Introduction
Primary malignant central nervous system (CNS)
tumors account for 2% of all cancers with an inconsistent rate of
morbidity and mortality. Malignant CNS tumors constitute the
leading cause of death from solid tumors in children and the third
leading cause of cancer-related death for adolescents and adults
aged 15–34 years (1).
Astrocytomas are tumors occurring in young adulthood
defined as CNS neoplasms originated in astrocytes, star-shaped
brain cells (2,3). The World Health Organization (WHO)
system classifies the astrocytic tumors into four grades: grade I
(pilocytic astrocytoma), grade II (diffuse astrocytoma) with
cytological atypia alone, grade III (anaplastic astrocytoma)
showing anaplasia and mitotic activity in addition and grade IV
(glioblastoma) presenting microvascular proliferation and/or
necrosis (4). Malignant gliomas are
those of grades III and IV (5).
Glioblastomas account for at least 80% of malignant gliomas
(6).
Rho GTPases are known to be involved in the
stimulation of cell cycle progression. The family of Rho GTPases
contains 20 small G proteins playing important roles in the
regulation of the cytoskeleton, the cell cycle, the cell migration
and the cell polarity (7). Rho
GTPases are guanine nucleotide binding proteins existing in two
forms: the active form which is GTP bound and the inactive one that
being GDP bound and it is important to note that only in the active
form, Rho GTPases can interact with other effectors mediating their
cellular functions (8). The most
important studied members of the Rho family are Rac 1, Cdc42 and
Rho A (8).
Genetic screens of many human cancers have revealed
altered expression of various Rho family GTPases (9). RhoC mRNA level is increased in
metastatic melanomas and Rac3 activity is increased in highly
proliferative breast cancer cell lines (9). Regulators of Rho GTPases also show
aberrant expression in human tumors, including Vav1 in
neuroblastomas (10). A comparison
of the gene expression pattern in a metastatic breast cancer cell
line compared to its non-metastatic counterpart revealed that many
genes encoding actin regulatory proteins are more highly expressed
in metastatic cells (11). All of
these processes are regulated by the proteins of the Rho GTPase
family making them with their regulators and effectors important in
controlling tumor formation and progression in humans (7).
The regulation of Rho GTPases is governed by three
classes of regulatory proteins: GAP (GTPase activating proteins),
GEF (guanine nucleotide exchange factors), which accelerate the
very slow intrinsic guanine nucleotide exchange and GTP hydrolysis
activity, and GDI (guanine nucleotide dissociation inhibitors)
(12,13). GAPs belong to a specific family of
GTPases that accelerate the rate of GTP hydrolysis by up to
105 times (14). Hence,
a tumor suppressor role has been suggested for GAPs counteracting
the oncogenic potential of Rho proteins (15). Since the identification of the first
RhoGAP (16) more than 50 RhoGAPs
in the human genome were characterized, three of which contain
START domain: DLC1, DLC2 known also as START-GAP2 or StarD13 and
DLC3 (17). The three DLC proteins
are characterized by the presence of three motifs: a sterile α
motif (SAM), a RhoGAP catalytic domain, and a START (Star-related
lipid transfer) domain (18). The
SAM domain consists of approximately 70 residues and has been shown
to play several roles particularly as protein interaction modules
because of its ability to interact with other SAM domains (19). SAM domains are located on the
N-terminus and may bind to DNA or RNA (20). DLC2-SAM shows only 15–30% homology
with other SAM domains and is considered as the prototype in the
family of the DLC2-related proteins (19).
When searching for additional candidate tumor
suppressor loci critical in hepatocellular carcinoma after the
well-known and established tumor suppressor genes p53, c-myc,
p16ink4 and β-catenin, Ching et
al identified a novel gene DLC2 on chromosome
13q12 which was found to be underexpressed in hepatocellular
carcinoma (21,22). DLC2 which is also known as
steroidogenic acute regulatory protein-related lipid transfer
(START) domain containing protein 13 (StarD13), display high level
of homology with DLC1 (deleted in liver cancer 1), a gene coding
for a Rho GTPase activating protein. These two proteins have 51%
identity and 64% similarity at the level of their amino acid
sequences sharing the same SAM-RhoGAP-START domain organization
(22,23). DLC1 is down-regulated in many types
of cancers including lung, breast, prostate, kidney, colon, uterus,
ovary, and stomach due to two major causes: genomic deletion and
promoter hypermethylation.
