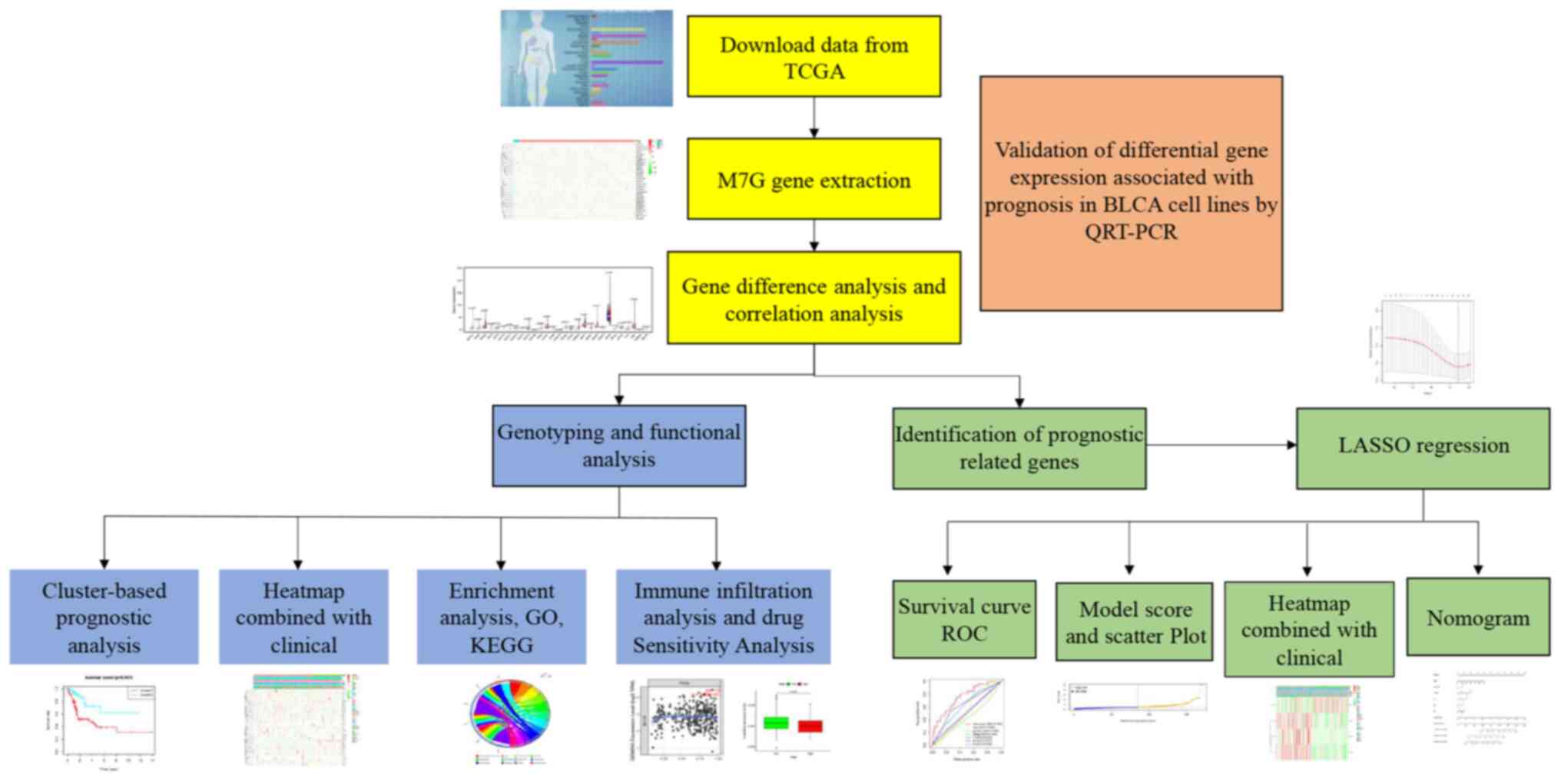
Introduction
Bladder urothelial carcinoma (BLCA) is a malignant
tumor of the bladder urothelium accounting for ~500,000 new cases
and 200,000 mortalities worldwide each year (1). Approximately 70% of patients with BLCA
have non-muscle-invasive bladder cancer (NMIBC), and usually
undergo transurethral resection of the bladder tumor and subsequent
radiotherapy, chemotherapy, or immunotherapy. However, ~80% of
patients suffer NMIBC recurrence within 5 years of diagnosis, with
tumor progression occurring in 30% of cases (2). For these patients, a combination of
surgery, radiotherapy, chemotherapy, and immunotherapy can be used
(3). However, these treatments
increase the surgical risk and treatment-related toxicity, and
patients with aggressive or advanced BLCA still face adverse
clinical outcomes. A remaining challenge is designing approaches to
filter patients who may benefit from aggressive treatment.
Therefore, novel biomarkers that can be used to predict prognosis
and treatment response are urgently needed.
Bioinformatics has become an integral tool in
biomedical research and treatment development in the past decade,
and it plays a vital role in deciphering genomes, transcriptomes,
and proteomes generated via high-throughput experimental techniques
or from tissues collected in traditional biological studies
(4). For example, sequence-based
methods used to analyze multiple genes or proteins have been
explored (5). The primary purpose
of bioinformatics analysis is to mine the association between
features according to the feature information based on
multidimensional data. Based on public databases, potential
associations among clinical patient parameters are analyzed, and
this can be used to the reveal risk factors of a disease, further
develop survival prediction models for a disease, and develop
precise diagnoses and treatment recommendations for patients
(6). Through bioinformatics
analysis, previous studies have identified molecules and phenotypes
associated with the diagnosis, treatment, and prognosis of BLCA,
such as pyroptosis (7), immune cell
infiltration (8–10), hypoxia (11), and inflammation (12). However, several other biological
features of BLCA remain to be identified.
The recent progress made in the understanding of
epitranscriptomics has led to the identification of numerous types
of post-transcriptional modifications of eukaryotic RNA, such as
5-methylcytosine, N6-methyladenosine, and N7-methylguanine (m7G)
(13). Methylated m7G RNA is one of
the most conserved modified nucleosides, and is commonly detected
in eubacteria, eukaryotes, (14)
and archaea (15). m7G
modification, which adds a methyl group to the seventh N of guanine
(G) in RNA via a methyltransferase, is an important form of base
modification involved in post-transcriptional regulation. Recently,
m7G-related genes have attracted widespread attention in oncology;
for example, a previous study has shown that methyltransferase 1
(METTL1)-mediated m7G transfer RNA (tRNA) modification can boost
oncogene translation and promote the progression of intrahepatic
cholangiocarcinoma (16). Based on
the association between tumors and m7G, a previous study identified
six genes associated with poor patient prognosis, thus showing
potential for evaluating gastric cancer prognosis (17). However, to the best of our
knowledge, there are no studies that have investigated the
expression pattern, molecular function, or prognostic value of
m7G-related regulatory genes in BLCA; thus, the present study aimed
to mine the differentially expressed genes between BLCA samples and
normal healthy tissues and to construct a prognostic model to
explore the potential function of m7G in this disease. Furthermore,
with the increasing use of immunotherapy and personalized medicine,
immune infiltration has become a prognostic factor for multiple
cancer types, and thus, the present study further predicted the
immune landscape, immune biomarkers, and drug sensitivity of the
risk-stratified population, aiming to provide new clinical
treatment options (Fig. 1).
Materials and methods
RNA-sequencing transcriptomic data
collection
RNA-sequencing transcriptomic data and corresponding
clinical information were obtained from The Cancer Genome Atlas
(TCGA; http://portal.gdc.cancer.gov/). These
data included 414 BLCA tissues and 19 normal adjacent tissues.
Clinical information, including age, sex, and TNM stage was
collected. In total, 29 m7G-related regulatory genes were
identified by screening the Gene Set Enrichment Analysis database
(GSEA; http://www.gsea-msigdb.org/gsea/index.jsp) and by
performing literature searches (18–23).
In the BLCA cohort from TCGA, the expression data of these 29
m7G-related genes were extracted for subsequent analysis. The
GSE48075 dataset from the Gene Expression Omnibus (GEO) database
(http://www.ncbi.nlm.nih.gov/geo) was
used as the validation set, and this dataset contained 142 BLCA
samples with corresponding survival information and gene expression
data.
Identification of m7G regulatory genes
in BLCA
Differentially expressed genes (DEGs) involved in
m7G methylation were screened between BLCA and normal adjacent
tissues using the Wilcoxon test in R (version 4.1.2) (24). Significant results were those with a
false discovery rate (FDR) <0.05 and an absolute
log2-fold change >1. Violin plots were drawn using
the R package ‘vioplot’ (version 0.3.7, http://github.com/TomKellyGenetics/vioplot) to
show the differential expression of DEGs in BLCA and normal
adjacent tissue samples. Spearman correlation analysis was
performed to determine associations among DEGs. A protein-protein
interaction (PPI) network was obtained through the STRING database
(https://string-db.org/) (25) to query the interaction of
DEG-related proteins. The Gene Expression Profiling Interactive
Analysis (GEPIA) 2 database (http://gepia2.cancer-pku.cn) (26) was also used to validate DEGs in
BLCA.
Identification of two clusters with
different clinical outcomes based on m7G gene consensus clustering
and functional enrichment analysis
Based on DEGs, the BLCA cohort was divided into two
distinct subgroups using the R package ‘ConsensusClusterPlus’
(version 1.36.0, http://rdocumentation.org/packages/ConsensusClusterPlus/versions/1.36.0).
Survival curves were drawn to compare the overall survival (OS)
between two groups based on Kaplan-Meier analysis. Differences in
clinical data (survival status, stage, pathological grade, sex, and
age) between the two groups were detected using a χ2
test. Gene Ontology (GO) and Kyoto Encyclopedia of Genes and
Genomes (KEGG) analyses were performed to functionally annotate
DEGs in the two subgroups.
Evaluation of the prognostic value of
m7G-related regulatory genes in patients with BLCA
Univariate Cox regression analysis was used to
estimate the correlation between m7G-related genes and OS. Based on
the R package ‘glmnet’ (version 4.1.3, http://rdocumentation.org/packages/glmnet), the least
absolute shrinkage and selection operator (LASSO) Cox regression
model was then utilized to narrow down the candidate genes and to
develop the prognostic model. Ultimately, the four genes and their
coefficients were retained, and the penalty parameter (λ) was
decided by the minimum criteria. The gene expression acquired from
the LASSO Cox regression and its coefficients were multiplied to
generate the risk score to divide patients with BLCA into low- and
high-risk groups, and the OS time was compared between the two
subgroups via Kaplan-Meier analysis. The ‘survival’ (version 3.3.0,
http://rdocumentation.org/packages/survival),
‘survminer’ (version 0.4.9, http://CRAN.R-project.org/package=survminer), and
‘timeROC’ (version 0.4, http://rdocumentation.org/packages/timeROC/versions/0.4)
R packages were used to perform receiver operating characteristic
(ROC) curve analysis. Differences in clinically relevant variables
between different risk groups were evaluated using a χ2
test and visualized with heatmaps. Moreover, univariate and
multivariate Cox regression analyses were performed by using the R
packages ‘survivalROC’ (version 1.0.3, http://rdocumentation.org/packages/survivalROC) and
‘survival’ to assess whether the risk score was an independent
predictor.
Validation of the m7G-related gene
prognostic model
To validate the prognostic value of these four
m7G-regulated genes, the GSE48075 cohort was used as a validation
set. The same risk score calculation formula and the same cutoff
value that could distinguish between high and low-risk groups were
used to classify patients in three cohorts, and Kaplan-Meier
survival and ROC curve analyses were performed to evaluate the
predictive performance.
Construction of prediction nomogram
and decision curve
Based on the R package ‘rms’ (version 6.2.0,
http://rdocumentation.org/packages/rms), clinically
relevant factors (histological grade, sex, stage, and age) and risk
scores were used to construct prognostic nomograms to predict OS in
patients with BLCA. Moreover, the R package ‘ggDCA’ (version 1.1,
http://rdocumentation.org/packages/ggDCA/versions/1.1)
was used to finish the decision curve analysis, which could assess
whether the present prognostic model could benefit clinical
patients.
Tumor immune infiltration
analysis
To evaluate the immune function of the
aforementioned four m7G-regulated genes, the ImmuCellAI database
(http://bioinfo.life.hust.edu.cn) was
first utilized to analyze the infiltration of immune cells in BLCA
and adjacent tissues. Next, Tumor IMmune Estimation Resource
(TIMER; http://cistrome.shinyapps.io/timer) was used to assess
the expression of four m7G-related genes with prognostic
significance in association with the infiltration of six different
immune cell types (B cells, CD4+ T cells,
CD8+ T cells, neutrophils, macrophages, and dendritic
cells). TIMER is a database that uses high-throughput sequencing
data to analyze the infiltration of immune cells in tumor tissues,
and mainly provides the infiltration of the above six immune cell
types (27). In addition, the
TIMER, CIBERSORT, QUANTISEQ, MCPcounter, XCELL, and EPIC algorithms
were used to assess immune component profiles, and the R package
‘limma’ was used for visualization. Furthermore, single sample
(ss)GSEA was used to quantify the infiltration and immune function
of immune cell subsets with the GSVA R software package (version
1.20.0, http://rdocumentation.org/packages/GSVA/versions/1.20.0).
