Introduction
Melanoma, originating in melanocytes and nevus
cells, is one of the most common cutaneous neoplasms. Melanoma only
represents a small subset of these tumors, yet it is the most
common skin tumor type, with increasing incidence and mortality
rates worldwide (1,2). Currently, the primary treatments of
malignant melanoma are surgical excision, immunotherapy, adjuvant
chemotherapy, targeted therapy drugs (3) and radiotherapies (2,4).
However, these therapeutic strategies do not facilitate the current
clinical practice requirements due to the high metastatic potential
and drug resistance (5,6). Moreover, long-term survival remains
poor, even after treatment with these therapies (7). Therefore, it is important to develop
novel drug candidates to overcome melanoma treatment
limitations.
Previous studies have shown that mitochondrial
uncoupling has become an effective antitumor treatment (8–10).
Triclosan is a widely used antibacterial and antifungal agent in
everyday personal care and consumer products, including
toothpastes, antiseptic soaps and plastics, and is also a mild
mitochondrial uncoupler (11–13).
Therefore, triclosan may have anticancer effects in melanoma cells.
Previous studies have focused on its antibacterial effects
(14–16), thus few studies have investigated its
anticancer properties and its effects in melanoma have not been
shown.
Mitochondria are highly dynamic organelles that are
involved in ATP generation, reactive oxygen species (ROS)
generation and Ca2+ signaling, which continually undergo
fusion and fission to maintain the balance between energy
production and cell death under physiological condition (17). However, dysfunction of this balance
has been recognized as an important factor for cancer progression.
Mitochondrial bioenergetic and biosynthetic requirements are
altered to resist cancer cell apoptosis, and promote tumor cell
proliferation and migration, for example in glioblastoma and
breast, lung and prostate cancer (18). In addition, ROS from mitochondria are
considered novel signal mediators, which are involved in cell
proliferation, tumor progression, differentiation and cell death
(19). Thus, these properties of
mitochondria contribute to this organelle becoming a promising
target in cancer therapy.
The present study aimed to investigate the effect of
triclosan on melanoma and the underlying mechanism. Thus, the
present results may facilitate the development of triclosan as a
potential treatment candidate against melanoma.
Materials and methods
Materials
Triclosan was obtained from Shanghai Baidi Biody-Bio
Co., Ltd. Hoechst, Cal-AM, Eth-1, Fluo-3/AM, mito-Tracker,
mito-SOX, tetramethylrhodamine methyl ester (TMRM) and DAPI were
purchased from Thermo Fisher Scientific, Inc. Dihydroethidium dye
was purchased from the Beyotime Institute of Biotechnology. Tempol,
3-MA and acetylcysteine (NAC) were purchased from Sigma Aldrich;
Merck KGaA. Tempol (0.5 and 1 mM) is a radical scavenger that was
used to test the effect of ROS levels on cytotoxicity induced by
triclosan (20 µM) in the lactate dehydrogenase (LDH) release assay.
S3I-201 was purchased from EMD Millipore. S3I-201 (10 and 20 µM) is
a STAT3 inhibitor that was used to detect the effect of STAT3
activity change on cytotoxicity induced by triclosan (20 µM) in the
LDH release assay. Anti-p-STAT3 (Y705, #9131, 1:1,000), anti-STAT3
(#4904, 1:1,000), anti-p-AMPK (Thr172, #2535, 1:1,000), anti-AMPK
(#2532, 1:1,000) and anti-p62 (#88588, 1:1,000) were purchased from
Cell Signaling Technology, Inc., Bcl-2 (ab196495, 1:1,000) antibody
was purchased from Abcam and LC3 (L7543, 1:1,000) antibody was
purchased from Sigma Aldrich; Merck KGaA.
Cell culture
A375 cells and HFF-1 cells were purchased from
Zhongqiaoxinzhou Biotech. Cells were maintained in high glucose
DMEM (HyClone; Cytiva) supplemented with 10% FBS (Biological
Industries), 100 µg/ml−1 penicillin and 100
µg/ml−1 streptomycin at 37°C in 5% CO2. The
time of treatment and concentration are shown in the figure
legends. Briefly, cells were treated with different concentrations
(0–200 µM) of drugs (triclosan, NAC, S3I-201, Tempol and 3-MA) for
24 h at 37°C.
Measurement of cell viability
Cell viability was measured using a colorimetric MTT
assay. A375 and HFF-1 cells were seeded into a 96-well
flat-bottomed plate at 5×103 per well and treated with
or without triclosan at the indicated concentrations (0–200 µM) for
24 h and then incubated with MTT (5 mg/ml−1) for 4 h at
37°C. After 4 h, the supernatant was removed and then the cells
were incubated with 200 µl DMSO for 3 min at room temperature to
dissolve the purple formazan. The optical density (OD) was read at
490 nm using an Infinite M200 microplate reader (Tecan Trading
AG).
LDH release assay
LDH release in culture medium was evaluated by using
LDH Cytotoxicity Assay kits (Beyotime Institute of Biotechnology)
following the manufacturer's instructions. A375 cells were seeded
(5,000 cells/well) in a 96-well plate and treated with triclosan
(0–40 µM), Tempol (0.5 and 1 mM), S3I-201 (10 and 20 µM), NAC (1
µM) or combinations of these compounds for 24 h. Subsequently,
after cells were centrifuged at 444 × g for 5 min at room
temperature, the supernatant was collected and transferred into
another 96-well plate for LDH assay test. The supernatant was
incubated with LDH release testing buffer at room temperature for
30 min. The OD was read at 490 nm by using an Infinite M200
microplate reader.
LIVE/DEAD cell staining
The live and dead cells were detected by using a
LIVE/DEAD® cell viability assay kit (Invitrogen; Thermo
Fisher Scientific, Inc.) according to the manufacturer's
instructions. After A375 cells were seeded (50,000 cells/well) and
adhered, they were treated with triclosan (0–40 µM) for 24 h and
then incubated with a mixture of 2 µM calcein AM and 4 µM EthD-1
for 15 min at 37°C. The labeled cells were randomly visualized
using a fluorescence microscope at 20× magnification and counted by
using Image Pro Plus image analysis software (version 5.0; Media
Cybernetics Inc.). The wavelength of fluorescence excitation was
488 nm for live cells and 594 nm for dead cells.
Measurement of mitochondrial membrane
potential (MMP, ΔΨm)
Cultured A375 cells were treated with TMRM (50 nM)
for 45 min and DAPI (10 µg/ml−1) for 15 min at 37°C and
were then washed 4 times with warm PBS (37°C). Fluorescence was
then measured using a confocal microscope FLUOVIEW FV10i
(magnification, ×20). Confocal microscope images were captured
using FV10-ASW version 3.1 Viewer software (Olympus Corporation).
Images of TMRM fluorescence were obtained using an excitation at
630 nm. Images of DAPI staining were obtained using an excitation
at 460 nm. The mitochondrial membrane potential was represented by
the relative intensity of the fluorescence.
Measurement of ATP concentration
The concentration of ATP was measured using the
luciferin-luciferase method following the protocol of the S0026 ATP
detection assay kit (Beyotime Institute of Biotechnology). The A375
cells were collected on ice within 5 min and immediately lysed with
40 µl lysis buffer from the ATP detection kit. After being
centrifuged at 12,000 g at 4°C for 5 min, the supernatant was
transferred to a new 1.5 ml tube for the ATP test. The luminescence
from a 20-µl sample was assayed using an Infinite M200 microplate
reader (luminometer mode) together with 100 µl ATP detection buffer
from the ATP assay kit. The samples' protein concentrations were
measured using a BCA assay kit (P0010, Beyotime Institute of
Biotechnology) at the same time and the ATP concentrations were
normalized to the amount of total protein in each sample (µM/mg,
protein).
Staining of mitochondrial
morphology
Cultured A375 cells were loaded with a mitochondrial
selective probe mito-Tracker Green (50 nM) at 37°C for 15 min.
Images were captured using confocal microscopy (magnification,
×60). Images of mito-tracker Green fluorescence were obtained with
an excitation at 490 nm and an emission at 516 nm.
Measurement of mitochondrial reactive
oxygen species (mito-ROS)
Cultured A375 cells were loaded with Mito-SOX (2 µM)
for 20 min and DAPI at 37°C for 15 min and then the fluorescence
was measured using confocal microscopy (magnification, ×60). Images
of Mito-SOX fluorescence were captured using an excitation at 630
nm. Images of DAPI staining were captured by using an excitation at
460 nm.
Measurement of intracellular
superoxide anion concentration
Cultured A375 cells were loaded with dihydroethidium
(2 µM) at 37°C for 30 min and DAPI for 15 min and then the
fluorescence was measured using a fluorescence microscope
(magnification, ×20). Images of dihydroethidium fluorescence were
captured using an excitation at 594 nm.
Western blotting
Protein samples from cultured A375 cells were
harvested with RIPA lysis buffer (Beyotime Institute of
Biotechnology) containing 1% protein inhibitor and 10% phosphatase
inhibitor. After being centrifuged at 12,000 × g at 4°C for 15 min,
the supernatant was transferred and protein concentrations were
assessed using a BCA assay kit (Bio-Rad Laboratories, Inc.). Equal
amounts of protein (80 µg/lane) were loaded onto a 8–15% gel,
resolved using SDS-PAGE and blotted onto a nitrocellulose membrane.
