and the heartbeat of
the mouse stopped and the spontaneous respiration ceased it was
considered that the mouse was dead and both kidneys were quickly
removed. One kidney was fixed with paraformaldehyde (4%) for
histological examination and the other was placed at −80°C for
western blot analysis.
Detection of body weight, FBG and
biochemical parameters
The body weight and FBG levels were observed to
evaluate the T2DM model. The body weight (n=8-10) was measured once
every two weeks. The FBG levels were measured every two weeks from
8–16 weeks by using a portable blood glucose meter (Sinocare Inc.).
The test was performed using a second drop of blood from the tail
vein, the total blood volume being ~40 µl. Following the sacrifice
of the mice, the serum creatinine (SCR) and blood urea nitrogen
(BUN) were measured using urea assay kit and creatinine assay kit
(Nanjing Jiancheng Bioengineering Institute; cat. nos. C013-2-1 and
C011-2-1). At 16 weeks, the 24 h urine samples were collected and
kits used as follows: the urinary albumin level (cat. no.
C035-2-1), serum total cholesterol (TC; cat. no. A111-1-1),
triglyceride (TG; cat. no. A110-1-1), free fatty acids (FFA; cat.
no. A042-2-1), low density lipoprotein (LDL-C; no. A113-1-1) and
high-density lipoprotein (HDL-C; cat. no. A112-1-1) (all Nanjing
Jiancheng Bioengineering Institute). All kit procedures were
conducted strictly according to the manufacturer instructions.
Histology examination
The morphological examination of the kidneys was
performed using hematoxylin and eosin (H&E), periodic
acid-Schiff (PAS; Beijing Solarbio Science & Technology Co.,
Ltd.; cat. no. G1208) and Masson's trichrome staining (Beijing
Solarbio Science & Technology Co., Ltd.; cat. no. G1340)
methods. H&E staining was conducted to observe the pathological
changes of the kidney (28).
Briefly, the kidneys (n=4) were fully fixed with paraformaldehyde
(4%) at 4°C for 24 h, dehydrated and embedded in paraffin, then cut
into 5-µm thick sections. The kidney sections were deparaffinized
in xylene and rehydrated in graded alcohol series (anhydrous
ethanol, 85% ethanol and 75% ethanol), and then stained with
hematoxylin for 4 min and eosin for 35 sec at room temperature
(RT). After being rinsed with running water, the tissues were
sealed using neutral resin and were imaged using an Intelligent
Tissue Section Imaging System (3DHISTECH, Ltd.) to observe renal
histopathological changes. Renal injury was scored in a blinded
manner with the following scoring criteria: no injury=0; <25%=1;
25–50%=2; 50–75%=3; and >75%=4.
Masson's staining was performed to observe tissue
fibrosis (29). For Masson
staining, the paraffin sections (n=4) were first dewaxed, hydrated
and stained with hematoxylin for 4 min, then underwent acidic
ethanol fractionation for 15 sec and were stained with Masson's
blue solution for 5 min. This was followed by fuchsin staining for
9 min, phosphomolybdic acid for 3 min, then aniline blue staining
for 6 min. All steps were performed at RT. The sections were then
sealed using neutral resin and imaged using the Intelligent Tissue
Section Imaging System (3DHISTECH, Ltd.). Finally, the mean density
of Masson staining from five random areas (magnification, ×400) per
section was analyzed by the Image-pro Plus 6.0 image software
(Media Cybernetics, Inc.) to evaluate renal fibrosis.
PAS staining was further performed to measure the
disposition of acidic glycoproteins in order to assess renal injury
(30). The renal paraffin sections
(n=4) were dewaxed and hydrated, then stained in the Schiff
solution for 8 min and the hematoxylin solution for 1 min at RT.
The sections were sealed using neutral resin. The results of
PAS-staining were imaged with the Intelligent Tissue Section
Imaging System (3DHISTECH, Ltd.). The positive areas were purplish
red and the cell nuclei were blue. To evaluate the extent of renal
fibrosis, the mean intensity of positive mesangial cells and the
ratio of mesangial area to glomerular (%) from five random areas
(magnification, ×400) per section were analyzed by the Image-Pro
Plus 6.0 software (Media Cybernetics, Inc.).
Reactive oxygen species (ROS)
measurement
Dihydroethidium (DHE) was used to measure ROS
production (31). Briefly, mice
(n=3) were injected via the tail vein with DHE (0.1 ml/10 g, 100
µM; Beyotime Institute of Biotechnology). The mouse was euthanized
by cervical dislocation after 30 min and the kidneys were quickly
removed and embedded in OCT composite media (Sakura Finetek USA,
Inc.) at −20°C for 1 h. The renal tissue was sectioned at 10 µm
using a cryostat (CM3050; Leica Microsystems GmbH) and the sections
rinsed three times with PBS buffer and stained with Hoechst
solution (5 mg/l; MilliporeSigma) at RT for 5 min. The section was
washed and sealed with an anti-fluorescent quenching agent, then
imaged using the Intelligent Tissue Section Imaging System
(3DHISTECH, Ltd.). Finally, the fluorescence intensity from three
random areas (magnification, ×400) per section was examined using
the Image-Pro Plus software (Media Cybernetics, Inc.) to detect the
levels of ROS production.
β-galactosidase (β-Gal) activity
The β-Gal activity was examined by β-Gal staining
for senescence-associated injury. The frozen sections (n=3) were
first warmed to RT, allowed to dry slightly, fixed with the β-Gal
fixative (Beyotime Institute of Biotechnology) for 15 min, then
washed three times with PBS. The PBS was then discarded and the
β-Gal staining working solution was added to the sections for
incubation at 37°C overnight. The staining solution was then
discarded, rinsed three times with PBS and imaged using the
Intelligent Tissue Section Imaging System (3DHISTECH, Ltd.).
Finally, the mean density (blue) from three random areas
(magnification, ×400) per tissue was examined with the Image-Pro
Plus software (Media Cybernetics, Inc.) to estimate β-Gal
activity.
Immunohistochemistry
The 5 µm paraffin sections of kidney tissue (n=4)
were dewaxed and rehydrated. For antigen retrieval, the section was
heated using a microwave oven for 8 min in sodium citrate buffer,
stopped for 7 min, then reheated for 8 min. The section was then
incubated in H2O2 (3%) at RT for 30 min to
block endogenous peroxidase and then sealed with blocking solution
at RT for 1 h. The sections were incubated in rabbit polyclonal
antibodies (Table SI) at 4°C
overnight. The sections were rinsed with PBS and incubated with
polymer-coupled peroxidase-labeled goat anti-rabbit IgG at RT for 1
h, then the section was stained with a DAB kit and hematoxylin.
Finally, at the end of the light-protected sealing, the sections
were imaged by the Intelligent Tissue Section Imaging System
(3DHISTECH, Ltd.). The mean intensity from five random renal cortex
regions (magnification, ×400) per tissue was quantified by
Image-Pro Plus software (Media Cybernetics, Inc.) to assess the
expression of fibronectin (FN) and collagen IV (COL4).
Western blotting
Total proteins from renal cortexes (n=4) were
isolated by the RIPA lysates buffer (cat. no. P0013B; Beyotime
Institute of Biotechnology) in a fully automated sample freeze
grinder (Shanghai Jingxin Industrial Development Co., Ltd.). After
lysing for 30 min on ice, the tissue lysate was centrifuged (12,000
× g) at 4°C for 20 min to extract the supernatant. The protein
concentration was determined using a BCA assay kit, and then the
protein (20 µg) was separated on 10% gels using SDS-PAGE and
transferred onto PVDF membranes. Next, the membrane was blocked
with the 5% defatted milk at RT for 1 h and was incubated in the
primary antibodies (Table SI) at
4°C overnight. The membrane was then washed three times in TBS with
0.05% Tween-20 (TBST) and was incubated in the corresponding
secondary antibodies of goat anti-mouse IgG (cat. no. S0002;
Affinity Biosciences; 1:10,000) or goat anti-rabbit IgG (cat. no.
S0001; Affinity Biosciences; 1:10,000) for 1 h at RT. After being
rinsed in TBST three times, the Western ECL kit (Bio-Rad
Laboratories, Inc.) was applied to visualize the results and the
Chemi Imaging System (Q4800 mini, Bioshine Chemi) was used to image
the bands. The intensity of protein bands was measured by an image
J v1.53 software (National Institutes of Health). The results were
normalized to β-actin to indicate the change in target protein.
Transmission electron microscopy
Briefly, the renal cortex specimen was sectioned
into 1 mm3 pieces and transferred to 2.5% glutaraldehyde
at 4°C for 2 h (n=3). After washing with PBS, the tissues were
fixed with osmium tetroxide (1%) at RT for 2 h and embedded in Epon
812 for 12 h at 45°C, and then heated at 72°C for 24 h after
dehydration. Ultrathin sections (70 nm) were sliced with an
ultra-microtome (Leica UC7; Leica Microsystems GmbH). The section
was double stained using lead citrate (stained for 30 min at RT)
and uranyl acetate (stained at 4°C overnight before embedding).
Ultrastructural images were taken by a transmission electron
microscope (HT7700; Hitachi High-Technologies Corporation).
Glomerular ultrastructural damage was determined by observing the
glomerular mesangial and foot processes.
Calcium imaging
The human mesangial cell (HMC) line was purchased
from the Modern Analysis and Testing Center of Central South
University. The HMC line was cultured with DMEM medium containing
10% FBS (Zhejiang Tianhang Biotechnology Co., Ltd.) and placed in a
37°C incubator. The HMCs were divided into four groups: control,
high glucose (HG; 25 mM) + palmitic acid (PA; 200 µM), vector + HG
+ PA and TRPC6-siRNA + HG + PA. The sequences of the short
interfering RNAs (siRNAs) were: TRPC6 siRNA sense,
5′-GAGCAUCAUUGACGCAAAUTT-3′ and antisense,
5′-AUUUGCGUCAAUGAUGCUCTT-3′; and negative control siRNA sense,
5′-UUCUCCGAACGUGUCACGUTT-3′ and antisense,
5′-ACGUGACACGUUCGGAGAATT-3′. First, the cells were cultured in the
dishes (35 mm) for 24 h and then transfected with Lipofectamine™
2000 (Thermo Fisher Scientific, Inc.) with an empty vector or siRNA
for 48 h at 37°C according to the manufacturer's instructions.
After treatment with PA and/or HG for 24 h, the HMCs were incubated
in a Fura-2 AM dye solution with F-127 for 20 min at 37°C and
rinsed with an extracellular solution three times. The fluorescence
density was detected every 3 sec under alternating excitation at
340 and 380 nm (F340 and F380) using a Digital Calcium Imaging
Analysis Platform (IX73, DG-4PLUS/OF30; Olympus Corporation). The
ratio (F340/F380) was measured with extracellular Ca2+
(1 mM) for 300 sec to estimate the basal level of
[Ca2+]i. Then, the BAPTA (1 mM,
MedChemExpress), a calcium chelator, or 2 mM CaCl2 were
added into the extracellular solution and the Δ ratio (F340/F380)
was used to estimate the change of calcium homeostasis after BAPTA
or high calcium stimulation. The experiment was repeated three
times independently.
