Metabolic disorders, particularly those concerning
glucose metabolism, play an important role in the proliferation and
development of tumors (1). In normal
cells, the energy provided for cell biological activity is
predominantly dependent on changes in glucose metabolism that can
transform glucose into pyruvate after several steps. Subsequently,
pyruvate is converted to oxaloacetate, resulting in the production
of citrate in the mitochondrial tricarboxylic acid cycle (TCA
cycle). The process of glucose metabolism and the mitochondrial TCA
cycle can generate energy in the form of adenosine triphosphate
(ATP) and other forms, such as NADPH and FADH2 (1). NADPH and FADH2 are subsequently
committed to the electron transport chain complexes to yield ATP,
which is known as oxidative phosphorylation (OXPHOS) (2,3).
There are significant differences between normal
cells and tumor cells regarding glucose metabolism. Warburg
suggested that glycolysis is the predominant metabolic mechanism
that produces the most ATP in cancer cells, which means that
pyruvate derived from glucose is converted to lactate to exert its
effects instead of being incorporated into the TCA cycle (2), and this is known as ‘the Warburg
effect’. Subsequently, the ‘reverse Warburg effect’ proposed that
tumor-associated fibroblasts can produce large amounts of lactic
acid via aerobic glycolysis, which is provided to adjacent cells in
a paracrine manner, causing the activation of mitochondria,
increasing OXPHOS in adjacent cells and promoting tumor activity
(3). Generally, ‘the Warburg effect’
and ‘reverse Warburg effect’ play an essential role in the
development of cancer.
The incidence and mortality rates associated with
cancer remain high, and despite the vast array of treatments
available, chemotherapy resistance remains a significant challenge
(4). There are several mechanisms of
resistance, such as the mutation on binding sites, the activation
of downstream effectors and the participation of alternative
survival pathways to bypass target inhibition (5). In addition, cancer cells are resistant
to immunotherapy via the Wnt-β-catenin signaling pathway,
mitogen-activated protein kinase signaling pathways, cell cycle
regulation signaling pathways and pathways activated based on the
absence of the tumor suppressor phosphoinositide phosphatase, PTEN
(6). Extensive research has been
performed on glycolysis, which is considered a core process in
tumor biological activity. Currently, increasing evidence suggests
that that increased aerobic glycolysis is closely associated with
chemotherapy resistance, even under O2-rich conditions
(7). For example, lapatinib and
tamoxifen can induce resistance in breast cancer cells by promoting
glycolysis (8–10). However, the clinical application of
glycolysis inhibition through medical approaches is limited. Thus,
it is essential to re-emphasize glycolysis in cancer cells to
overcome therapy resistance. The present review summarizes some of
the most common targets in drug-resistant tumor cells, investigates
their role in the acquisition of drug resistance, and summarizes
corresponding drugs against these targets to suppress chemotherapy
resistance in tumor cells.
Increasing evidence suggests that glycolysis in
cancer is associated with drug resistance (11,12).
Aberrant expression of glycolysis-related enzymes, as regulators of
glycolysis, induces glycolysis dysregulation, which contributes to
tumorigenesis, tumor development and tumor therapy resistance
(13).
HK2 is a critical enzyme that promotes breast cancer
progression and resistance via tumor glycolysis (14). HK2 blocks apoptosis by binding to the
voltage-dependent anion channel (VDAC), which contributes to
chemoresistance (15). Upregulation
of HK2 expression can also induce chemotherapy resistance. Liu
et al (14) demonstrated that
by suppressing the mTOR-S6K signaling pathway, upregulation of HK2
promotes autophagy, subsequently conferring tamoxifen resistance to
MCF-7 breast cancer cells.
In addition to upregulation of HK2 expression, its
phosphorylation on Thr473 can also induce drug resistance. Proviral
insertion in murine lymphomas 2 increases HK2 enzyme activity and
enhances glycolysis by phosphorylating HK2 on Thr473, contributing
to paclitaxel resistance (16).
Conversely, SMI-4a can re-sensitize paclitaxel-resistant cells by
dephosphorylating HK2 on Thr473 (17). In addition, an increase in HK2 dimers
can also promote gemcitabine resistance. Fan et al (18) reported that in pancreatic cancer,
reactive oxygen species (ROS) derived from gemcitabine promote HK2
dimerization and bind to VDAC, which inhibits apoptosis by
suppressing the formation of mitochondrial permeability transition
pores, ultimately resulting in gemcitabine resistance (15,18).
Given the vital role of HK2 in tumor resistance, it
can be used as a valuable target in investigating chemoresistance
inhibition. HK2 inhibitor 3-bromopyruvate facilitates the
dissociation of HK2 from the mitochondrial complex, potentiating
daunorubicin-induced apoptosis and promoting leukemia cell
sensitivity to daunorubicin (19)
(Fig. 1). Furthermore, in ovarian
cancer, the tyrosine analog, NK007, can overcome taxol resistance
by degrading HK2 (20). In breast
cancer, curcumin overcomes resistance to 4-hydroxytamoxifen by
inhibiting snail family transcriptional repressor 2 (SLUG or SNAI
2) and subsequently downregulating HK2 expression (21). In a clinical study, the combination
of docetaxel and curcumin for the treatment of patients with
metastatic castration tolerant prostate cancer resulted in a high
response rate, good tolerance and patient acceptability (22) (Table
I). In another clinical study, lonidamine (LND), which inhibits
aerobic glycolytic activity by influencing HK2 (23), was used with high dose epidoxorubicin
for refractory epithelial ovarian cancer. The results indicated
that this therapeutic strategy had an excellent second-line
therapeutic activity for patients (23). Furthermore, the addition of LND to
the carboplatin/cisplatin-paclitaxel standard regimen for advanced
ovarian cancer was demonstrated to overcome cisplatin resistance in
patients (24).
PGAM1 facilitates the transformation of
3-phosphoglycerate to 2-phosphoglycerate in glycolysis. Its
metabolic activity facilitates cancer metabolism and chemotherapy
resistance (25). Previous studies
have reported that PGAM1 is upregulated in different types of
cancer, such as hepatocellular carcinoma (26), colorectal cancer (27,28) and
lung cancer (29). The allosteric
regulation of PGAM1 is an important mechanism to change the
activity of PGAM1 (25). HKB99, a
novel allosteric inhibitor of PGAM1, overcomes erlotinib resistance
in non-small cell lung cancer (NSCLC) by enhancing oxidative stress
and altering several signaling pathways, including JNK/c-jun
activation, and AKT and ERK inhibition (25) (Fig.
1). However, Chen et al (30) discovered that the protein and mRNA
expression levels of PGAM1 are downregulated in
methotrexate-resistant cells. This phenomenon indicated that
aberrant expression of PGAM1 may be associated with multidrug
resistance (MDR) in breast cancer. Further studies are required to
determine the molecular mechanism underlying drug resistance caused
by PGAM1. In addition, few clinical studies have emphasized on
exploiting the effect of PGAM1 inhibitors on tumor resistance.
PKM2 can translocate to the nucleus where it serves
as a transcription coactivator, and this process can induce
chemotherapy resistance (37). Ge
et al (37) assessed PKM2
expression from nuclear and cytoplasmic extracts in breast cancer.
The results demonstrated that PKM2 protein accumulated in the
nucleus instead of the cytoplasm in response to overexpression of
nicotinamide phosphoribosyl transferase, and induced resistance
against tamoxifen in breast cancer cells. Furthermore, PKM2
translocation to the nucleus also leads to resistance to sorafenib
and enzalutamide in liver and prostate cancers, respectively
(31,34). Polypyrimidine tract-binding protein
(PTBP1) is the primary regulator that affects PKM2 expression, and
it can influence drug resistance in tumor cells by regulating PKM2
expression (35,38). In colon cancer, PTBP1 knockdown
decreases PKM2 expression, inhibits glycolysis and increases cell
sensitivity to vincristine and oxaliplatin (35). In addition, upregulation of PTBP1
expression in pancreatic ductal adenocarcinoma cells induces PKM
alternative splicing, which leads to resistance against gemcitabine
(38).
It has been demonstrated that a series of drugs
combined with chemotherapy drugs can increase the effectiveness of
chemotherapy. Shikonin, an inhibitor of PKM2, combined with
cisplatin exhibits a more significant cytotoxic effect by inducing
necroptosis and ROS production compared with when either one is
used alone (39). In osteosarcoma,
treatment combined with metformin leads to the inhibition of
glucose uptake, lactate production and ATP production by
downregulating PKM2 expression. It can also diminish cisplatin
resistance in osteosarcoma cancer stem cells (36,40)
(Fig. 1). Metformin increases the
antitumor effect of other chemotherapy drugs on osteosarcoma stem
cells, such as adriamycin and 5-fluorouracil (36). Furthermore, high expression of the
ATP binding cassette subfamily B member 1 (ABCB1) gene in patients
with acute lymphoblastic leukemia (ALL) is associated with drug
resistance and affects prognosis (41). In a clinical study, metformin
combined with chemotherapy was particularly effective in patients
with elevated ABCB1 expression (clinicaltrials.gov, NCT03118128) (41). In addition, ROS derived from NADPH
oxidase 4 (NOX4) can suppress the P300/CBP-associated
factor-dependent acetylation and lysosomal degradation of PKM2,
leading to an increase in PKM2 expression and the occurrence of
chemotherapy resistance (42).
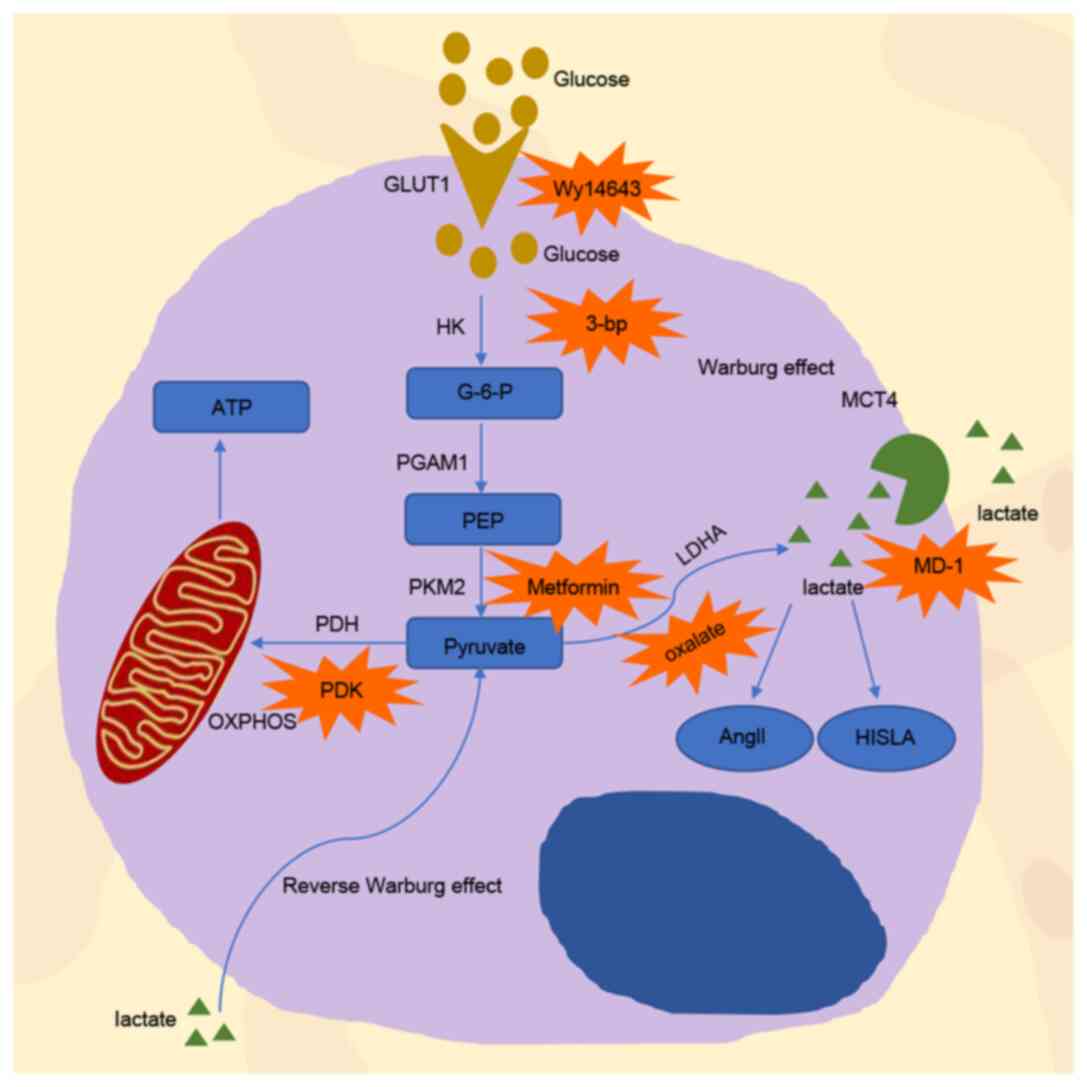
The PDH complex is composed of three enzymes that
serve catalytic functions, named E1, E2 and E3. PDH is an E1 enzyme
that can catalyze pyruvate conversion to acetyl coenzyme A in a
rate-limiting reaction (43).