Finally, according to the four-gene expression, boxplots were used
to reveal the differential expression of 24 immune checkpoints in
the two subgroups of risk stratification, including T cell immune
receptor with Ig and ITIM domains (TIGIT), programmed cell death 1
(PD-1), programmed cell death ligand 1 (PD-L1), tumor necrosis
factor receptor superfamily, and cytotoxic T lymphocyte-associated
protein 4 (CTLA4).
Potential drug prediction
To explore potential compounds associated with
four-gene therapy, the four-gene list was input into a connectivity
map (Cmap; http://clue.io/), which included gene
expression signatures derived from 9 cancer cell lines treated with
2,429 well-annotated compounds. Data from the Cmap were compared
with the four-gene signatures to assign connectivity scores. The
scores were inversely correlated with the compound's therapeutic
effect. Next, IC50 values were predicted for standard
chemotherapeutics in the low- and high-risk groups with the R
package ‘pRRophetic’ (28).
Validation of the expression of four
prognosis-relevant proteins
Immunohistochemistry data from the Human Protein
Atlas (HPA; http://www.proteinatlas.org/) was used to validate the
protein expression of the four aforementioned prognosis-relevant
genes between BLCA and normal bladder tissues from TCGA.
Cell culture and small interfering RNA
transfection
Human bladder epithelial cells SV-HUC-1 (cat. no.
CRL-9520™), human bladder carcinoma cells 5637 (cat. no. HTB-9™),
and T24 (cat. no. HTB-4™) were obtained from ATCC. SV-HUC-1 cells
were cultured in Ham's F-12K medium (cat. no. PM150910; Procell
Life Science & Technology Co., Ltd.), while 5637 and T24 cells
were cultured in RPMI 1640 medium (cat. no. C11875500BT; Gibco;
Thermo Fisher Scientific, Inc.). All media were supplemented with
10% heat-inactivated FBS (cat. no. B-9™) and T2 and 1%
penicillin-streptomycin (cat. no. 15070063; Gibco; Thermo Fisher
Scientific, Inc.). The cells were kept in a 37°C incubator with 5%
CO2. Small interfering (si) gem nuclear
organelle-associated protein 5 (GEMIN5) was designed and
synthesized by Guangzhou RiboBio Co., Ltd. and transfected with
Lipofectamine® 3000 (Thermo Fisher Scientific, Inc.). At
48 h post-transfection, the cells were collected for functional
experiments. The interference efficiency was detected by reverse
transcription-quantitative PCR (RT-qPCR).
RT-qPCR
Total RNA was extracted from the three cell types
using TRIzol® reagent (cat. no. 15596-026; Ambion;
Thermo Fisher Scientific, Inc.). cDNA was synthesized using a cDNA
reverse transcription kit (cat. no. EP0751; Thermo Fisher
Scientific, Inc.). cDNA was used as a template, and the β-actin
gene was used as the internal reference for normalizing the RT-qPCR
data. A two-step standard PCR amplification procedure was conducted
according to the manufacturer's instructions (cat. no. 10222ES60;
Shanghai Yeasen Biotechnology Co., Ltd.). The relative expression
of the target gene was calculated using the 2−ΔΔCq
method (29). The primers used are
shown in Table SI.
Cell viability and colony formation
assays
The viability of T24 and 5637 cells was detected
using a Cell Counting Kit-8 (CCK-8) assay kit (cat. no. BS350B;
Beijing Labgic Technology Co., Ltd.) according to the
manufacturer's instructions. The optical density was measured with
a microplate reader (Thermo Fisher Scientific, Inc.) at a
wavelength of 450 nm. The cell viability values of each group were
detected after 24, 48, and 72 h. For the colony formation assays,
cells in the logarithmic growth stage were collected, and 1,000
cells were plated in six-well plates. The cells were cultured for
10 days, fixed with methanol at room temperature for 20 min, and
stained with 0.1% crystal violet at room temperature for 15 min.
The colonies were imaged and counted using light microscopy. The
number of colonies consisting of ≥50 cells was counted.
Statistical analysis
All data were statistically analyzed using R. FDR
was used in GO and KEGG analyses. Gene expression and immune
infiltration levels were calculated using a paired Student's t-test
or Wilcoxon test. Differences in OS between groups were compared
using Kaplan-Meier analysis. Independent predictors were evaluated
using Cox regression analysis. A two-tailed P<0.05 was
considered to indicate a statistically significant difference.
Results
Identification of differentially
expressed m7G-related genes in BLCA
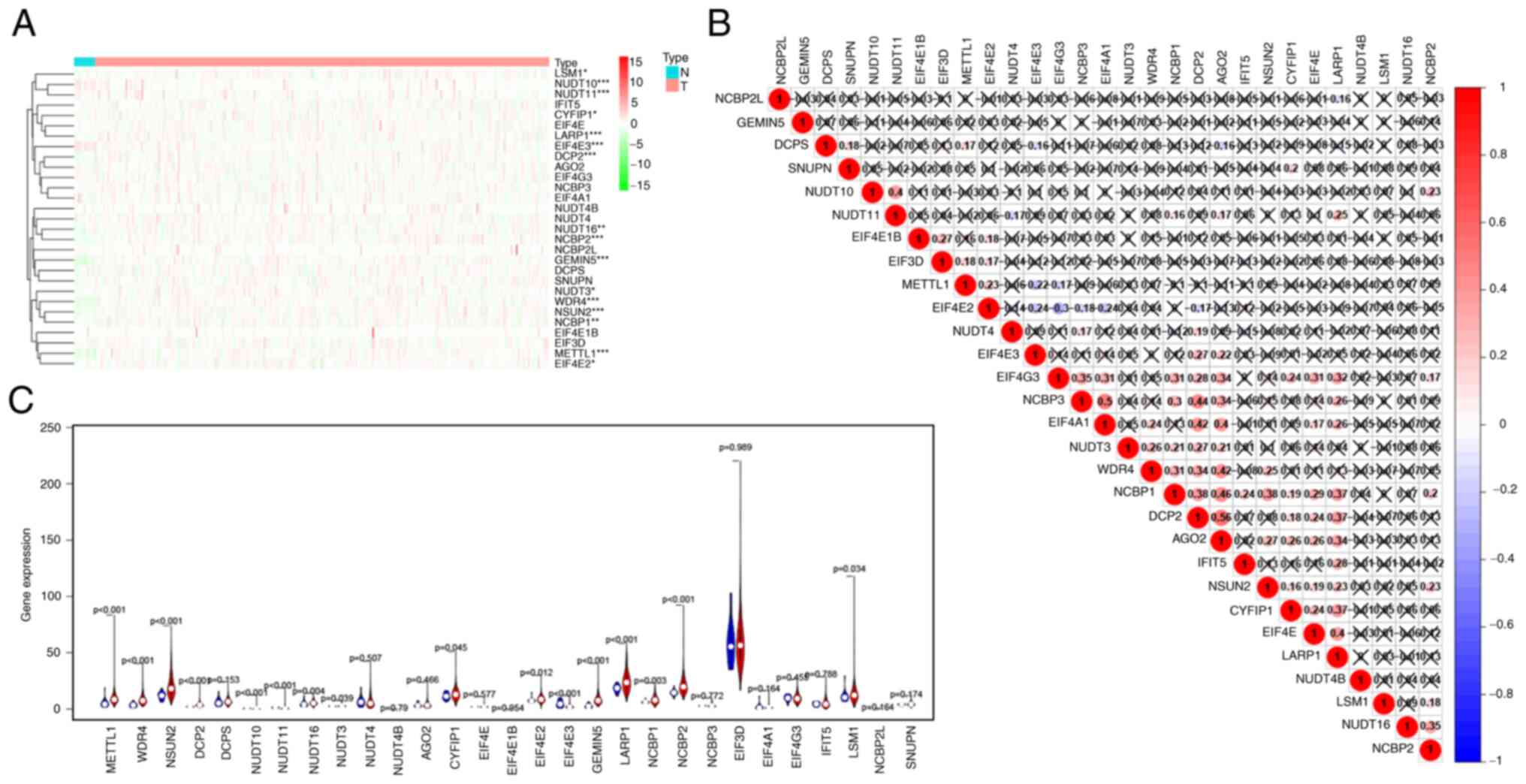
Differential expression analysis of 29 m7G-regulated
genes was performed in BLCA (n=414) and adjacent normal tissues
(n=19). The heatmap showed that 17 of these m7G modification
regulators were differentially expressed between BLCA and normal
tissues (Fig. 2A). The expression
levels of METTL1, WDR4, NSUN2, DCP2, nudix hydrolase (NUDT)10,
NUDT11, NUDT16, NUDT3, AGO2, cytoplasmic FMR1 interacting protein 1
(CYFIP1), EIF4E2, EIF4E3, GEMIN5, LARP1, NCBP1, NCBP2, and LSM1
were higher in BLCA tissues than in normal tissues (Fig. 2B). Fig.
2C showed that the correlation between GEMIN5 and LARP1 was the
most significant (correlation coefficient, 0.71). The GEPIA2
database results showed that the expression of EIF4E3, NUDT10,
NUDT11 and METTL1 significantly differed between the two groups
(Fig. S1). RT-qPCR was performed
to confirm the expression of prognostic m7G-related genes in
bladder cancer cells. A total of 17 gene symbols were imported into
the STRING database to generate the PPI network. The genes with the
highest clustering coefficient were LARP1, GEMIN5, NUDT11, NUDT10,
and WDR4 (Fig. S2).