Nile red staining
Nile red staining was used to evaluate the
production of lipids in the tissue sections. All steps were
performed at room temperature. The frozen sections (n=3) were
incubated in Nile red dilution (1 µg/ml) for 20 min. After rinsing
three times, the sections were stained using Hoechst33258 for 4 min
and were sealed using an anti-fluorescence quencher. All steps were
performed at RT. The sections were imaged by the Intelligent Tissue
Section Imaging System (3DHISTECH, Ltd.). The red fluorescence
intensity from three renal cortex regions (magnification, ×400) per
section was quantified using Image-Pro Plus software (Media
Cybernetics, Inc.) to evaluate the lipid production in mouse kidney
cortical regions.
Statistical analysis
The experimental results in the present study are
shown as mean ± SD of ≥3 independent experiments. For statistical
analysis, the data was analyzed using the GraphPad Prism 9.0
statistical software (GraphPad; Dotmatics). Data from each group
were analyzed first by one-way ANOVA, then the difference between
the two groups was performed by Tukey's test. P<0.05 was
considered to indicate a statistically significant difference.
Results
Trpc6 knockout improves renal function
and blood lipid metabolism in T2DM mice
The results revealed that body weight increased
slowly in both WT and Trpc6−/− groups, suggesting
that Trpc6 knockout had no obvious influence on body weight
of normal mice. However, in Trpc6−/− + HFD mice,
body weight significantly increased from 2–16 weeks compared with
the WT control mice (Fig. S2A;
P<0.01). The T2DM mice showed significant weight loss in both WT
+ HFD + STZ and Trpc6−/− + HFD + STZ groups after
STZ injection, compared with the WT mice (Fig. S2A; P<0.01). Compared with the
WT + HFD + STZ mice, Trpc6 knockout visibly delayed body
weight loss (Fig. S2A;
P<0.01) in Trpc6−/− T2DM mice. The FBG
results indicated that the FBG was raised in the
Trpc6−/− + HFD mice compared with the WT control
mice, but within the normal range. After STZ modeling, the FBG
levels were raised in both WT + HFD + STZ and
Trpc6−/− + HFD + STZ mice compared with the WT
mice (Fig. S2B;
P<0.01). However, compared with the WT + HFD + STZ mice,
Trpc6 knockout decreased the FBG levels (Fig. S2B; P<0.01) in T2DM mice,
but the levels were still >20 mM. Additional renal function
indexes showed that the levels of BUN, SCR and urine protein were
clearly raised in WT + HFD + STZ mice compared with the WT mice
(Fig. S2C-E; P<0.05 or
P<0.01); however, compared with the WT + HFD + STZ mice,
knockout Trpc6 markedly decreased the levels of BUN, SCR and
urine protein in T2DM mice (Fig.
S2C-E; P<0.05). Furthermore, HFD-treatment alone had no
influence on renal function in Trpc6−/− mice
compared with the WT control mice. The results revealed that
Trpc6 knockout significantly improved renal function, but
had only a weak hypoglycemic effect in T2DM mice.
The present study further evaluated the change of
lipid metabolism-related parameters of FFA, TC, TG, LDL-C and HDL-C
in serum and FFA in renal tissue. The results showed no significant
difference in these parameters in Trpc6−/− mice
compared with the WT mice, indicating that knockout Trpc6
had no influence on lipid metabolism in normal mice. However, the
levels of these parameters, were markedly raised in the WT + HFD +
STZ mice, with the exception of HDL-C (Fig. S2F-K; P<0.05 or P<0.01).
Compared with WT + HFD + STZ mice, Trpc6 knockout clearly
decreased the levels of TG, TC and LDL-C (Fig. S2F, J and I; P<0.05 or
P<0.01) but there was no statistical difference in HDL-C and FFA
in T2DM mice. The data indicated that Trpc6 knockout may
ameliorate blood lipid metabolism, but had no effect on FFA in
renal tissues of T2DM mice.
Trpc6 knockout significantly protects
against kidney injury and renal fibrosis in T2DM mice
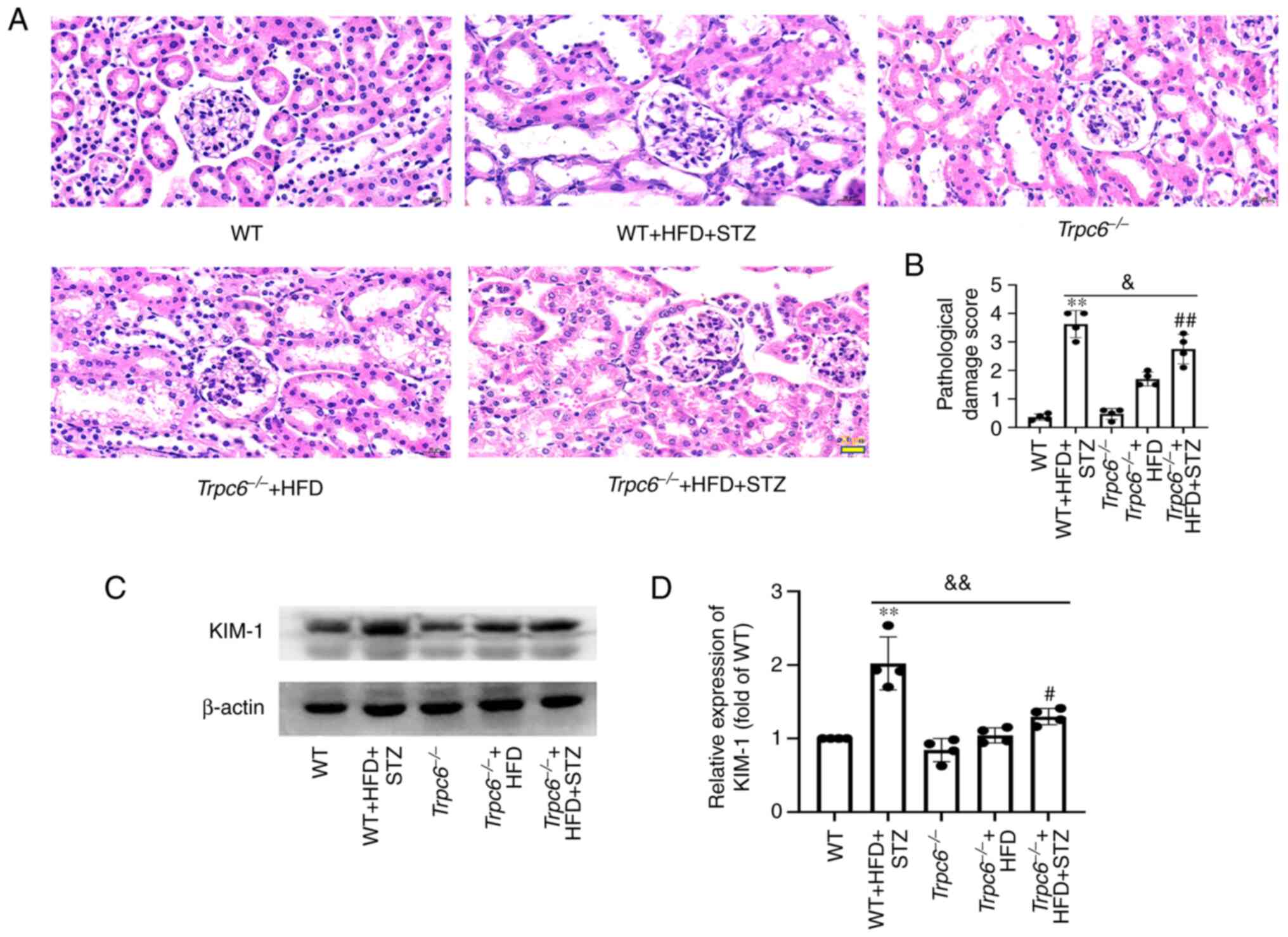
The present study further explored renal injury by
using H&E staining, immunoblotting and PAS staining in
Trpc6−/− T2DM mice. The H&E staining
indicated that there was no obvious renal injury in WT,
Trpc6−/− and Trpc6−/− + HFD
group non-T2DM mice. When compared with the WT mice, the renal
tissue showed evident damages in WT + HFD + STZ mice; the
glomerular mesangial cells were expanded and proliferated and a
number of renal tubular cells were vacuolated and detached.
However, these pathological changes were improved in
Trpc6−/− + HFD + STZ mice (Fig. 1A and B; P<0.05). The present
study further evaluated the biomarker, kidney injury molecule-1
(KIM-1), by western blotting (32). The results revealed that the
expression of KIM-1 in the kidney of WT + HFD + STZ mice was
clearly raised compared with WT mice (Fig. 1C and D; P<0.01), but was
markedly reduced in Trpc6−/− + HFD + STZ mice
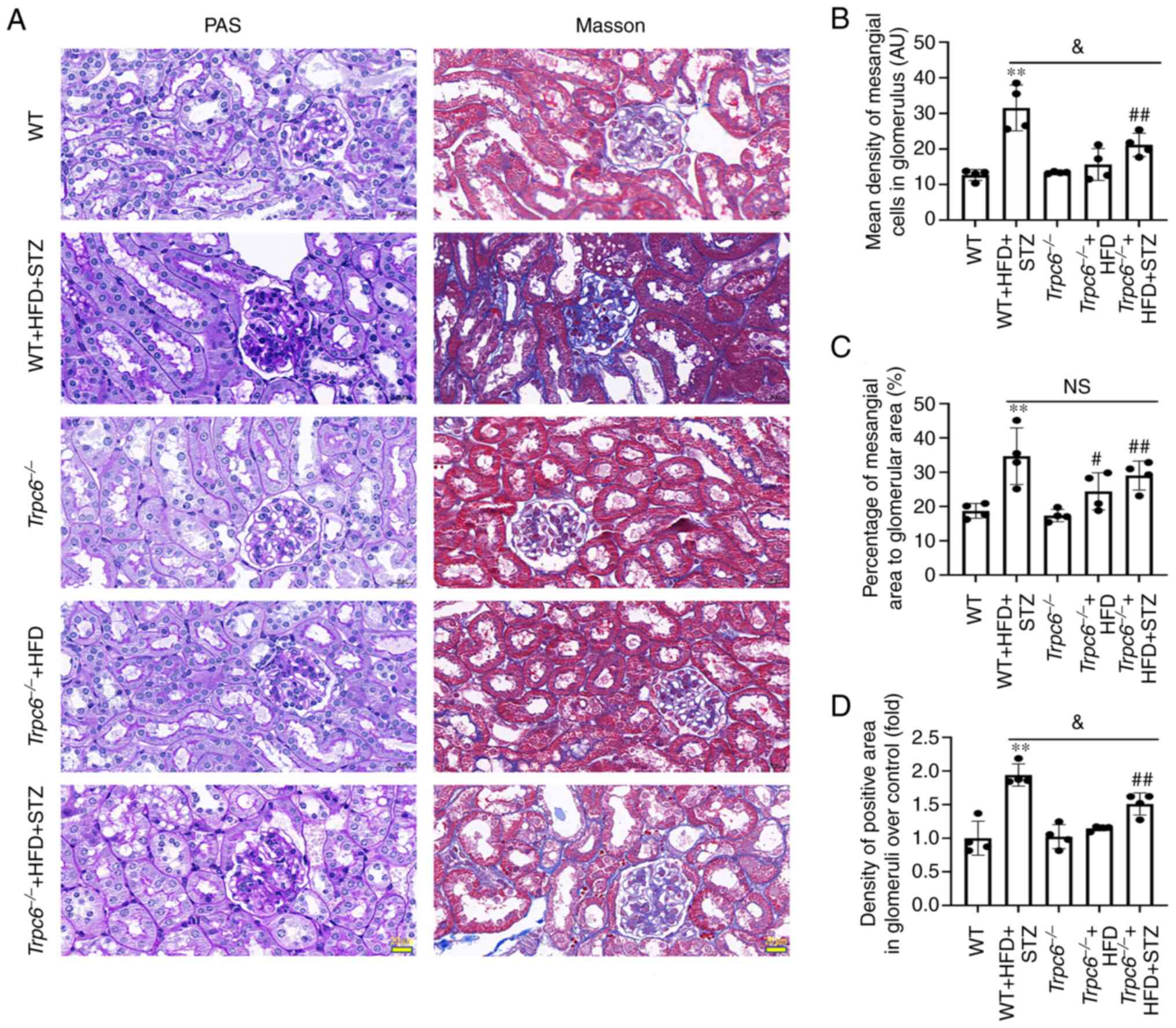
compared with WT + HFD + STZ mice (Fig. 1C and D; P<0.01). PAS staining
was used to assess glomerulosclerosis and renal damage (33). The PAS staining results revealed
that more PAS-positive substances were found in the glomeruli and
renal tubulointerstitial in WT + HFD + STZ mice compared with the
WT mice (Fig. 2A-C; P<0.01).