Pyruvate dehydrogenase kinase (PDK) and pyruvate dehydrogenase
phosphatase mainly regulate PDH activity (43). PDK can inhibit PDH activity by
phosphorylating PDH, whereas pyruvate dehydrogenase phosphatase can
activate PDH by reversing the phosphorylation of this protein
(43) (Fig. 1).
There are four subtypes of PDKs that participate in
glycolysis and exert their effects on chemoresistance in tumor
response, including PDK1-4 (43). In
ovarian cancer cells, overexpression of PDK1 promotes cisplatin
resistance (44). Overexpression of
PDK1 increases epidermal growth factor receptor (EGFR)
phosphorylation and promotes chemotherapy resistance in ovarian
cancer (44). Through the
transcriptional regulation of cyclin and CBS domain divalent metal
cation transporter 3, PDK2 promotes lung adenocarcinoma cell
proliferation and cisplatin resistance (45). Several studies have demonstrated that
hypoxia-inducible factor (HIF)-1α regulates the expression of
pyruvate dehydrogenase kinase 3 (PDK3) and further induces
chemotherapy resistance under hypoxic conditions (43,46).
Nucleus accumbens-1 mediates the inhibition of mitochondrial
function via HIF-1α-mediated PDK3 overexpression, the inhibition of
pyruvate dehydrogenase function and the repression of mitochondrial
respiration (47). This process can
protect cancer cells from apoptosis under hypoxic conditions
(47). Upregulation of PDK4
increases resistance to chemotherapy in hepatocytes and colon
cancer cells (48). In addition,
PDK4 expression increases in tamoxifen-resistant MCF-7 cells,
resulting in augmented PDH activity and resistance to tamoxifen
mediated by the phosphorylation of PDH (49).
Dichloroacetate (DCA) is a small molecule that
promotes the entry of pyruvate into the mitochondria (50). By decreasing the expression of EGFR,
DCA can sensitize MCF7 breast cancer cells to cell death induced by
tamoxifen (51). The primary
molecular mechanism underlying the antitumor effect of DCA involves
the conversion of glycolysis into the oxidative metabolism of
glucose, which decreases lactic acid production, promotes the
production of cytotoxic reactive oxygen intermediates (52) and stimulates the Krebs cycle, and
results in chemoresistance and radiotherapy resistance (53). Currently, several studies have
combined DCA with some chemotherapy drugs and achieved remarkable
results. For example, DCA plus cetuximab notably promotes tumor
regression, whereas the use of either drug alone does not induce
tumor regression (54). In addition,
DCA combined with erlotinib or gefitinib significantly decreases
EGFR activity and decreases resistance against tamoxifen (55). Other antitumor drugs have been
developed based on the role of DCA. Mitaplatin, a synthetic drug
based on cisplatin and DCA, not only destroys nuclear DNA through
the action of cisplatin but also attacks mitochondria based on DCA
in cancer cells. Under the influence of mitaplatin, the
mitochondrial membrane gradient potential is altered in cancer
cells, resulting in the release of cytochrome c, translocation of
apoptosis-inducing factors from the mitochondria to the nucleus,
and apoptosis (56). Due to these
properties, mitaplatin can selectively kill tumor cells that are
cultured with normal fibroblasts and partially overcome the
resistance to cisplatin (56).
2,2-Dichloro-1-(4-isopropoxy-3-nitrophenyl)ethan-1-one(Cpd64) is a
novel PDK1 inhibitor that is more efficient and specific than DCA
and enhances the anticancer effect of EGFR-TKi (57). In a phase III clinical trial,
devimistat (CPI-613), a PDH inhibitor, was combined with large
doses of cytarabine and mitoxantrone to treat refractory acute
myeloid leukemia and this combination achieved more favorable
results (clinicaltrials.gov, NCT03504410)
(58).
Several factors can change the expression of LDHA
and therefore affect drug resistance. When cancer cells are in an
anoxic environment, LDH plays a considerable part in anaerobic
metabolism, which is inseparable from HIF-1α (65). It has been reported that LDH-5, an
isozyme of LDH-1, can be induced by hypoxia, and the transcription
of LDH-5 is directly regulated by HIF1 (65). Through the upregulation of LDHA,
HIF-1α-overexpressing mutants (HIF-1α/∆ODD) are resistant to
G1 phase cell cycle arrest induced by cetuximab, and
acquire cetuximab resistance in head and neck squamous cell
carcinoma cell (66). ATP-binding
cassette, subfamily C, member 3 (ABCC3), a member of the
ATP-binding cassette (ABC) transporter family, is another factor
that can alter LDHA levels (67). In
human urinary bladder cancer (UBC) cells that lack ABCC3, the
blockade of LDHA signaling increases the sensitivity of UBC cells
to cis-diamminedichloroplatinum (67).
Due to the critical role of LDHA in drug resistance,
a combination of the LDHA inhibitor oxalate and paclitaxel can
provide a synergistic inhibitory effect and clinical benefit
against paclitaxel-resistant breast cancer due to the enhancement
of apoptosis (60) (Fig. 1). Recently, galloflavin was
identified as a novel LDH inhibitor that induces human breast
cancer cell death by blocking different glycolytic pathways
(68).
Changes in glucose can significantly affect the rate
of glycolysis, leading to the occurrence of multiple drug
resistance (69). Several studies
have demonstrated that high glucose intake can induce cisplatin
resistance in ovarian and bladder cancer cells, DOX resistance in
breast cancer cells, and gemcitabine resistance in pancreatic
cancer cells (18,70,71).
In terms of the molecular mechanism, when glucose is
severely deficient, glucose regulated protein 78 (GRP78) expression
is induced, leading to etoposide resistance and cisplatin
susceptibility (71,72). GRP78 and B-cell lymphoma 2 (Bcl-2)
competitively associate with Bcl-2 interacting killer (BIK), and
upregulated GRP78 expression decreased the association between BIK
and Bcl-2, subsequently inhibiting apoptosis and promoting drug
resistance in breast cancer cells (73). Lee et al (72) performed a retrospective study and
demonstrated a significant association between GRP78 and recurrence
time in patients, suggesting that GRP78, which can predict
chemotherapy outcomes, deserves further investigation. In a high
glucose microenvironment, glucose promotes growth factor receptor
signaling through the acetylation of acetyl-CoA-dependent Rictor,
which activates rapamycin complex 2 to facilitate resistance to
EGFR-, PI3K- or AKT-targeted therapy in glioblastoma (74). Furthermore, 2-DG combined with
5-fluorouracil can significantly improve its therapeutic effect in
a high glucose microenvironment (75). 18F-fluorodeoxyglucose positron
emission tomography (18F-FDG PET) is a metabolic imaging tool used
to detect lesions with increased glycolysis based on the glucose
analog, fluorine-18 fluorodeoxyglucose (76). 18F-FDG PET can predict overall tumor
behavior and sensitivity to treatment. In a clinical study,
researchers successfully predicted the sensitivity to preoperative
chemotherapy in patients with gastroesophageal cancer (77). Another clinical study demonstrated
that 18F-FDG PET can predict treatment outcomes for 103/108 (95%)
patients following two courses of conventional standard-dose
chemotherapy for advanced Hodgkin's disease (78).
GLUT1 affects tumor cell resistance though several
pathways. Activation of the yes-associated protein 1/TEA domain
transcription factor 1 pathway may result in increased GLUT1
expression, thereby increasing cell viability in cisplatin-treated
cancer cells, decreasing cell death and causing cisplatin
resistance (86). The role of GLUT1
in chemotherapy resistance is associated with HIF-1α. Altered
expression of GLUT-1 induced by HIF-1α is associated with augmented
proliferation, chemotherapy resistance and metastasis (87). Genistein, a natural isoflavone, can
re-sensitize aerobic glycolytic hepatocellular carcinoma cells to
apoptosis by downregulating HIF-1α expression, inactivating GLUT1
and inhibiting aerobic glycolysis, resulting in decreased
resistance to sorafenib (87).
Given the important role of GLUT1 in drug
resistance, researchers have demonstrated several ways to overcome
chemotherapy resistance by inhibiting this protein. In colorectal
cancer cells, Wy14,643, a PPARα agonist, inhibits GLUT1
transcriptional activity, decreases glucose uptake and blocks the
mTOR pathway, which in turn decrease tumor growth and
chemoresistance (88). GLUT1 can
also react with other metabolites to potentiate anti-drug
resistance. AG-PEG-SS-PLA, an aminoglucose (AG)-conjugated,
redox-responsive nanomicelle from a single disulfide bond-bridged
block polymer of polyethylene glycol and polylactic acid, can be
formed by GLUT-1 and glutathione polymerization (89). Paclitaxel-loaded AG-PEG-SS-PLA
nanomicelles activate the caspase-9 and caspase-3 cascade by
upregulating pro-apoptotic proteins, such as Bcl2 associated X and
BH3 interacting domain death agonist, and inhibiting Bcl-2, leading
to apoptosis and improvement in MDR (89).
The Warburg effect implies that the main mechanism
of glucose metabolism in cancer cells is aerobic glycolysis,
whereas mitochondrial OXPHOS is inhibited, which enhances lactic
acid production and consequently promotes the occurrence of drug
resistance (60,90–92).
Apicella et al (12)
demonstrated that MET- or EGFR-addicted cancer cells exhibit
increased glycolysis and lactic acid production following long-term
utilization of tyrosine kinase inhibitors (TKIs). In cancer cells,
lactic acid can promote the production of hepatocyte growth factor
(HGF), in a nuclear factor kb-dependent manner. Overexpression of
HGF upregulates MET expression by promoting signal transduction,
which results in continuous resistance to TKIs. Decreased lactate
production attenuates TKI resistance by inducing alterations to HGF
and MET activity. In cervical cancer, chemotherapy resistance may
be associated with the presence of L- and D-lactic acid in the
cervix (93).