Similar to DLC1, DLC2 is down-regulated in several
types of cancer including lung, ovarian, renal, breast, uterine,
gastric, colon and rectal tumors (23). Through its RhoGAP activity, the DLC2
protein acts on RhoA-C and Cdc42 but not on Rac1 (13,21,24).
Xiaorong et al reported significant correlations between
underexpression of DLC2 and cell differentiation. In addition, a
negative correlation was established between DLC2 and RhoA. DLC2
seemed to inhibit hepatocarcinogenesis by suppressing RhoA activity
(25). On the other hand, a study
conducted by Yau et al investigated the role of DLC2 by
generating DLC2-deficient mice. The mice that were defective in
DLC2 were able to survive to adulthood unlike the knockout of DLC1
which led to embryonic lethality (15).
In this study, we examine the role of StarD13 as a
potential tumor suppressor in astrocytoma to compare it with the
newly described role of StarD13 as a tumor suppressor for
hepatocellular carcinoma.
Materials and methods
Cell culture
The human astrocytoma cell line T98G was cultured in
DMEM medium supplemented with 10% FBS and 100 U
penicillin/streptomycin at 37°C and 5% CO2 in a
humidified chamber.
Antibodies and reagents
Goat polyclonal anti-StarD13 antibody was obtained
from Santa Cruz Biotechnology. Mouse monoclonal anti-Rho, mouse
monoclonal anti-Rac1 antibodies were purchased from Upstate
Biotechnology (Lake Placid, NY). The anti-Cdc42 antibody (Sc-87)
was obtained from Santa Cruz Biotechnology. Anti-goat and
anti-mouse HRP-conjugated secondary antibodies were obtained from
Promega. Fluorescent secondary antibodies (AlexaFluor 488) and
rhodamin phalloidin were obtained from Invitrogen.
The full length GFP-StarD13 construct was a generous
gift from Dr Hitoshi Yagisawa from the University of Hyogo, Japan
[mammalian expression plasmids for GFP fusion proteins,
pEGFPSTART-GAP1(wt)] (13).
Cell transfection and small interfering
RNA
Cells were transfected with 5 μg GFP-StarD13 or
control empty GFP alone vectors using Lipfectamine LTX with Plus
reagent (Invitrogen) as described by the manufacturer. Goat
FlexiTube siRNA for StarD13 was obtained from Qiagen. The siRNAs
used had the following target sequence: StarD13: 5′-CCCGCAATACGCTC
AGTTATA-3′. The cells were transfected with the siRNA at a final
concentration of 10 nM using HiPerfect (Qiagen) as described by the
manufacturer. Control cells were transfected with siRNA sequences
targeting GL2 luciferase (Qiagen). After 72 h, protein levels in
total cell lysates were analyzed by western blotting using the
appropriate antibodies or the effect of the corresponding knockdown
was assayed.
Pull down assays and western blot
analyses
For experiments, cells were starved in low-glucose
Opti-MEM medium without FBS for 3 h, and stimulated with a final
concentration of 10 nM recombinant human epidermal growth factor
(Hu EGF) for various times (Invitrogen). Cells were then lysed and
the pull-down assay performed using the RhoA/Rac1/Cdc42 Activation
Assay Combo kit (Cell BioLabs) following the manufacturer's
instructions. Briefly, cell lysates were incubated with GST-RBD
(for Rho pull down) or GST-CRIB (for the Rac and Cdc42 pull down)
for 1 h at 4°C with gentle agitation. Then, the samples were
centrifuged, and the pellet washed several times. After the last
wash, the pellets were resuspended with sample buffer and boiled
for 5 min. GTP-RhoA or GST-Rac/Cdc42 were detected by western
blotting using anti-RhoA, anti-Rac or anti-Cdc42, respectively.
Total RhoA/Rac and Cdc42 were collected prior to the incubation
with GST-RBD/GST-CRIB and used as a loading control.