Trpc6 knockout significantly alleviated PAS-positive
substances in Trpc6−/− + HFD + STZ mice compared
with WT + HFD + STZ mice (Fig.
2A-C; P<0.05). The results revealed that Trpc6
knockout clearly protected against renal injury in T2DM mice.
Renal fibrosis is another important marker of renal
damages. The present study used Masson staining to examine whether
Trpc6 knockout had protective effects on glomerular fibrosis
in T2DM mice. The results indicated that little fibrosis was found
in the renal tubules and glomeruli in the WT,
Trpc6−/− and Trpc6−/− + HFD
groups. Compared with the WT mice, the tubular interstitial and
glomerular fibrosis (blue) was markedly increased in WT + HFD + STZ
mice (Fig. 2A and D; P<0.01).
By contrast, Trpc6 knockout clearly alleviated renal
fibrosis in Trpc6−/− + HFD + STZ mice compared
with WT + HFD + STZ mice (Fig. 2A and
D; P<0.05). The results revealed that knockout Trpc6
may protect against renal fibrosis in T2DM mice and that HFD
treatment alone had no influence on renal injury in
Trpc6−/− mice.
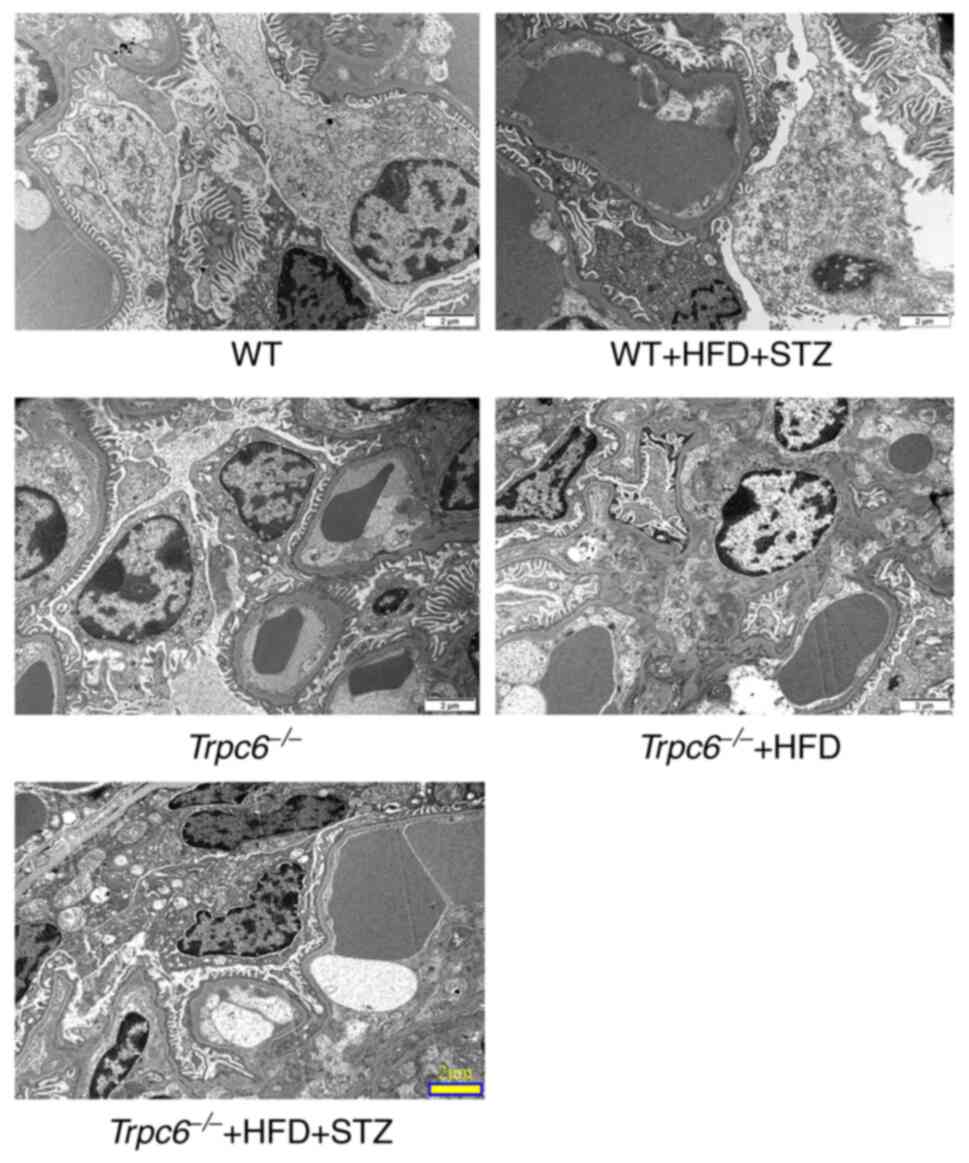
Trpc6 knockout clearly improves the
glomerular ultrastructure in T2DM mice
Transmission electron microscopy was performed to
detect the effect of Trpc6 knockout on the glomerular
microstructure in T2DM mice. The results showed that there was
almost no glomerular damage in WT, Trpc6−/− and
Trpc6−/− + HFD group non-T2DM mice (Fig. 3). Compared with the WT control
mice, the WT + HFD + STZ mice showed obvious glomerular
ultrastructure damages. The foot processes showed obvious fusion
and disappearance (Fig. 3).
However, Trpc6 knockout clearly improved these glomerular
ultrastructure damages in Trpc6−/− + HFD + STZ
mice when compared with WT + HFD + STZ mice (Fig. 3). The data indicate that
Trpc6 knockout clearly improves the glomerular
ultrastructure in T2DM mice.
Trpc6 knockout has no influence on
renal ROS generation in T2DM mice
ROS-induced oxidative stress injury plays a main
role in DKD progression (34);
therefore, ROS accumulation in the renal cortex was assessed using
DHE staining. The results revealed that there was almost no ROS
production in the renal cortex in the WT and
Trpc6−/− mice. Compared with the WT mice, ROS
production was clearly increased in both WT + HFD + STZ mice and
Trpc6−/− + HFD + STZ mice (Fig. 4A and B; P<0.01). Similarly, ROS
production was also slightly raised in Trpc6−/− +
HFD mice (Fig. 4A and B;
P<0.01). These data indicated that knockout Trpc6 showed
no influence on the production of ROS in the renal cortex of T2DM
mice and that ROS may be an upstream regulator of TRPC6 in
T2DM.
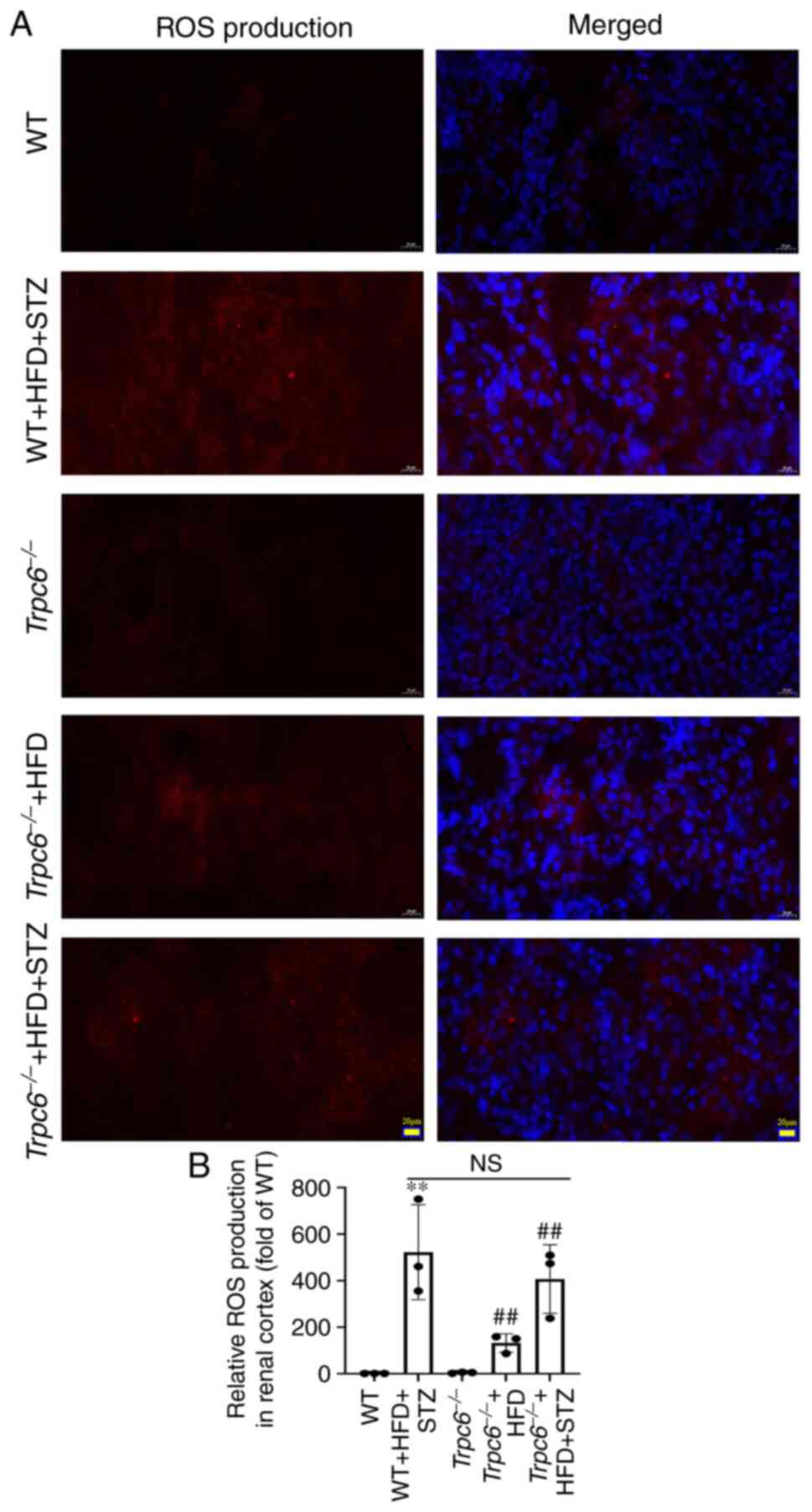 | Figure 4.Trpc6 knockout has no
influence on renal ROS generation in T2DM mice. (A) The production
of ROS in the renal cortex (DHE staining, magnification, ×400;
scale bar, 20 µm); (B) Relative ROS production in renal cortex.