After blocking with 5% non-fat milk at 4°C for 2 h, the membranes
were incubated with primary antibodies at 4°C overnight. The
primary antibodies were anti-p-STAT3 (Y705, #9131, 1:1,000),
anti-STAT3 (#4904, 1:1,000), anti-p-AMPK (Thr172, #2535, 1:1,000),
anti-AMPK (#2532, 1:1,000), anti-p62 (#88588, 1:1,000), anti-Bcl-2
(ab196495, 1:1,000) and anti-LC3 (L7543, 1:1,000). After being
rinsed with TBS-0.1% Tween 20 (5 min for 3 times), the membranes
were subsequently incubated with fluorescence-conjugated goat
anti-mouse IgG or goat anti-rabbit IgG secondary antibody
(1:10,000;LI-COR Biosciences) for 1 h at room temperature. Images
were captured using an Odyssey infrared imaging system and Odyssey
version 3.0 software (LI-COR Biosciences).
siRNA transfection
A375 cells were seeded into 6-well flat-bottom plate
at 5×103 per well. After cells adhered, the medium was
replaced with serum-free DMEM and then the cells were transfected
with small interfering (si)RNA AMPKα1/2 (80 nM) or scrambled siRNA
used as a negative control (NC) (Shanghai GenePharma Co., Ltd.)
using Lipofectamine® 2000 (Invitrogen; Thermo Fisher
Scientific, Inc.). After 6 h of incubation at 37°C, the medium was
removed and the cells were cultured for another 48 h before
subsequent experiments. RNA oligo sequence were listed (5′-3′) as
follows: AMPKα1 forward, GCACGAGUUGACUGGACAUTT and reverse,
AUGUCCAGUCAACUCGUGCTT; AMPKα2 forward, GCUGACUUCGGACUCUCUATT and
reverse, UAGAGAGUCCGAAGUCAGCTT; and NC forward,
UUCUCCGAACGUGUCACGUTT and reverse, ACGUGACACGUUCGGAGAATT.
Measurement of
[Ca2+]i
Cultured A375 cells were treated with Fluo-3/AM (5
µM) and incubated at 37°C for 15 min. Then the cells were incubated
with DAPI for 15 min at 37°C. The fluorescence intensity reflecting
[Ca2+]i was measured using confocal
microscopy (magnification, ×60). Images of Fluo-3/AM fluorescence
were obtained with an excitation at 488 nm and an emission at 518
nm, and measured using Image-Pro Plus version 5.0 (Media
Cybernetics, Inc.).
Statistical analysis
Data were expressed by mean ± standard error of the
mean (SEM) (unless otherwise shown) and analyzed by using Sigma
Plot version 12.5 (Systat Software, Inc.). Statistical significance
of two groups was determined using an unpaired Student's t-test.
For >2 groups, one-way ANOVA followed by Tukey's post hoc test
was used. For the data with control value of 1 and no SEM, the
randomized block ANOVA was used (20). P<0.05 was considered to indicate a
statistically significant difference.
Results
Triclosan induces cytotoxicity in A375
cells
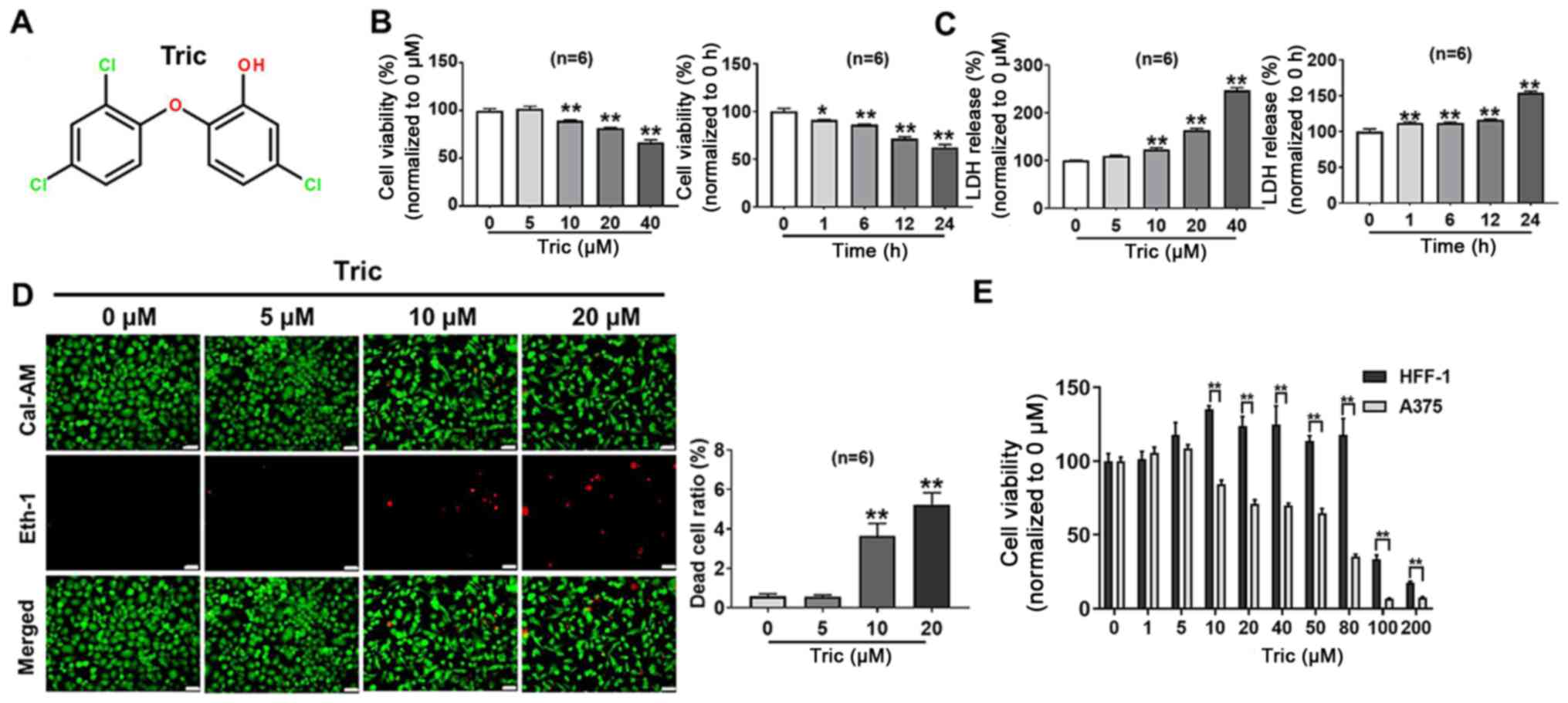
The chemical structure of triclosan is shown in
Fig. 1A. To evaluate the
cytotoxicity of triclosan on melanoma cells, A375 cells were
treated with various concentrations of Triclosan for 24 h and then
cell viability was measured using a MTT and LDH release assays. In
addition, cell death was detected using the LIVE/DEAD cell
viability assay kit. It was demonstrated that triclosan inhibited
A375 cell survival in a dose- and time-dependent manner (Fig. 1B-D). Fig.
1B demonstrated that 40 µM triclosan could significantly induce
a decrease in cell viability (P<0.01). In addition, the result
of LD50 of triclosan is presented in Fig. S1, with the LD50 of
triclosan in A375 cells being 52.72 µM. To further examine whether
triclosan had beneficial selective effects between melanoma cells
and other cell types, the same treatment was used on skin
fibroblast HFF-1 cells. It was demonstrated that compared with
HFF-1 cells, A375 cells were more sensitive to triclosan (Fig. 1E); triclosan significantly decreased
A375 cell viability at 10, 20, 40, 50, 80, 100 and 200 µM (all
P<0.01). Therefore, the present results suggested that triclosan
inhibited cell viability in a dose- and time-dependent manner in
A375 cells and these effects have tumor cell selectivity.