Immunohistochemistry
Permission for tissue collection was granted by the
Committee on Human Subjects in Research (CHSR) at the Lebanese
American University (approval given on March 26 2010, CHSR tracking
no. NSMS26032010-1). Tissues were collected from Rizk Medical
Center with the help of Dr Noha Bejjani and from Hammoud Hospital
with the help of Dr Najla Fakhreddine. Frozen human astrocytoma
tissues of grades I and IV were sectioned to 8 μm sections using a
refrigerated microtome. Tissues were fixed with 4% paraformaldehyde
for 10 min, and permeabilized with 0.5% Triton X-100 for 10 min. To
decrease background fluorescence, tissues were rinsed with 0.1 M
glycine then incubated with 0.1 M glycine for 10 min. For blocking,
tissues were incubated 4 times with 1% BSA, 1% FBS in PBS for 5
min. Samples were stained with the StarD13 primary antibody for 2 h
and with a fluorophore-conjugated secondary antibody for 2 h.
Tissue fluorescent images were taken using a 10× objective on a
Zeiss LSM confocal microscope. For image analysis, all digital
images were imported in ImageJ software (National Institutes of
Health, MA). The total fluorescence intensity of a fixed area from
at least 10 different frames from each tissue was determined.
Proliferation
Depending on the type of experiment, cells were
seeded either in 12-well plate or in 96-well plate. After 24 h of
seeding, cells were transfected with StarD13 siRNA or overexpressed
with GFP-StarD13. At the end of each treatment period, cell
viability was determined using the trypan blue exclusion method or
the cell proliferation reagent (WST-1; Roche, Mannheim, Germany) as
recommended by the manufacturer. Briefly, at the end of the
treatment period, water-soluble tetrazolim salt (WST-1 was added to
the cells and kept in a humidified incubator (37°C) at 95% air and
5% CO2 for 4 h. The formazan dye was then measured
colorimetrically at 450 nm. The results were expressed as percent
of control.
Flow cytometry
Treated cells were placed into 15 ml Falcon tubes
and centrifuged at 1500 rpm for 5 min. The pellet was then washed
by resuspending it in 1 ml of ice-cold 1× phosphate buffered saline
(PBS) followed by 4 ml of 70% ethanol. Cells were then treated with
100 μl of RNase and incubated for 1 h at 37°C. The cells were then
pelleted at 2000 rpm for 5 min, and the pellets were washed with
500 μl of 1× PBS, transferred to labeled 6 ml polystyrene round
bottom falcon tube, and stained with 30 μl propidium iodide for 10
min in the dark. Cells were analyzed using a FACScan and the DNA
content determined by CellQuest software.
Annexin staining
Cells were trypsinized and centrifuged at 1500 rpm
for 5 min. The pellet was then washed by resuspending it in 1 ml of
ice-phosphate buffered saline (PBS). Cells were centrifuged again
under the same conditions as before. Cells were then stained with 5
μl of Annexin V-FITC and 10 μl of propidium iodide cells and
incubated at room temperature for 10 min and protected from light.
The fluorescence of the cells was determined immediately with a
flow cytometer.
REMBRANDT database
To determine the expression of StarD13 in human
gliomas, we mined the publicly available Repository for Molecular
Brain Neoplasia Data (REMBRANDT) gene expression microarray
database containing 452 clinically annotated brain tumor specimens
(National Cancer Institute, 2005; REMBRANDT home page:
<http://rembrandt.nci.nih.gov>;
accessed December 20, 2010). We specifically examined the gene
expression data from nonneoplastic brain (NB, n=28), low-grade
astrocytomas (LGGs, n=148), and glioblastoma multiformes (GBMs,
n=226).