Results are expressed as mean ± SD, n=4. **P<0.01 vs. WT group;
##P<0.01 vs. Trpc6−/− group; NS,
not significant. ROS, reactive oxygen species; T2DM, type-2
diabetes mellitus; DHE, dihydroethidium; WT, wild type; HFD, high
fat diet; STZ, streptozotocin. |
Trpc6 knockout decreases β-Gal
activity in T2DM mice
β-Gal is one of the essential markers of cellular
senescence and is also involved in the pathogenesis of renal
fibrosis (35). The present study
developed a β-Gal staining to assess renal senescence in T2DM mice.
The results revealed that there was only a small amount of β-Gal
expression in the WT and Trpc6−/− groups.
Compared with WT mice, the expression of β-Gal was clearly
increased in WT + HFD + STZ mice (Fig. S3A and B; P<0.01). The
activities of β-Gal were also increased in
Trpc6−/− + HFD mice when compared with
Trpc6−/− mice (Fig.
S3A and B; P<0.01). However, the activities of β-Gal were
clearly reduced in Trpc6−/− + HFD + STZ mice when
compared with WT + HFD + STZ mice (Fig. S3A and B; P<0.01). These results
indicated that knockout of Trpc6 improved renal aging injury
in T2DM mice.
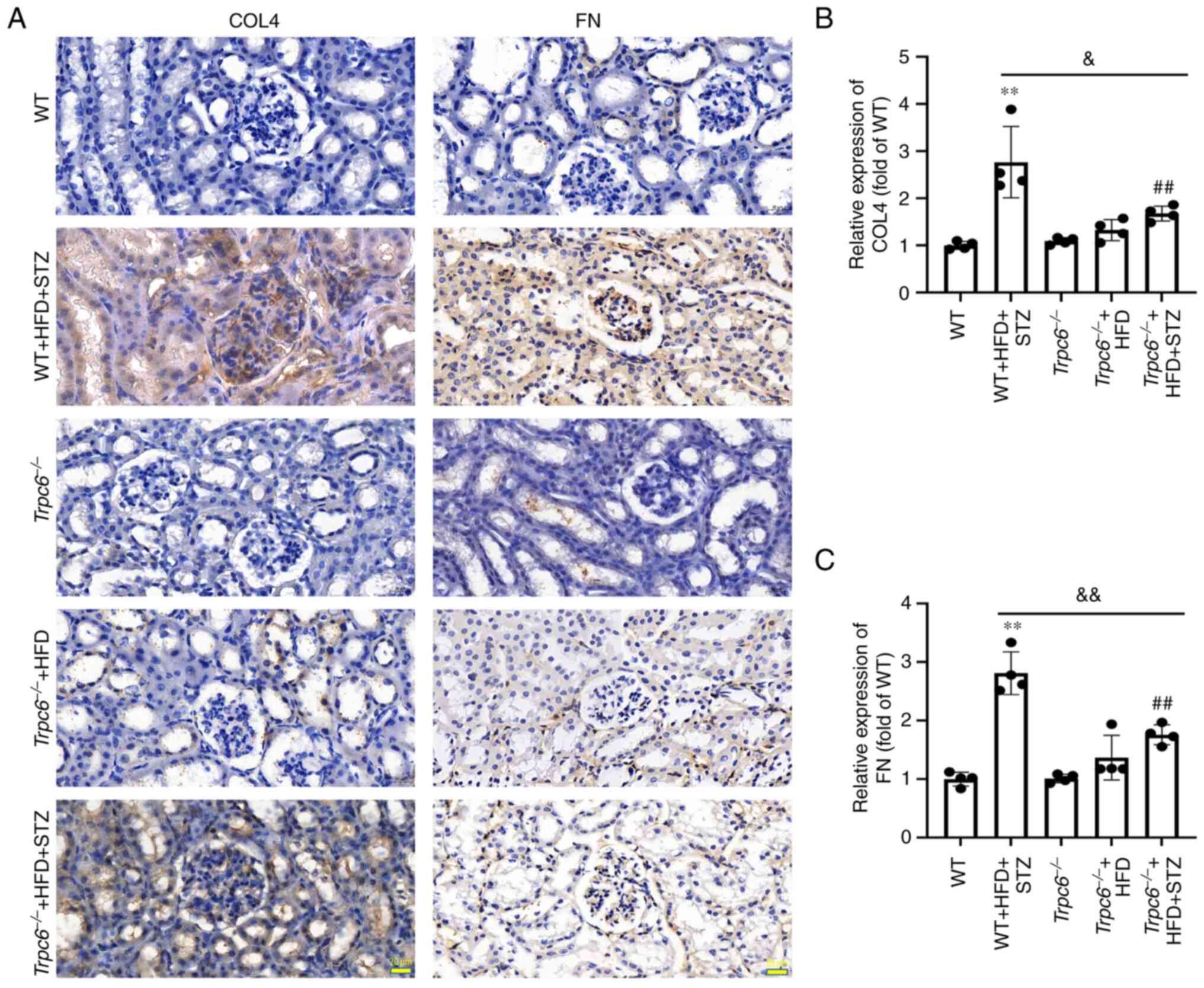
Trpc6 knockout decreases renal
fibrosis-related protein expression in T2DM mice
To confirm the role of Trpc6 knockout in
renal fibrosis in T2DM mice, the expression of the COL4 and FN
genes were measured using immunohistochemistry (36). The results demonstrated that the
expression of COL4 and FN were clearly increased in the kidney of
WT + HFD + STZ mice when compared with WT mice (Fig. 5A-C; P<0.01) and the expression
of COL4 and FN were clearly reduced in the
Trpc6−/− + HFD + STZ mice compared with WT + HFD
+ STZ mice (Fig. 5A-C; P<0.05
or P<0.01). The results indicated that knockout Trpc6 can
alleviate renal fibrosis in T2DM-induced DKD.
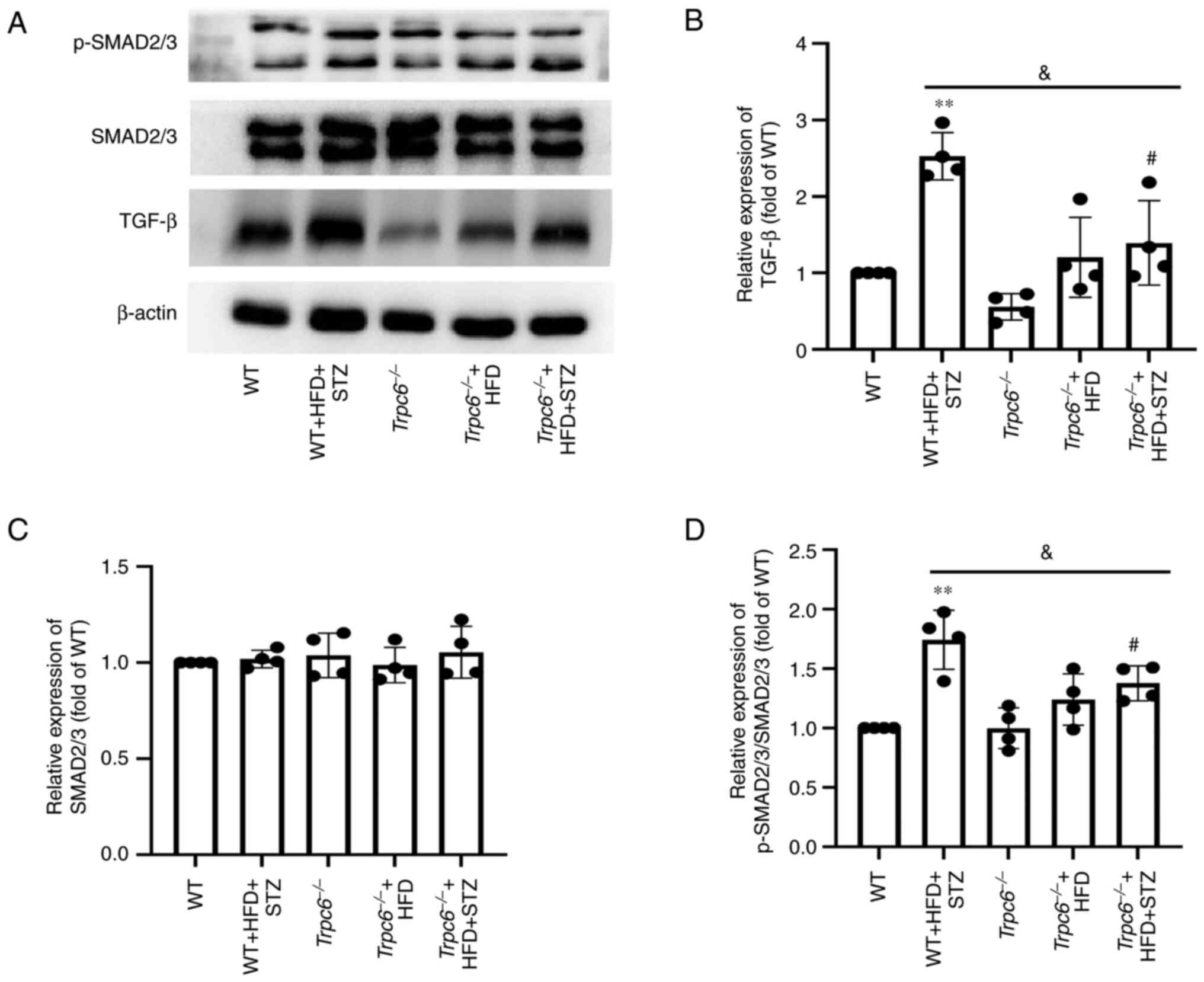
In addition, the levels of TGF-β and p-SMAD2/3
proteins were examined in T2DM mice. The results showed that the
levels of TGF-β and p-SMAD2/3 were clearly raised in WT + HFD + STZ
mice when compared with WT control mice (Fig. 6A, B and D; P<0.01); however, the
expression of p-SMAD2/3 and TGF-β were clearly decreased in the
Trpc6−/− + HFD + STZ mice when compared with WT +
HFD + STZ mice (Fig. 6A, B and D;
P<0.05). The results indicated that knockout of Trpc6 may
attenuate renal fibrosis by inhibiting the pathway of
TGF-β/SMAD2/3in T2DM mice.