When the production of lactic acid increases, the
acidic microenvironment changes. Cancer cells express several
families of plasma membrane pH regulators to protect them and
maintain normal physiological activities; these families are
co-expressed and redundant on the plasma membrane, including
carbonic anhydrase IX (CAIX), sodium-hydrogen antiporter 1 (NHE1)
and the monocarboxylic transporters (MCT), particularly MCT1 and
MCT4. These families can cause acidic by-products to be leaked from
the cytoplasm, resulting in the dysregulation of pH in the tumor
microenvironment, that is, alkalized intracellular fluid and
acidified extracellular fluid (94).
It has been reported that this form of metabolism can enhance
resistance to radiation and chemotherapy (94–102).
The monocarboxylic transporters, MCT1 and MCT4, are
mainly involved in the transport of lactic acid (Fig. 1). MCT1 is the most common
monocarboxylic transporter expressed in p53-deficient tumors
(103), whereas MCT4 expression is
upregulated under hypoxic conditions and elevates with HIF
induction (104). MCT1 is
responsible for lactic acid uptake via oxidative cells, and MCT4 is
responsible for the release of lactic acid from hypoxic cells
(105). Abnormal expression of the
MCT family is associated with drug resistance. Apicella et
al (12) demonstrated that
intratumoral lactic acid increases caused by high MCT1 expression
are associated with poor prognosis. In addition, MCT1 is a major
transporter that assists 3-bromopyruvate (3-BrPA) (106). Overexpression of MCT1 in cancer
cells can sensitize tumor xenografts in response to 3-BrPA
treatment in vivo (106).
Conversely, downregulation of MCT4 can overcome anti-angiogenic
therapy resistance (107). Based on
the effects of MCT1 and MCT4, it is reasonable that the
monocarboxylate transporter MCT1 and MCT4 dual inhibitor MD-1 can
significantly inhibit oral squamous cell carcinoma, an invasive and
therapeutic-resistant malignancy (108) (Fig.
1).
Carbonic anhydrase IX (CAIX or CA9) is a
tumor-related metalloenzyme that can convert H2O and
CO2 to HCO3− and H+
ions reversibly, and is also induced by HIF (109). It has been reported that transient
and long-term exposure to an extracellular acidic microenvironment
(pH 6.7±0.1) increases CAIX expression in melanoma, breast cancer
and colorectal cancer cells (109).
Extracellular acidosis can cause chemotherapy resistance, which
indicates that there may be an association between CAIX and
chemotherapy resistance (110,111).
Based on carbonic anhydrase CAIX staining in 188 microarray tumors,
it was demonstrated that this protein is upregulated in basal
cell-like breast tumors and is associated with chemotherapy
resistance (110). Simultaneously,
overexpression of CAIX in tongue cancer cells can promote
chemotherapy resistance (111).
SLC-0111, which inhibits CAIX, enhances the toxic effect of
temozolomide and dacarbazine, and is currently being used for the
treatment of advanced melanoma. SLC-0111 also increases the
response of breast cancer cells to DOX and enhances the inhibitory
effects of 5-fluorouracil on colon cancer (109).
NHE-1 is a plasma membrane glycoprotein composed of
815 amino acids, and it is a member of the elevated Na/H exchanger
gene family, and is also known as a PH regulator. Abnormal NHE-1
expression has been associated with drug resistance (112,113).
For example, increased NHE1 expression can promote T cell-ALL
resistance to DOX (113). In
addition, cariporide, a NHE1 inhibitor, significantly increases
breast cancer cell sensitivity to adriamycin by inducing apoptosis,
promoting intracellular DOX accumulation and blocking the
G0/G1 phase (114). Recently, cariporide, zoniporide and
eniporide, which are effective and selective NHE-1 inhibitors, were
demonstrated to be well tolerated in humans. However, only a few
clinical trials have been performed in the field of oncology
(115).
Due to the high rate of glycolysis, glucose levels
within the tumor are deficient, and additional energy is in demand
for normal physiological cancer cell activity (116); thus, other sources of energy are
required. Excess pyruvate and lactate produced by glycolysis in
cancer-associated fibroblasts can be delivered to adjacent cancer
cells to enhance the mitochondrial activity, resulting in cancer
cell resistance to many clinically used drugs, such as tamoxifen,
used in endocrine therapy, Herceptin, used in Her-2-targeted
therapy, and ebithromycin, used in chemotherapy (117). In breast cancer cells, this process
can assist cancer cell survival in the presence of a lack of
glucose for a long time, leading to PI3K/mTOR inhibitor resistance
(116).
Recently, immunotherapy has been considered a
milestone for cancer therapy. Programmed cell death 1 (PD1), PD1
ligand 1 (PD-L1), and checkpoint molecules, such as cytotoxic T
lymphocyte antigen 4, have been identified, and drugs targeting
them have been used on patients (118). Cancer immunotherapy has achieved
significant breakthroughs and success. However, drug resistance has
deterred clinical progression for several years. With a deeper
understanding of immune mechanisms and immunotherapy efficacy,
previous studies have acknowledged that tumors are resistant to
immunotherapy via interferon (IFN) signaling and antigen
presentation, the PI3K-AKT-mTOR axis, Wnt-β-catenin signaling, and
deletion of the tumor suppressor phosphoinositide phosphatase,
PTEN, which can activate different pathways (118–121).
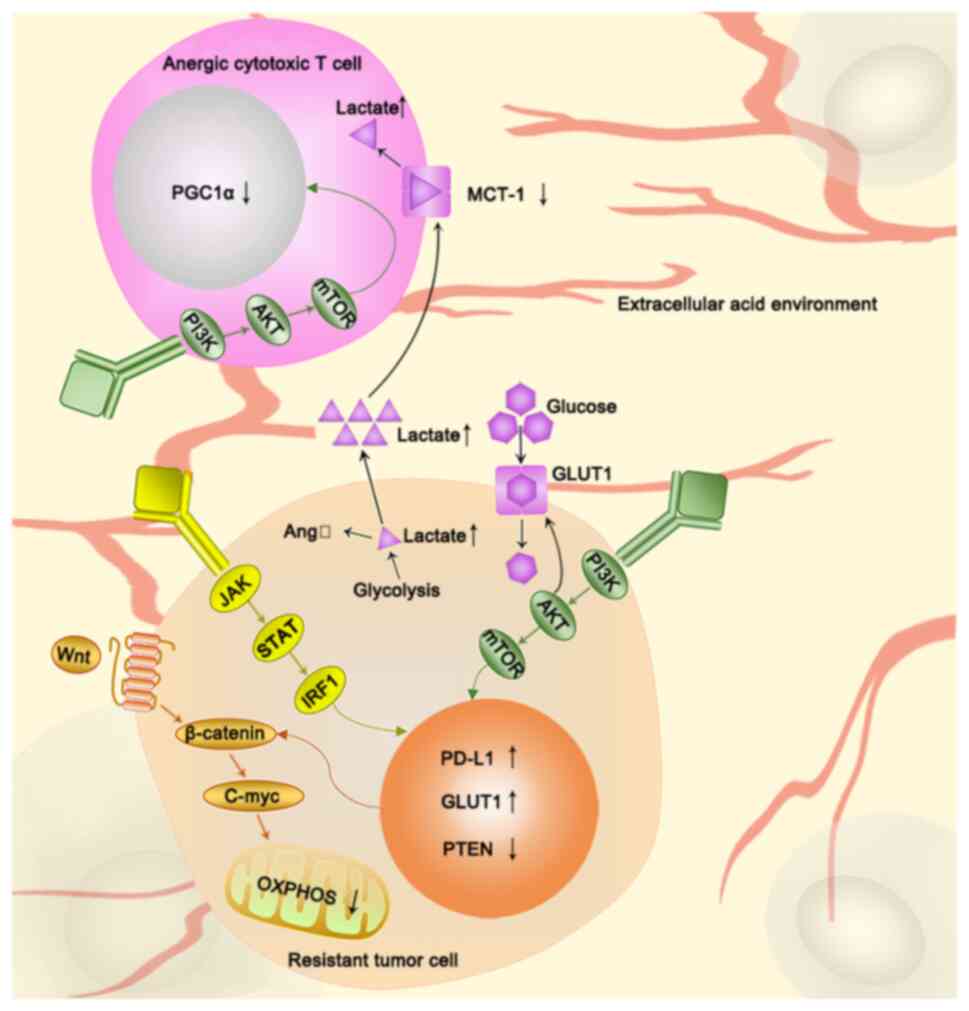
Interferon binds IRF1 and the PD-L1 promoter via the
Janus kinase 1 (JAK1)/JAK2-signal transducer and activators of
transcription 1 (STAT1)/STAT2/STAT3-interferon regulatory factor 1
(IRF1) axis, thereby regulating the expression of PD-L1 and causing
resistance to immune checkpoint inhibitors (119). The loss of copies of IFN-γ-mediated
genes, such as IFNGR1, IRF-1, JAK2 and IFNGR2 can
cause metastatic melanoma resistance to ipilimumab (anti-CTLA-4
therapy) (122). The loss of
JAK1 and JAK2, IFN-γ pathway genes, is associated
with resistance to PD-1 therapy (122). Notably, the IFN signaling pathway
can also alter glycolysis to cause chemotherapy resistance.
Sustained STAT1 signaling causes chemotherapy resistance by
increasing the expression of genes associated with glycolysis and
OXPHOS (123) (Table II). STAT3 induces chemotherapy
resistance by protecting mitochondrial oxidative phosphorylation
and controlling the opening of mitochondrial permeability
transition pores (124). In
addition, IFN-γ signaling is downregulated in highly glycolytic
tumor cells, and the disruption of tumor IFN-γ signaling is an
essential cause of tumor immunotherapy resistance (125). In clinical studies, the chimeric
anti-CD20 antibody rituximab and IFN-alpha 2a combined
immunotherapy has been proven effective (126,127).
In another clinical study, the combination of interleukin-2 and
α-IFN, based on a dose-increasing experiment, resulted in an
increased response rate (128).
These results indicate that the IFN signaling pathway is a good
potential target for combination therapy (Fig. 2).
The metabolic reprogramming of tumor cells consumes
a lot of energy and nutrients, which means that immune cells are in
a state of nutrient depletion. The immune response to the tumor
also has a significant negative effect (129). There is considerable evidence that
the Wnt-β-catenin signaling pathway connects glycolysis to the
immune response to the tumor. First, Wnt-β-catenin signaling is
closely associated with glycolysis (130). The Wnt-β-catenin target gene,
c-Myc, regulates and controls cancer cell metabolism (130). Wnt5B, a Wnt ligand, has been
demonstrated to inhibit mitochondrial functions in triple negative
breast cancer cells through c-Myc (131). Secondly, Wnt-β-catenin signaling is
also associated with immunity. The Wnt ligand increases the
expression of β-catenin in dendritic cells, and subsequently, the
functions of Tregs and CD8+ T cells are activated, and
antitumor immunity is suppressed (120). Thus, targeting the Wnt-β-catenin
signaling pathway has become a strategy that can both target
glycolysis and decrease immune resistance. Upon Wnt inhibition, the
Wnt-β-catenin-target gene, MCT1, is also downregulated,
leading to a reduction in tumor microenvironment acidity,
maintaining antitumor immunity, and preventing cell migration and
metastasis (132). Accordingly,
there is evidence that a combination of PD-1 and Wnt inhibitors can
increase PD-1 inhibitor efficacy (133) (Fig.
2).
Overactivation of the pathological PI3K-AKT-mTOR
pathway is the primary mechanism underlying immune checkpoint
inhibitor resistance (134). AKT
plays a vital role in the tumor microenvironment, whereby it
decreases the expression of peroxisome proliferator-activated
receptor gamma coactivator 1-α (PGC1α) in tumor-infiltrating
lymphocytes (TILs) within the tumor microenvironment. PGC1α can
also regulate the biological function of mitochondria. Thus, a lack
of this protein can cause TIL energy exhaustion and decrease
antitumor immunity in the tumor microenvironment (135). Based on in vitro
experiments, the addition of PGC1 inhibits tumor growth and
increases overall survival (135)
(Fig. 2).