Identification of two clusters of
patients with BLCA with distinct clinical outcomes using consensus
clustering based on m7G RNA-modification regulators
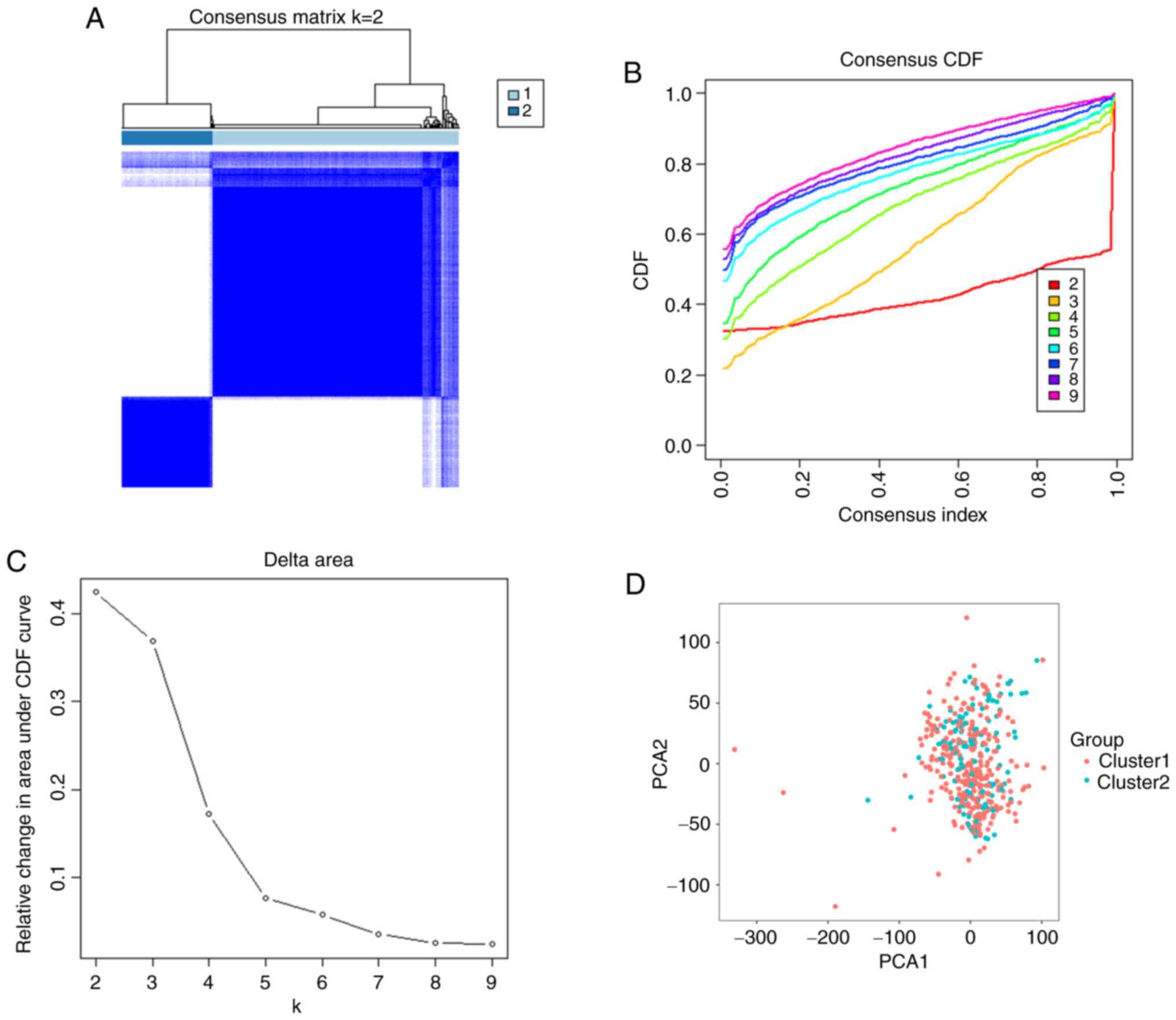
To further explore the clinical relevance of m7G
RNA-modifying modulators, patients with BLCA were clustered into
subgroups according to the differential gene expression. Based on
the similarity of m7G-related genes, k=2 produced the best
clustering, and patients with BLCA could be divided into two
distinct and non-overlapping groups (Fig. 3A-C). Principal component analysis
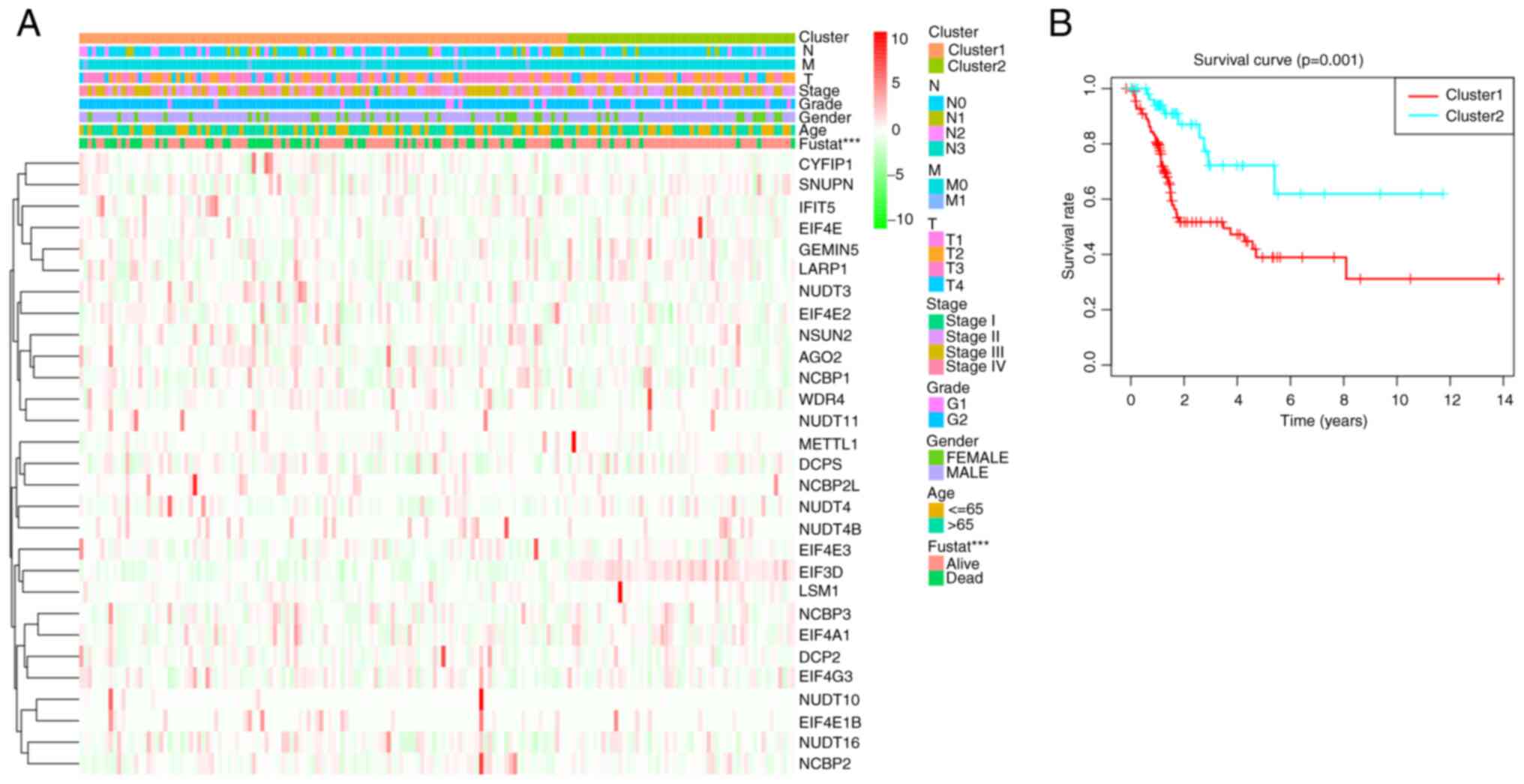
was used to validate the results of clustering (Fig. 3D). Subsequently, whether there were
significant differences in OS, stage, age, grade, or sex between
these two clusters was evaluated. The results showed that the
prognosis in cluster 2 was significantly better (P<0.01) than
that of cluster 1 (Fig. 4B), but no
significant differences were observed in age, sex, histological
grade, or pathological stage between the two clusters (Fig. 4A). Next, the m7G genes associated
with prognosis were screened out for in-depth analysis based on a
heat map. The results of consensus clustering showed a strong
association between the expression patterns of m7G-related genes
and clinical parameters.
Functional enrichment analysis of m7G
RNA-modification regulators
GO and KEGG enrichment analyses were performed on
the DEGs between clusters 1 and 2 to investigate the results of
clustering from the perspective of associated pathways and
biological processes. According to the results of the GO enrichment
analysis, the upregulated genes were primarily enriched in
‘Extracellular matrix organization’, ‘Extracellular structure
organization’, ‘External encapsulating structure organization’,
‘Phagocytosis, recognition’, ‘Complement activation, Classical
pathway’, ‘Humoral immune response mediated by circulating
immunoglobulin’, ‘Phagocytosis, engulfment’, ‘Complement
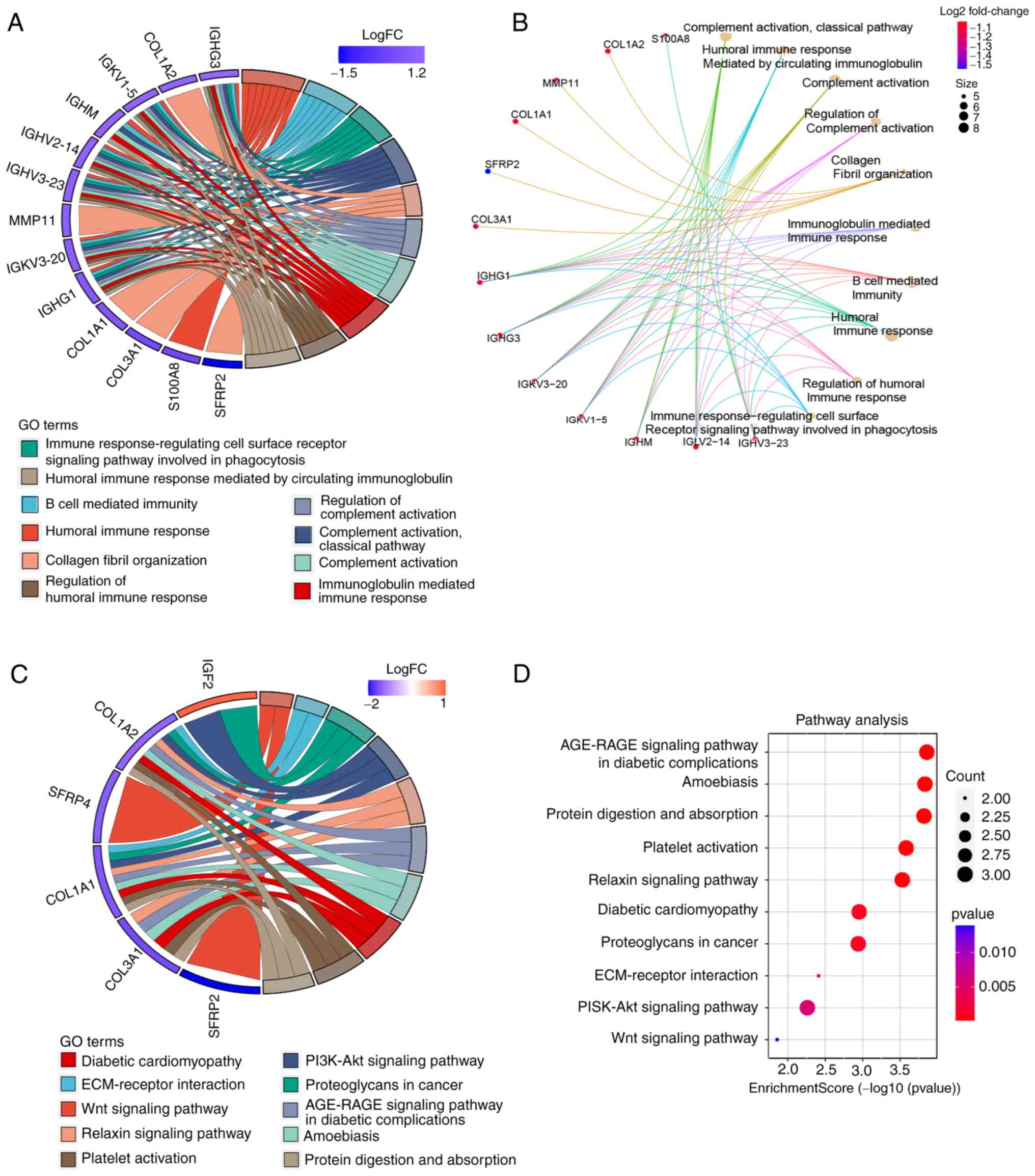
activation’, and ‘B cell receptor signaling pathway’ (Fig. 5A and B). KEGG enrichment analysis
results showed that these upregulated genes were significantly
enriched in ‘Proteoglycans in cancer’, ‘Wnt signaling pathway’,
‘ECM-receptor interaction’, and ‘PI3K-Akt signaling pathway’
(Fig. 5C and D).
Construction of a prognostic model
based on four m7G-regulated genes
Based on the association between m7G RNA methylation
regulators and the OS of patients with BLCA, Univariate Cox
regression analysis was performed on the expression levels of the
29 important regulators to explore the clinical relevance. The
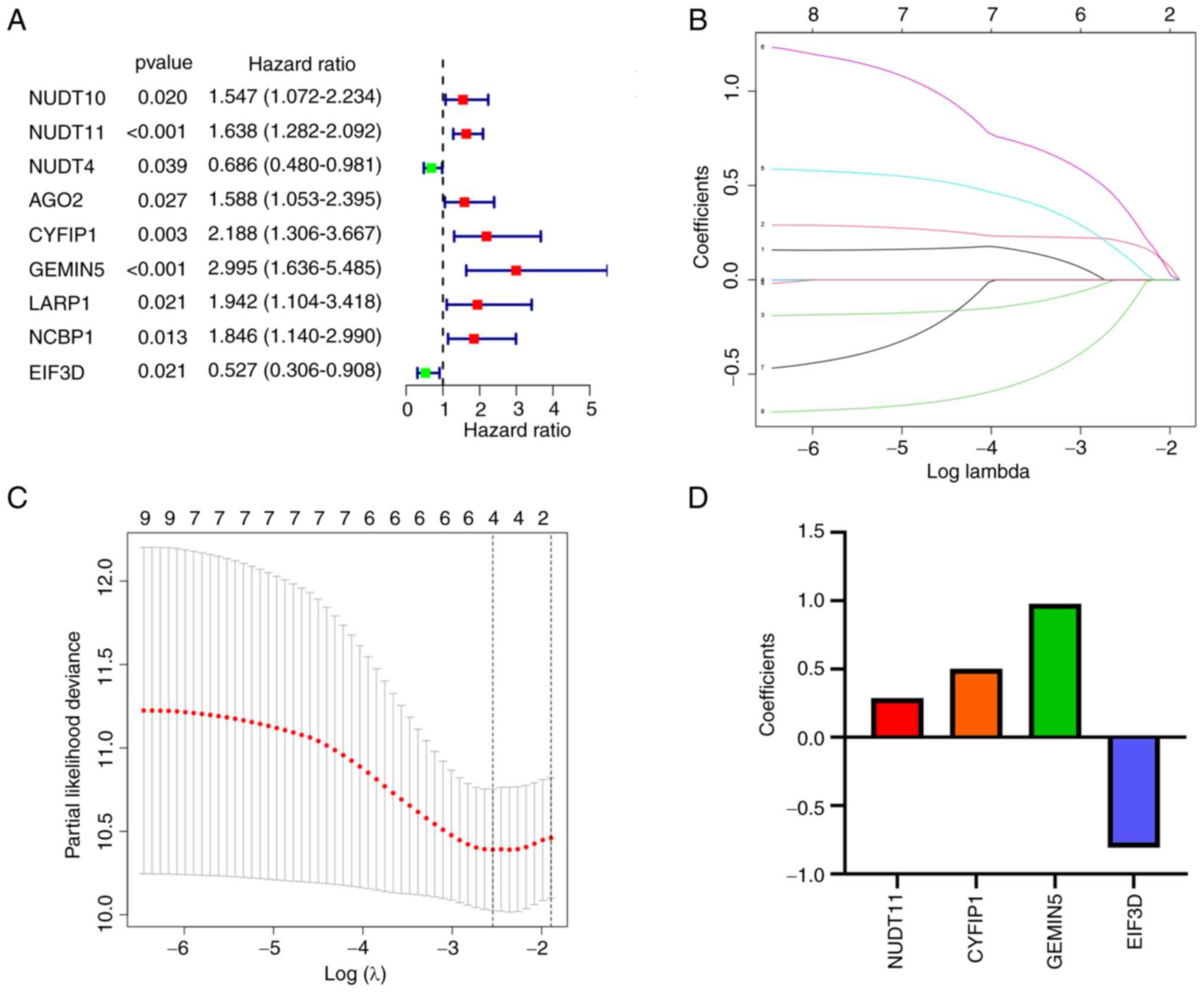
results showed that 9 of the 29 genes were notably associated with
OS (P<0.05). As shown in Fig.