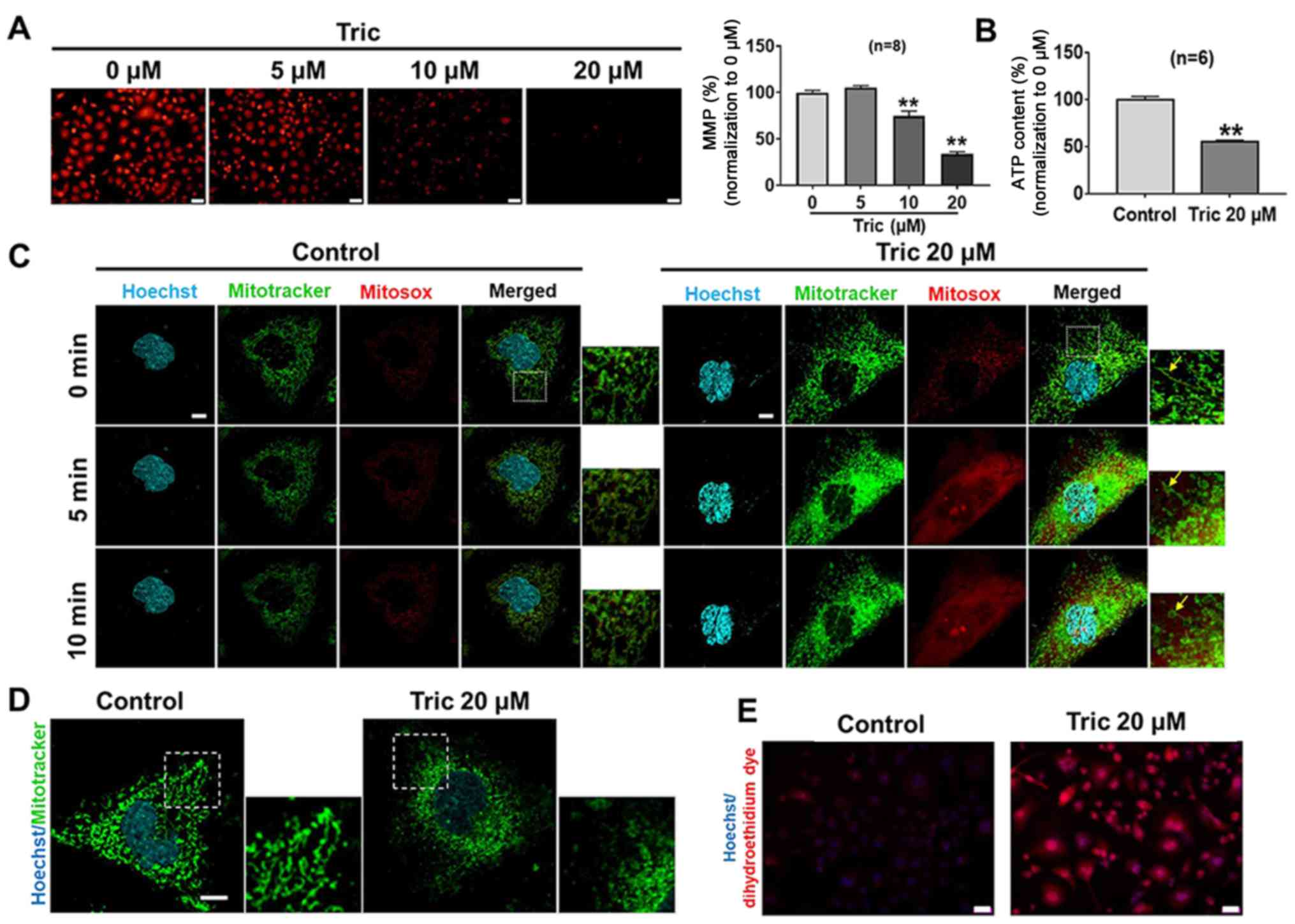
Triclosan induces mitochondrial
morphology and function changes in melanoma cells
Triclosan induces mitochondrial uncoupling in human
mast cells and keratinocytes and has been reported to disrupt
mitochondrial function (13,21). Therefore, in order to investigate
whether triclosan could induce mitochondrial uncoupling of melanoma
cells, A375 cells were exposed to different concentrations of
Triclosan for 24 h and MMP (ΔΨm) was measured using TMRM. It was
revealed that compared with the control group, triclosan treatment
depolarized MMP in dose-dependent manner (Fig. 2A). The present study also
investigated whether triclosan-induced MMP could cause ATP
depletion in A375 cells. It was demonstrated that ATP content was
decreased after 20 µM triclosan treatment (Fig. 2B). Collectively, the present results
suggested that triclosan induced mitochondrial uncoupling in
melanoma cells.
The mitochondria of A375 cells were stained using
the mitochondria-specific probes mito-Tracker and mito-SOX. It was
shown that acute triclosan treatment induced mitochondrial swelling
and fission, accompanied by prominent ROS production within 10 min
(Fig. 2C). The present study also
examined the long-term effect of triclosan on mitochondrial
morphology, showing that triclosan induced excessive mitochondrial
fission and elevated production of ROS at 5–10 min, but
mitochondrial swelling was not observed at 24 h (Fig. 2D and E). Mitochondrial swelling
begins with changes in ion homeostasis of the matrix, which induces
an osmotic imbalance between the cytosol and the matrix (22). As a result, increased colloidal
osmotic pressure enhances the water influx leading to matrix
swelling and mitochondrial size volume increased (22). Mitochondrial fission is shown as
granular fragmentation (23).
Therefore, the present results indicated that triclosan induced
mitochondrial fission and ROS increase. Moreover, the acute effect
of triclosan induced reversible mitochondrial swelling.
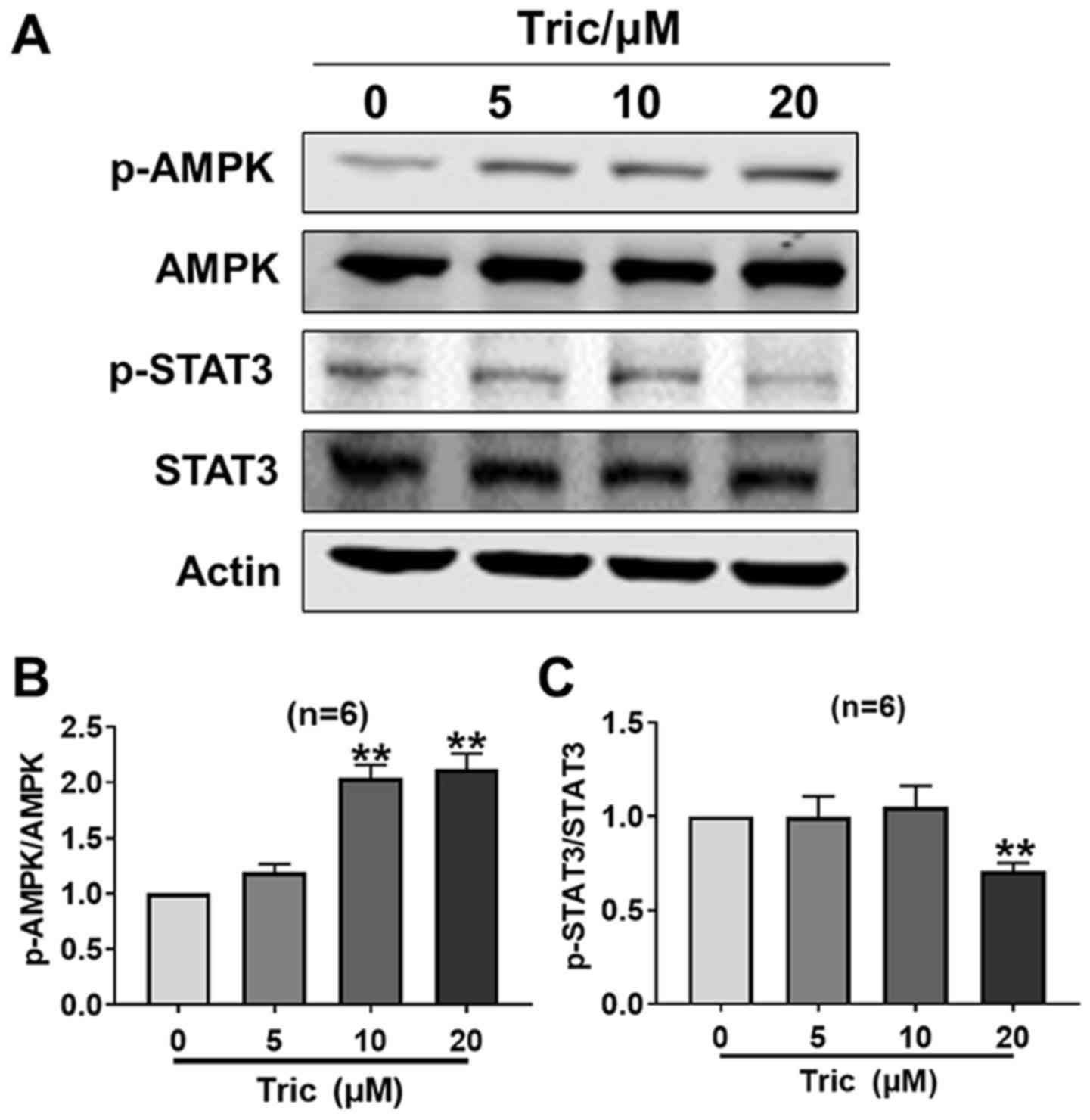
Triclosan activates AMPK and inhibits
STAT3 activity in melanoma cells
As a mitochondrial uncoupler, triclosan decreases
ATP content and induces AMP generation (13). AMPK is a downstream signaling protein
of AMP and is subsequently activated (24). It has been previously reported that a
cross-talk occurs between AMPK and STAT3 (25,26),
thus the present study investigated whether this cross-talk exists
during triclosan-induced melanoma cell death. It was demonstrated
that triclosan significantly increased p-AMPK protein expression at
10 and 20 µM, and decreased p-STAT3 protein expression at 20 µM
(both P<0.01), so that the p-AMPK/AMPK ratio increased and the
p-STAT3/STAT3 ratio decreased, indicating that triclosan activated
AMPK and inhibited STAT3 activity in melanoma cells after 24 h
treatment (Fig. 3). Therefore, the
present results suggested that triclosan may induce melanoma cell
death and mitochondria dysfunction via AMPK activation and STAT3
activity inhibition.