AKT can promote glucose uptake by increasing the
membrane localization of facilitative GLUT1 and GLUT4. AKT also
contributes to the phosphorylation of HK-2 and stimulates its
translocation to the mitochondria (135). All of these changes can affect
glycolysis and induce chemotherapy resistance (135). Accordingly, PI3K-AKT inhibitors
have become a focus of research because of their dual
anti-chemotherapy and immunotherapy resistance effects (136). In phase I clinical studies, the AKT
inhibitor afuresertib combined with carboplatin and paclitaxel
exhibited promising results for the treatment of recurrent
platinum-resistant ovarian cancer (137). However, clinical trials of AKT
inhibitors for the immunotherapy-resistant disease remain to be
performed (Fig. 2).
Glycolysis is also associated with immunotherapy
resistance through PTEN-deficiency. The abnormal accumulation of
β-catenin, caused by tumor-specific mutations, such as the specific
loss of PTEN from melanocytes in lung cancer, leads to activation
of the Wnt-β-catenin signaling pathway (138) (Fig.
2). PTEN-deficiency-associated and activated pathways can cause
immune resistance. PTEN deficiency facilitates the immune escape of
melanoma by restricting T cell access to tumor cells and preventing
T cells from killing cancer cells (121). PI3Kβ inhibitors can effectively
reverse the immunotherapy resistance caused by the loss of PTEN
(121). In PTEN-deficient melanoma
cells, some substrates upregulation of glycolysis, such as pyruvate
and lactate, provoke overactivation, which is characterized by
adoptive T cell therapy resistance (125). A clinical study has demonstrated
that in PTEN-deficient tumors, following treatment of metastatic
castration-resistant prostate cancer with the Akt inhibitor
ipatasertib coupled with the CYP17 inhibitor abiraterone,
radiographic progression-free survival was extended compared with
tumors without PTEN deficiency (139). This study further demonstrated that
AKT inhibitors combined with antitumor drugs can play an important
role in treating PTEN-deficient tumors.
Lactic acid levels are also associated with immune
resistance. Lactic acid can be derived from tumor-associated
glycolysis and from activated immune cells and macrophages
(140). Lactate release in tumor
cells also increases the expression of a myeloid-specific lncRNA,
HIF-1α-stabilizing long non-coding RNA (HISLA) in macrophages, and
elevated HISLA expression promotes aerobic glycolysis in
tumor-associated macrophages through extracellular vesicles (EV)
transport, which forms a pre-feedback loop (141) (Fig.
1). Blocking EV-mediated HISLA in vivo has been
demonstrated to inhibit glycolysis and drug resistance in breast
cancer (141). Furthermore, the
excessive accumulation of lactic acid can cause immunosuppression,
leading to resistance to immunotherapy. First, hypoxic tumor cells
produce angiotensin II (AngII) through an anoxia-lactic
acid-chymase-dependent mechanism (142). In the tumor microenvironment, local
AngII is associated with cancer cell evasion of immune surveillance
(143). In addition, the inhibition
of AngII signaling may enhance tumor sensitivity to checkpoint
immunotherapy (143) (Fig. 2). Secondly, lactic acid can impair
the cytotoxic function of T cells. The activation of T cells uses
glycolysis and relies on lactic acid secretion. The accumulation of
intracellular lactic acid in the tumor cell causes an extracellular
acidic environment, leading to the inhibition of MCT-1 (144). T cells cannot effectively secret
lactate, and under these conditions, their metabolism is
disordered, contributing to a significant reduction in cytotoxic
activity, which severely affects T cell functionality (144) (Fig.
2). In addition, lactic acid can increase
L-arginine-metabolizing enzyme arginase-1 (ARG1) expression in
macrophages and inhibit the antitumor immune response (145). Following treatment with DCA, the
declined ARG1 mRNA expression can effectively reactivate the
immune state regulated by lactic acid and improve antitumor
immunotherapy benefits (145).
The Warburg effect indicates that tumor cells tend
to undergo glycolysis regardless of aerobic and anaerobic
conditions, which implies that mitochondrial dysfunction is a
feature of tumor cells (146).
However, recently, this statement has been challenged (147). Several tumor cells have been
reported to have metabolic plasticity, indicating a transformation
from glycolysis to mitochondrial OXPHOS, leading to the production
of vast amounts of energy and resistance to drugs (148). Recent evidence suggests that cancer
cells can obtain glycolysis/OXPHOS mixed phenotypes, in which the
ATP production is an outcome from both glycolysis and OXPHOS to
support physiological activity of cells (149). In addition, cells with this
characteristic are more likely to acquire drug resistance (150). When lactic acid increases, it can
be used as an energy source by adjacent cancer cells to activate
mitochondria and stimulate OXPHOS (151) (Fig.
1).
Such changes in OXPHOS can affect the drug
resistance of tumor cells. Following chemotherapy, Farge et
al (150) described a new
method to identify and study acute myeloid leukemia (AML) cells
remaining in the bone marrow. The results demonstrated that OXPHOS
is increased in AML cells remaining in the bone marrow of mice
following cytarabine therapy, and that the inhibition of OXPHOS can
re-sensitize AML cells to cytarabine. In epithelial ovarian cancer,
the oxygen consumption rate, mitochondrial respiration and
oxidative phosphorylation in cisplatin-resistant cells were higher
than those in cisplatin-sensitive cells (152). The activation of OXPHOS is a
typical feature of hepatocellular carcinoma cell resistance to DOX
(153). Generally, increased OXPHOS
can promote the occurrence of chemotherapy resistance (150).
OXPHOS affects the treatment of tumors in several
ways. As it results in the production of a large amount of ATP,
this is bound to stimulate the activity of some transporters, one
of which is drug transporters. In breast cancer cells, the
continuous supply of ATP derived from OXPHOS is utilized by ABC
transporters, leading to the outflow of DOX and the induction of an
MDR phenotype (148). Tumor stem
cells are also associated with drug resistance caused by OXPHOS.
Increased OXPHOS mediated by mitochondria can stimulate tumor stem
cells to expand, conferring resistance to tumor cells (154). Furthermore, NANOG, a stem cell
marker, inhibits mitochondrial OXPHOS genes and promotes sorafenib
resistance (155).
Several drugs inhibit the occurrence and
development of tumors, and they can also affect OXPHOS. Some drugs,
like cytarabine, 5-fluorouracil, TKIs, MAPKi and BRAFi can promote
OXPHOS activity and increase drug resistance in the mitochondria,
whereas others, such as anthracyclines, etoposide, sorafenib,
paclitaxel and staurosporine, significantly decrease OXPHOS
activity in the mitochondria (147). For those drugs that promote OXPHOS
activity in mitochondria, it is necessary to find a better way to
solve the associated drug resistance. Metformin is a type of
mitochondrial inhibitor and combining it with cisplatin can
attenuate cisplatin resistance in epithelial ovarian cancer cells
(152). Metformin can also be
combined with TKIs to overcome tumor chemotherapy resistance
(147). In addition, targeting
mitochondrial respiration and HIF-1α may reverse tumor cell
chemotherapeutic resistance (154).
When the anoxic environment is destroyed and the HIF1α pathway is
blocked, sex-determining region Y (SRY)-Box2 drives OXPHOS
reprogramming, which helps tumor cells obtain an invasive oxidative
tumor phenotype and enhance drug resistance and metastatic ability
(156).
Changes in glycolysis, particularly increases in
key enzymes and intermediates of this pathway, can affect the
sensitivity of tumors to chemotherapeutic reagents, resulting in an
increase in ATP production, and providing sufficient energy for the
biological activity of tumor cells (14,69).
This process enhances the repair of DNA damage, increases the
phosphorylation, translocation into the nucleus, and
autophagy-associated activity of enzymes, and causes drug
resistance (157). In addition,
several clinical trials have been performed to investigate the
therapeutic effect of glycolysis-targeting therapy combined with
clinical first-and second-line chemotherapeutic drugs on tumors
(Table I).
Recently, several studies have demonstrated that
the role of mitochondria in tumor metabolism is becoming essential.
The reverse Warburg effect emerged, indicating that the increase in
lactic acid as an energy material can be converted into pyruvate in
the mitochondria and enhance mitochondrial activity and OXPHOS in
adjacent cells (117). In addition,
tumor cells also have metabolic plasticity. Glycolysis can be
moderately transformed into OXPHOS when the external environment
changes or there is plenty of oxygen around the tumor cells. This
transformation is closely associated with chemotherapy (118,148).
Increased mitochondrial activity and OXPHOS results in the
production of higher levels of ATP and NADPH. High levels of ATP
provide a vast amount of energy to tumor cells, and NADPH is a key
antioxidant that can decrease ROS damage to tumor cells (158).
With an increase in studies on tumor immunity,
immune checkpoint inhibitors have been developed for clinical
applications (149). However,
immunotherapy resistance is remains a major challenge. Based on a
broadened understanding of tumor immunity, it is apparent that
immunotherapy resistance is closely associated with glucose
metabolism (125). The
PI3K-AKT-mTOR axis, PTEN deficiency, IFN signaling and the
Wnt-β-catenin signaling pathway can affect corresponding enzymes
involved in glycolysis and enhance immunotherapy resistance
(123,125,131,135).
Conversely, the release of lactic acid, a glycolysis intermediate
metabolite, can promote the occurrence of immunotherapy resistance.
The interaction between glucose metabolism and immunotherapy
resistance forms a positive feedback pathway and constitutes an
important factor in tumor drug resistance (141). Currently, there are a few studies
on the effect of the PI3K-AKT-mTOR axis, PTEN deficiency, IFN
signaling, Wnt-β-catenin signaling pathway targeting agents on
immunotherapy drug resistance, and chemotherapy resistance. The
interdisciplinary study on tumor glycolysis and immunotherapy
resistance should endeavor to receive more attention to proceed to
understand the molecular mechanisms involved.
Regarding these glucose metabolic processes,
targeted inhibitors may be used in combination with chemotherapy
reagents or immune checkpoint inhibitors in the future. However,
due to the lack of tumor drug resistance markers, targeted
inhibitor prognostic indexes, and the specificity of these
inhibitors, their clinical application is profoundly limited. In
general, altered glycolysis, as a ubiquitous feature of
drug-resistant tumor cells, represents a promising target, and
novel strategy to overcome drug resistance clinically.
Not applicable.
The present review was supported by grants from the
National Natural Science Foundation of China (grant nos. 81672612
and 81572607) and the Project of Invigorating Health Care through
Science, Technology and Education (The Project of Jiangsu
Provincial Medical Youth Talent; grant no. QNRC2016095).
Not applicable.
JP and YC designed the present review and drafted
the initial manuscript. XW and YH analyzed the data. SX, WZ and SW
revised the review for important intellectual content. ZF reviewed
and critiqued the review following revisions. HX designed the
present review, edited, reviewed and critiqued the manuscript. All
authors have read and approved the final manuscript.
Not applicable.
Not applicable.
The authors declare that they have no competing
interests.
|
1
|
Zaal EA and Berkers CR: The influence of
metabolism on drug response in cancer. Front Oncol. 8:5002018.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Warburg O, Wind F and Negelein E: The
metabolism of tumors in the body. J Gen Physiol. 8:519–530. 1927.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Bonuccelli G, Whitaker-Menezes D,
Castello-Cros R, Pavlides S, Pestell RG, Fatatis A, Witkiewicz AK,
Vander Heiden MG, Migneco G, Chiavarina B, et al: The reverse
Warburg effect: Glycolysis inhibitors prevent the tumor promoting
effects of caveolin-1 deficient cancer associated fibroblasts. Cell
Cycle. 9:1960–1971. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Holohan C, Van Schaeybroeck S, Longley DB
and Johnston PG: Cancer drug resistance: An evolving paradigm. Nat
Rev Cancer. 13:714–726. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Boumahdi S and de Sauvage FJ: The great
escape: Tumour cell plasticity in resistance to targeted therapy.