6A, NUDT10, NUDT11, AGO2, CYFIP1, GEMIN5, LARP1, and NCBP1 were
considered risk genes with a hazard ratio (HR) >1, whereas NUDT4
and eukaryotic translation initiation factor 3 subunit D (EIF3D)
were considered protective genes with HR<1.
LASSO Cox regression analysis identified 9 genes
with the strongest predictive ability (Fig. 6B and C). Based on the corresponding
coefficients in the LASSO algorithm, four optimal genes (NUDT11,
CYFIP1, GEMIN5, and EIF3D) were selected to establish the BLCA risk
model (Fig. 6D). The risk score was
calculated as follows: Risk score=(0.289× expression value of
NUDT11) + (0.503× expression value of CYFIP1) + (0.979× expression
value of GEMIN5)-(0.807× expression value of EIF3D).
To explore the prognostic role of these four-gene
signature models, patients with BLCA were divided into low- and
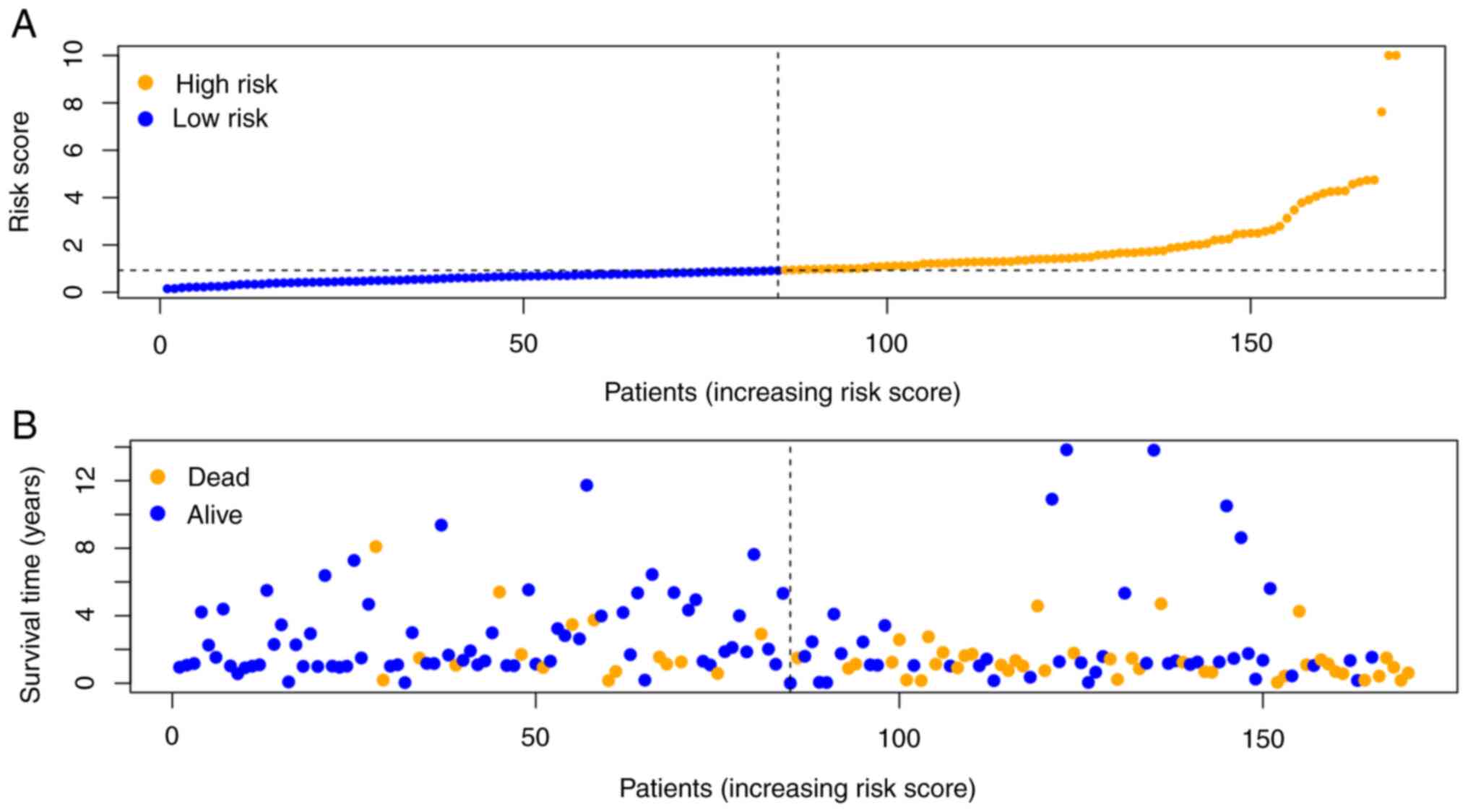
high-risk groups according to the median risk score. The risk score
distribution of patients with BLCA was plotted (Fig. 7A), and the survival status of
patients with BLCA was evaluated using a dot matrix (Fig. 7B). Survival analysis showed that
patients with high-risk scores had a poorer OS than those with
low-risk scores (Fig. 8A;
P<0.001). The 5-year OS rate was 67.3% in the low-risk group and
24.5% in the high-risk group. The ROC curve analysis results showed
that the area under the curve (AUC) at 1-, 3- and 5-year OS was
0.755, 0.745, and 0.758, respectively, thus showing good predictive
power for survival outcome (Fig.
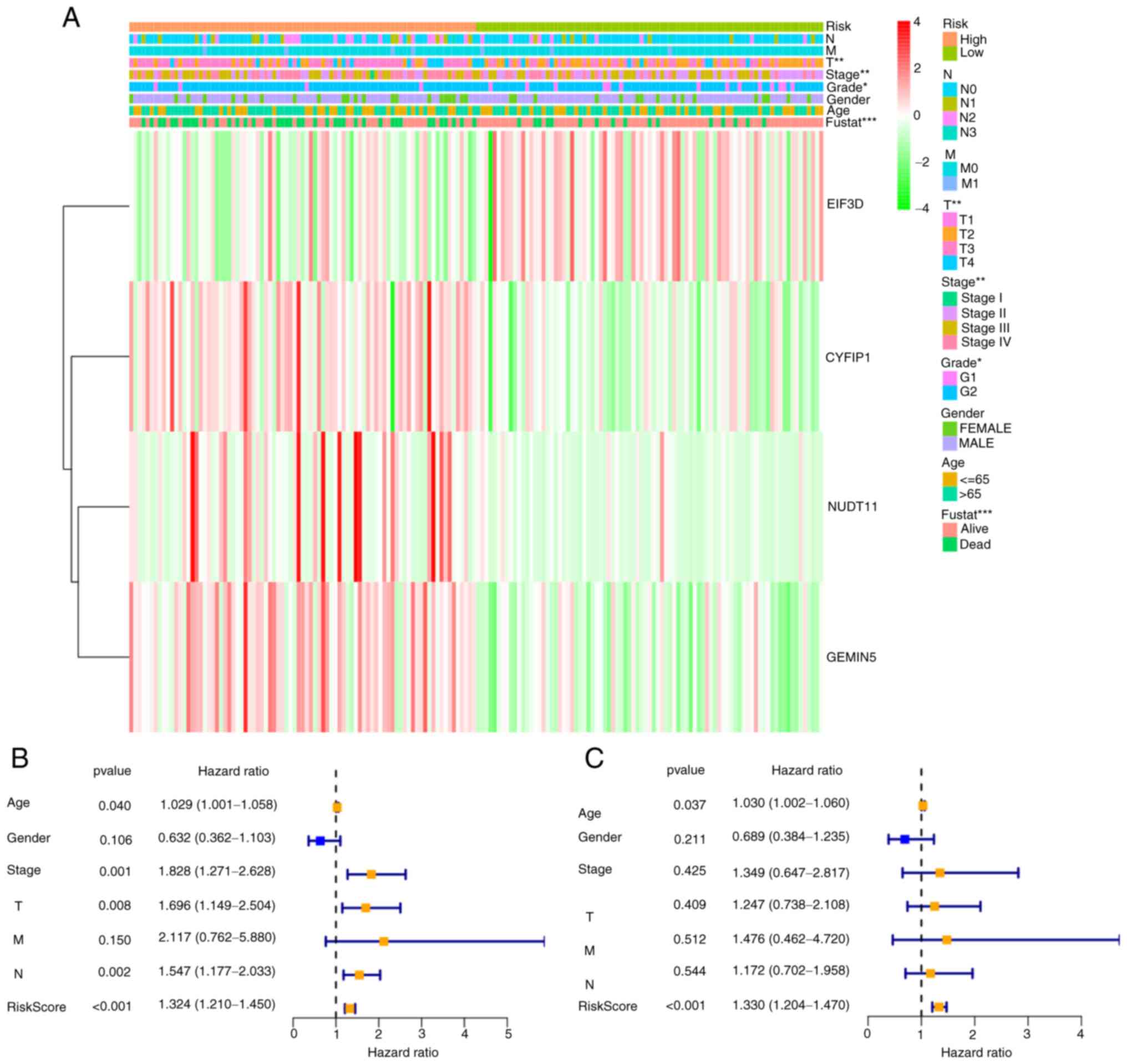
8B). A heatmap with clinical relevance showed the differential
expression of the four prognostic genes in the high- and low-risk
groups (Fig. 9A). Of note,
significant differences in clinical parameters such as fustat
(P<0.001), stage (P<0.01) and grade (P<0.05) were
observed.