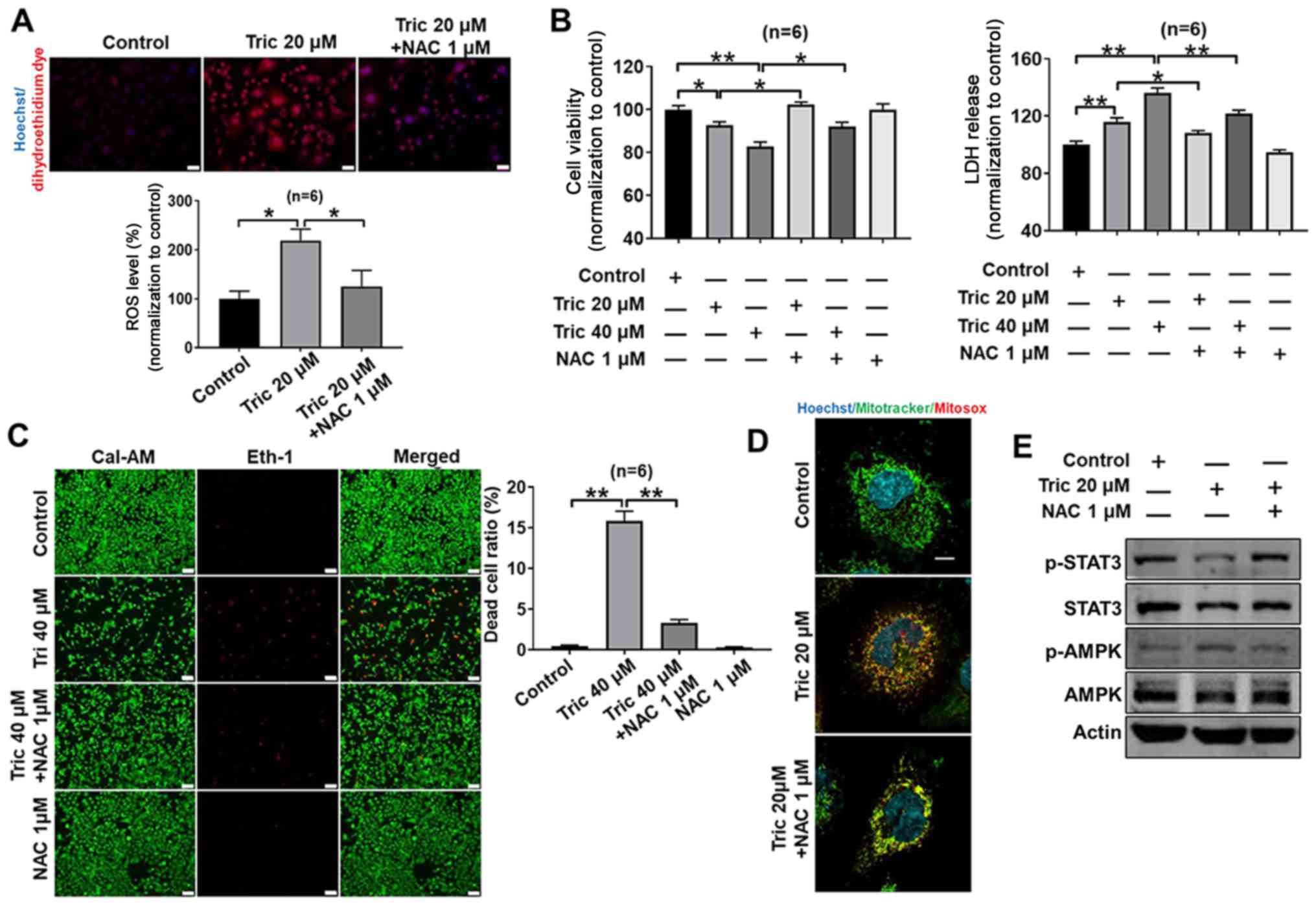
ROS generation is involved in
triclosan-induced cell death and mitochondrial dysfunction in A375
cells
ROS levels increase in tumor cells during
proliferation and cell death (27–29).
Triclosan elevated ROS production in A375 cells and the present
study investigated whether triclosan-induced cell death resulted
from a ROS increase. It was demonstrated that 20 µM triclosan
increased ROS production in melanoma cells, which was inhibited by
the antioxidant NAC (Fig. 4A).
Furthermore, triclosan-induced decrease in cell viability and
increased LDH release was inhibited by co-treatment with NAC in
A375 cells (Fig. 4B). The protective
effects of NAC against triclosan-induced cell death were further
demonstrated by the LIVE/DEAD cell viability assay (Fig. 4C), as 40 µM triclosan induced
significant cell death (showed by Eth-1), which was then reversed
using 1 µM NAC. Moreover, 20 µM triclosan induced a marked
mitochondrial fission, and NAC reversed triclosan-induced
mitochondrial fission (Fig. 4D).
Oxidative stress is a trigger for AMPK activity and
a repressor for STAT3 activity (30,31). It
was reported that triclosan activated AMPK activity, inhibited
STAT3 activity (Fig. 3) and induced
ROS levels (Fig. 4A and D), thus
indicating that triclosan-induced ROS generation may contribute to
triclosan-induced AMPK activation and STAT3 activity inhibition.
Moreover, these triclosan-induced effects were reversed by NAC in
A375 cells (Fig. 4E), thus
suggesting that ROS are upstream molecules of the AMPK and STAT3
signaling pathways.
STAT3 activity inhibition is involved
in triclosan-induced cell death in melanoma cells
STAT3 serves an important role in tumor cell
survival (32,33) and Bcl-2 is a downstream signaling
molecule of STAT3 (34).
Triclosan-induced STAT3 inhibition (Fig.
3) significantly inhibited Bcl-2 expression levels in A375
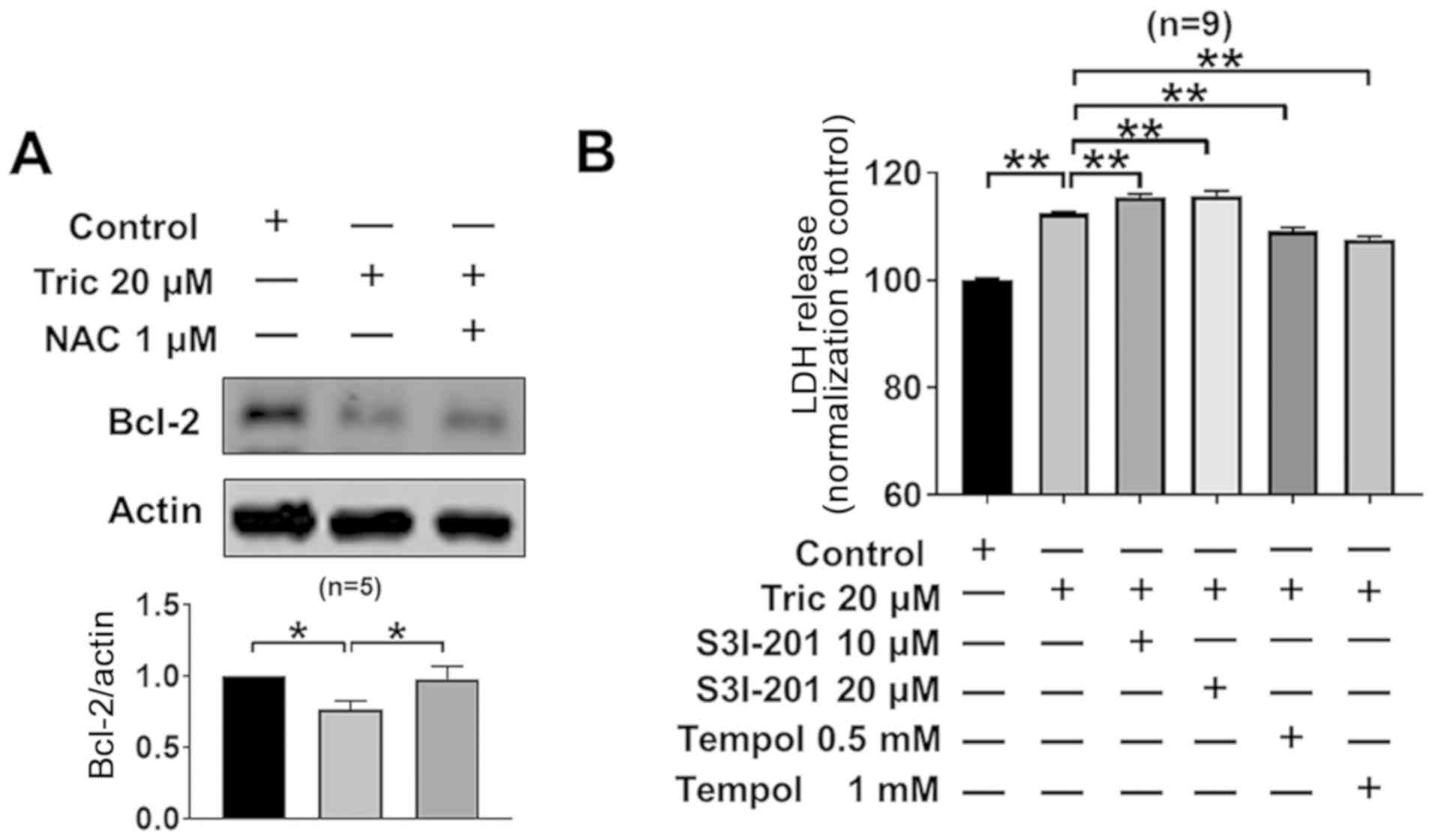
cells (P<0.05; Fig. 5A).