Results
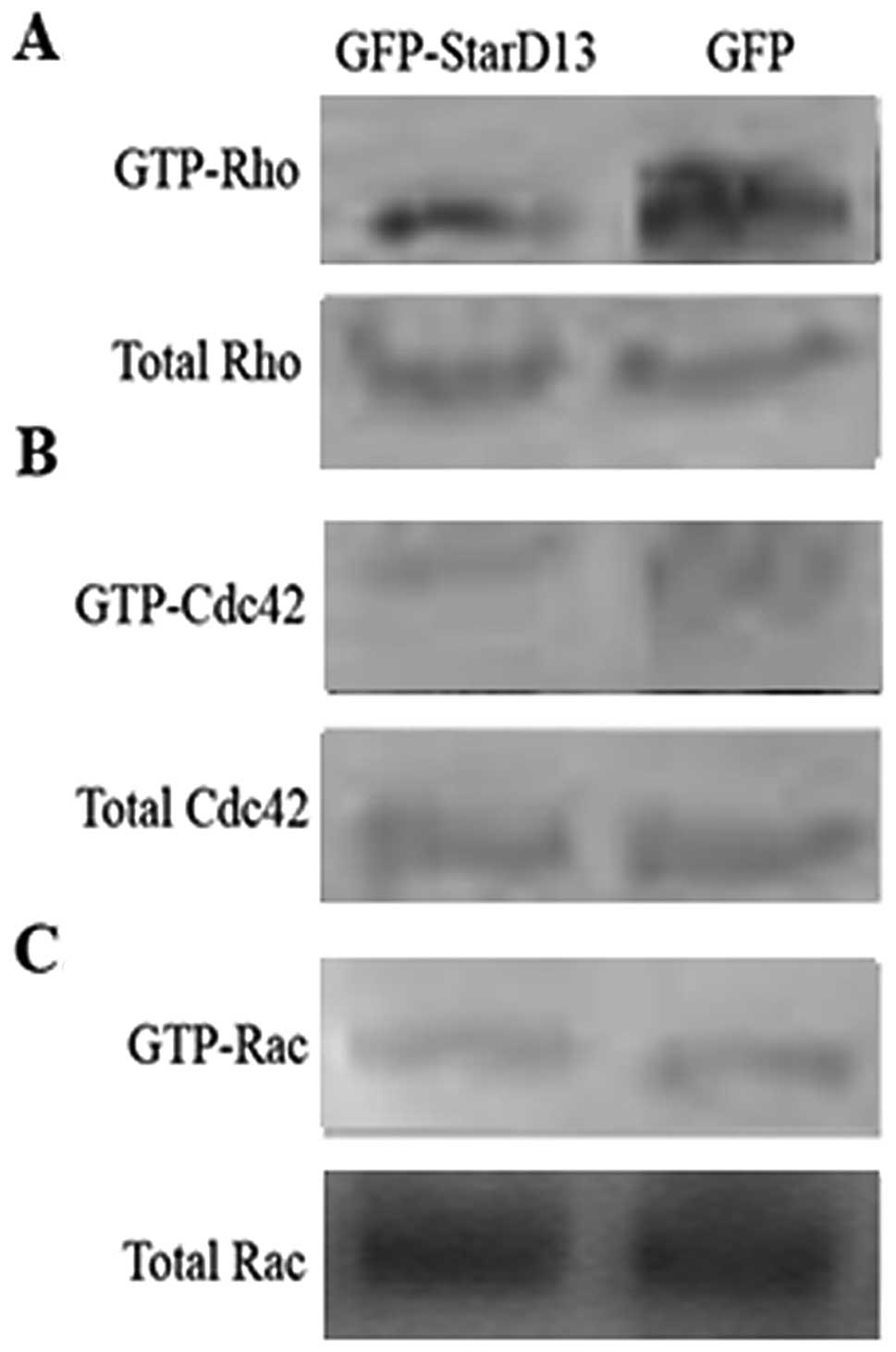
StarD13 is a GAP for RhoA and Cdc42 not
for Rac1
In order to study the role of StarD13 in astrocytoma
malignancy, we started by verifying that StarD13 is a GAP for RhoA
and Cdc42 and not for Rac1. This was achieved by studying the
activation of the three Rho GTPases in T98 cells following the
transfection by GFP-StarD13. Using a GST-RBD (Rho binding domain
from Rhotekin) and GST-CRIB pull down assay, we looked for
GTP-loaded Rho and Cdc42/Rac, respectively. We found that the
levels of the active Rho and Cdc42 were lower in cells transfected
with GFP-StarD13 (Fig. 1A and B) as
compared to the controls. On the other hand, the overexpression of
StarD13 did not affect the active Rac1 (Fig. 1C). This confirmed that StarD13 is a
specific GAP for Rho and Cdc42.