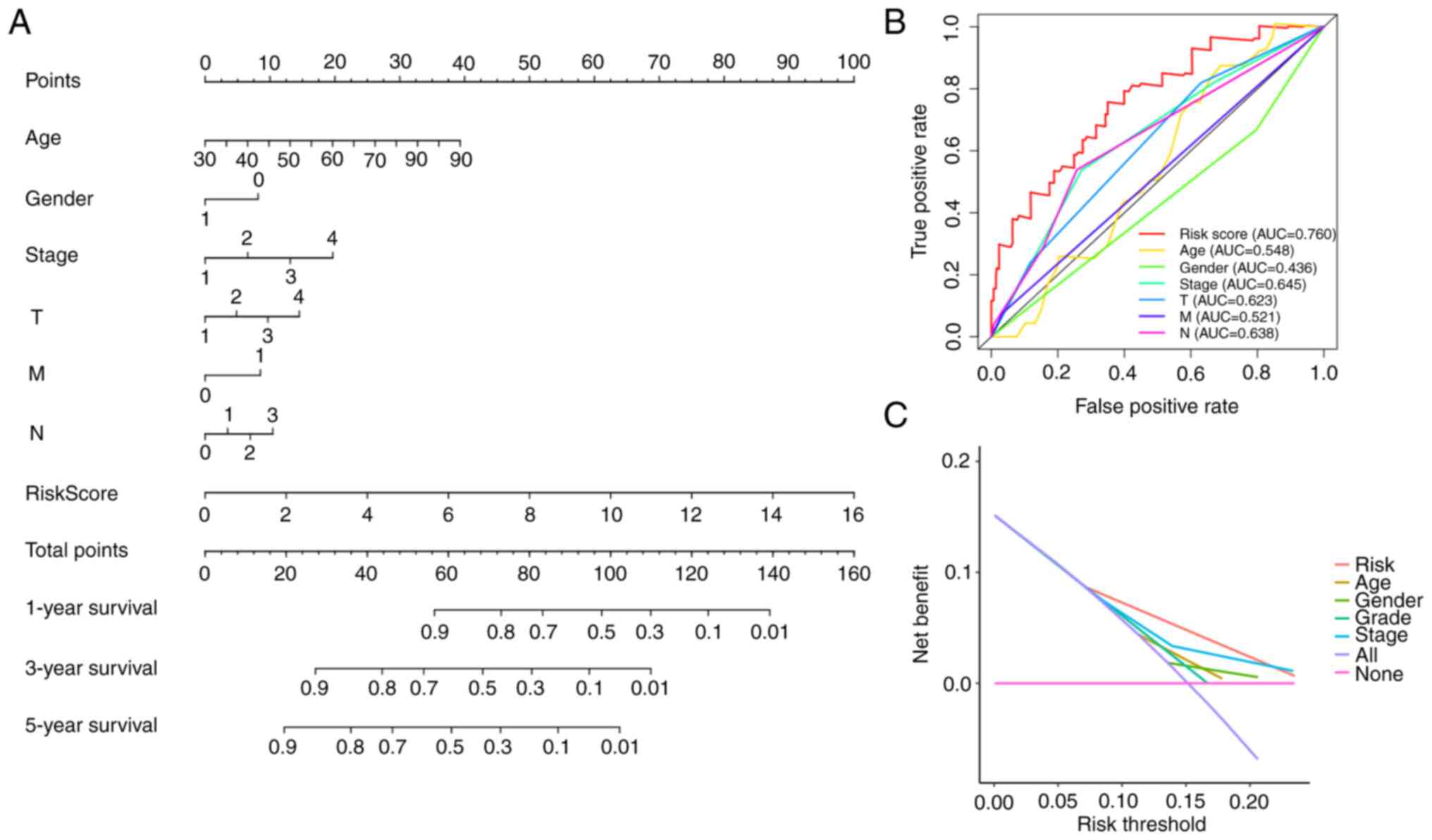
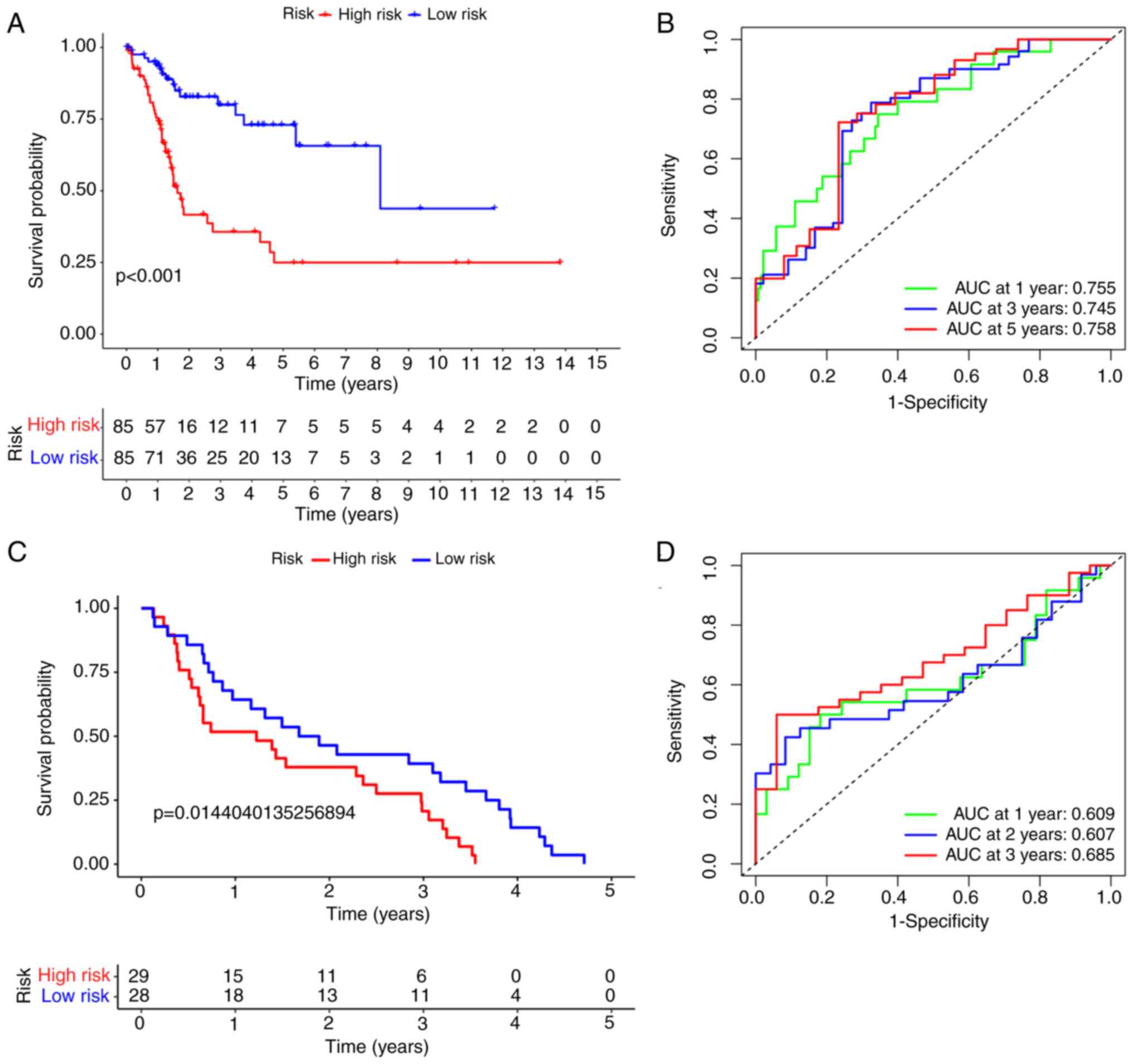 | Figure 8.Kaplan-Meier survival analysis of the
prognostic model. Patients in both datasets were assigned to
low-risk (blue) or high-risk (red) groups, using the median risk
score as the cutoff value. (A) Survival curve analysis of TCGA
cohort. In TCGA cohort, the low-risk group had a higher probability
of survival than that of the high-risk group (P<0.001). (B) ROC
curve analysis of TCGA cohort. The 1-, 3-, and 5-year AUCs were
0.755, 0.745, and 0.758, respectively. (C) Survival curve analysis
of the GEO cohort. The low-risk group had a higher probability of
survival than that of the high-risk group (P=0.014). (D) ROC curve
analysis of the GEO cohort. The 1-, 2-, and 3-year AUCs were 0.609,
0.607, and 0.685, respectively. TCGA, The Cancer Genome Atlas; ROC,
receiver operating characteristic; AUC, area under the curve; GEO,
Gene Expression Omnibus. |
GEO database validates the predictive
performance of the risk model
To estimate the predictive capability of four-gene
signatures in GEO datasets, the GSE48075 dataset was used for
validation. A total of 57 patients with BLCA in the GSE48075 cohort
were divided into low- (n=29) and high-risk (n=28) groups according
to the cutoff value of TCGA cohort. The results of survival
analysis showed that patients with BLCA in the low-risk group had a
significantly better OS compared with that of patients in the
high-risk group (P=0.014; Fig. 8C),
which was consistent with the results on the training set. The AUCs
for 3-year OS was 0.685, suggesting that the risk model had a good
predictive ability in BLCA (Fig.
8D). Since this cohort did not survive >5 years, no 5-year
ROC curves were drawn.
Four-gene risk signature independently
predicts prognosis in patients with BLCA
Upon removing samples without complete clinical
information, 170 samples were qualified for Cox regression
analysis. Cox univariate analysis indicated that stage and the
four-gene risk score were significantly associated with OS in
patients with BLCA (P<0.001; Fig.
9B). Multivariate Cox regression analysis was performed to
assess whether risk score was independent of other
clinicopathological features as a predictor for BLCA, and the
results showed that the risk score of patients with BLCA was
independently associated with OS (P<0.001; Fig. 9C). In conclusion, these results
indicated that the four-gene risk signature could be used to
predict the prognosis of patients with BLCA independently of other
clinicopathological features such as sex, histological grade, age,
or pathological stage.
Construction of a nomogram
To facilitate the clinical application of four-gene
risk signature, a detailed prognostic nomogram based on
histological grade, sex, pathological stage, age, and risk score
was established (Fig. 10A). The
nomogram could precisely predict 1-, 3-, and 5-year OS in patients
with BLCA. The AUC for risk score was 0.76, which indicated that
this model had good predictive value for patients with BLCA
(Fig. 10B). Nomogram-based
clinical decisions had a higher net benefit compared with that of
individual predictors (Fig.
10C).
Correlation of prognosis-related m7G
genes with immune infiltration
GO and KEGG enrichment analyses indicated that
m7G-related gene functions primarily involve the activation of
cancer-related signaling pathways, affecting complement and B cell
activity. Further immune infiltration analysis revealed significant
differences in normal adjacent and BLCA tissues. Differential
expression of immune cells (Fig.
11A-Q) indicated that tumor immune infiltration is important in
patients with BLCA. According to the TIMER, CIBERSORT, QUANTISEQ,
and MCPCOUNTER algorithms, the expression levels of CD8+
T cells, CD4+ T cells, neutrophils, macrophages, and
monocytes in the high-risk group were higher than those in the
low-risk group (Fig. 11R),
suggesting that immune infiltration may influence patient outcomes.
Quantifying enrichment fractions implied that the prognosis in the
high-risk groups may be affected by antigen-presenting cells'
function, chemokine receptor, immune checkpoint expression, and
parainflammation (Fig. 11S).
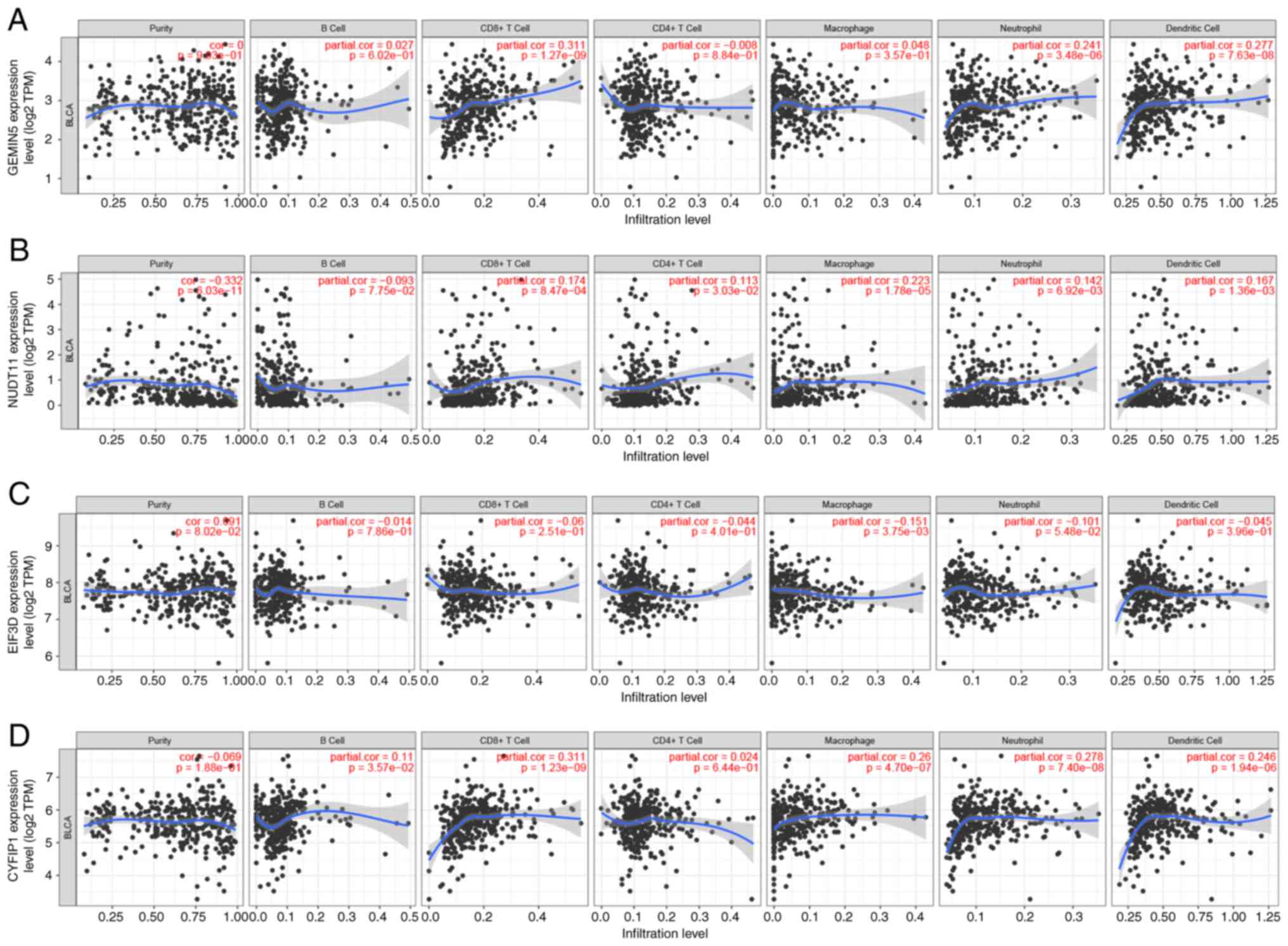
The TIMER database was used to investigate the link
between the expression levels of four genes and immune
infiltration. After adjusting for purity, the expression of GEMIN5
(Fig. 12A) was positively
correlated with CD8+ T cells (P=1.27×10−9),
dendritic cells (P=7.63×10−8), and neutrophils
(P=3.48×10−6). NUDT11 expression levels were correlated
with the number of CD8+ T cells
(P=8.47×10−4), CD4+ T cells
(P=3.03×10−2), dendritic cells (P=1.36×10−3),
macrophages (P=1.78×10−5) and neutrophils
(P=6.92×10−3) (Fig.
12B). EIF3D expression levels were negatively correlated with
macrophage levels (P=1.78×10−5) (Fig. 12C). CYFIP1 expression levels were
significantly correlated with the number of B cells
(P=3.57×10−2), CD8+ T cells
(P=1.23×10−9), dendritic cells (P=1.94×10−6),
macrophages (P=4.7×10−7) and neutrophils
(P=7.4×10−8) (Fig.
12D). As shown in Fig. S3,
copy number loss of NUDT11 resulted in reduced B cell and
neutrophil infiltration, while loss of GEMIN5 and CYFIP1 copy
number reduced the infiltration of CD4+ T cells. Loss of
EIF3D copy number resulted in increased levels of dendritic cells.
Kaplan-Meier curve results indicated that enrichment of the four
genes in CD8+ T cells was associated with a poorer prognosis in
patients with BLCA.