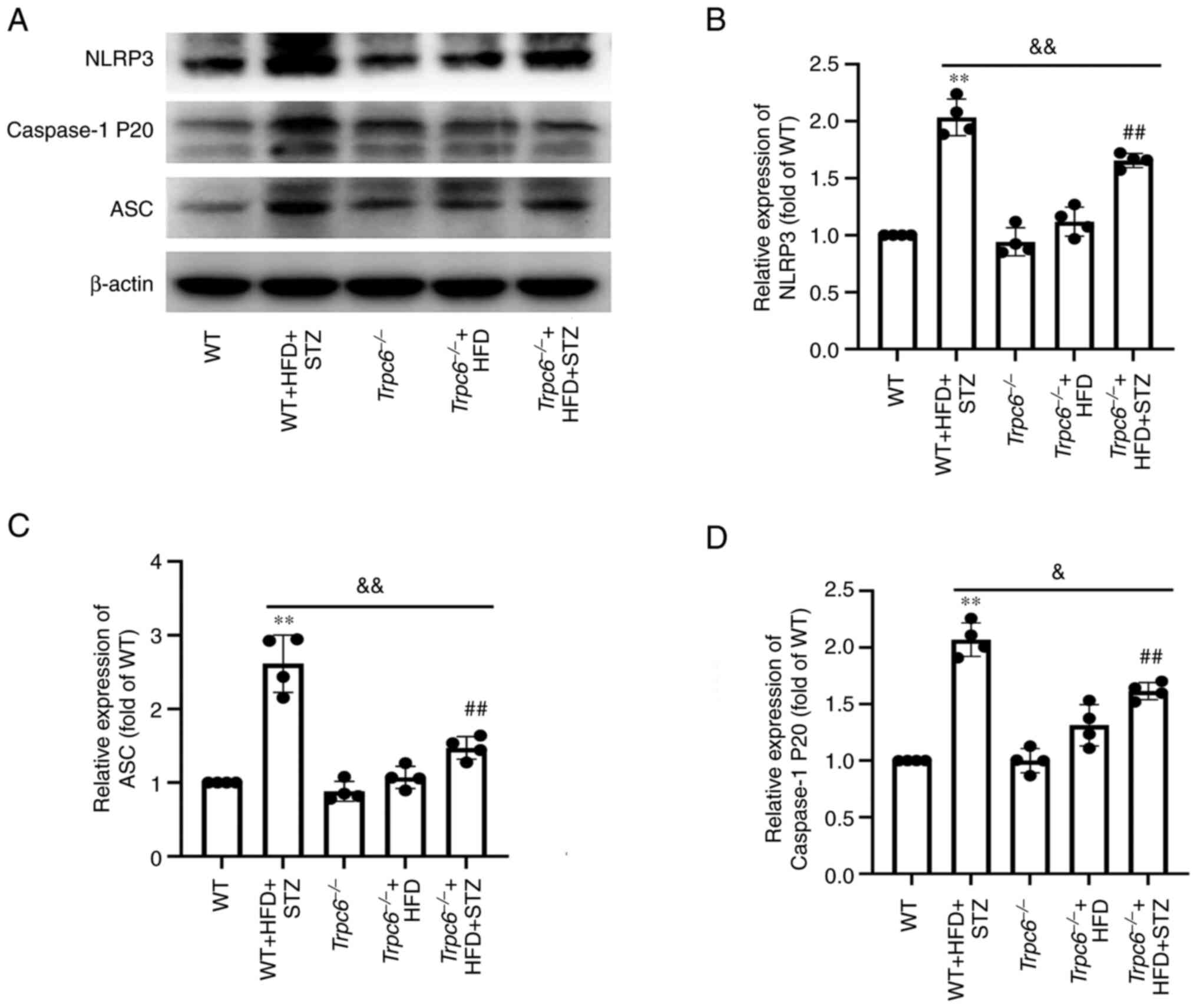
Trpc6 knockout decreases renal NLRP3
inflammasomes in T2DM mice
Next, NLRP3 inflammasomes in the kidney were
measured to investigate whether Trpc6 knockout can attenuate
renal inflammation in T2DM mice. The results revealed that
expression of NLRP3, cleaved-caspase-1 (P20) and ASC were clearly
raised in the renal cortex of WT + HFD + STZ mice when compared
with WT mice (Fig. 7A-D;
P<0.01). However, Trpc6 knockout clearly decreased the
expression of NLRP3, caspase-1 and ASC in
Trpc6−/− + HFD + STZ mice when compared with WT +
HFD + STZ mice (Fig. 7A-D;
P<0.01 or P<0.05). The results suggested that T2DM promoted
renal inflammation and that Trpc6 knockout attenuated renal
inflammation by inhibiting NLRP3 inflammasomes in T2DM.
Trpc6 knockout has no influence on
CD36 and p-phospholipase C (PLC) expression and renal lipid
deposition in T2DM mice
The effect of Trpc6 knockout on CD36 and
p-PLC expression in T2DM mice was evaluated. The results revealed
that CD36 and p-PLC expression was markedly upregulated in the
kidney of WT + HFD + STZ mice and Trpc6−/− + HFD
+ STZ mice when compared with WT control mice (Fig. S4A-D; P<0.01 or P<0.05).
Compared with the WT + HFD + STZ mice, knockout Trpc6 had no
significant influence on the levels of CD36 and p-PLC/PLC in renal
cortex tissues of T2DM mice (Fig.
S4A-D; P>0.05). The results suggest that the CD36 and PLC
signaling may be the upstream regulator of TRPC6 in T2DM mice.
Nile red staining was used to assess the amount of
lipid deposition in kidney. The results revealed that the renal
lipid deposition in WT + HFD + STZ mice and
Trpc6−/− + HFD + STZ groups was markedly
increased when compared with WT control mice (Fig. S5A and B; P<0.01) and that
knockout Trpc6 shows no significant influence on renal lipid
deposition in T2DM mice. Similarly, HFD-treatment alone also
increased renal lipid deposition in Trpc6−/− +
HFD mice (Fig. S5A and B;
P<0.01). These data indicated that knockout Trpc6 has no
significant effect on the renal lipid deposition in T2DM mice and
that lipid deposition may be the upstream regulator of TRPC6 in
T2DM.
Trpc6 knockout inhibits the renal
CN-NFAT2 signaling in T2DM mice
Next, changes in CN-NFAT2 signaling in
Trpc6−/− T2DM was detected mice. The results
revealed that there was a slight expression of TRPC6 in the kidney
cortex of WT mice, but that TRPC6 expression was markedly increased
in the kidneys of WT + HFD + STZ mice when compared with WT control
mice (Fig. 8A and B; P<0.01).
Additionally, TRPC6 was almost absent in kidney tissues of
Trpc6−/− mice, Trpc6−/− + HFD
mice and Trpc6−/− + HFD + STZ mice. Furthermore,
the results indicated that CN and NFAT2 expression was markedly
increased in the kidneys of WT + HFD + STZ mice when compared with
WT control mice (Fig. 8A and B;
P<0.01); however, Trpc6 knockout clearly decreased CN and
NFAT2 expression in the kidney of Trpc6−/− + HFD
+ STZ mice when compared with WT + HFD + STZ mice (Fig. 8A and B; P<0.05). The results
indicated that TRPC6-CN-NFAT2 signaling may play a key role in
promoting DKD in T2DM and that Trpc6 knockout can partially
inhibit the CN-NFAT2 signaling pathway in mice of T2DM.
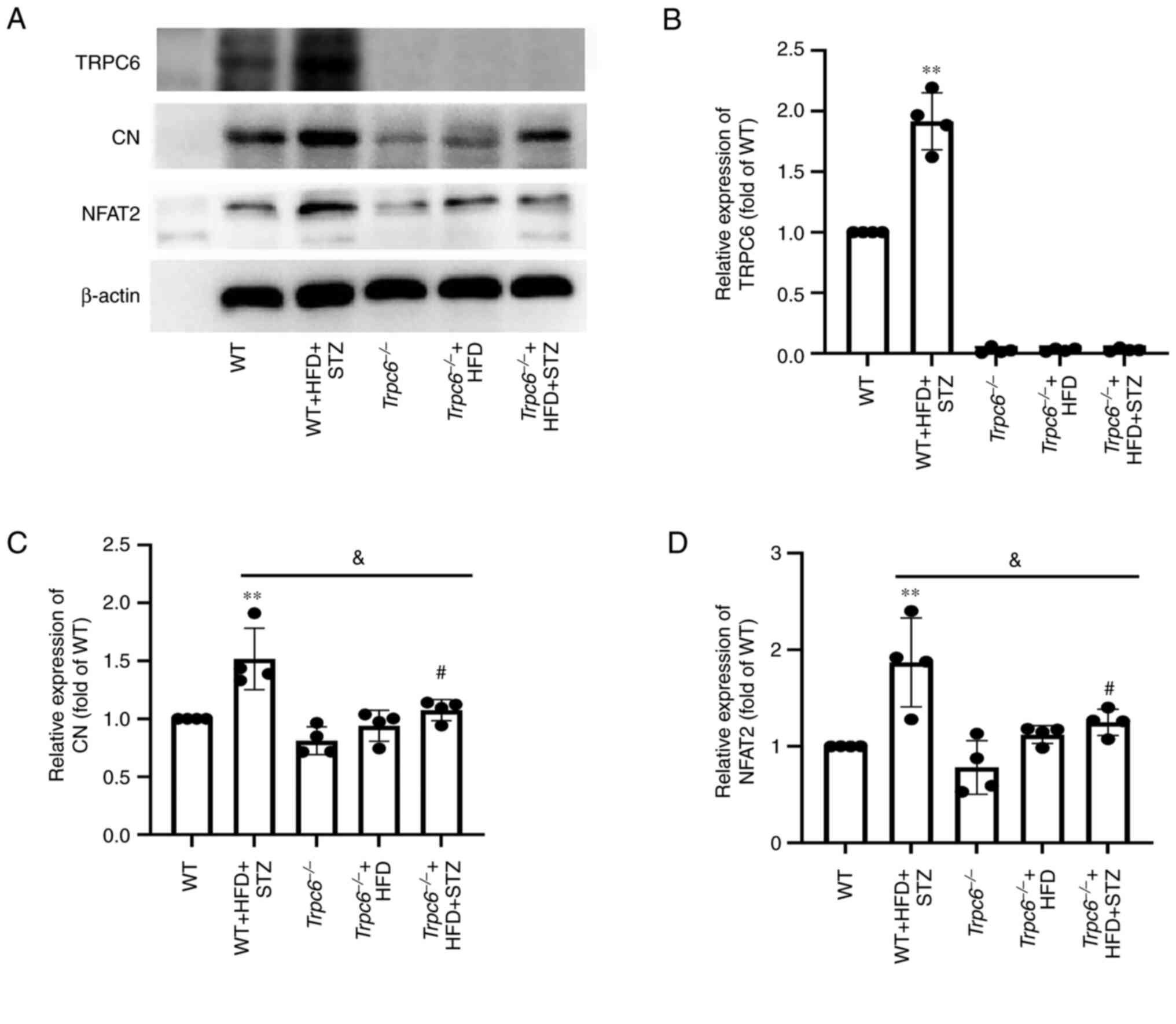 | Figure 8.Trpc6 knockout inhibits the
renal CN-NFAT2 signaling in T2DM mice. (A) The bands of NFAT2, CN,
TRPC6 and β-actin; (B) Relative expression of TRPC6; (C) Relative
expression of CN; (D) Relative expression of NFAT2. Results are
expressed as mean ± SD, n=4. **P<0.01 vs. WT group;
#P<0.05 vs. Trpc6−/− group;
&P<0.05 vs. WT + HFD + STZ group. CN,
calcineurin; NFAT, nuclear factor of activated T cells; T2DM,
type-2 diabetes mellitus; TRPC6, transient receptor potential
channel 6; WT, wild type; HFD, high fat diet; STZ,
streptozotocin. |
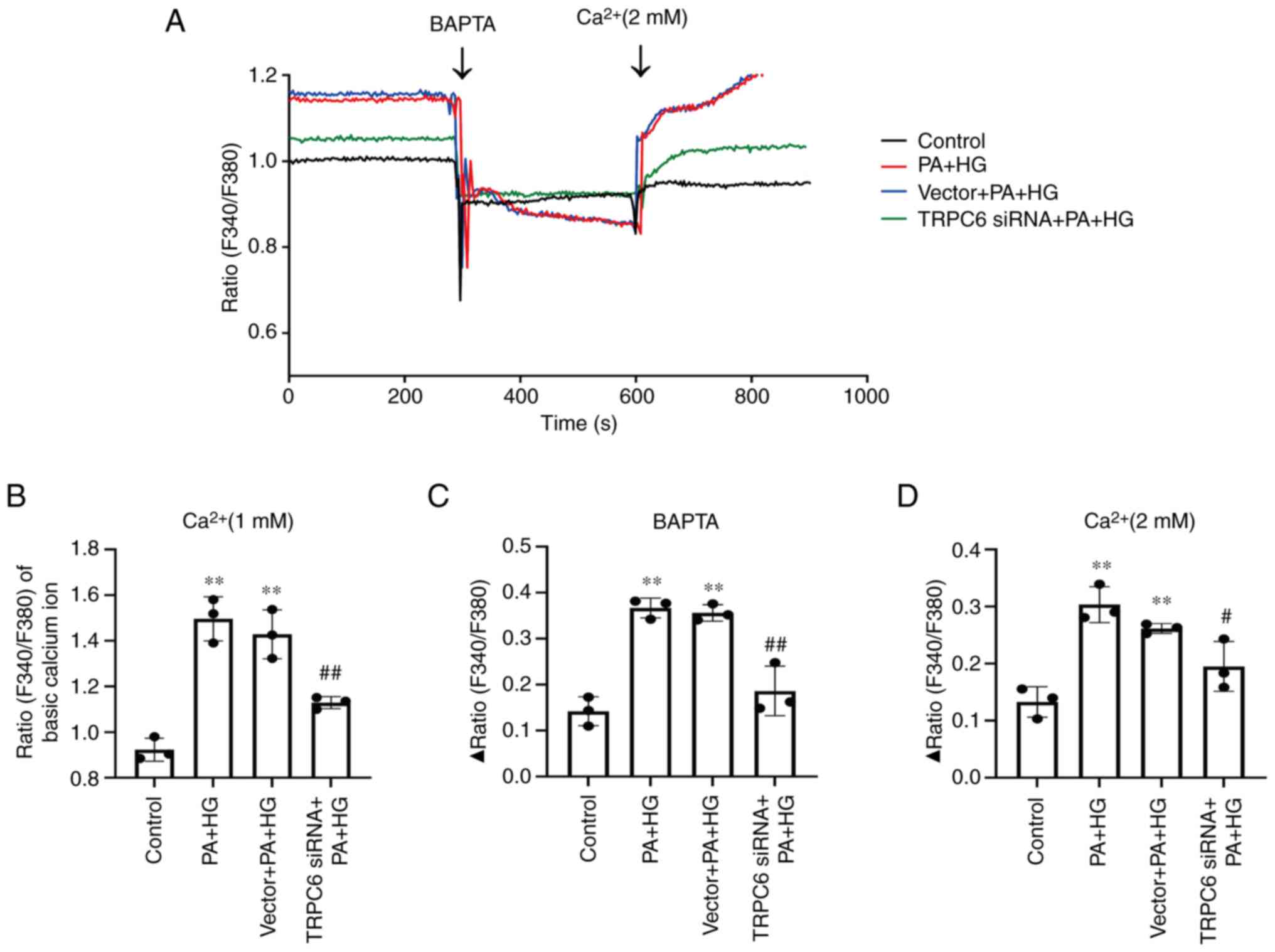
Trpc6 knockout alleviates calcium
homeostasis disorder in PA + HG-induced HMCs
Finally, the effects of Trpc6 knockdown on
[Ca2+]i was evaluated by using calcium
imaging in PA + HG-induced HMCs. The results revealed that the HMCs
exhibited a significant calcium overload after PA + HG stimulation
with [Ca2+]i increase and calcium homeostasis
imbalance after BAPTA and CaCl2 stimulation, compared
with the control group (Fig. 9A-D;
P<0.01 or P<0.05). As compared with the PA + HG group,
treatment with TRPC6-siRNA decreased the
[Ca2+]i and reduced the Δ Ratio F340/F380
induced by BAPTA and CaCl2 stimulation (Fig. 9A-D; P<0.01 or P<0.05). These
results indicated that PA + HG treatment can activate the TRPC6
channels in HMCs and that Trpc6 knockdown can ameliorate the
dysregulation of calcium homeostasis induced by PA + HG.
Discussion
T2DM is a common metabolic disease that is
characterized by chronic disorder of glycolipid metabolism
(37). DKD is the most dangerous
complication caused by DM disorders. However, the pathogenesis of
T2DM-induced DKD has not been fully elucidated and there are still
no effective therapeutic methods for it. Some studies have reported
that DKD involves multiple mechanisms, such as disruption of
glucolipid metabolism, oxidative stress, inflammation, apoptosis,
necrosis and autophagy (38–42).
Recent evidence suggests that alterations in Ca2+
signaling can directly or indirectly influence the onset and
progression of DKD (43). The
TRPC6 channel is reported as a critical contributor in a number of
renal diseases, including DKD (44,45),
therefore, it was hypothesized that upregulation of TRPC6 in T2DM
may regulate CN-NFAT2 signaling and activate NLRP3 inflammasomes
and the TGF-β/p-SMAD2/3 pathway, resulting in renal damage and