Nat Rev Drug Discov. 19:39–56. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Gershon O, Ezenwa NE and Osabohien R:
Implications of oil price shocks on net oil-importing African
countries. Heliyon. 5:e022082019. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Woo YM, Shin Y, Lee EJ, Lee S, Jeong SH,
Kong HK, Park EY, Kim HK, Han J, Chang M and Park JH: Inhibition of
aerobic glycolysis represses Akt/mTOR/HIF-1α axis and restores
tamoxifen sensitivity in antiestrogen-resistant breast cancer
cells. PLoS One. 10:e01322852015. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Komurov K, Tseng JT, Muller M, Seviour EG,
Moss TJ, Yang L, Nagrath D and Ram PT: The glucose-deprivation
network counteracts lapatinib-induced toxicity in resistant
ErbB2-positive breast cancer cells. Mol Syst Biol. 8:5962012.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Ruprecht B, Zaal EA, Zecha J, Wu W,
Berkers CR, Kuster B and Lemeer S: Lapatinib resistance in breast
cancer cells is accompanied by phosphorylation-mediated
reprogramming of glycolysis. Cancer Res. 77:1842–1853. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
He M, Jin Q, Chen C, Liu Y, Ye X, Jiang Y,
Ji F, Qian H, Gan D, Yue S, et al: The miR-186-3p/EREG axis
orchestrates tamoxifen resistance and aerobic glycolysis in breast
cancer cells. Oncogene. 38:5551–5565. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Icard P, Shulman S, Farhat D, Steyaert JM,
Alifano M and Lincet H: How the Warburg effect supports
aggressiveness and drug resistance of cancer cells? Drug Resist
Updat. 38:1–11. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Apicella M, Giannoni E, Fiore S, Ferrari
KJ, Fernández-Pérez D, Isella C, Granchi C, Minutolo F, Sottile A,
Comoglio PM, et al: Increased lactate secretion by cancer cells
sustains non-cell-autonomous adaptive resistance to MET and EGFR
targeted therapies. Cell Metab. 28:848–865 e6. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Varghese E, Samuel SM, Liskova A, Samec M,
Kubatka P and Busselberg D: Targeting glucose metabolism to
overcome resistance to anticancer chemotherapy in breast cancer.
Cancers (Basel). 12:22522020. View Article : Google Scholar
|
|
14
|
Liu X, Miao W, Huang M, Li L, Dai X and
Wang Y: Elevated hexokinase II expression confers acquired
resistance to 4-hydroxytamoxifen in breast cancer cells. Mol Cell
Proteomics. 18:2273–2284. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Krasnov GS, Dmitriev AA, Lakunina VA,
Kirpiy AA and Kudryavtseva AV: Targeting VDAC-bound hexokinase II:
A promising approach for concomitant anti-cancer therapy. Expert
Opin Ther Targets. 17:1221–1233. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Yang T, Ren C, Qiao P, Han X, Wang L, Lv
S, Sun Y, Liu Z, Du Y and Yu Z: Correction: PIM2-mediated
phosphorylation of hexokinase 2 is critical for tumor growth and
paclitaxel resistance in breast cancer. Oncogene. 39:720–721. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Yang T, Ren C, Qiao P, Han X, Wang L, Lv
S, Sun Y, Liu Z, Du Y and Yu Z: PIM2-mediated phosphorylation of
hexokinase 2 is critical for tumor growth and paclitaxel resistance
in breast cancer. Oncogene. 37:5997–6009. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Fan K, Fan Z, Cheng H, Huang Q, Yang C,
Jin K, Luo G, Yu X and Liu C: Hexokinase 2 dimerization and
interaction with voltage-dependent anion channel promoted
resistance to cell apoptosis induced by gemcitabine in pancreatic
cancer. Cancer Med. 8:5903–5915. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Rai Y, Yadav P, Kumari N, Kalra N and
Bhatt AN: Hexokinase II inhibition by 3-bromopyruvate sensitizes
myeloid leukemic cells K-562 to anti-leukemic drug, daunorubicin.
Biosci Rep. 39:BSR201908802019. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Li Z, Tang X, Luo Y, Chen B, Zhou C, Wu X,
Tang Z, Qi X, Cao G, Hao J, et al: NK007 helps in mitigating
paclitaxel resistance through p38MAPK activation and HK2
degradation in ovarian cancer. J Cell Physiol. Feb 20–2019.(Epub
ahead of print).
|
|
21
|
Geng C, Li J, Ding F, Wu G, Yang Q, Sun Y,
Zhang Z, Dong T and Tian X: Curcumin suppresses 4-hydroxytamoxifen
resistance in breast cancer cells by targeting SLUG/Hexokinase 2
pathway. Biochem Biophys Res Commun. 473:147–153. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Mahammedi H, Planchat E, Pouget M, Durando
X, Curé H, Guy L, Van-Praagh I, Savareux L, Atger M, Bayet-Robert
M, et al: The new combination docetaxel, prednisone and curcumin in
patients with castration-resistant prostate cancer: A pilot phase
II study. Oncology. 90:69–78. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Gadducci A, Brunetti I, Muttini MP,
Fanucchi A, Dargenio F, Giannessi PG and Conte PF: Epidoxorubicin
and lonidamine in refractory or recurrent epithelial ovarian
cancer. Eur J Cancer. 30A:1432–1435. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
De Lena M, Lorusso V, Latorre A, Fanizza
G, Gargano G, Caporusso L, Guida M, Catino A, Crucitta E, Sambiasi
D and Mazzei A: Paclitaxel, cisplatin and lonidamine in advanced
ovarian cancer. A phase II study. Eur J Cancer. 37:364–368. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Huang K, Liang Q, Zhou Y, Jiang LL, Gu WM,
Luo MY, Tang YB, Wang Y, Lu W, Huang M, et al: A novel allosteric
inhibitor of phosphoglycerate mutase 1 suppresses growth and
metastasis of non-small-cell lung cancer. Cell Metab.
30:1107–1119.e8. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Ren F, Wu H, Lei Y, Zhang H, Liu R, Zhao
Y, Chen X, Zeng D, Tong A, Chen L, et al: Quantitative proteomics
identification of phosphoglycerate mutase 1 as a novel therapeutic
target in hepatocellular carcinoma. Mol Cancer. 9:812010.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Usuba T, Ishibashi Y, Okawa Y, Hirakawa T,
Takada K and Ohkawa K: Purification and identification of
monoubiquitin-phosphoglycerate mutase B complex from human
colorectal cancer tissues. Int J Cancer. 94:662–668. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Liu L, Wang S, Zhang Q and Ding Y:
Identification of potential genes/proteins regulated by Tiam1 in
colorectal cancer by microarray analysis and proteome analysis.
Cell Biol Int. 32:1215–1222. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Chen G, Gharib TG, Wang H, Huang CC, Kuick
R, Thomas DG, Shedden KA, Misek DE, Taylor JM, Giordano TJ, et al:
Protein profiles associated with survival in lung adenocarcinoma.
Proc Natl Acad Sci USA. 100:13537–13542. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Chen SY, Cai JX, Zhang WP, Zhang XW, Hu
SS, Lu J, Xing JF and Dong YL: Proteomic identification of
differentially expressed proteins associated with the multiple drug
resistance in methotrexate-resistant human breast cancer cells. Int
J Oncol. 45:448–458. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Wang HJ, Pochampalli M, Wang LY, Zou JX,
Li PS, Hsu SC, Wang BJ, Huang SH, Yang P, Yang JC, et al:
KDM8/JMJD5 as a dual coactivator of AR and PKM2 integrates AR/EZH2
network and tumor metabolism in CRPC. Oncogene. 38:17–32. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Qian Y, Bi L, Yang Y and Wang D: Effect of
pyruvate kinase M2-regulating aerobic glycolysis on chemotherapy
resistance of estrogen receptor-positive breast cancer. Anticancer
Drugs. 29:616–627. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Tian S, Li P, Sheng S and Jin X:
Upregulation of pyruvate kinase M2 expression by fatty acid
synthase contributes to gemcitabine resistance in pancreatic
cancer. Oncol Lett. 15:2211–2217. 2018.PubMed/NCBI
|
|
34
|
Wong TL, Ng KY, Tan KV, Chan LH, Zhou L,
Che N, Hoo RLC, Lee TK, Richard S, Lo CM, et al: CRAF methylation
by PRMT6 regulates aerobic glycolysis driven hepatocarcinogenesis
via ERK-dependent PKM2 nuclear relocalization and activation.
Hepatology. 71:1279–1296. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Cheng C, Xie Z, Li Y, Wang J, Qin C and
Zhang Y: PTBP1 knockdown overcomes the resistance to vincristine
and oxaliplatin in drug-resistant colon cancer cells through
regulation of glycolysis. Biomed Pharmacother. 108:194–200. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Shang D, Wu J, Guo L, Xu Y, Liu L and Lu
J: Metformin increases sensitivity of osteosarcoma stem cells to
cisplatin by inhibiting expression of PKM2. Int J Oncol.
50:1848–1856. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Ge X, Zhao Y, Dong L, Seng J, Zhang X and
Dou D: NAMPT regulates PKM2 nuclear location through 14-3-3 ζ:
Conferring resistance to tamoxifen in breast cancer. J Cell
Physiol. 234:23409–23420. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Calabretta S, Bielli P, Passacantilli I,
Pilozzi E, Fendrich V, Capurso G, Fave GD and Sette C: Modulation
of PKM alternative splicing by PTBP1 promotes gemcitabine
resistance in pancreatic cancer cells. Oncogene. 35:2031–2039.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Wang X, Zhang F and Wu XR: Inhibition of
pyruvate kinase M2 markedly reduces chemoresistance of advanced
bladder cancer to cisplatin. Sci Rep. 7:459832017. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Gatti M, Solari A, Pattarozzi A,
Campanella C, Thellung S, Maniscalco L, De Maria R, Würth R,
Corsaro A, Bajetto A, et al: In vitro and in vivo characterization
of stem-like cells from canine osteosarcoma and assessment of drug
sensitivity. Exp Cell Res. 363:48–64. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Ramos-Penafiel C, Olarte-Carrillo I,
Ceron-Maldonado R, Rozen-Fuller E, Kassack-Ipiña JJ, Meléndez-Mier
G, Collazo-Jaloma J and Martínez-Tovar A: Effect of metformin on
the survival of patients with ALL who express high levels of the
ABCB1 drug resistance gene. J Transl Med. 16:2452018. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Shanmugasundaram K, Nayak BK, Friedrichs
WE, Kaushik D, Rodriguez R and Block K: NOX4 functions as a
mitochondrial energetic sensor coupling cancer metabolic
reprogramming to drug resistance. Nat Commun. 8:9972017. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Lu CW, Lin SC, Chen KF, Lai YY and Tsai
SJ: Induction of pyruvate dehydrogenase kinase-3 by
hypoxia-inducible factor-1 promotes metabolic switch and drug
resistance. J Biol Chem. 283:28106–28114. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Zhang M, Cong Q, Zhang XY, Zhang MX, Lu YY
and Xu CJ: Pyruvate dehydrogenase kinase 1 contributes to cisplatin
resistance of ovarian cancer through EGFR activation. J Cell
Physiol. 234:6361–6370. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Hu T, Yu S, Li Y, Ren H, Ning Q, Wang J,
Liang X and Li M: PDK2 induces cisplatin-resistance in lung
adenocarcinoma via transcriptional regulation of CNNM3. J Drug
Target. 27:460–465. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Lu CW, Lin SC, Chien CW, Lin SC, Lee CT,
Lin BW, Lee JC and Tsai SJ: Overexpression of pyruvate
dehydrogenase kinase 3 increases drug resistance and early
recurrence in colon cancer. Am J Pathol. 179:1405–1414. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Ren YJ, Wang XH, Ji C, Guan YD, Lu XJ, Liu
XR, Zhang HH, Guo LC, Xu QH, Zhu WD, et al: Silencing of NAC1
expression induces cancer cells oxidative stress in hypoxia and
potentiates the therapeutic activity of elesclomol. Front
Pharmacol. 8:8042017. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Strowitzki MJ, Radhakrishnan P, Pavicevic
S, Scheer J, Kimmer G, Ritter AS, Tuffs C, Volz C, Vondran F,
Harnoss JM, et al: High hepatic expression of PDK4 improves
survival upon multimodal treatment of colorectal liver metastases.