Furthermore, co-treatment with NAC reversed triclosan-induced Bcl-2
downregulation. In addition, treatment with the STAT3 inhibitor
S3I-201 increased triclosan-induced LDH release (Fig. 5B). Moreover, the ROS scavenger Tempol
(35) also partially reversed
triclosan-induced increase of LDH release (0.5 and 1 mM; Fig. 5B). Collectively, the present results
suggested that triclosan led to cell death via the ROS/STAT3/Bcl-2
signaling pathway.
AMPK activation is not involved in
Triclosan-induced cell death, but does affect
[Ca2+]i transport in melanoma cells
To further investigate the effect of AMPK in
triclosan-induced cell death, AMPK was knocked down in A375 cells
using siRNA transfection. It was demonstrated that treatment with
si-AMPK significantly decreased the protein expression levels of
AMPK compared with the NC group (P<0.01; Fig. 6A). In addition, in the
si-AMPK+Triclosan+NAC group, knockdown of AMPK decreased the
protective effect of NAC on triclosan-induced LDH release compared
with in the NC+Triclosan+NAC group in A375 cells (Fig. 6C), while knockdown of AMPK did not
significantly affect cell death and LDH release following triclosan
treatment (Fig. 6B and C).
Therefore, the present results indicated that AMPK activation was
not involved in triclosan-induced cell death and AMPK inhibition
may contribute to cell damage. It has been previously reported that
AMPK activity regulates mitochondrial function (36,37).
Therefore, the present study hypothesized that triclosan may affect
mitochondrial function in A375 cells by activating AMPK. It was
demonstrated that triclosan induced mitochondrial swelling, but
this effect was attenuated in AMPK-knockdown in A375 cells
(Fig. 6D), indicating that AMPK
could affect mitochondrial swelling. AMPK activation increases
intracellular Ca2+ (38)
and Ca2+-induced mitochondrial swelling and cytochrome C
release in isolated mitochondria (39–41). The
present results suggested that triclosan increased
[Ca2+]i (Fig.
6E) and this effect was decreased with si-AMPK or NAC
treatment. Thus, AMPK-knockdown may inhibit Ca2+
transport, which could decrease Ca2+ release to the
mitochondria and limit mitochondrial swelling.
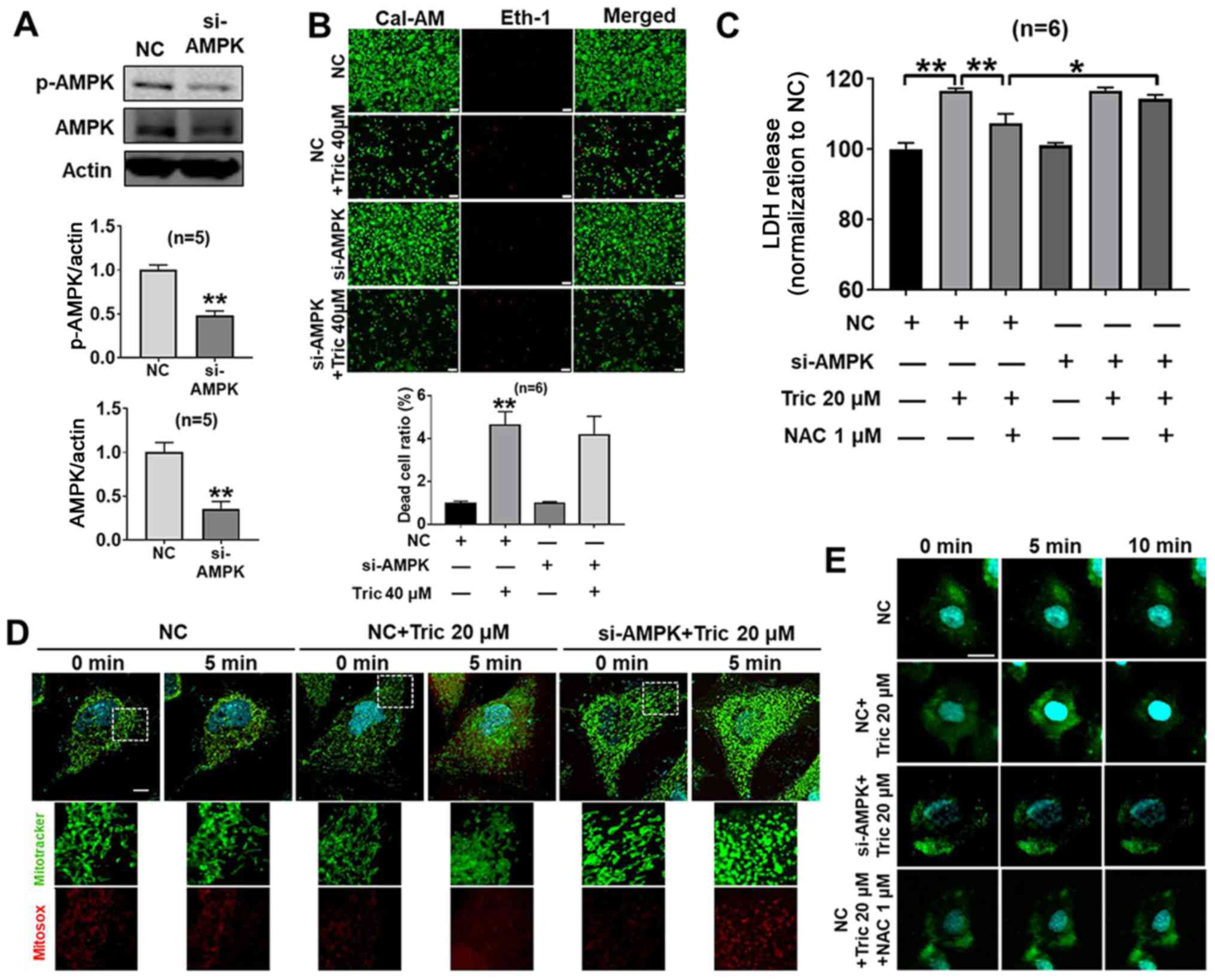 | Figure 6.AMPK activation does not affect
triclosan-induced cell death but induces mitochondrial dynamics by
[Ca2+]i transport in A375 cells. (A) si-AMPK
inhibited the protein expression levels of AMPK in A375 cells.
Statistical significance of two groups was determined using
unpaired Student's t-test. (B) Knockdown of AMPK had no significant
effect on cell death. Scale bar, 50 µm. (C) NAC decreased the
triclosan-induced increase of LDH release, but this effect was
attenuated by knockdown of AMPK. (D) Triclosan induced
mitochondrial swelling, but this effect was attenuated in knockdown
of AMPK in A375 cells. Scale bar, 10 µm. (E) Triclosan increased
[Ca2+]I, but this effect was decreased with
si-AMPK treatment or NAC treatment. Scale bar, 10 µm. *P<0.05,
**P<0.01. NC, negative control; NAC, acetylcysteine; AMPK,
AMP-activated protein kinase; si, small interfering RNA. |
Triclosan induces autophagy in A375
cells
Previous studies have revealed an association
between AMPK and autophagy (42,43) and
that cell death is accompanied by autophagy (44,45). As
triclosan induced cell death and AMPK activation, the effects of
triclosan on the levels of the marker protein of autophagy
microtubule-associated protein 1A/1B-light chain 3 II (LC3-II) in
A375 cells were examined. It was reported that triclosan increased
LC3-II protein expression levels and this was reversed by NAC
treatment (Fig. 7A). Furthermore,
the autophagy inhibitor 3-MA increased triclosan-induced LDH
release compared with triclosan alone (Fig. 7B), indicating that autophagy serves a
protective role during triclosan-induced cell death. In order to
identify the role of AMPK in triclosan-induced autophagy, the
present study detected the expression levels of LC3 and p62 after
AMPK-knockdown. It was found that the protein expression levels of
AMPK were decreased after si-AMPK treatment, indicating that
si-AMPK was successfully transfected into the A375 cells (Fig. 7C). Moreover, triclosan did not
increase the protein expression levels of LC3-II after si-AMPK
treatment, and thus did not recruit p62 (Fig. 7C). Collectively, the present results
indicated that AMPK may be involved in triclosan-induced autophagy.
While si-AMPK treatment increased p-STAT3 expression levels in A375
cells, si-AMPK treatment did not prevent triclosan-induced
inhibition of STAT3 (Fig. 7C) and
cell death was observed (Fig. 6C).