Exploring a potential treatment for
patients with BLCA based on four genes
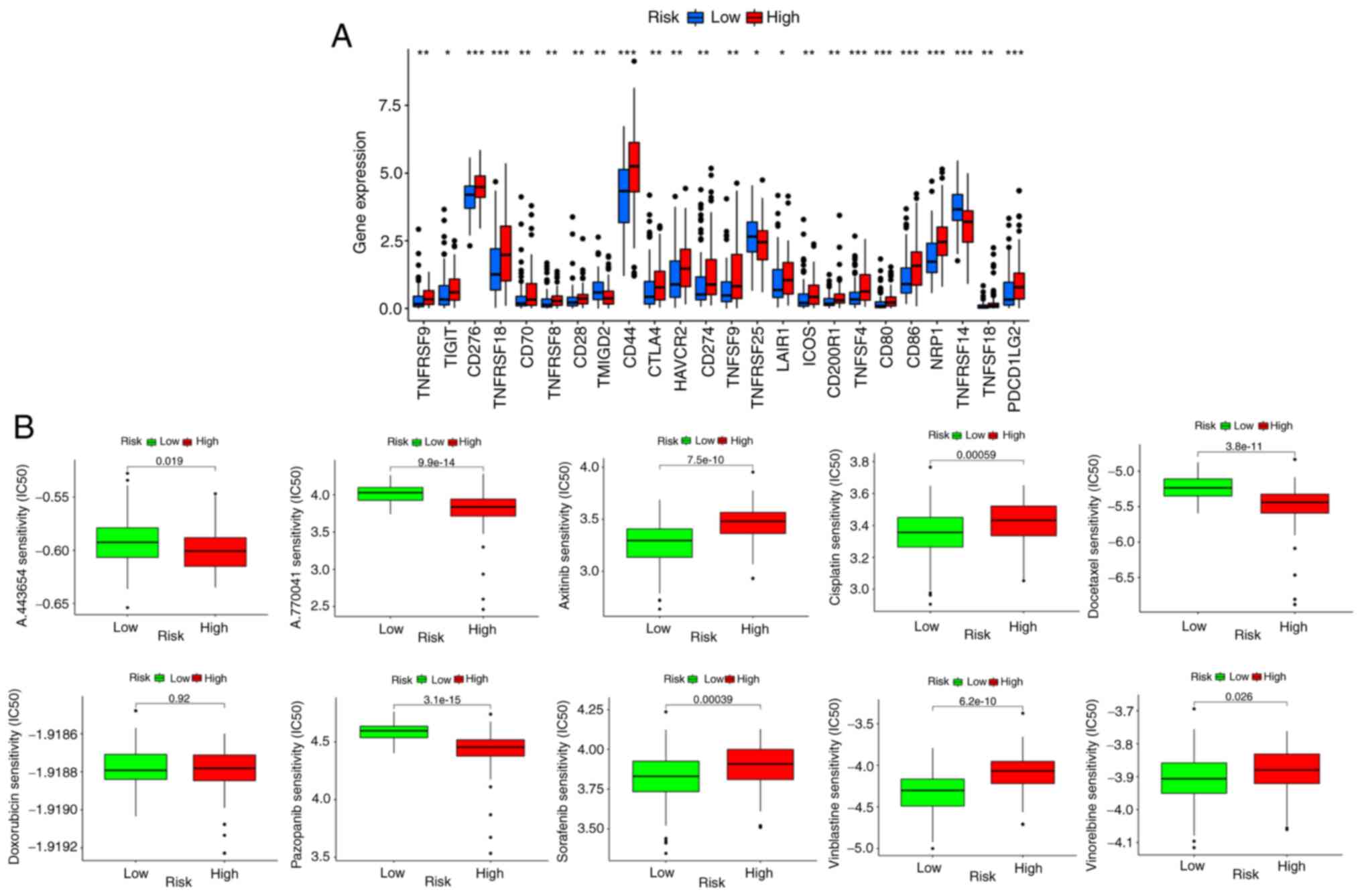
A clear association was found between high-risk
patients stratified based on four genes and the expression of
various immune checkpoints, such as CTLA4, PD-1, PD-L1, and PD-L2
(Fig. 13A). Immune checkpoint
inhibitors may be effective in these patients. Sensitivity analysis
of chemotherapeutic drugs showed that A.443654, A.770041,
docetaxel, and pazopanib were effective in low-risk groups, while
axitinib, cisplatin, sorafenib, vinblastine, and vinorelbine may be
more effective in high-risk groups. There was no difference in the
sensitivity to doxorubicin between the two groups (Fig. 13B). All DEGs were divided into up
and downregulated groups and uploaded to the CMap database to
identify candidate small molecule drugs for treating BLCA. With
P<0.01 and n>2 as the screening criteria, 10 small-molecule
drugs with treatment effects on BLCA were identified:
BRD-K50174388, I-BET-762, beclomethasone-dipropionate,
BRD-K64233461, KU-C104131, BRD-K41668190, erlotinib, BRD-K53120552,
BRD-K23021002, and heptaminol (Table
I). A negative enrichment score represented an inhibitory
effect.
 | Table I.The top 10 small molecule drugs in
the connectivity map dataset. |
Table I.
The top 10 small molecule drugs in
the connectivity map dataset.
| Compounds | Enrichment | P-value | n | Percent non-null
(%) | Type |
|---|
| BRD-K50174388 | −0.6759 | 0.0081a | 3 | 100 | NA |
| I-BET-762 | −0.6758 | 0.0025a | 3 | 100 | Bromodomain
inhibitor |
|
Beclomethasone-dipropionate | −0.6758 | 0.0025a | 3 | 100 |
Immunosuppressant |
| BRD-K64233461 | −0.6757 |
<0.001b | 3 | 100 | NA |
| KU-C104131 | −0.6757 |
<0.001b | 3 | 100 | NA |
| BRD-K41668190 | −0.6757 |
<0.001b | 3 | 100 | NA |
| Erlotinib | −0.6756 |
<0.001b | 3 | 100 | EGFR inhibitor |
| BRD-K53120552 | −0.6755 |
<0.001b | 3 | 100 | NA |
| BRD-K23021002 | −0.6755 |
<0.001b | 3 | 100 | NA |
| Heptaminol | −0.6754 |
<0.001b | 3 | 100 |
Vasoconstrictor |
Immunohistochemistry validation based
on the HPA database
As shown in Fig.
S4, the protein expression levels of GEMIN5 and CYFIP1 were
elevated in BLCA compared with those in normal bladder tissues.
NUDT11 expression was more evident in normal bladder tissues than
in cancer tissues. Of note, EFI3D was not included in the figure as
it was not included in the aforementioned database.
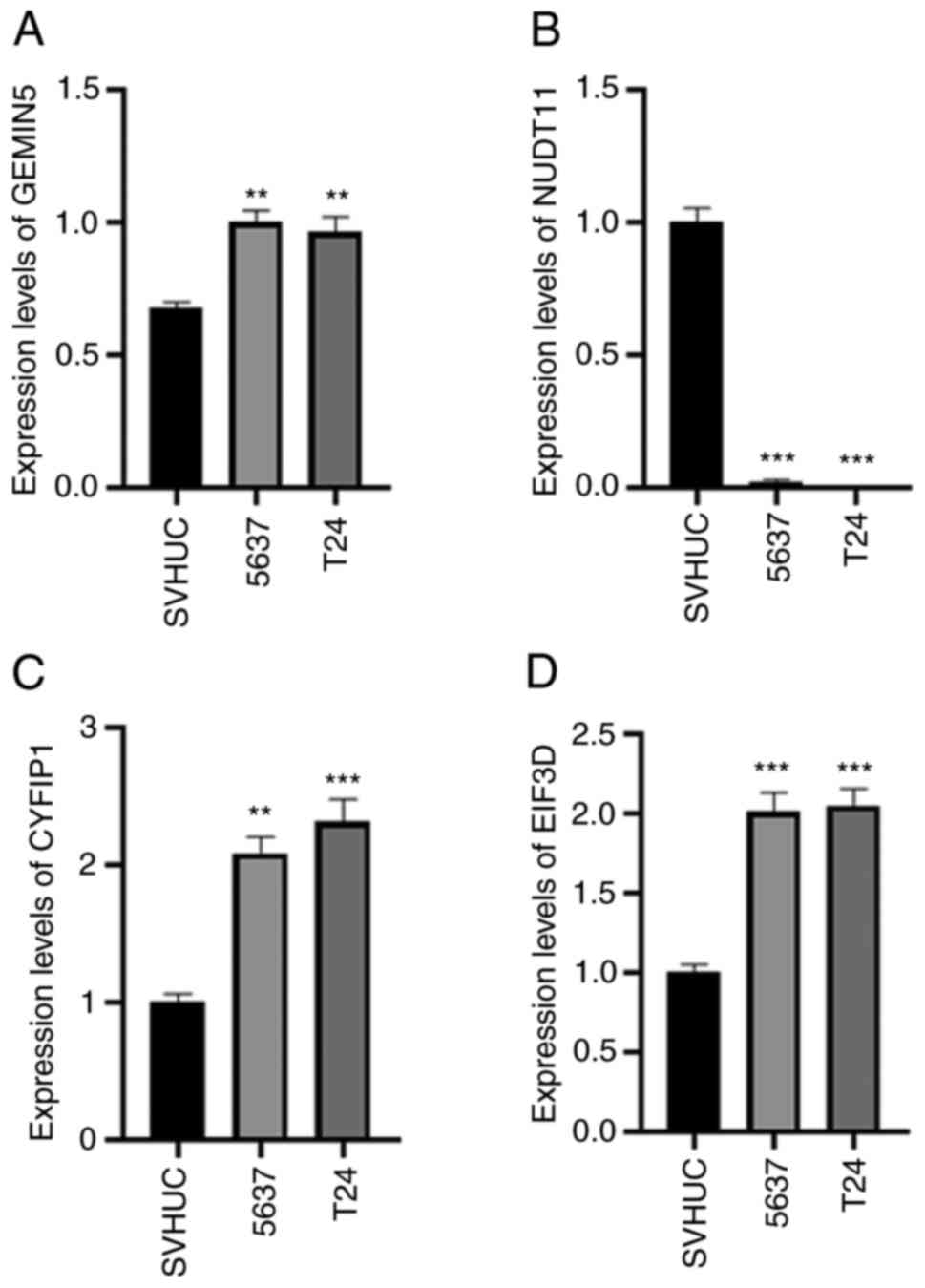
RT-qPCR-mediated verification of the
mRNA expression of four genes in BLCA cell lines
The RT-qPCR results showed that GEMIN5, CYFIP1, and
EIF3D exhibited high levels of expression in T24 and 5637 cells,
while NUDT11 exhibited a high expression level in SV-HUC-1 cells.
The results were statistically significant (Fig. 14).
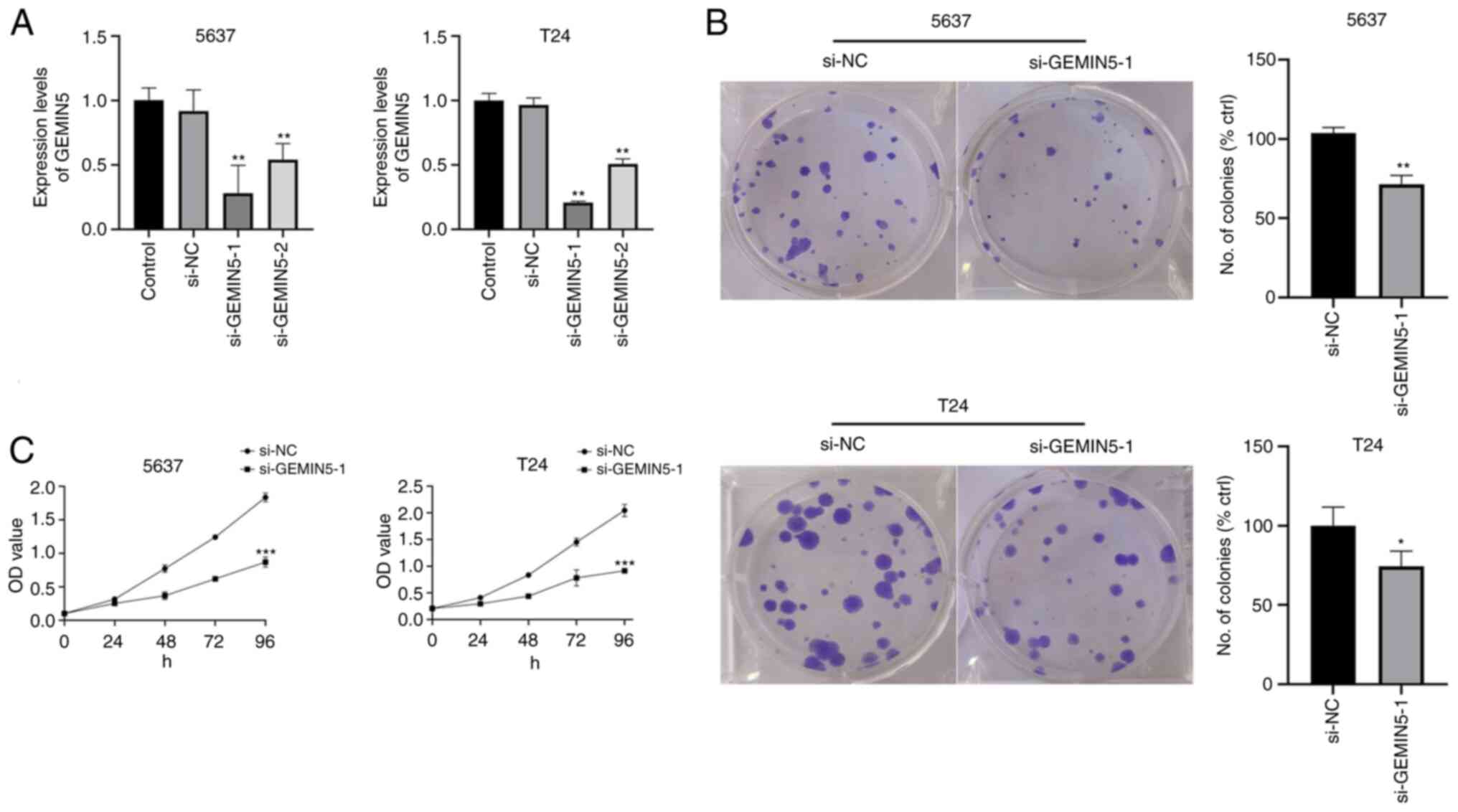
Knockdown of GEMIN5 suppresses bladder
cancer cell proliferation
To validate the BLCA proliferation-promoting ability
of m7G-associated genes, GEMIN5 was selected, since it had the
highest HR value in the risk model, to perform CCK-8 and colony
formation assays. si-GEMIN5 was used to knock down the expression
of GEMIN5 in T24 and 5637 cells, and si-GEMIN5-1 had the best
knockdown effect (Fig. 15A). CCK-8
(Fig. 15B) and colony formation
(Fig. 15C) assays revealed the
that knockdown of GEMIN5 inhibited the proliferation of T24 and
5637 cells.