Br J Cancer. 120:675–688. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Walter W, Thomalla J, Bruhn J, Fagan DH,
Zehowski C, Yee D and Skildum A: Altered regulation of PDK4
expression promotes antiestrogen resistance in human breast cancer
cells. Springerplus. 4:6892015. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Michelakis ED, Webster L and Mackey JR:
Dichloroacetate (DCA) as a potential metabolic-targeting therapy
for cancer. Br J Cancer. 99:989–994. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Woo SH, Seo SK, Park Y, Kim EK, Seong MK,
Kim HA, Song JY, Hwang SG, Lee JK, Noh WC and Park IC:
Dichloroacetate potentiates tamoxifen-induced cell death in breast
cancer cells via downregulation of the epidermal growth factor
receptor. Oncotarget. 7:59809–59819. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Xuan Y, Hur H, Ham IH, Yun J, Lee JY, Shim
W, Kim YB, Lee G, Han SU and Cho YK: Dichloroacetate attenuates
hypoxia-induced resistance to 5-fluorouracil in gastric cancer
through the regulation of glucose metabolism. Exp Cell Res.
321:219–230. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Clayton: Case of ulceration of the larynx.
Med Phys J. 32:25–26. 1814.PubMed/NCBI
|
|
54
|
Lu H, Lu Y, Xie Y, Qiu S, Li X and Fan Z:
Rational combination with PDK1 inhibition overcomes cetuximab
resistance in head and neck squamous cell carcinoma. JCI Insight.
4:e1311062019. View Article : Google Scholar
|
|
55
|
Yang Z and Tam KY: Anti-cancer synergy of
dichloroacetate and EGFR tyrosine kinase inhibitors in NSCLC cell
lines. Eur J Pharmacol. 789:458–467. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Xue X, You S, Zhang Q, Wu Y, Zou GZ, Wang
PC, Zhao YL, Xu Y, Jia L, Zhang X and Liang XJ: Mitaplatin
increases sensitivity of tumor cells to cisplatin by inducing
mitochondrial dysfunction. Mol Pharm. 9:634–644. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Yang Z, Zhang SL, Hu X and Tam KY:
Inhibition of pyruvate dehydrogenase kinase 1 enhances the
anti-cancer effect of EGFR tyrosine kinase inhibitors in non-small
cell lung cancer. Eur J Pharmacol. 838:41–52. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Pardee TS, Luther S, Buyse M, Powell BL
and Cortes J: Devimistat in combination with high dose cytarabine
and mitoxantrone compared with high dose cytarabine and
mitoxantrone in older patients with relapsed/refractory acute
myeloid leukemia: ARMADA 2000 Phase III study. Future Oncol.
15:3197–3208. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Ding J, Karp JE and Emadi A: Elevated
lactate dehydrogenase (LDH) can be a marker of immune suppression
in cancer: Interplay between hematologic and solid neoplastic
clones and their microenvironments. Cancer Biomark. 19:353–363.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Zhou M, Zhao Y, Ding Y, Liu H, Liu Z,
Fodstad O, Riker AI, Kamarajugadda S, Lu J, Owen LB, et al: Warburg
effect in chemosensitivity: Targeting lactate dehydrogenase-A
re-sensitizes taxol-resistant cancer cells to taxol. Mol Cancer.
9:332010. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Muramatsu H, Sumitomo M, Morinaga S,
Kajikawa K, Kobayashi I, Nishikawa G, Kato Y, Watanabe M, Zennami
K, Kanao K, et al: Targeting lactate dehydrogenase-A promotes
docetaxel-induced cytotoxicity predominantly in
castration-resistant prostate cancer cells. Oncol Rep. 42:224–230.
2019.PubMed/NCBI
|
|
62
|
Nagamine A, Araki T, Nagano D, Miyazaki M
and Yamamoto K: L-Lactate dehydrogenase B may be a predictive
marker for sensitivity to anti-EGFR monoclonal antibodies in
colorectal cancer cell lines. Oncol Lett. 17:4710–4716.
2019.PubMed/NCBI
|
|
63
|
Feng L, E LL, Soloveiv MM, Wang DS, Zhang
BO, Dong YW and Liu HC: Synergistic cytotoxicity of cisplatin and
Taxol in overcoming Taxol resistance through the inhibition of LDHA
in oral squamous cell carcinoma. Oncol Lett. 9:1827–1832. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Hua G, Liu Y, Li X, Xu P and Luo Y:
Targeting glucose metabolism in chondrosarcoma cells enhances the
sensitivity to doxorubicin through the inhibition of lactate
dehydrogenase-A. Oncol Rep. 31:2727–2734. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Koukourakis MI, Giatromanolaki A, Sivridis
E, Bougioukas G, Didilis V, Gatter KC and Harris AL: Lactate
dehydrogenase-5 (LDH-5) overexpression in non-small-cell lung
cancer tissues is linked to tumour hypoxia, angiogenic factor
production and poor prognosis. Br J Cancer. 89:877–885. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Lu H, Li X, Luo Z, Liu J and Fan Z:
Cetuximab reverses the Warburg effect by inhibiting HIF-1-regulated
LDH-A. Mol Cancer Ther. 12:2187–2199. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Liu X, Yao D, Liu C, Cao Y, Yang Q, Sun Z
and Liu D: Overexpression of ABCC3 promotes cell proliferation,
drug resistance, and aerobic glycolysis and is associated with poor
prognosis in urinary bladder cancer patients. Tumour Biol.
37:8367–8374. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Aggarwal A and Keluskar V: Epithelioid
hemangioma (angiolymphoid hyperplasia with eosinophilia) in the
oral mucosa. Indian J Dent Res. 23:271–274. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Hamadneh L, Abuarqoub R, Alhusban A and
Bahader M: Upregulation of PI3K/AKT/PTEN pathway is correlated with
glucose and glutamine metabolic dysfunction during tamoxifen
resistance development in MCF-7 cells. Sci Rep. 10:219332020.
View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Sun H, Wang H and Wang X, Aoki Y and Wang
X, Yang Y, Cheng X, Wang Z and Wang X: Aurora-A/SOX8/FOXK1
signaling axis promotes chemoresistance via suppression of cell
senescence and induction of glucose metabolism in ovarian cancer
organoids and cells. Theranostics. 10:6928–6945. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Dong D, Ko B, Baumeister P, Swenson S,
Costa F, Markland F, Stiles C, Patterson JB, Bates SE and Lee AS:
Vascular targeting and antiangiogenesis agents induce drug
resistance effector GRP78 within the tumor microenvironment. Cancer
Res. 65:5785–5791. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
72
|
Lee E, Nichols P, Spicer D, Groshen S, Yu
MC and Lee AS: GRP78 as a novel predictor of responsiveness to
chemotherapy in breast cancer. Cancer Res. 66:7849–7853. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
73
|
Zhou H, Zhang Y, Fu Y, Chan L and Lee AS:
Novel mechanism of anti-apoptotic function of 78-kDa
glucose-regulated protein (GRP78): Endocrine resistance factor in
breast cancer, through release of B-cell lymphoma 2 (BCL-2) from
BCL-2-interacting killer (BIK). J Biol Chem. 286:25687–25696. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
74
|
Cristanziano VD, Bottcher S, Diedrich S,
Timmen-Wego M, Knops E, Lübke N, Kaiser R, Pfister H, Kaboré Y and
D'Alfonso R: Detection and characterization of enteroviruses and
parechoviruses in healthy people living in the South of Cote
d'Ivoire. J Clin Virol. 71:40–43. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
75
|
Cheng Y, Diao D, Zhang H, Guo Q, Wu X,
Song Y and Dang C: High glucose-induced resistance to
5-fluorouracil in pancreatic cancer cells alleviated by
2-deoxy-D-glucose. Biomed Rep. 2:188–192. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
76
|
Panizo C, Perez-Salazar M, Bendandi M,
Rodríguez-Calvillo M, Boán JF, García-Velloso MJ, Richter J and
Rocha E: Positron emission tomography using 18F-fluorodeoxyglucose
for the evaluation of residual Hodgkin's disease mediastinal
masses. Leuk Lymphoma. 45:1829–1833. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
77
|
Weber WA, Ott K, Becker K, Dittler HJ,
Helmberger H, Avril NE, Meisetschläger G, Busch R, Siewert JR,
Schwaiger M and Fink U: Prediction of response to preoperative
chemotherapy in adenocarcinomas of the esophagogastric junction by
metabolic imaging. J Clin Oncol. 19:3058–3065. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
78
|
Gallamini A, Rigacci L, Merli F, Nassi L,
Bosi A, Capodanno I, Luminari S, Vitolo U, Sancetta R, Iannitto E,
et al: The predictive value of positron emission tomography
scanning performed after two courses of standard therapy on
treatment outcome in advanced stage Hodgkin's disease.
Haematologica. 91:475–481. 2006.PubMed/NCBI
|
|
79
|
Chen LQ, Cheung LS, Feng L, Tanner W and
Frommer WB: Transport of sugars. Annu Rev Biochem. 84:865–894.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
80
|
Reckzeh ES and Waldmann H: Small-Molecule
inhibition of glucose transporters GLUT-1-4. Chembiochem. 21:45–52.
2020. View Article : Google Scholar : PubMed/NCBI
|
|
81
|
Mayer A, Hockel M, Wree A and Vaupel P:
Microregional expression of glucose transporter-1 and oxygenation
status: Lack of correlation in locally advanced cervical cancers.
Clin Cancer Res. 11:2768–2773. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
82
|
Suzuki S, Okada M, Takeda H, Kuramoto K,
Sanomachi T, Togashi K, Seino S, Yamamoto M, Yoshioka T and
Kitanaka C: Involvement of GLUT1-mediated glucose transport and
metabolism in gefitinib resistance of non-small-cell lung cancer
cells. Oncotarget. 9:32667–32679. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
83
|
Wang T, Ning K, Sun X, Zhang C, Jin LF and
Hua D: Glycolysis is essential for chemoresistance induced by
transient receptor potential channel C5 in colorectal cancer. BMC
Cancer. 18:2072018. View Article : Google Scholar : PubMed/NCBI
|
|
84
|
Guan DG, Chen HM, Liao SF and Zhao TZ:
Combination of temozolomide and Taxol exerts a synergistic
inhibitory effect on Taxolresistant glioma cells via inhibition of
glucose metabolism. Mol Med Rep. 12:7705–7711. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
85
|
Liu PJ, Ma F, Lou HP, Zhu YN and Chen Y:
Relationship between serum uric acid levels and metabolic syndrome
in Chinese postmenopausal women. Climacteric. 17:148–154. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
86
|
Wang L, Sun J, Gao P, Su K, Wu H, Li J and
Lou W: Wnt1-inducible signaling protein 1 regulates laryngeal
squamous cell carcinoma glycolysis and chemoresistance via the
YAP1/TEAD1/GLUT1 pathway. J Cell Physiol. Feb 25–2019.(Epub ahead
of print).
|
|
87
|
Li S, Li J, Dai W, Zhang Q, Feng J, Wu L,
Liu T, Yu Q, Xu S, Wang W, et al: Genistein suppresses aerobic
glycolysis and induces hepatocellular carcinoma cell death. Br J
Cancer. 117:1518–1528. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
88
|
Gou Q, Dong C, Jin J, Liu Q, Lu W, Shi J
and Hou Y: PPARα agonist alleviates tumor growth and
chemo-resistance associated with the inhibition of glucose
metabolic pathway. Eur J Pharmacol. 863:1726642019. View Article : Google Scholar : PubMed/NCBI
|
|
89
|
Zhou Y, Wen H, Gu L, Fu J, Guo J, Du L,
Zhou X, Yu X, Huang Y and Wang H: Aminoglucose-functionalized,
redox-responsive polymer nanomicelles for overcoming
chemoresistance in lung cancer cells. J Nanobiotechnology.