Thus, the present results suggested that STAT3 serves a role in
triclosan-induced cell death. The mechanism of triclosan-induced
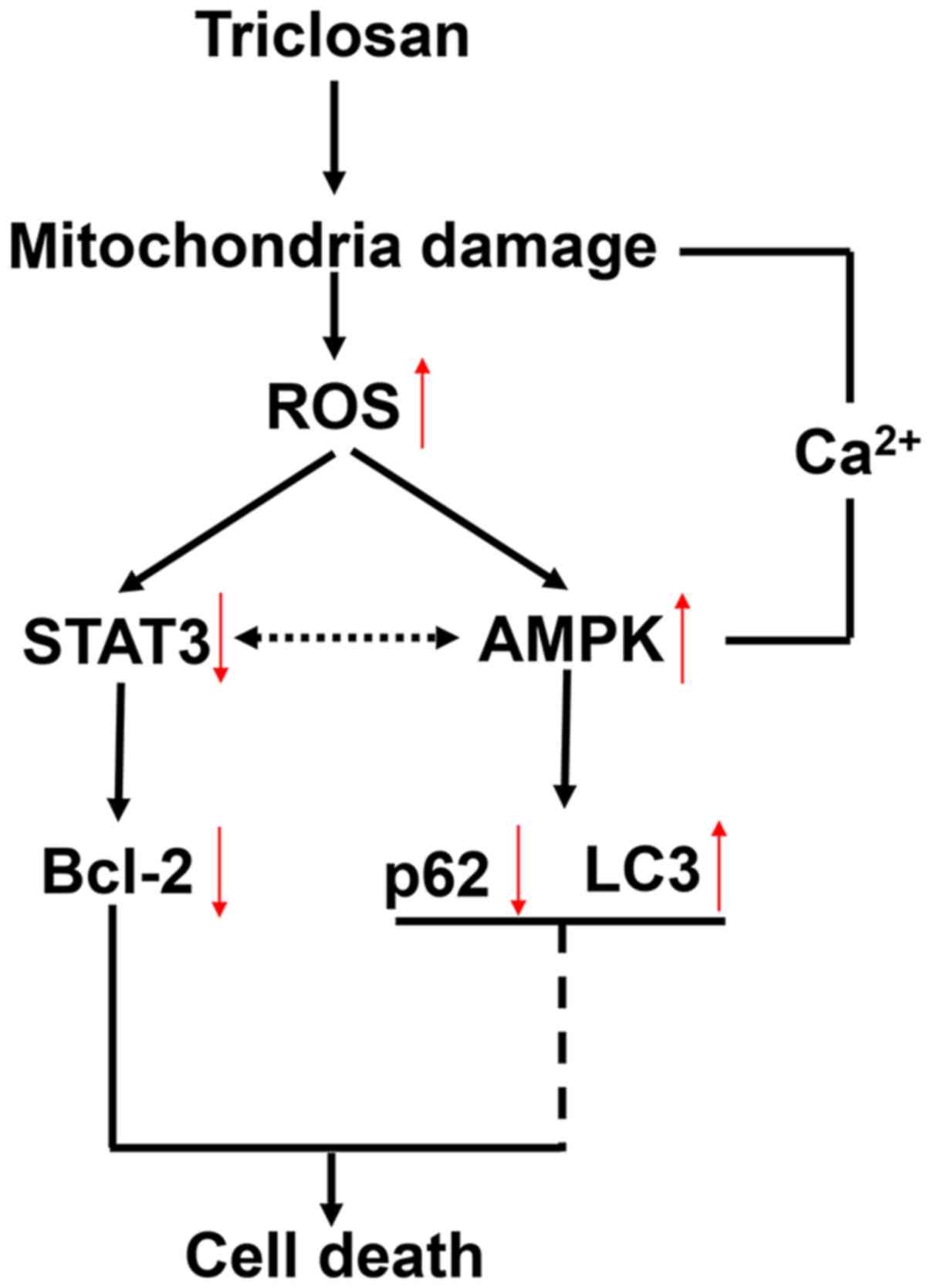
cell death and autophagy was summarized in Fig. 8.
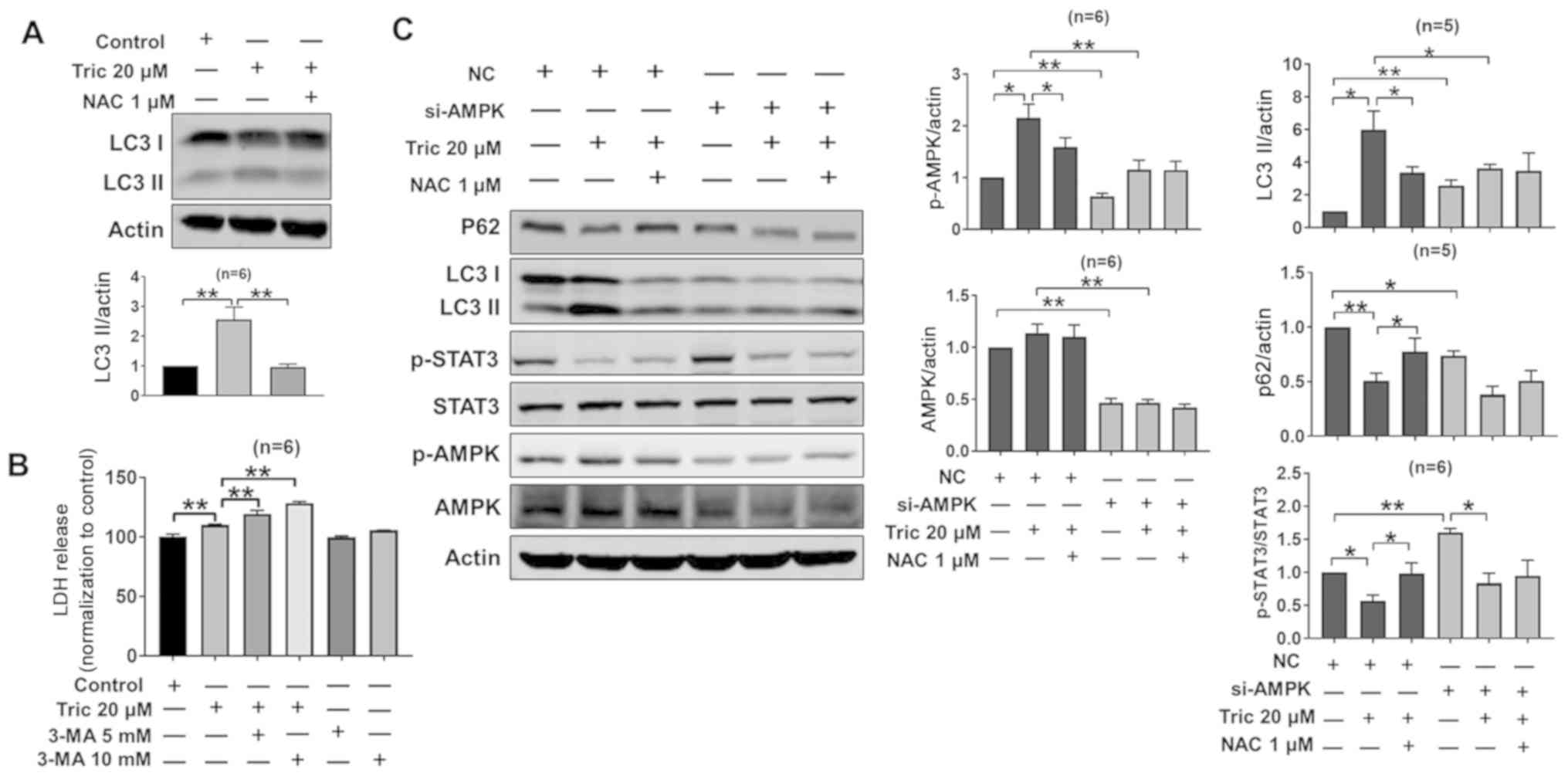 | Figure 7.Triclosan induces AMPK-dependent
autophagy in A375 cells. (A) NAC treatment attenuated the
triclosan-induced autophagy response. (B) Triclosan-induced LDH
release was increased by treatment with the autophagy inhibitor
3-MA. (C) Western blotting results indicated that triclosan
inhibited p62 and p-STAT3 protein expression levels, and increased
LC3II and p-AMPK expression levels, which were reversed by NAC.
AMPK-knockdown increased STAT3 in A375 cells, but LC3II was
decreased after triclosan treatment. *P<0.05, **P<0.01. NC,
negative control; NAC, acetylcysteine; AMPK, AMP-activated protein
kinase; si, small interfering RNA; p-, phosphorylated; LC3II,
microtubule-associated protein 1A/1B-light chain 3; LDH, lactate
dehydrogenase. |
Discussion
Triclosan, a widely used antibacterial and
antifungal agent, is present in everyday household personal care
and consumer products (11,12). Moreover, previous studies have
focused on its antibacterial effect (14–16). To
the best of our knowledge, the present study is the first to
identify that triclosan induces cell death and autophagy in A375
cells. Furthermore, the present results suggested the importance of
triclosan in cancer cell death as a novel antitumor drug. The
present study primarily used the human melanoma cell line A375 to
investigate underlying mechanism of action of triclosan.