Discussion
BLCA is a lethal solid tumor with complex molecular
and cellular heterogeneity (30).
Molecular markers associated with BLCA presently used in the clinic
include urinary nuclear matrix protein 22, bladder tumor antigen,
and fibrin degradation product. However, these tumor markers have
low specificity and poor predictive ability. Thus, there is an
urgent need to identify novel BLCA biomarkers that have i)
predictive and prognostic value, ii) diagnostic value, and iii) can
be used to support the development of personalized treatment plans.
With the development of bioinformatics-based techniques, numerous
studies have confirmed the predictive role of novel biomarkers in
BLCA (31–33).
As a relatively more recently identified mechanism
of gene regulation, RNA m7G modification has attracted large
interest recently. m7G is a general tRNA modification in
prokaryotes and eukaryotes, and is catalyzed by METTL1/WDR4, an
integral complex involved in epigenetic regulation (34). m7G at position 46 of tRNA is present
in the variable loop, and it is likely that METTL1 reduces tRNA
stability by affecting this structure (35). In addition to tRNAs, m7G regulates
the translation of mRNA. Different methyltransferases incorporate
m7G onto different mRNAs and/or into secondary structure motifs,
thus exerting effects on mRNAs, including translation (36). A general mechanism has been found to
be involved in m7G-mediated cancer, in which impaired m7G
selectively suppresses the translation efficiency of oncogenic
mRNAs with a higher frequency of m7G tRNA cognate codons by
prolonging ribosome pausing periods (37). As evidence of the regulatory role of
m7G in mRNA and microRNA regulation is continuously being
uncovered, it is necessary to investigate the role of RNAs other
than tRNA.
Although the study of m7G modification in the field
of cancer is still in its infancy, combined with bioinformatics
analysis, it may still provide clues for subsequent basic and
clinical research. The present study demonstrated that the majority
of m7G-related genes were expressed at higher levels in BLCA than
in non-BLCA tissues, and identified four genes associated with BLCA
prognosis (namely GEMIN5, NUDT11, CYFIP1, and EIF3D), which were
shown to be differentially expressed in bladder cancer cell lines
based on RT-qPCR validation. Through KEGG and GO enrichment
analyses, the potential biological functions of m7G-related genes
in BLCA were explored. Cell experiments also demonstrated that the
viability and proliferation of T24 and 5637 cells were
significantly inhibited by knocking down GEMIN5. Next, the training
set (TCGA cohort) was used to construct the predictive model, and
the validation set (GEO cohort) was used to test the reliability of
the model. The risk score calculated based on the four-gene model
could forecast BLCA patient prognosis independently. Predictive
nomograms that considered both clinicopathological features and
risk scores were also built. Consequently, the risk signature
reported on the present study could help clinicians make precise
personalized survival predictions. Moreover, the current study
found that m7G-related genes were closely associated with the
immune microenvironment of BLCA, and immune infiltration in
patients with BLCA could also be predicted based in the present
risk model. Together, this prognostic model was strongly associated
with clinicopathological factors, immune cells and immune-related
functions.
As an essential regulator of m7G, GEMIN5 expression
was found to be significantly upregulated in BLCA and associated
with a poor patient prognosis. GEMIN5 can regulate translation, and
has been reported to specifically bind the m7G cap (38). Other studies have shown that its
expression is increased in gastric cancer tissue, acute myeloid
leukemia, and liver cancer (17,39,40).
Furthermore, GEMIN5 can exert oncogenic effects by regulating mRNA
splicing and tumor cell motility, and by reprogramming cellular
translation (41,42). CYFIP1 is a newly identified tumor
suppressor. CYFIP1/2, WASF1/2/3, NCKAP1/1 L, Abi1/2/3, and BRK1
form a heteropentameric complex called the WASF regulatory complex
(43). Abnormal distribution of
CYFIP1 has been observed with cancer metastasis and invasion, and a
previous study managed to inhibit breast cancer metastasis by
blocking CYFIP1 and Rac1 protein interactions, actin
polymerization, and β1-integrin/FAK/Src signaling (44). Moreover, a stapled peptide targeting
NCKAP1 and CYFIP1 was designed to destabilize the WASF3 complex to
inhibit invasion (45). NUDT11 is a
phosphoinositide phosphohydrolase, and the turnover of
bisphosphonates can affect cancer cell apoptosis (46). The present results suggested that
NUDT11 was expressed at a lower level in BLCA than in normal tissue
cells, but high levels of its expression implied a worse prognosis.
A previous study showed that, when NUDT11 was suppressed, the
proliferation and viability of prostate cancer cells were affected,
and colony formation was significantly reduced (47). EIF3 is the largest and most complex
ribosomal EIF complex (48); it
binds to the 40S ribosome and maintains the dissociation of the 40S
and 60S ribosomal subunits. The function of EIF3 in promoting or
inhibiting tumor progression is controversial. For example, EIF3D
has been reported to promote colon cancer (49), melanoma (50), and breast cancer (51), but it is expressed at lower levels
in liver cancer than in normal tissues. and is associated with a
better prognosis (52). An
examination of EIF3E mRNA levels in non-small cell lung carcinoma
and breast cancer showed low levels in ~1/3 of the samples
(53). However, the EIF3E levels in
human glioblastoma cells were high, and siRNA-mediated knockdown
inhibited cell proliferation (54).
The present results suggested that EIF3D was a suppressor of BLCA,
and that increased expression was associated with a better
prognosis for patients. However, various claims about the
association between the EIF3 subunit and cancer lack strength and
appear to be based on experiments devoid of strict controls
(55,56). Changes in EIF3 in other parts of the
cell or in functions such as the activation of transcription
factors or protein kinases can influence the status of cancer
(57). In conclusion, the role of
EIF3 is complex, and the same EIF3 subunit could have opposite
roles in different cancer types. The biological processes
associated with the effect of EIF3D on BLCA need to be further
investigated.
Cancer immunity plays a key role in cancer
progression, and increasing evidence suggests that the tumor immune
microenvironment is vital for the initiation and progression of
bladder cancer (10,58). Notably, the current results showed
that, although immune cells such as CD8+ T cells
markedly infiltrated BLCA, they did not appear to be effective in
killing cancer cells in the high-risk group; instead, high levels
of CD8+ T cell infiltration led to a worse prognosis.
Immune functional analysis revealed that the high-risk group had
higher levels of cancer-associated fibroblasts, which could produce
fibrous cocoons to protect cancer cells. Excessive parainflammation
can transform M1-type macrophages into M2-type macrophages, thus
inhibiting immune functions and promoting tumor progression
(59). Chemokines released by tumor
cells can recruit T cells to reach the tumor tissue and be
activated by tumor antigens. However, only 10% of T cells recruited
to the tumor microenvironment can recognize tumor cells; the rest
act as ‘bystanders’ and have no cytotoxic effect on tumor cells,
such as exhausted T cells that express high levels of the immune
checkpoint molecules PD-1, CTLA4, and TIGIT (60). A recent study defined tumor-derived
lactate as an inhibitor of CD8+ T cell cytotoxicity
(61). Therefore, in high-risk
patients defined based on the aforementioned risk model, the tumor
could have strong immune evasion abilities. Importantly, the
present study found that immune checkpoint molecules were
significantly expressed in the high-risk group, and immunotherapy
may thus be effective for these patients. Designing anti-BLCA drugs
against these targets could be one future approach. Moreover, the
current study found 10 compounds and 9 drugs to potentially treat
BLCA. However, the present study has certain limitations. As the
study was based on information within public databases, real-world
prospective cohort studies are required to validate the risk score
formula. In addition, the differential expression and
cancer-promoting ability of related genes were only verified
through cellular experiments, and the biological mechanism
underlying the effects of m7G-related RNAs in BLCA remains unclear.
Detailed in vitro and in vivo experiments need to be
performed to explore the important functions of the aforementioned
four genes with prognostic properties in BLCA.
In conclusion, the present study screened four genes
related to patient prognosis based on the correlation between BLCA
and m7G modification regulators, and a risk scoring model with good
predictive accuracy was established. Moreover, the relationship
between risk score and tumor immunity was evaluated, which provided
new ideas and methods for the treatment of BLCA.
Supplementary Material
Supporting Data
Supporting Data
Acknowledgements
Not applicable.
Funding
Funding: No funding was received.
Availability of data and materials
Publicly available datasets were analyzed in the
present study. These data can be found in the following URLs: The
Cancer Genome Atlas database (https://portal.gdc.cancer.gov/) and Gene Expression
Omnibus database (https://www.ncbi.nlm.nih.gov/geo/). The data used
and/or generated in the present study are available from the
corresponding author on reasonable request.
Authors' contributions
CZ, JX, and KH conceived and designed the
experiments, and contributed reagents/materials/analysis tools. SZ,
TZ, KH and JL made contributions to the methodology and statistical
analysis, and provided supervision. CZ, JX, SZ, JL and TZ performed
the data collection and analyzed the data. TZ and KH confirm the
authenticity of all the raw data. CZ, JX, TZ and KH contributed to
the writing of the manuscript. All authors read and approved the
final manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Glossary
Abbreviations
Abbreviations:
|
BLCA
|
bladder urothelial carcinoma
|
|
GEMIN5
|
gem nuclear organelle-associated
protein 5
|
|
CYFIP1
|
cytoplasmic FMR1 interacting protein
1
|
|
NUDT11
|
nudix hydrolase 11
|
|
EIF3D
|
eukaryotic translation initiation
factor 3 subunit D
|
|
PD-1
|
programmed cell death 1
|
|
PD-L1
|
programmed cell death ligand 1
|
|
CTLA4
|
cytotoxic T lymphocyte-associated
protein 4
|
|
ROC
|
receiver operating characteristic
|
References
|
1
|
Richters A, Aben KKH and Kiemeney L: The
global burden of urinary bladder cancer: An update. World J Urol.
38:1895–1904. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Zhu CZ, Ting HN, Ng KH and Ong TA: A
review on the accuracy of bladder cancer detection methods. J
Cancer. 10:4038–4044. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Cathomas R, Lorch A, Bruins HM, Compérat
EM, Cowan NC, Efstathiou JA, Fietkau R, Gakis G, Hernández V,
Espinós EL, et al: The 2021 updated European association of urology
guidelines on metastatic urothelial carcinoma. Eur Urol. 81:95–103.
2022. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Kanehisa M and Bork P: Bioinformatics in
the post-sequence era. Nat Genet. 33 (Suppl):S305–S310. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Foulkes AC, Watson DS, Griffiths CEM,
Warren RB, Huber W and Barnes MR: Research techniques made simple:
Bioinformatics for genome-scale biology. J Invest Dermatol.
137:e163–e168. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Zafeiris D, Rutella S and Ball GR: An
artificial neural network integrated pipeline for biomarker
discovery using Alzheimer's disease as a case study. Comput Struct
Biotechnol J. 16:77–87. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Xie R, Xie M, Zhu L, Chiu JWY, Lam W and
Yap DYH: The relationship of pyroptosis-related genes, patient
outcomes, and tumor-infiltrating cells in bladder urothelial
carcinoma (BLCA). Front Pharmacol. 13:9309512022. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Xu C, Song L, Peng H, Yang Y, Liu Y, Pei
D, Guo J, Liu N, Liu J, Li X, et al: Clinical eosinophil-associated
genes can serve as a reliable predictor of bladder urothelial
cancer. Front Mol Biosci. 9:9634552022. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Jin K, Qiu S, Jin D, Zhou X, Zheng X, Li
J, Liao X, Yang L and Wei Q: Development of prognostic signature
based on immune-related genes in muscle-invasive bladder cancer:
Bioinformatics analysis of TCGA database. Aging (Albany NY).