15:872017. View Article : Google Scholar : PubMed/NCBI
|
|
90
|
Warburg O: On respiratory impairment in
cancer cells. Science. 124:269–270. 1956.PubMed/NCBI
|
|
91
|
Kim JW and Dang CV: Cancer's molecular
sweet tooth and the Warburg effect. Cancer Res. 66:8927–8930. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
92
|
Chen Z, Lu W, Garcia-Prieto C and Huang P:
The Warburg effect and its cancer therapeutic implications. J
Bioenerg Biomembr. 39:267–274. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
93
|
Wagner W, Ciszewski WM and Kania KD: L-
and D-lactate enhance DNA repair and modulate the resistance of
cervical carcinoma cells to anticancer drugs via histone
deacetylase inhibition and hydroxycarboxylic acid receptor 1
activation. Cell Commun Signal. 13:362015. View Article : Google Scholar : PubMed/NCBI
|
|
94
|
Granja S, Tavares-Valente D, Queiros O and
Baltazar F: Value of pH regulators in the diagnosis, prognosis and
treatment of cancer. Semin Cancer Biol. 43:17–34. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
95
|
Doyen J, Parks SK, Marcie S, Pouyssegur J
and Chiche J: Knock-down of hypoxia-induced carbonic anhydrases IX
and XII radiosensitizes tumor cells by increasing intracellular
acidosis. Front Oncol. 2:1992013. View Article : Google Scholar : PubMed/NCBI
|
|
96
|
Dubois L, Peeters S, Lieuwes NG, Geusens
N, Thiry A, Wigfield S, Carta F, McIntyre A, Scozzafava A, Dogné
JM, et al: Specific inhibition of carbonic anhydrase IX activity
enhances the in vivo therapeutic effect of tumor irradiation.
Radiother Oncol. 99:424–431. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
97
|
Betof AS, Rabbani ZN, Hardee ME, Kim SJ,
Broadwater G, Bentley RC, Snyder SA, Vujaskovic Z, Oosterwijk E,
Harris LN, et al: Carbonic anhydrase IX is a predictive marker of
doxorubicin resistance in early-stage breast cancer independent of
HER2 and TOP2A amplification. Br J Cancer. 106:916–922. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
98
|
Aomatsu N, Yashiro M, Kashiwagi S,
Kawajiri H, Takashima T, Ohsawa M, Wakasa K and Hirakawa K:
Carbonic anhydrase 9 is associated with chemosensitivity and
prognosis in breast cancer patients treated with taxane and
anthracycline. BMC Cancer. 14:4002014. View Article : Google Scholar : PubMed/NCBI
|
|
99
|
Rauch C: On the relationship between
drug's size, cell membrane mechanical properties and high levels of
multi drug resistance: A comparison to published data. Eur Biophys
J. 38:537–546. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
100
|
Perez-Sayans M, Somoza-Martin JM,
Barros-Angueira F, Diz PG, Rey JM and Garcia-Garcia A: Multidrug
resistance in oral squamous cell carcinoma: The role of vacuolar
ATPases. Cancer Lett. 295:135–143. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
101
|
Larsen AK, Escargueil AE and Skladanowski
A: Resistance mechanisms associated with altered intracellular
distribution of anticancer agents. Pharmacol Ther. 85:217–229.
2000. View Article : Google Scholar : PubMed/NCBI
|
|
102
|
Le Floch R, Chiche J, Marchiq I, Naiken T,
Ilc K, Murray CM, Critchlow SE, Roux D, Simon MP and Pouysségur J:
CD147 subunit of lactate/H+ symporters MCT1 and
hypoxia-inducible MCT4 is critical for energetics and growth of
glycolytic tumors. Proc Natl Acad Sci USA. 108:16663–16668. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
103
|
Boidot R, Vegran F, Meulle A, Le Breton A,
Dessy C, Sonveaux P, Lizard-Nacol S and Feron O: Regulation of
monocarboxylate transporter MCT1 expression by p53 mediates inward
and outward lactate fluxes in tumors. Cancer Res. 72:939–948. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
104
|
Saenz-de-Santa-Maria I,
Bernardo-Castineira C, Secades P, Bernaldo-de-Quirós S, Rodrigo JP,
Astudillo A and Chiara MD: Clinically relevant HIF-1α-dependent
metabolic reprogramming in oropharyngeal squamous cell carcinomas
includes coordinated activation of CAIX and the miR-210/ISCU
signaling axis, but not MCT1 and MCT4 upregulation. Oncotarget.
8:13730–13746. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
105
|
Draoui N, Schicke O, Seront E, Bouzin C,
Sonveaux P, Riant O and Feron O: Antitumor activity of
7-aminocarboxycoumarin derivatives, a new class of potent
inhibitors of lactate influx but not efflux. Mol Cancer Ther.
13:1410–1418. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
106
|
Li QX, Zhang P, Liu F, Wang XZ, Li L, Wang
ZK, Jiang CC, Zheng HL and Liu H: Monocarboxylate transporter 1
enhances the sensitivity of breast cancer cells to 3-bromopyruvate
in vitro. Nan Fang Yi Ke Da Xue Xue Bao. 37:588–593. 2017.(In
Chinese). PubMed/NCBI
|
|
107
|
Pisarsky L, Bill R, Fagiani E, Dimeloe S,
Goosen RW, Hagmann J, Hess C and Christofori G: Targeting metabolic
symbiosis to overcome resistance to anti-angiogenic therapy. Cell
Rep. 15:1161–1174. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
108
|
Khammanivong A, Saha J, Spartz AK,
Sorenson BS, Bush AG, Korpela DM, Gopalakrishnan R, Jonnalagadda S,
Mereddy VR, O'Brien TD, et al: A novel MCT1 and MCT4 dual inhibitor
reduces mitochondrial metabolism and inhibits tumour growth of
feline oral squamous cell carcinoma. Vet Comp Oncol. 8:324–341.
2020. View Article : Google Scholar
|
|
109
|
Andreucci E, Ruzzolini J, Peppicelli S,
Bianchini F, Laurenzana A, Carta F, Supuran CT and Calorini L: The
carbonic anhydrase IX inhibitor SLC-0111 sensitises cancer cells to
conventional chemotherapy. J Enzyme Inhib Med Chem. 34:117–123.
2019. View Article : Google Scholar : PubMed/NCBI
|
|
110
|
Tan EY, Yan M, Campo L, Han C, Takano E,
Turley H, Candiloro I, Pezzella F, Gatter KC, Millar EK, et al: The
key hypoxia regulated gene CAIX is upregulated in basal-like breast
tumours and is associated with resistance to chemotherapy. Br J
Cancer. 100:405–411. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
111
|
Zheng G, Peng C, Jia X, Gu Y, Zhang Z,
Deng Y, Wang C, Li N, Yin J, Liu X, et al: ZEB1 transcriptionally
regulated carbonic anhydrase 9 mediates the chemoresistance of
tongue cancer via maintaining intracellular pH. Mol Cancer.
14:842015. View Article : Google Scholar : PubMed/NCBI
|
|
112
|
Takahashi E, Abe J and Berk BC:
Angiotensin II stimulates p90rsk in vascular smooth muscle cells. A
potential Na(+)-H+ exchanger kinase. Circ Res.
81:268–273. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
113
|
Altaf E, Huang X, Xiong J, Yang X, Deng X,
Xiong M, Zhou L, Pan S, Yuan W, Li X, et al: NHE1 has a notable
role in metastasis and drug resistance of T-cell acute
lymphoblastic leukemia. Oncol Lett. 14:4256–4262. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
114
|
Chen Q, Liu Y, Zhu XL, Feng F, Yang H and
Xu W: Increased NHE1 expression is targeted by specific inhibitor
cariporide to sensitize resistant breast cancer cells to
doxorubicin in vitro and in vivo. BMC Cancer. 19:2112019.
View Article : Google Scholar : PubMed/NCBI
|
|
115
|
Paskeviciute M and Petrikaite V:
Overcoming transporter-mediated multidrug resistance in cancer:
Failures and achievements of the last decades. Drug Deliv Transl
Res. 9:379–393. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
116
|
Park S, Chang CY, Safi R, Liu X, Baldi R,
Jasper JS, Anderson GR, Liu T, Rathmell JC, Dewhirst MW, et al:
ERRα-Regulated lactate metabolism contributes to resistance to
targeted therapies in breast cancer. Cell Rep. 15:323–335. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
117
|
Yu T, Yang G, Hou Y, Tang X, Wu C, Wu XA,
Guo L, Zhu Q, Luo H, Du YE, et al: Cytoplasmic GPER translocation
in cancer-associated fibroblasts mediates cAMP/PKA/CREB/glycolytic
axis to confer tumor cells with multidrug resistance. Oncogene.
36:2131–2145. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
118
|
Kalbasi A and Ribas A: Tumour-intrinsic
resistance to immune checkpoint blockade. Nat Rev Immunol.
20:25–39. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
119
|
Garcia-Diaz A, Shin DS, Moreno BH, Saco J,
Escuin-Ordinas H, Rodriguez GA, Zaretsky JM, Sun L, Hugo W, Wang X,
et al: Interferon receptor signaling pathways regulating PD-L1 and
PD-L2 expression. Cell Rep. 19:1189–1201. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
120
|
Liang X, Fu C, Cui W, Ober-Blöbaum JL,
Zahner SP, Shrikant PA, Clausen BE, Flavell RA, Mellman I and Jiang
A: β-catenin mediates tumor-induced immunosuppression by inhibiting
cross-priming of CD8+ T cells. J Leukoc Biol.
95:179–190. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
121
|
Peng W, Chen JQ, Liu C, Malu S, Creasy C,
Tetzlaff MT, Xu C, McKenzie JA, Zhang C, Liang X, et al: Loss of
PTEN promotes resistance to T cell-mediated immunotherapy. Cancer
Discov. 6:202–216. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
122
|
Gao J, Shi LZ, Zhao H, Chen J, Xiong L, He
Q, Chen T, Roszik J, Bernatchez C, Woodman SE, et al: Loss of IFN-ү
pathway genes in tumor cells as a mechanism of resistance to
anti-CTLA-4 therapy. Cell. 167:397–404.e9. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
123
|
Pitroda SP, Wakim BT, Sood RF, Beveridge
MG, Beckett MA, MacDermed DM, Weichselbaum RR and Khodarev NN:
STAT1-dependent expression of energy metabolic pathways links
tumour growth and radioresistance to the warburg effect. BMC Med.