fibrosis. The present study found that knockout Trpc6
clearly alleviated renal dysfunction, renal injury and fibrosis and
inhibited CN-NFAT2 signaling and NLRP3 inflammasomes in T2DM mice.
The present study suggested that Trpc6 knockout can
attenuate T2DM-induced DKD progression and that the TRPC6-CN-NFAT2
signaling pathway may be an important therapeutic target in
DKD.
The typical symptoms of T2DM are polydrinking,
polyphagia, polyuria, weight loss and elevated fasting glucose
(46), consistent with the results
of body weight and FBG in the present study, demonstrating the
success of the T2DM mice model. The SCR and BUN levels are
important indicators for renal dysfunction assessment (47). In the present study, Trpc6
knockout only had a slight hypoglycemic effect, but significantly
reduced the serum BUN and SCR in T2DM mice, suggesting that
Trpc6 knockout can improve DKD in T2DM, but that it might
not be caused by a hypoglycemic effect. Abnormal lipid metabolism
is also an important feature of T2DM,therefore, the lipid
metabolism in Trpc6−/− T2DM mice was further
observed. The data indicated that the serum TG, TC, LDL-C, FFA and
renal lipid deposition were markedly increased in WT T2DM mice.
However, Trpc6 knockout markedly reduced the serum TG, TC
and LDL-C levels, but had no significant difference in FFA levels
and lipid deposition in the renal cortex, suggesting that
Trpc6 knockout may have a mild regulation of lipid
metabolism in the body, but has no influence on abnormal lipid
metabolism in the kidney in T2DM. Mesangial cell expansion and the
excessive deposition of acidic glycoprotein are important
pathological features of DKD (48). In addition, the kidney injury
factor, KIM-1, is often used to estimate renal injury (47). Moreover, the senescence-associated
β-Gal, a marker of cellular senescence in response to cellular
stress, is also often used to evaluate renal damages in DKD
(49). It has been reported that
diabetes and cellular senescence can induce a vicious cycle,
resulting in the development of T2DM (50). In the present study, H&E and
PAS staining results further confirmed that Trpc6 knockout
can attenuate renal injury and excessive deposition of acidic
glycoprotein in T2DM mice. The KIM-1 and β-Gal results also
confirmed that knockout of Trpc6 clearly alleviated
T2DM-induced renal injury and cellular senescence. Additionally,
the electron microscope results revealed that Trpc6 knockout
significantly attenuated glomerular ultrastructure damages. These
data suggested that knockout Trpc6 can alleviate
T2DM-induced renal injury.
Renal fibrosis, the most common pathological
process in DKD, is a result of ongoing renal tissue damage and
inflammation induced by hyperglycemic and hyperlipidemic
environments in T2DM. Renal fibrosis, similar to other organs, is
characterized by excessive extracellular matrix deposition,
resulting in destruction of renal structure and reduction of renal
function (51,52). Increased expression of FN and COL4
is largely involved in renal fibrosis (36); therefore, Masson staining is
commonly used to examine collagen deposition in renal tissues
(53). As shown by Masson staining
and IHC results, a significant increase of glomerular and
interstitial fibrosis and FN and COL4 expression was observed in
the T2DM mice, which was significantly attenuated by Trpc6
knockout. The present study further indicated that Trpc6
knockout significantly inhibited TGF-β/p-SMAD2/3 signaling, the
primary pathogenic mechanism of glomerular fibrosis in DKD, which
is highly activated in T2DM mice (54–57).
Furthermore, HFD-treatment alone could not induce renal fibrosis in
Trpc6−/− mice. These data suggested that
Trpc6 knockout can ameliorate renal fibrosis in T2DM.
Additionally, the results indicated that knockout of Trpc6
had no influence on lipid deposition and FFA in renal tissue and
only had a slight hypoglycemic effect in T2DM mice. Accordingly, it
was hypothesized that Trpc6 knockout might improve
T2DM-induced DKD through other mechanisms.
Previous studies suggested that Ca2+
homeostasis dysregulation is strongly associated with renal
inflammation and injury (58) and
that hyperglycemic and hyperlipidemic environments may
significantly increase Ca2+ levels because of increased
calcium entry into the cells (43,59).
As the primary mechanism of Ca2+ influx, TRPC constitute
a nonselective cation channel superfamily with Ca2+
permeability. Among the TRP family, TRPC6 is the most apparent
calcium-selective channel and acts as an important calcium entry
mechanism in the kidney. It has been reported that activation of
TRPC6 due to increased gene expression or functionally acquired
mutations can lead to heart and kidney disease (60,61).
Under stress conditions, extracellular signaling molecules, in
conjunction with G protein-coupled receptors, can activate PLCs on
the plasma membrane, thereby cleaving phosphatidylinositol
(4,5) biphosphate (PIP2) to generate the IP3
and DAG, which can increase intracellular Ca2+ by
activating TRPC6 in the cell membrane, leading to intracellular
calcium overload (62). In T2DM,
abnormal lipid metabolism can lead to excessive FFA entering cells
through CD36, a specific transport receptor, resulting in the
activation of PLC and increase of DAG and IP3 (63). Calcium overload further activates
CN-NFAT signaling, which triggers the pathological renal fibrotic
disease. It has been reported that TRPC6 cation conductance is
closely associated with calcium-dependent activation of CN and
stimulates transcriptional regulation of NFAT2 (64–67).