Triclosan is a mild mitochondrial uncoupler
(13). A previous study has
demonstrated that triclosan induces mitochondrial depolarization in
a dose-dependent manner in HaCaT cells, induces no significant
mitochondrial swelling at 10 µM and prevents mitochondrial swelling
after subsequent CaCl2 treatment, which alone resulted
in mitochondrial swelling (41). The
present results indicated that triclosan induced significant
mitochondrial swelling. Unlike the previous study that focused on
healthy human skin cells (41), the
present study used skin cancer cells. Hence, the present study
hypothesized that triclosan may induce cell death by regulating
mitochondrial function. Consistent with this hypothesis, it was
demonstrated that triclosan decreased MMP and ATP content, induced
reversible mitochondrial swelling and mitochondrial fission.
Mitochondria is a major source of ROS (46) and the present study investigated
whether triclosan affected the production of ROS. It was
demonstrated that triclosan increased the production of
intracellular and mito-ROS.
Mito-ROS are physiological activators of AMPK
(30). As a known target for
treating type 2 diabetes and metabolic syndrome, AMPK has been
regarded as a novel target for cancer prevention and treatment
(47). The major mechanism
activating AMPK is increasing the AMP/ATP ratio (48). The present results revealed that
triclosan increased the AMP/ATP ratio. Furthermore, a previous
study has shown that AMPK serves an important role in regulating
mitochondrial function (30).
Similarly, mitochondrial damage leads to ROS production, activating
AMPK, and AMPK affects mitochondrial function in return (49).
AMPK promotes autophagy (43), which is the primary protective
process in cells and also serves a role in cell death (50). The effect of autophagy on the
anticancer action of AMPK is complicated; inducing or preventing
autophagy both contribute to cancer therapy (51,52). As
triclosan induces cell death, the present study investigated the
effects of triclosan on autophagic responses. It was demonstrated
that triclosan increased autophagic responses, but the autophagy
inhibitor 3-MA increased triclosan-induced LDH release, thus
indicating that autophagy serves a protective role during
triclosan-induced cell death. Furthermore, triclosan inhibited
autophagy after si-AMPK transfection, which indicated that AMPK may
be involved in triclosan-induced autophagy. Some studies have
showed that AMPK activation suppresses the STAT3 signaling pathway
(26,53,54);
however, genetic or pharmacological inhibition of STAT3
significantly increases the ADP/ATP ratio and activates AMPK
signaling (55,56). Therefore, there is a dual directional
regulation mechanism between AMPK and STAT3. In the present study,
while si-AMPK increased STAT3 expression levels, si-AMPK did not
prevent the triclosan-induced decrease in STAT3 expression levels
and cell death still occurred. Therefore, the present results
suggested that STAT3 may serve an role in triclosan-induced cell
death.
A limitation of the present study is that
comparative tests of triclosan and other anticancer drugs were not
performed. Therefore, future studies should investigate this.
However, the present study measured the effects of triclosan on the
viability of normal cultured skin HFF-1 cells, indicating that the
cytotoxic effects of triclosan are cancer-specific, which may
contribute to its clinical application. Moreover, triclosan
treatment, an ingredient of antiseptic soaps, may be applied to the
skin. Thus, the ability to treat melanoma via application of
therapeutic agents to the skin may be of clinical benefit.
Supplementary Material
Supporting Data
Acknowledgements
Not applicable.
Funding
This project was supported by The Jiaxing Science
and Technology Project (grant nos. 2017AY33004 and 2018AD32083),
the Medical Scientific Research Foundation of Zhejiang Province,
China (grant nos. 2019KY694) and a Starting Research Fund from
Jiaxin University (grant no. CD70519041).
Availability of data and materials
The datasets used and/or analyzed during the present
study are available from the corresponding author on reasonable
request.
Authors' contributions
JJ designed the project, performed the experiments,
interpreted the data and wrote the paper. JG designed the project,
interpreted the data and wrote the paper. NC, HP and WX performed
the experiments and interpreted the data. HX, SL, ZG and RD
performed the experiments. YH designed the project, revised the
manuscript and gave final approval of the version to be published.
All authors read and approved the final manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Torre LA, Bray F, Siegel RL, Ferlay J,
Lortet-Tieulent J and Jemal A: Global cancer statistics, 2012. CA
Cancer J Clin. 65:87–108. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Miller KD, Siegel RL, Lin CC, Mariotto AB,
Kramer JL, Rowland JH, Stein KD, Alteri R and Jemal A: Cancer
treatment and survivorship statistics, 2016. CA Cancer J Clin.
66:271–289. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Abbas Q, Raza H, Hassan M, Phull AR, Kim
SJ and Seo SY: Acetazolamide inhibits the level of tyrosinase and
melanin: An enzyme kinetic, in vitro, in vivo, and in silico
studies. Chem Biodivers. 14:2017. View Article : Google Scholar
|
|
4
|
Lee CW, Yen FL, Ko HH, Li SY, Chiang YC,
Lee MH, Tsai MH and Hsu LF: Cudraflavone C induces apoptosis of
A375.S2 melanoma cells through mitochondrial ROS production and
MAPK activation. Int J Mol Sci. 18:15082017. View Article : Google Scholar
|
|
5
|
Trunzer K, Pavlick AC, Schuchter L,
Gonzalez R, McArthur GA, Hutson TE, Moschos SJ, Flaherty KT, Kim
KB, Weber JS, et al: Pharmacodynamic effects and mechanisms of
resistance to vemurafenib in patients with metastatic melanoma. J
Clin Oncol. 31:1767–1774. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Tuong W, Cheng LS and Armstrong AW:
Melanoma: Epidemiology, diagnosis, treatment, and outcomes.
Dermatol Clin. 30113–124. (ix)2012. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Eggermont AM, Spatz A and Robert C:
Cutaneous melanoma. Lancet. 383:816–827. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Nowinski SM, Solmonson A, Rundhaug JE, Rho
O, Cho J, Lago CU, Riley CL, Lee S, Kohno S, Dao CK, et al:
Mitochondrial uncoupling links lipid catabolism to Akt inhibition
and resistance to tumorigenesis. Nat Commun. 6:81372015. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Samudio I, Fiegl M and Andreeff M:
Mitochondrial uncoupling and the Warburg effect: Molecular basis
for the reprogramming of cancer cell metabolism. Cancer Res.
69:2163–2166. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Baffy G, Derdak Z and Robson SC:
Mitochondrial recoupling: A novel therapeutic strategy for cancer?
Br J Cancer. 105:469–474. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Weatherly LM and Gosse JA: Triclosan
exposure, transformation, and human health effects. J Toxicol
Environ Health B Crit Rev. 20:447–469. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Ribado JV, Ley C, Haggerty TD, Tkachenko
E, Bhatt AS and Parsonnet J: Household triclosan and triclocarban
effects on the infant and maternal microbiome. EMBO Mol Med.
9:1732–1741. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Weatherly LM, Shim J, Hashmi HN, Kennedy
RH, Hess ST and Gosse JA: Antimicrobial agent triclosan is a proton
ionophore uncoupler of mitochondria in living rat and human mast
cells and in primary human keratinocytes. J Appl Toxicol.
36:777–789. 2016. View
Article : Google Scholar : PubMed/NCBI
|
|
14
|
Campbell L and Zirwas MJ: Triclosan.
Dermatitis. 17:204–207. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Moretro T, Hoiby-Pettersen GS, Habimana O,
Heir E and Langsrud S: Assessment of the antibacterial activity of
a triclosan-containing cutting board. Int J Food Microbiol.
146:157–162. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Wu HX, Tan L, Tang ZW, Yang MY, Xiao JY,
Liu CJ and Zhuo RX: Highly efficient antibacterial surface grafted
with a triclosan-decorated poly(N-hydroxyethylacrylamide) brush.
ACS Appl Mater Interfaces. 7:7008–7015. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Maycotte P, Marin-Hernandez A,
Goyri-Aguirre M, Anaya-Ruiz M, Reyes-Leyva J and Cortes-Hernandez
P: Mitochondrial dynamics and cancer. Tumour Biol.
39:10104283176983912017. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Senft D and Ronai ZA: Regulators of
mitochondrial dynamics in cancer. Curr Opin Cell Biol. 39:43–52.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Phull AR, Nasir B, Haq IU and Kim SJ:
Oxidative stress, consequences and ROS mediated cellular signaling
in rheumatoid arthritis. Chem Biol Interact. 281:121–136. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Lew M: Good statistical practice in
pharmacology. Problem 2. Br J Pharmacol. 152:299–303. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Weatherly LM, Nelson AJ, Shim J, Riitano
AM, Gerson ED, Hart AJ, de Juan-Sanz J, Ryan TA, Sher R, Hess ST
and Gosse JA: Antimicrobial agent triclosan disrupts mitochondrial
structure, revealed by super-resolution microscopy, and inhibits
mast cell signaling via calcium modulation. Toxicol Appl Pharmacol.
349:39–54. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Park JY, Jang SY, Shin YK, Koh H, Suh DJ,
Shinji T, Araki T and Park HT: Mitochondrial swelling and
microtubule depolymerization are associated with energy depletion
in axon degeneration. Neuroscience. 238:258–269. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Liu MY, Jin J, Li SL, Yan J, Zhen CL, Gao
JL, Zhang YH, Zhang YQ, Shen X, Zhang LS, et al: Mitochondrial
fission of smooth muscle cells is involved in artery constriction.