7:682009. View Article : Google Scholar : PubMed/NCBI
|
|
124
|
Poli V and Camporeale A: STAT3-mediated
metabolic reprograming in cellular transformation and implications
for drug resistance. Front Oncol. 5:1212015. View Article : Google Scholar : PubMed/NCBI
|
|
125
|
Cascone T, McKenzie JA, Mbofung RM, Punt
S, Wang Z, Xu C, Williams LJ, Wang Z, Bristow CA, Carugo A, et al:
Increased tumor glycolysis characterizes immune resistance to
adoptive T cell therapy. Cell Metab. 27:977–987.e4. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
126
|
Sacchi S, Federico M, Vitolo U, Boccomini
C, Vallisa D, Baldini L, Petrini M, Rupoli S, Di Raimondo F, Merli
F, et al: Clinical activity and safety of combination immunotherapy
with IFN-alpha 2a and Rituximab in patients with relapsed low grade
non-Hodgkin's lymphoma. Haematologica. 86:951–958. 2001.PubMed/NCBI
|
|
127
|
Davis TA, Maloney DG, Grillo-Lopez AJ,
White CA, Williams ME, Weiner GJ, Dowden S and Levy R: Combination
immunotherapy of relapsed or refractory low-grade or follicular
non-Hodgkin's lymphoma with rituximab and interferon-alpha-2a. Clin
Cancer Res. 6:2644–2652. 2000.PubMed/NCBI
|
|
128
|
Rosenberg SA, Lotze MT, Yang JC, Linehan
WM, Seipp C, Calabro S, Karp SE, Sherry RM, Steinberg S and White
DE: Combination therapy with interleukin-2 and alpha-interferon for
the treatment of patients with advanced cancer. J Clin Oncol.
7:1863–1874. 1989. View Article : Google Scholar : PubMed/NCBI
|
|
129
|
Magalhaes I, Yogev O, Mattsson J and
Schurich A: The metabolic profile of tumor and virally infected
cells shapes their microenvironment counteracting T cell immunity.
Front Immunol. 10:23092019. View Article : Google Scholar : PubMed/NCBI
|
|
130
|
Pate KT, Stringari C, Sprowl-Tanio S, Wang
K, TeSlaa T, Hoverter NP, McQuade MM, Garner C, Digman MA, Teitell
MA, et al: Wnt signaling directs a metabolic program of glycolysis
and angiogenesis in colon cancer. EMBO J. 33:1454–1473. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
131
|
Yang L, Perez AA, Fujie S, Warden C, Li J,
Wang Y, Yung B, Chen YR, Liu X, Zhang H, et al: Wnt modulates MCL1
to control cell survival in triple negative breast cancer. BMC
Cancer. 14:1242014. View Article : Google Scholar : PubMed/NCBI
|
|
132
|
Sprowl-Tanio S, Habowski AN, Pate KT,
McQuade MM, Wang K, Edwards RA, Grun F, Lyou Y and Waterman ML:
Lactate/pyruvate transporter MCT-1 is a direct Wnt target that
confers sensitivity to 3-bromopyruvate in colon cancer. Cancer
Metab. 4:202016. View Article : Google Scholar : PubMed/NCBI
|
|
133
|
Feng M, Jin JQ, Xia L, Xiao T, Mei S, Wang
X, Huang X, Chen J, Liu M, Chen C, et al: Pharmacological
inhibition of β-catenin/BCL9 interaction overcomes resistance to
immune checkpoint blockades by modulating Treg cells.
Sci Adv. 5:eaau52402019. View Article : Google Scholar : PubMed/NCBI
|
|
134
|
George S, Miao D, Demetri GD, Adeegbe D,
Rodig SJ, Shukla S, Lipschitz M, Amin-Mansour A, Raut CP, Carter
SL, et al: Loss of PTEN is associated with resistance to Anti-PD-1
checkpoint blockade therapy in metastatic uterine leiomyosarcoma.
Immunity. 46:197–204. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
135
|
Ramapriyan R, Caetano MS, Barsoumian HB,
Mafra ACP, Zambalde EP, Menon H, Tsouko E, Welsh JW and Cortez MA:
Altered cancer metabolism in mechanisms of immunotherapy
resistance. Pharmacol Ther. 195:162–171. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
136
|
Jabbarzadeh Kaboli P, Salimian F, Aghapour
S, Xiang S, Zhao Q, Li M, Wu X, Du F, Zhao Y, Shen J, et al:
Akt-targeted therapy as a promising strategy to overcome drug
resistance in breast cancer-A comprehensive review from
chemotherapy to immunotherapy. Pharmacol Res. 156:1048062020.
View Article : Google Scholar : PubMed/NCBI
|
|
137
|
Blagden SP, Hamilton AL, Mileshkin L, Wong
S, Michael A, Hall M, Goh JC, Lisyanskaya AS, DeSilvio M, Frangou
E, et al: Phase IB dose escalation and expansion study of AKT
inhibitor afuresertib with carboplatin and paclitaxel in recurrent
platinum-resistant ovarian cancer. Clin Cancer Res. 25:1472–1478.
2019. View Article : Google Scholar : PubMed/NCBI
|
|
138
|
Tammela T, Sanchez-Rivera FJ, Cetinbas NM,
Wu K, Joshi NS, Helenius K, Park Y, Azimi R, Kerper NR, Wesselhoeft
RA, et al: A Wnt-producing niche drives proliferative potential and
progression in lung adenocarcinoma. Nature. 545:355–359. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
139
|
de Bono JS, De Giorgi U, Rodrigues DN,
Massard C, Bracarda S, Font A, Arranz Arija JA, Shih KC, Radavoi
GD, Xu N, et al: Randomized phase II study evaluating Akt blockade
with ipatasertib, in combination with abiraterone, in patients with
metastatic prostate cancer with and without PTEN loss. Clin Cancer
Res. 25:928–936. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
140
|
Kunkl M, Sambucci M, Ruggieri S, Amormino
C, Tortorella C, Gasperini C, Battistini L and Tuosto L: CD28
autonomous signaling Up-regulates C-Myc expression and promotes
glycolysis enabling inflammatory T cell responses in multiple
sclerosis. Cells. 8:5752019. View Article : Google Scholar
|
|
141
|
Chen F, Chen J, Yang L, Liu J, Zhang X,
Zhang Y, Tu Q, Yin D, Lin D, Wong PP, et al: Extracellular
vesicle-packaged HIF-1α-stabilizing lncRNA from tumour-associated
macrophages regulates aerobic glycolysis of breast cancer cells.
Nat Cell Biol. 21:498–510. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
142
|
Xie G, Liu Y, Yao Q, Zheng R, Zhang L, Lin
J, Guo Z, Du S, Ren C, Yuan Q and Yuan Y: Hypoxia-induced
angiotensin II by the lactate-chymase-dependent mechanism mediates
radioresistance of hypoxic tumor cells. Sci Rep. 7:423962017.
View Article : Google Scholar : PubMed/NCBI
|
|
143
|
Xie G, Cheng T, Lin J, Zhang L, Zheng J,
Liu Y, Xie G, Wang B and Yuan Y: Local angiotensin II contributes
to tumor resistance to checkpoint immunotherapy. J Immunother
Cancer. 6:882018. View Article : Google Scholar : PubMed/NCBI
|
|
144
|
Fischer K, Hoffmann P, Voelkl S,
Meidenbauer N, Ammer J, Edinger M, Gottfried E, Schwarz S, Rothe G,
Hoves S, et al: Inhibitory effect of tumor cell-derived lactic acid
on human T cells. Blood. 109:3812–3819. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
145
|
Ohashi T, Akazawa T, Aoki M, Kuze B,
Mizuta K, Ito Y and Inoue N: Dichloroacetate improves immune
dysfunction caused by tumor-secreted lactic acid and increases
antitumor immunoreactivity. Int J Cancer. 133:1107–1118. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
146
|
Warburg O: On the origin of cancer cells.
Science. 123:309–314. 1956. View Article : Google Scholar : PubMed/NCBI
|
|
147
|
Bosc C, Selak MA and Sarry JE: Resistance
is futile: Targeting mitochondrial energetics and metabolism to
overcome drug resistance in cancer treatment. Cell Metab.
26:705–707. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
148
|
Alexa-Stratulat T, Pesic M, Gasparovic AC,
Trougakos IP and Riganti C: What sustains the multidrug resistance
phenotype beyond ABC efflux transporters? Looking beyond the tip of
the iceberg. Drug Resist Updat. 46:1006432019. View Article : Google Scholar : PubMed/NCBI
|
|
149
|
Roth KG, Mambetsariev I, Kulkarni P and
Salgia R: The mitochondrion as an emerging therapeutic target in
cancer. Trends Mol Med. 26:119–134. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
150
|
Farge T, Saland E, de Toni F, Aroua N,
Hosseini M, Perry R, Bosc C, Sugita M, Stuani L, Fraisse M, et al:
Chemotherapy-resistant human acute myeloid leukemia cells are not
enriched for leukemic stem cells but require oxidative metabolism.
Cancer Discov. 7:716–735. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
151
|
Migneco G, Whitaker-Menezes D, Chiavarina
B, Castello-Cros R, Pavlides S, Pestell RG, Fatatis A, Flomenberg
N, Tsirigos A, Howell A, et al: Glycolytic cancer associated
fibroblasts promote breast cancer tumor growth, without a
measurable increase in angiogenesis: Evidence for
stromal-epithelial metabolic coupling. Cell Cycle. 9:2412–2422.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
152
|
Ricci F, Brunelli L, Affatato R, Chilà R,
Verza M, Indraccolo S, Falcetta F, Fratelli M, Fruscio R,
Pastorelli R and Damia G: Overcoming platinum-acquired resistance
in ovarian cancer patient-derived xenografts. Ther Adv Med Oncol.
11:17588359198395432019. View Article : Google Scholar : PubMed/NCBI
|
|
153
|
Wu L, Zhao J, Cao K, Liu X, Cai H, Wang J,
Li W and Chen Z: Oxidative phosphorylation activation is an
important characteristic of DOX resistance in hepatocellular
carcinoma cells. Cell Commun Signal. 16:62018. View Article : Google Scholar : PubMed/NCBI
|
|
154
|
Lee KM, Giltnane JM, Balko JM, Schwarz LJ,
Guerrero-Zotano AL, Hutchinson KE, Nixon MJ, Estrada MV, Sánchez V,
Sanders ME, et al: MYC and MCL1 cooperatively promote
chemotherapy-resistant breast cancer stem cells via regulation of
mitochondrial oxidative phosphorylation. Cell Metab. 26:633–647.e7.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
155
|
Chen CL, Uthaya Kumar DB, Punj V, Xu J,
Sher L, Tahara SM, Hess S and Machida K: NANOG metabolically
reprograms tumor-initiating stem-like cells through tumorigenic
changes in oxidative phosphorylation and fatty acid metabolism.
Cell Metab. 23:206–219. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
156
|
Andreucci E, Pietrobono S, Peppicelli S,
Ruzzolini J, Bianchini F, Biagioni A, Stecca B and Calorini L: SOX2
as a novel contributor of oxidative metabolism in melanoma cells.
Cell Commun Signal. 16:872018. View Article : Google Scholar : PubMed/NCBI
|
|
157
|
Zhao Y, Butler EB and Tan M: Targeting
cellular metabolism to improve cancer therapeutics. Cell Death Dis.
4:e5322013. View Article : Google Scholar : PubMed/NCBI
|
|
158
|
Choi HJ, Jhe YL, Kim J, Lim JY, Lee JE,
Shin MK and Cheong JH: FoxM1-dependent and fatty acid
oxidation-mediated ROS modulation is a cell-intrinsic drug
resistance mechanism in cancer stem-like cells. Redox Biol.
36:1015892020. View Article : Google Scholar : PubMed/NCBI
|