Meanwhile, NFAT2 is a downstream signaling molecule of the
Ca2+ signaling pathway, but activation of NFAT2 itself
can enhance the expression of the Trpc6 gene to form a
positive feedback loop (68). In
addition, the damage and fibrosis of MCs are important pathological
features of DKD. HMCs are often used as a cellular model for DKD
(69). Our previous findings
suggested that inhibition of Trpc6 by SKF96365 (TRPC6
inhibitor) or siRNA could significantly suppress calcium
homeostasis imbalance and significantly inhibit NFAT2 expression in
the nucleus of PA-induced HMCs (68). In the present study, the results
revealed that the CD36 and p-PLC levels were markedly increased in
the T2DM mice. However, knockout of Trpc6 had no influence
on CD36 and p-PLC expression, which may be the upstream regulator
of TRPC6 signaling in the T2DM mice. Furthermore, the results
revealed that T2DM could increase the expression of TRPC6, CN and
NFAT2 in renal tissue, which were clearly downregulated in
Trpc6−/− T2DM mice. Calcium imaging results
showed that knockdown of Trpc6 by siRNA also significantly
inhibited calcium homeostasis imbalance in PA + HG-induced HMCs.
The results suggested that TRPC6-mediated activation of CN-NFAT2
signaling can facilitate the process of DKD and that Trpc6
knockout may ameliorate renal injury and glomerular fibrosis by
inhibiting calcium overload and the CN-NFAT2 pathway in T2DM
mice.
Activation of CN-NFAT2 is closely related to the
inflammatory cascade response (70), which is also one of the important
pathogeneses of DKD. Accumulating evidence indicates that
activation of NLRP3 inflammasomes can recruit the ASC and
procaspase-1 to cleave the pro-IL-1β to the bioactive form,
promoting the inflammatory cascade response in a number of
diseases, including DKD (15).
Studies revealed that activation of NLRP3 inflammasomes could
exacerbate renal fibrosis in a number of kidney diseases (71,72).
Inhibition of NLRP3 displayed anti-inflammatory effects and
decreased renal injury and fibrosis in DKD (15). In the present study, the results
suggested a significant activation of NLRP3 inflammasomes in T2DM
mice and that knockout of Trpc6 significantly inhibited the
NLRP3 activation in the kidney of T2DM mice. These data suggested
that NLRP3 signaling may be the downstream process of
TRPC6-CN-NFAT2 signaling in DKD, however, the regulatory mechanism
of NFAT2 on NLRP3 needs to be further studied.
Additionally, ROS plays a key role in the
pathogenesis of renal injury and fibrosis, as excessive ROS
production is associated with a number of pathophysiological
processes, such as inflammation, cellular structure and
Ca2+ homeostasis imbalance (73). The present study also demonstrated
that ROS production in renal cortex was clearly elevated in T2DM
mice; however, Trpc6 knockout did not alleviate ROS
production in T2DM mice, suggesting that the TRPC6-associated
calcium homeostasis dysregulation might be the downstream signaling
pathway of ROS oxidative stress.
In summary, T2DM can activate the CN-NFAT2
signaling pathway through a TRPC6-mediated calcium overload,
leading to NLRP3 inflammasome activation and ultimately resulting
in renal inflammation injury and renal fibrosis. Trpc6
knockout inhibited the TGF-β/p-SMAD2/3 by suppressing renal calcium
overload and CN-NFAT2 signaling and attenuating glomerular fibrosis
in T2DM. However, the present study only explored the role of
Trpc6 knockout in improving DKD in vivo. It is
remains to be elucidated what the relationship of TRPC6 and NLRP3
is and future studies are needed to investigate the mechanism of
TRPC6 in DKD progression.
Supplementary Material
Supporting Data
Supporting Data
Acknowledgements
The authors would like to thank Miss Hanyang Xu,
Mr. Bo Li and Mr. Dake Huang (School of Basic Medical Sciences,
Anhui Medical University, Anhui, China) for providing technical
assistance.
Funding
The present study was supported by the National Natural Science
Foundation of China (grant nos. 81970630 and 82204703), the Natural
Science Foundation of Anhui Province (grant no. 2208085MH219) and
the Major Projects of the Anhui Provincial Department of Education
(grant no. KJ2020ZD14).
Availability of data and materials
The datasets used and analyzed during the present
study are available from the corresponding author on reasonable
request.
Authors' contributions
RS performed the experiments, and prepared the
manuscript. MH and YL contributed to the immunoblot analysis and
interpretation of the results. QFS and YS collated the data. LH and
LLK were mainly responsible for the histological experiments. WPL
and WZL designed the study, critically revised the manuscript for
intellectually important content and supervised the study. WPL and
WZL confirm the authenticity of all the raw data. All authors read
and approved the final manuscript.
Ethics approval and consent to
participate
All experiments involving animals were approved by
the Anhui Medical University Ethics Committee (approval no.
LLSC20190302).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Toi PL, Anothaisintawee T, Chaikledkaew U,
Briones JR, Reutrakul S and Thakkinstian A: Preventive role of diet
interventions and dietary factors in type 2 diabetes mellitus: An
umbrella review. Nutrients. 12:27222020. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Iatcu CO, Steen A and Covasa M: Gut
microbiota and complications of type-2 diabetes. Nutrients.
14:1662021. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
KDOQI clinical practice guideline for
diabetes and CKD: 2012 update. Am J Kidney Dis. 60:850–886. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Jia Y, Guan M, Zheng Z, Zhang Q, Tang C,
Xu W, Xiao Z, Wang L and Xue Y: miRNAs in urine extracellular
vesicles as predictors of early-Stage diabetic nephropathy. J
Diabetes Res. 2016:79327652016. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Ma R, Wang Y, Xu Y, Wang R, Wang X, Yu N,
Li M and Zhou Y: Tacrolimus protects podocytes from apoptosis via
downregulation of TRPC6 in diabetic nephropathy. J Diabetes Res.
2021:88321142021. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Adeshara KA, Diwan AG and Tupe RS:
Diabetes and complications: Cellular signaling pathways, current
understanding and targeted therapies. Curr Drug Targets.
17:1309–1328. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Gregg EW, Sattar N and Ali MK: The
changing face of diabetes complications. Lancet Diabetes
Endocrinol. 4:537–547. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Ali MK, Pearson-Stuttard J, Selvin E and
Gregg EW: Interpreting global trends in type 2 diabetes
complications and mortality. Diabetologia. 65:3–13. 2022.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Caro JJ, Ward AJ and O'Brien JA: Lifetime
costs of complications resulting from type 2 diabetes in the U.S.
Diabetes Care. 25:476–481. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Rayego-Mateos S, Rodrigues-Diez RR,
Fernandez-Fernandez B, Mora-Fernández C, Marchant V, Donate-Correa
J, Navarro-González JF, Ortiz A and Ruiz-Ortega M: Targeting
inflammation to treat diabetic kidney disease: The road to 2030.
Kidney Int. 103:282–296. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Karunasagara S, Hong GL, Park SR, Lee NH,
Jung DY, Kim TW and Jung JY: Korean red ginseng attenuates
hyperglycemia-induced renal inflammation and fibrosis via
accelerated autophagy and protects against diabetic kidney disease.
J Ethnopharmacol. 254:1126932020. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Gong Z, Zhao S, Zhou J, Wang L, Du X, Li
H, Chen Y, Cai W and Wu J: Curcumin alleviates DSS-induced colitis
via inhibiting NLRP3 inflammsome activation and IL-1β production.
Mol Immunol. 104:11–19. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Sharma BR and Kanneganti TD: NLRP3
inflammasome in cancer and metabolic diseases. Nat Immunol.
22:550–559. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Hutton HL, Ooi JD, Holdsworth SR and
Kitching AR: The NLRP3 inflammasome in kidney disease and
autoimmunity. Nephrology (Carlton). 21:736–744. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Wu M, Han W, Song S, Du Y, Liu C, Chen N,
Wu H, Shi Y and Duan H: NLRP3 deficiency ameliorates renal
inflammation and fibrosis in diabetic mice. Mol Cell Endocrinol.
478:115–125. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Kim Y, Lim JH, Kim MY, Kim EN, Yoon HE,
Shin SJ, Choi BS, Kim YS, Chang YS and Park CW: The adiponectin
receptor agonist adiporon ameliorates diabetic nephropathy in a
model of type 2 diabetes. J Am Soc Nephrol. 29:1108–1127. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Dietrich A and Gudermann T: TRPC6. Handb
Exp Pharmacol. 125–141. 2007.doi: 10.1007/978-3-540-34891-7_7.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Kong L, Sun R, Zhou H, Huang D, Xing W, Wu
B, Li H, Hu W, Song S and Xu Y: Trpc6 knockout improves behavioral
dysfunction and reduces Aβ production by inhibiting CN-NFAT1
signaling in T2DM mice. Exp Neurol. 363:1143502023. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Staruschenko A, Spires D and Palygin O:
Role of TRPC6 in progression of diabetic kidney disease. Curr
Hypertens Rep. 21:482019. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Eder P: Cardiac remodeling and disease:
SOCE and TRPC signaling in cardiac pathology. Adv Exp Med Biol.
993:505–521. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Ma R, Chaudhari S and Li W: Canonical
transient receptor potential 6 channel: A new target of reactive
oxygen species in renal physiology and pathology. Antioxid Redox
Signal. 25:732–748. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Lin BL, Matera D, Doerner JF, Zheng N, Del
Camino D, Mishra S, Bian H, Zeveleva S, Zhen X, Blair NT, et al: In
vivo selective inhibition of TRPC6 by antagonist BI 749327
ameliorates fibrosis and dysfunction in cardiac and renal disease.
Proc Natl Acad Sci USA. 116:10156–10161. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Nie B, Liu C, Bai X, Chen X, Wu S, Zhang
S, Huang Z, Xie M, Xu T, Xin W, et al: AKAP150 involved in
paclitaxel-induced neuropathic pain via inhibiting CN/NFAT2 pathway
and downregulating IL-4. Brain Behav Immun. 68:158–168. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Rusnak F and Mertz P: Calcineurin: Form
and function. Physiol Rev. 80:1483–1521. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Rao A, Luo C and Hogan PG: Transcription
factors of the NFAT family: Regulation and function. Annu Rev
Immunol. 15:707–747. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Mognol GP, Carneiro FR, Robbs BK, Faget DV
and Viola JP: Cell cycle and apoptosis regulation by NFAT
transcription factors: New roles for an old player. Cell Death Dis.
7:e21992016. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Ling H, Zhu Z, Yang J, He J, Yang S, Wu D,
Feng S and Liao D: Dihydromyricetin improves type 2
diabetes-induced cognitive impairment via suppressing oxidative
stress and enhancing brain-derived neurotrophic factor-mediated
neuroprotection in mice. Acta Biochim Biophys Sin (Shanghai).
50:298–306. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Wick MR: The hematoxylin and eosin stain
in anatomic pathology-An often-neglected focus of quality assurance
in the laboratory. Semin Diagn Pathol. 36:303–311. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Yang L, Guo J, Yu N, Song H, Niu J and Gu
Y: Tocilizumab mimotope alleviates kidney injury and fibrosis by
inhibiting IL-6 signaling and ferroptosis in UUO model. Life Sci.
261:1184872020. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Lefkowitch JH: Special stains in
diagnostic liver pathology. Semin Diagn Pathol. 23:190–198. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Qiao S, Liu R, Lv C, Miao Y, Yue M, Tao Y,
Wei Z, Xia Y and Dai Y: Bergenin impedes the generation of
extracellular matrix in glomerular mesangial cells and ameliorates
diabetic nephropathy in mice by inhibiting oxidative stress via the
mTOR/β-TrcP/Nrf2 pathway. Free Radic Biol Med. 145:118–135. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Han WK, Bailly V, Abichandani R, Thadhani
R and Bonventre JV: Kidney Injury Molecule-1 (KIM-1): A novel
biomarker for human renal proximal tubule injury. Kidney Int.