Hypertension. 68:1245–1254. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Yan Y, Zhou XE, Xu HE and Melcher K:
Structure and physiological regulation of AMPK. Int J Mol Sci.
19:35342018. View Article : Google Scholar
|
|
25
|
Vasamsetti SB, Karnewar S, Kanugula AK,
Thatipalli AR, Kumar JM and Kotamraju S: Metformin inhibits
monocyte-to-macrophage differentiation via AMPK-mediated inhibition
of STAT3 activation: Potential role in atherosclerosis. Diabetes.
64:2028–2041. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Wang M, Xin H, Tang W, Li Y, Zhang Z, Fan
L, Miao L, Tan B, Wang X and Zhu YZ: AMPK serves as a therapeutic
target against anemia of inflammation. Antioxid Redox Signal.
27:251–268. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Xu Z, Zhang F, Bai C, Yao C, Zhong H, Zou
C and Chen X: Sophoridine induces apoptosis and S phase arrest via
ROS-dependent JNK and ERK activation in human pancreatic cancer
cells. J Exp Clin Cancer Res. 36:1242017. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Chen W, Zou P, Zhao Z, Chen X, Fan X,
Vinothkumar R, Cui R, Wu F, Zhang Q, Liang G and Ji J: Synergistic
antitumor activity of rapamycin and EF24 via increasing ROS for the
treatment of gastric cancer. Redox Biol. 10:78–89. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Chen X, Dai X, Zou P, Chen W, Rajamanickam
V, Feng C, Zhuge W, Qiu C, Ye Q, Zhang X and Liang G: Curcuminoid
EF24 enhances the anti-tumour activity of Akt inhibitor MK-2206
through ROS-mediated endoplasmic reticulum stress and mitochondrial
dysfunction in gastric cancer. Br J Pharmacol. 174:1131–1146. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Rabinovitch RC, Samborska B, Faubert B, Ma
EH, Gravel SP, Andrzejewski S, Raissi TC, Pause A, St-Pierre J and
Jones RG: AMPK maintains cellular metabolic homeostasis through
regulation of mitochondrial reactive oxygen species. Cell Rep.
21:1–9. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Chen W, Li P, Liu Y, Yang Y, Ye X, Zhang F
and Huang H: Isoalantolactone induces apoptosis through
ROS-mediated ER stress and inhibition of STAT3 in prostate cancer
cells. J Exp Clin Cancer Res. 37:3092018. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Kryczek I, Lin Y, Nagarsheth N, Peng D,
Zhao L, Zhao E, Vatan L, Szeliga W, Dou Y, Owens S, et al:
IL-22(+)CD4(+) T cells promote colorectal cancer stemness via STAT3
transcription factor activation and induction of the
methyltransferase DOT1L. Immunity. 40:772–784. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Chai EZ, Shanmugam MK, Arfuso F,
Dharmarajan A, Wang C, Kumar AP, Samy RP, Lim LH, Wang L, Goh BC,
et al: Targeting transcription factor STAT3 for cancer prevention
and therapy. Pharmacol Ther. 162:86–97. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Min K, Lawan A and Bennett AM: Loss of
MKP-5 promotes myofiber survival by activating STAT3/Bcl-2
signaling during regenerative myogenesis. Skelet Muscle. 7:212017.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Ye S, Xu P, Huang M, Chen X, Zeng S, Wang
Q, Chen J, Li K, Gao W, Liu R, et al: The heterocyclic compound
Tempol inhibits the growth of cancer cells by interfering with
glutamine metabolism. Cell Death Dis. 11:3122020. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Lantier L, Fentz J, Mounier R, Leclerc J,
Treebak JT, Pehmoller C, Sanz N, Sakakibara I, Saint-Amand E,
Rimbaud S, et al: AMPK controls exercise endurance, mitochondrial
oxidative capacity, and skeletal muscle integrity. FASEB J.
28:3211–3224. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Toyama EQ, Herzig S, Courchet J, Lewis TL
Jr, Loson OC, Hellberg K, Young NP, Chen H, Polleux F, Chan DC and
Shaw RJ: Metabolism. AMP-activated protein kinase mediates
mitochondrial fission in response to energy stress. Science.
351:275–281. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Vlachaki Walker JM, Robb JL, Cruz AM,
Malhi A, Weightman Potter PG, Ashford MLJ, McCrimmon RJ, Ellacott
KLJ and Beall C: AMP-activated protein kinase (AMPK) activator
A-769662 increases intracellular calcium and ATP release from
astrocytes in an AMPK-independent manner. Diabetes Obes Metab.
19:997–1005. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Javadov S, Chapa-Dubocq X and Makarov V:
Different approaches to modeling analysis of mitochondrial
swelling. Mitochondrion. 38:58–70. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Abe T, Takagi N, Nakano M, Tanonaka K and
Takeo S: The effects of monobromobimane on calcium and
phenylarsineoxide-induced mitochondrial swelling and cytochrome C
release in isolated brain mitochondria. Biol Pharm Bull.
27:524–527. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Teplova VV, Belosludtsev KN and Kruglov
AG: Mechanism of triclosan toxicity: Mitochondrial dysfunction
including complex II inhibition, superoxide release and uncoupling
of oxidative phosphorylation. Toxicol Lett. 275:108–117. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Dite TA, Ling NXY, Scott JW, Hoque A,
Galic S, Parker BL, Ngoei KRW, Langendorf CG, O'Brien MT, Kundu M,
et al: The autophagy initiator ULK1 sensitizes AMPK to allosteric
drugs. Nat Commun. 8:5712017. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Bujak AL, Crane JD, Lally JS, Ford RJ,
Kang SJ, Rebalka IA, Green AE, Kemp BE, Hawke TJ, Schertzer JD and
Steinberg GR: AMPK activation of muscle autophagy prevents
fasting-induced hypoglycemia and myopathy during aging. Cell Metab.
21:883–890. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Sun L, Hu L, Cogdell D, Lu L, Gao C, Tian
W, Zhang Z, Kang Y, Fleming JB and Zhang W: MIR506 induces
autophagy-related cell death in pancreatic cancer cells by
targeting the STAT3 pathway. Autophagy. 13:703–714. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Sutton MN, Yang H, Huang GY, Fu C,
Pontikos M, Wang Y, Mao W, Pang L, Yang M, Liu J, et al:
RAS-related GTPases DIRAS1 and DIRAS2 induce autophagic cancer cell
death and are required for autophagy in murine ovarian cancer
cells. Autophagy. 14:637–653. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Bolisetty S and Jaimes EA: Mitochondria
and reactive oxygen species: Physiology and pathophysiology. Int J
Mol Sci. 14:6306–6344. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Luo Z, Zang M and Guo W: AMPK as a
metabolic tumor suppressor: Control of metabolism and cell growth.
Future Oncol. 6:457–470. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Ke R, Xu Q, Li C, Luo L and Huang D:
Mechanisms of AMPK in the maintenance of ATP balance during energy
metabolism. Cell Biol Int. 42:384–392. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Yang YF, Wang YY, Hsiao M, Lo S, Chang YC,
Jan YH, Lai TC, Lee YC, Hsieh YC and Yuan SF: IMPAD1 functions as
mitochondrial electron transport inhibitor that prevents ROS
production and promotes lung cancer metastasis through the
AMPK-Notch1-HEY1 pathway. Cancer Lett. 485:27–37. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Mizushima N, Levine B, Cuervo AM and
Klionsky DJ: Autophagy fights disease through cellular
self-digestion. Nature. 451:1069–1075. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Lu L, Shen X, Tao B, Lin C, Li K, Luo Z
and Cai K: The nanoparticle-facilitated autophagy inhibition of
cancer stem cells for improved chemotherapeutic effects on
glioblastomas. J Mater Chem B. 7:2054–2062. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Wang RX, Xu XE, Huang L, Chen S and Shao
ZM: eEF2 kinase mediated autophagy as a potential therapeutic
target for paclitaxel-resistant triple-negative breast cancer. Ann
Transl Med. 7:7832019. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Ge A, Wang S, Miao B and Yan M: Effects of
metformin on the expression of AMPK and STAT3 in the spinal dorsal
horn of rats with neuropathic pain. Mol Med Rep. 17:5229–5237.
2018.PubMed/NCBI
|
|
54
|
Li H, Lee J, He C, Zou MH and Xie Z:
Suppression of the mTORC1/STAT3/Notch1 pathway by activated AMPK
prevents hepatic insulin resistance induced by excess amino acids.
Am J Physiol Endocrinol Metab. 306:E197–E209. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Gao JL, Zhao J, Zhu HB, Peng X, Zhu JX, Ma
MH, Fu Y, Hu N, Tai Y, Xuan XC and Dong DL: Characterizations of
mitochondrial uncoupling induced by chemical mitochondrial
uncouplers in cardiomyocytes. Free Radic Biol Med. 124:288–298.
2018. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Zhang W, Xia D, Li Z, Zhou T, Chen T, Wu
Z, Zhou W, Li Z, Li L and Xu J: Aurora-A/ERK1/2/mTOR axis promotes
tumor progression in triple-negative breast cancer and
dual-targeting Aurora-A/mTOR shows synthetic lethality. Cell Death
Dis. 10:6062019. View Article : Google Scholar : PubMed/NCBI
|