13:1859–1871. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Chen X, Xu R, He D, Zhang Y, Chen H, Zhu
Y, Cheng Y, Liu R, Zhu R, Gong L, et al: CD8(+) T effector and
immune checkpoint signatures predict prognosis and responsiveness
to immunotherapy in bladder cancer. Oncogene. 40:6223–6234. 2021.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Liu Z, Tang Q, Qi T, Othmane B, Yang Z,
Chen J, Hu J and Zu X: A robust hypoxia risk score predicts the
clinical outcomes and tumor microenvironment immune characters in
bladder cancer. Front Immunol. 12:7252232021. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Olkhov-Mitsel E, Hodgson A, Liu SK,
Vesprini D, Bayani J, Bartlett JMS, Xu B and Downes MR:
Upregulation of IFNγ-mediated chemokines dominate the immune
transcriptome of muscle-invasive urothelial carcinoma. Sci Rep.
12:7162022. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Boccaletto P, Magnus M, Almeida C, Zyla A,
Astha A, Pluta R, Baginski B, Jankowska E, Dunin-Horkawicz S,
Wirecki TK, et al: RNArchitecture: A database and a classification
system of RNA families, with a focus on structural information.
Nucleic Acids Res. 46:D202–D205. 2018.PubMed/NCBI
|
|
14
|
Jühling F, Mörl M, Hartmann RK, Sprinzl M,
Stadler PF and Pütz J: tRNAdb 2009: Compilation of tRNA sequences
and tRNA genes. Nucleic Acids Res. 37:D159–D162. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Edmonds CG, Crain PF, Gupta R, Hashizume
T, Hocart CH, Kowalak JA, Pomerantz SC, Stetter KO and McCloskey
JA: Posttranscriptional modification of tRNA in thermophilic
archaea (Archaebacteria). J Bacteriol. 173:3138–3148. 1991.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Luo Y, Yao Y, Wu P, Zi X, Sun N and He J:
The potential role of N(7)-methylguanosine (m7G) in cancer. J
Hematol Oncol. 15:632022. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Li XY, Wang SL, Chen DH, Liu H, You JX, Su
LX and Yang XT: Construction and validation of a m7G-related
gene-based prognostic model for gastric cancer. Front Oncol.
12:8614122022. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Tomikawa C: 7-methylguanosine
modifications in transfer RNA (tRNA). Int J Mol Sci. 19:40802018.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Yang Z, Zhang S, Xia T, Fan Y, Shan Y,
Zhang K, Xiong J, Gu M and You B: RNA modifications meet tumors.
Cancer Manag Res. 14:3223–3243. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Rong D, Sun G, Wu F, Cheng Y, Sun G, Jiang
W, Li X, Zhong Y, Wu L, Zhang C, et al: Epigenetics: Roles and
therapeutic implications of non-coding RNA modifications in human
cancers. Mol Ther Nucleic Acids. 25:67–82. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Zhang M, Song J, Yuan W, Zhang W and Sun
Z: Roles of RNA methylation on tumor immunity and clinical
implications. Front Immunol. 12:6415072021. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Furuichi Y: Discovery of m(7)G-cap in
eukaryotic mRNAs. Proc Jpn Acad Ser B Phys Biol Sci. 91:394–409.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Reddy R, Singh R and Shimba S: Methylated
cap structures in eukaryotic RNAs: Structure, synthesis and
functions. Pharmacol Ther. 54:249–267. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
R Core Team R, . A language and
environment for statistical computing. R Foundation for Statistical
Computing; Vienna: 2012, http://www.R-project.org/
|
|
25
|
Szklarczyk D, Gable AL, Lyon D, Junge A,
Wyder S, Huerta-Cepas J, Simonovic M, Doncheva NT, Morris JH, Bork
P, et al: STRING v11: Protein-protein association networks with
increased coverage, supporting functional discovery in genome-wide
experimental datasets. Nucleic Acids Res. 47:D607–D613. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Tang Z, Li C, Kang B, Gao G, Li C and
Zhang Z: GEPIA: A web server for cancer and normal gene expression
profiling and interactive analyses. Nucleic Acids Res. 45:W98–W102.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Li T, Fan J, Wang B, Traugh N, Chen Q, Liu
JS, Li B and Liu XS: TIMER: A web server for comprehensive analysis
of tumor-infiltrating immune cells. Cancer Res. 77:e108–e110. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Geeleher P, Cox N and Huang RS:
pRRophetic: An R package for prediction of clinical
chemotherapeutic response from tumor gene expression levels. PLoS
One. 9:e1074682014. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(−Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Lai H, Cheng X, Liu Q, Luo W, Liu M, Zhang
M, Miao J, Ji Z, Lin GN, Song W, et al: Single-cell RNA sequencing
reveals the epithelial cell heterogeneity and invasive
subpopulation in human bladder cancer. Int J Cancer. 149:2099–2115.
2021. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Huang Z, Yan Y, Wang T, Wang Z, Cai J, Cao
X, Yang C, Zhang F, Wu G and Shen B: Identification of ENO1 as a
prognostic biomarker and molecular target among ENOs in bladder
cancer. J Transl Med. 20:3152022. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Liu J, Zhou Z, Jiang Y, Lin Y, Yang Y,
Tian C, Liu J, Lin H and Huang B: EPHA3 could be a novel prognosis
biomarker and correlates with immune infiltrates in bladder cancer.
Cancers (Basel). 15:6212023. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Yu X, Luo B, Lin J and Zhu Y: Alternative
splicing event associated with immunological features in bladder
cancer. Front Oncol. 12:9660882023. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Chen Z, Zhu W, Zhu S, Sun K, Liao J, Liu
H, Dai Z, Han H, Ren X, Yang Q, et al: METTL1 promotes
hepatocarcinogenesis via m(7) G tRNA modification-dependent
translation control. Clin Transl Med. 11:e6612021. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Zhu J, Liu X, Chen W, Liao Y, Liu J, Yuan
L, Ruan J and He J: Association of RNA m7G modification gene
polymorphisms with pediatric glioma risk. Biomed Res Int.
2023:36783272023. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Han H, Yang C, Ma J, Zhang S, Zheng S,
Ling R, Sun K, Guo S, Huang B and Liang Y: N(7)-methylguanosine
tRNA modification promotes esophageal squamous cell carcinoma
tumorigenesis via the RPTOR/ULK1/autophagy axis. Nat Commun.
13:14782022. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Xia P, Zhang H, Xu K, Jiang X, Gao M, Wang
G, Liu Y, Yao Y, Chen X, Ma W, et al: MYC-targeted WDR4 promotes
proliferation, metastasis, and sorafenib resistance by inducing
CCNB1 translation in hepatocellular carcinoma. Cell Death Dis.
12:6912021. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Xu C, Ishikawa H, Izumikawa K, Li L, He H,
Nobe Y, Yamauchi Y, Shahjee HM, Wu XH, Yu YT, et al: Structural
insights into Gemin5-guided selection of pre-snRNAs for snRNP
assembly. Genes Dev. 30:2376–2390. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Li XY, Zhao ZJ, Wang JB, Shao YH, Hui-Liu,
You JX and Yang XT: m7G methylation-related genes as biomarkers for
predicting overall survival outcomes for hepatocellular carcinoma.
Front Bioeng Biotechnol. 10:8497562022. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Shao J, Wang S, West-Szymanski D, Karpus
J, Shah S, Ganguly S, Smith J, Zu Y, He C, Li Z, et al: Cell-free
DNA 5-hydroxymethylcytosine is an emerging marker of acute myeloid
leukemia. Sci Rep. 12:124102022. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Lee JH, Horak CE, Khanna C, Meng Z, Yu LR,
Veenstra TD and Steeg PS: Alterations in Gemin5 expression
contribute to alternative mRNA splicing patterns and tumor cell
motility. Cancer Res. 68:639–644. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Wollen KL, Hagen L, Vågbø CB, Rabe R,
Iveland TS, Aas PA, Sharma A, Sporsheim B, Erlandsen HO, Palibrk V,
et al: ALKBH3 partner ASCC3 mediates P-body formation and selective
clearance of MMS-induced 1-methyladenosine and 3-methylcytosine
from mRNA. J Transl Med. 19:2872021. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Limaye AJ, Whittaker MK, Bendzunas GN,
Cowell JK and Kennedy EJ: Targeting the WASF3 complex to suppress
metastasis. Pharmacol Res. 182:1063022022. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Chang JW, Kuo WH, Lin CM, Chen WL, Chan
SH, Chiu MF, Chang IS, Jiang SS, Tsai FY, Chen CH, et al: Wild-type
p53 upregulates an early onset breast cancer-associated gene GAS7
to suppress metastasis via GAS7-CYFIP1-mediated signaling pathway.
Oncogene. 37:4137–4150. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Teng Y, Qin H, Bahassan A, Bendzunas NG,
Kennedy EJ and Cowell JK: The WASF3-NCKAP1-CYFIP1 complex is
essential for breast cancer metastasis. Cancer Res. 76:5133–5142.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Morrison BH, Bauer JA, Kalvakolanu DV and
Lindner DJ: Inositol hexakisphosphate kinase 2 mediates growth
suppressive and apoptotic effects of interferon-beta in ovarian
carcinoma cells. J Biol Chem. 276:24965–24970. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Grisanzio C, Werner L, Takeda D, Awoyemi
BC, Pomerantz MM, Yamada H, Sooriakumaran P, Robinson BD, Leung R,
Schinzel AC, et al: Genetic and functional analyses implicate the
NUDT11, HNF1B, and SLC22A3 genes in prostate cancer pathogenesis.
Proc Natl Acad Sci U S A. 109:11252–11257. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Bandyopadhyay A, Lakshmanan V, Matsumoto
T, Chang EC and Maitra U: Moe1 and spInt6, the fission yeast
homologues of mammalian translation initiation factor 3 subunits
p66 (eIF3d) and p48 (eIF3e), respectively, are required for stable
association of eIF3 subunits. J Biol Chem. 277:2360–2367. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Yu X, Zheng B and Chai R:
Lentivirus-mediated knockdown of eukaryotic translation initiation
factor 3 subunit D inhibits proliferation of HCT116 colon cancer
cells. Biosci Rep. 34:e001612014. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Sudo H, Tsuji AB, Sugyo A, Kohda M, Sogawa
C, Yoshida C, Harada YN, Hino O and Saga T: Knockdown of COPA,
identified by loss-of-function screen, induces apoptosis and
suppresses tumor growth in mesothelioma mouse model. Genomics.
95:210–216. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Fan Y and Guo Y: Knockdown of eIF3D
inhibits breast cancer cell proliferation and invasion through
suppressing the Wnt/β-catenin signaling pathway. Int J Clin Exp
Pathol. 8:10420–10427. 2015.PubMed/NCBI
|
|
52
|
Golob-Schwarzl N, Krassnig S, Toeglhofer
AM, Park YN, Gogg-Kamerer M, Vierlinger K, Schröder F, Rhee H,
Schicho R, Fickert P and Haybaeck J: New liver cancer biomarkers:
PI3K/AKT/mTOR pathway members and eukaryotic translation initiation
factors. Eur J Cancer. 83:56–70. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Hershey JW: The role of eIF3 and its
individual subunits in cancer. Biochim Biophys Acta. 1849:792–800.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Sesen J, Cammas A, Scotland SJ, Elefterion
B, Lemarié A, Millevoi S, Mathew LK, Seva C, Toulas C, Moyal ECJ
and Skuli N: Int6/eIF3e is essential for proliferation and survival
of human glioblastoma cells. Int J Mol Sci. 15:2172–2190. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Feng X, Li J and Liu P: The biological
roles of translation initiation factor 3b. Int J Biol Sci.
14:1630–1635. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Wolf DA, Lin Y, Duan H and Cheng Y:
eIF-Three to Tango: Emerging functions of translation initiation
factor eIF3 in protein synthesis and disease. J Mol Cell Biol.
12:403–409. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Yin Y, Long J, Sun Y, Li H, Jiang E, Zeng
C and Zhu W: The function and clinical significance of eIF3 in
cancer. Gene. 673:130–133. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Luo Y, Chen L, Zhou Q, Xiong Y, Wang G,
Liu X, Xiao Y, Ju L and Wang X: Identification of a prognostic gene
signature based on an immunogenomic landscape analysis of bladder
cancer. J Cell Mol Med. 24:13370–13382. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Undi RB, Filiberti A, Ali N and Huycke MM:
Cellular carcinogenesis: Role of polarized macrophages in cancer
initiation. Cancers (Basel). 14:28112022. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Scheper W, Kelderman S, Fanchi LF,
Linnemann C, Bendle G, de Rooij MAJ, Hirt C, Mezzadra R, Slagter M,
Dijkstra K, et al: Low and variable tumor reactivity of the
intratumoral TCR repertoire in human cancers. Nat Med. 25:89–94.
2019. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Elia I, Rowe JH, Johnson S, Joshi S,
Notarangelo G, Kurmi K, Weiss S, Freeman GJ, Sharpe AH and Haigi
MC: Tumor cells dictate anti-tumor immune responses by altering
pyruvate utilization and succinate signaling in CD8(+) T cells.
Cell Metab. 34:1137–1150.e1136. 2022. View Article : Google Scholar : PubMed/NCBI
|