62:237–244. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Glastras SJ, Chen H, Teh R, McGrath RT,
Chen J, Pollock CA, Wong MG and Saad S: Mouse models of diabetes,
obesity and related kidney disease. PLoS One. 11:e01621312016.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Jha JC, Banal C, Chow BS, Cooper ME and
Jandeleit-Dahm K: Diabetes and kidney disease: Role of oxidative
stress. Antioxid Redox Signal. 25:657–684. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Gong W, Luo C, Peng F, Xiao J, Zeng Y, Yin
B, Chen X, Li S, He X, Liu Y, et al: Brahma-related gene-1 promotes
tubular senescence and renal fibrosis through
Wnt/β-catenin/autophagy axis. Clin Sci (Lond). 135:1873–1895. 2021.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Yan Q, Sui W, Xie S, Chen H, Xie S, Zou G,
Guo J and Zou H: Expression and role of integrin-linked kinase and
collagen IV in human renal allografts with interstitial fibrosis
and tubular atrophy. Transpl Immunol. 23:1–5. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Kharroubi AT and Darwish HM: Diabetes
mellitus: The epidemic of the century. World J Diabetes. 6:850–867.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Chen XC, Li ZH, Yang C, Tang JX, Lan HY
and Liu HF: Lysosome depletion-triggered autophagy impairment in
progressive kidney injury. Kidney Dis (Basel). 7:254–267. 2021.
View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Duan JY, Lin X, Xu F, Shan SK, Guo B, Li
FX, Wang Y, Zheng MH, Xu QS, Lei LM, et al: Ferroptosis and its
potential role in metabolic diseases: A curse or revitalization?
Front Cell Dev Biol. 9:7017882021. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Al Mamun A, Ara Mimi A, Wu Y, Zaeem M,
Abdul Aziz M, Aktar Suchi S, Alyafeai E, Munir F and Xiao J:
Pyroptosis in diabetic nephropathy. Clin Chim Acta. 523:131–143.
2021. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Sifuentes-Franco S, Padilla-Tejeda DE,
Carrillo-Ibarra S and Miranda-Díaz AG: Oxidative stress, apoptosis,
and mitochondrial function in diabetic nephropathy. Int J
Endocrinol. 2018:18758702018. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Samsu N: Diabetic Nephropathy: Challenges
in pathogenesis, diagnosis, and treatment. Biomed Res Int.
2021:14974492021. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Luo Y, Lu Z, Waaga-Gasser AM, Yang H, Liu
J, Wu J, Lu J, Liu X and Zhang L: Modulation of calcium homeostasis
may be associated with susceptibility to renal cell carcinoma in
diabetic nephropathy rats. Cancer Manag Res. 12:9679–9689. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Wang Q, Tian X, Wang Y, Wang Y, Li J, Zhao
T and Li P: Role of transient receptor potential canonical channel
6 (TRPC6) in diabetic kidney disease by regulating podocyte actin
cytoskeleton rearrangement. J Diabetes Res. 2020:68973902020.
View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Yu J, Li C, Ma L, Zhai B, Xu A and Shao D:
Transient receptor potential canonical 6 knockdown ameliorated
diabetic kidney disease by inhibiting nuclear factor of activated T
cells 2 expression in glomerular mesangial cells. Ren Fail.
44:1780–1790. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Zimmet P, Shi Z, El-Osta A and Ji L:
Epidemic T2DM, early development and epigenetics: Implications of
the Chinese Famine. Nat Rev Endocrinol. 14:738–746. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Vaidya VS, Ozer JS, Dieterle F, Collings
FB, Ramirez V, Troth S, Muniappa N, Thudium D, Gerhold D, Holder
DJ, et al: Kidney injury molecule-1 outperforms traditional
biomarkers of kidney injury in preclinical biomarker qualification
studies. Nat Biotechnol. 28:478–485. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Adeva-Andany MM, Adeva-Contreras L,
Fernández-Fernández C, Carneiro-Freire N and Domínguez-Montero A:
Histological Manifestations of diabetic kidney disease and its
relationship with insulin resistance. Curr Diabetes Rev.
19:e2803222027052023. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Lee BY, Han JA, Im JS, Morrone A, Johung
K, Goodwin EC, Kleijer WJ, DiMaio D and Hwang ES:
Senescence-associated beta-galactosidase is lysosomal
beta-galactosidase. Aging Cell. 5:187–195. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Bellary S, Kyrou I, Brown JE and Bailey
CJ: Type 2 diabetes mellitus in older adults: Clinical
considerations and management. Nat Rev Endocrinol. 17:534–548.
2021. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Bülow RD and Boor P: Extracellular matrix
in kidney fibrosis: More than just a scaffold. J Histochem
Cytochem. 67:643–661. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Yuan Q, Tan RJ and Liu Y: Myofibroblast in
kidney fibrosis: Origin, activation, and regulation. Adv Exp Med
Biol. 1165:253–283. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Chen PS, Li YP and Ni HF: Morphology and
evaluation of renal fibrosis. Adv Exp Med Biol. 1165:17–36. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Song MK, Lee JH, Ryoo IG, Lee SH, Ku SK
and Kwak MK: Bardoxolone ameliorates TGF-β1-associated renal
fibrosis through Nrf2/Smad7 elevation. Free Radic Biol Med.
138:33–42. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Fukasawa H, Yamamoto T, Suzuki H, Togawa
A, Ohashi N, Fujigaki Y, Uchida C, Aoki M, Hosono M, Kitagawa M and
Hishida A: Treatment with anti-TGF-beta antibody ameliorates
chronic progressive nephritis by inhibiting Smad/TGF-beta
signaling. Kidney Int. 65:63–74. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Ka SM, Huang XR, Lan HY, Tsai PY, Yang SM,
Shui HA and Chen A: Smad7 gene therapy ameliorates an autoimmune
crescentic glomerulonephritis in mice. J Am Soc Nephrol.
18:1777–1788. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Ka SM, Yeh YC, Huang XR, Chao TK, Hung YJ,
Yu CP, Lin TJ, Wu CC, Lan HY and Chen A: Kidney-targeting Smad7
gene transfer inhibits renal TGF-β/MAD homologue (SMAD) and nuclear
factor κB (NF-κB) signalling pathways, and improves diabetic
nephropathy in mice. Diabetologia. 55:509–519. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Szabó C: Nitric oxide, intracellular
calcium overload, and cytotoxicity. Shock. 6:25–26. 1996.
View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Han Y, Su Y, Han M, Liu Y, Shi Q, Li X,
Wang P and Li W and Li W: Ginsenoside Rg1 attenuates glomerular
fibrosis by inhibiting CD36/TRPC6/NFAT2 signaling in type 2
diabetes mellitus mice. J Ethnopharmacol. 302:1159232023.
View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Seo K, Rainer PP, Shalkey Hahn V, Lee DI,
Jo SH, Andersen A, Liu T, Xu X, Willette RN, Lepore JJ, et al:
Combined TRPC3 and TRPC6 blockade by selective small-molecule or
genetic deletion inhibits pathological cardiac hypertrophy. Proc
Natl Acad Sci USA. 111:1551–1556. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Reiser J, Polu KR, Möller CC, Kenlan P,
Altintas MM, Wei C, Faul C, Herbert S, Villegas I, Avila-Casado C,
et al: TRPC6 is a glomerular slit diaphragm-associated channel
required for normal renal function. Nat Genet. 37:739–744. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Hofmann T, Obukhov AG, Schaefer M,
Harteneck C, Gudermann T and Schultz G: Direct activation of human
TRPC6 and TRPC3 channels by diacylglycerol. Nature. 397:259–263.
1999. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Kobayashi M, Mutharasan RK, Feng J,
Roberts MF and Lomasney JW: Identification of hydrophobic
interactions between proteins and lipids: Free fatty acids activate
phospholipase C delta1 via allosterism. Biochemistry. 43:7522–7533.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Makarewich CA, Zhang H, Davis J, Correll
RN, Trappanese DM, Hoffman NE, Troupes CD, Berretta RM, Kubo H,
Madesh M, et al: Transient receptor potential channels contribute
to pathological structural and functional remodeling after
myocardial infarction. Circ Res. 115:567–580. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Koitabashi N, Aiba T, Hesketh GG, Rowell
J, Zhang M, Takimoto E, Tomaselli GF and Kass DA: Cyclic
GMP/PKG-dependent inhibition of TRPC6 channel activity and
expression negatively regulates cardiomyocyte NFAT activation Novel
mechanism of cardiac stress modulation by PDE5 inhibition. J Mol
Cell Cardiol. 48:713–724. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Chung HS, Kim GE, Holewinski RJ,
Venkatraman V, Zhu G, Bedja D, Kass DA and Van Eyk JE: Transient
receptor potential channel 6 regulates abnormal cardiac
S-nitrosylation in Duchenne muscular dystrophy. Proc Natl Acad Sci
USA. 114:E10763–E10771. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Wang L, Jirka G, Rosenberg PB, Buckley AF,
Gomez JA, Fields TA, Winn MP and Spurney RF: Gq signaling causes
glomerular injury by activating TRPC6. J Clin Invest.
125:1913–1926. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Su Y, Chen Q, Ju Y and Li W and Li W:
Palmitate induces human glomerular mesangial cells fibrosis through
CD36-mediated transient receptor potential canonical channel
6/nuclear factor of activated T cell 2 activation. Biochim Biophys
Acta Mol Cell Biol Lipids. 1865:1587932020. View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Bian Y, Shi C, Song S, Mu L, Wu M, Qiu D,
Dong J, Zhang W, Yuan C, Wang D, et al: Sestrin2 attenuates renal
damage by regulating Hippo pathway in diabetic nephropathy. Cell
Tissue Res. 390:93–112. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Mena MP, Papiewska-Pajak I, Przygodzka P,
Kozaczuk A, Boncela J and Cierniewski CS: NFAT2 regulates COX-2
expression and modulates the integrin repertoire in endothelial
cells at the crossroads of angiogenesis and inflammation. Exp Cell
Res. 324:124–136. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Zhang D, Ji P, Sun R, Zhou H, Huang L,
Kong L and Li W and Li W: Ginsenoside Rg1 attenuates LPS-induced
chronic renal injury by inhibiting NOX4-NLRP3 signaling in mice.
Biomed Pharmacother. 150:1129362022. View Article : Google Scholar : PubMed/NCBI
|
|
72
|
Shahzad K, Fatima S, Khawaja H, Elwakiel
A, Gadi I, Ambreen S, Zimmermann S, Mertens PR, Biemann R and
Isermann B: Podocyte-specific Nlrp3 inflammasome activation
promotes diabetic kidney disease. Kidney Int. 102:766–779. 2022.
View Article : Google Scholar : PubMed/NCBI
|
|
73
|
Li S, Zheng L, Zhang J, Liu X and Wu Z:
Inhibition of ferroptosis by up-regulating Nrf2 delayed the
progression of diabetic nephropathy. Free Radic Biol Med.
162:435–449. 2021. View Article : Google Scholar : PubMed/NCBI
|