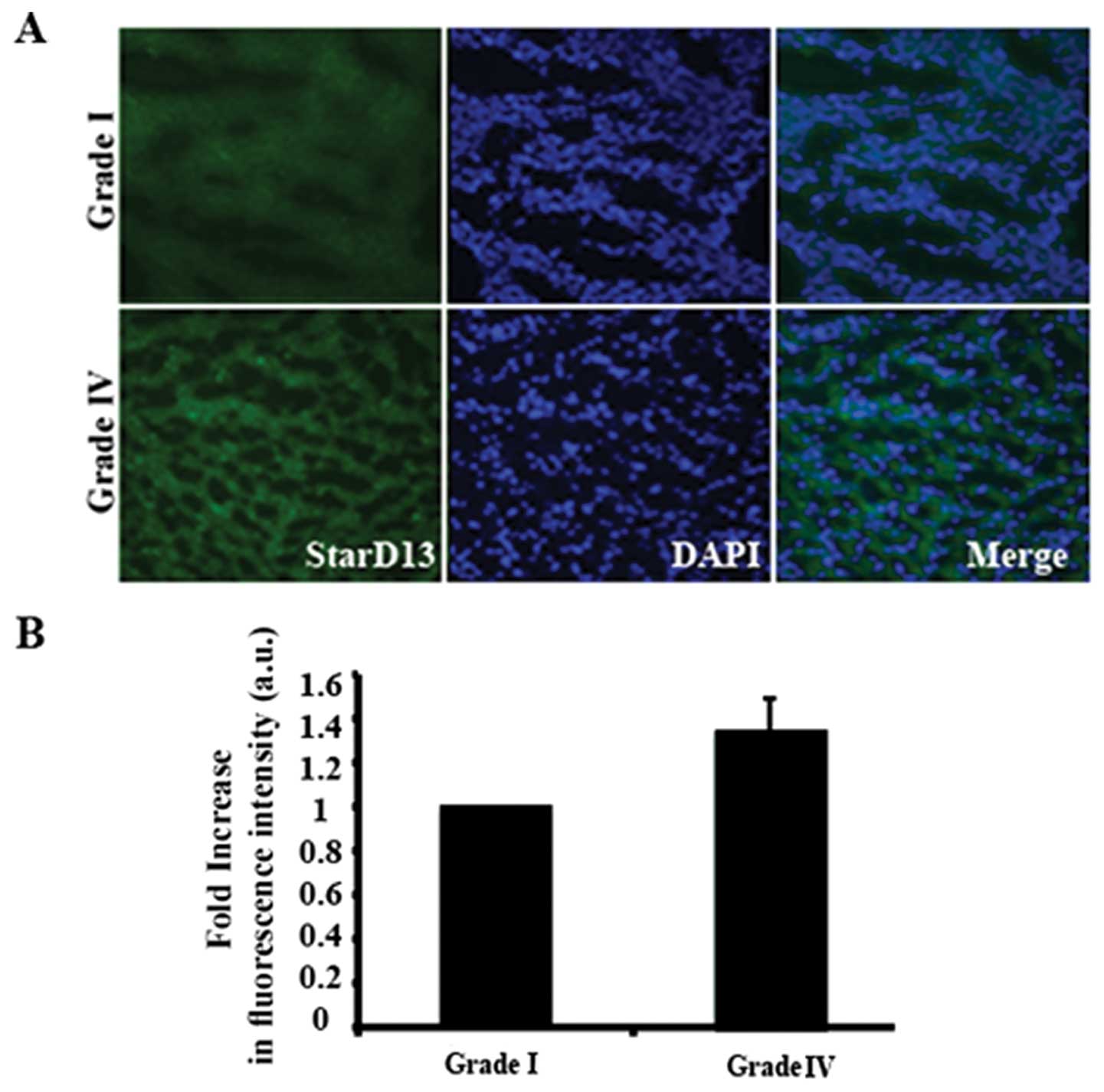
StarD13 is underexpressed in
astrocytoma
Then we investigated the expression levels of
StarD13 in astrocytoma malignancy using immunohistochemistry.
Tissues of grades I and IV were stained with anti-StarD13 antibody
(Fig. 2A) and the intensity of the
signal was measured using ImageJ software. Our results showed that
there was ~30% increase in the expression of StarD13 in grade IV
tumors as compared to grade I (Fig.
2B).
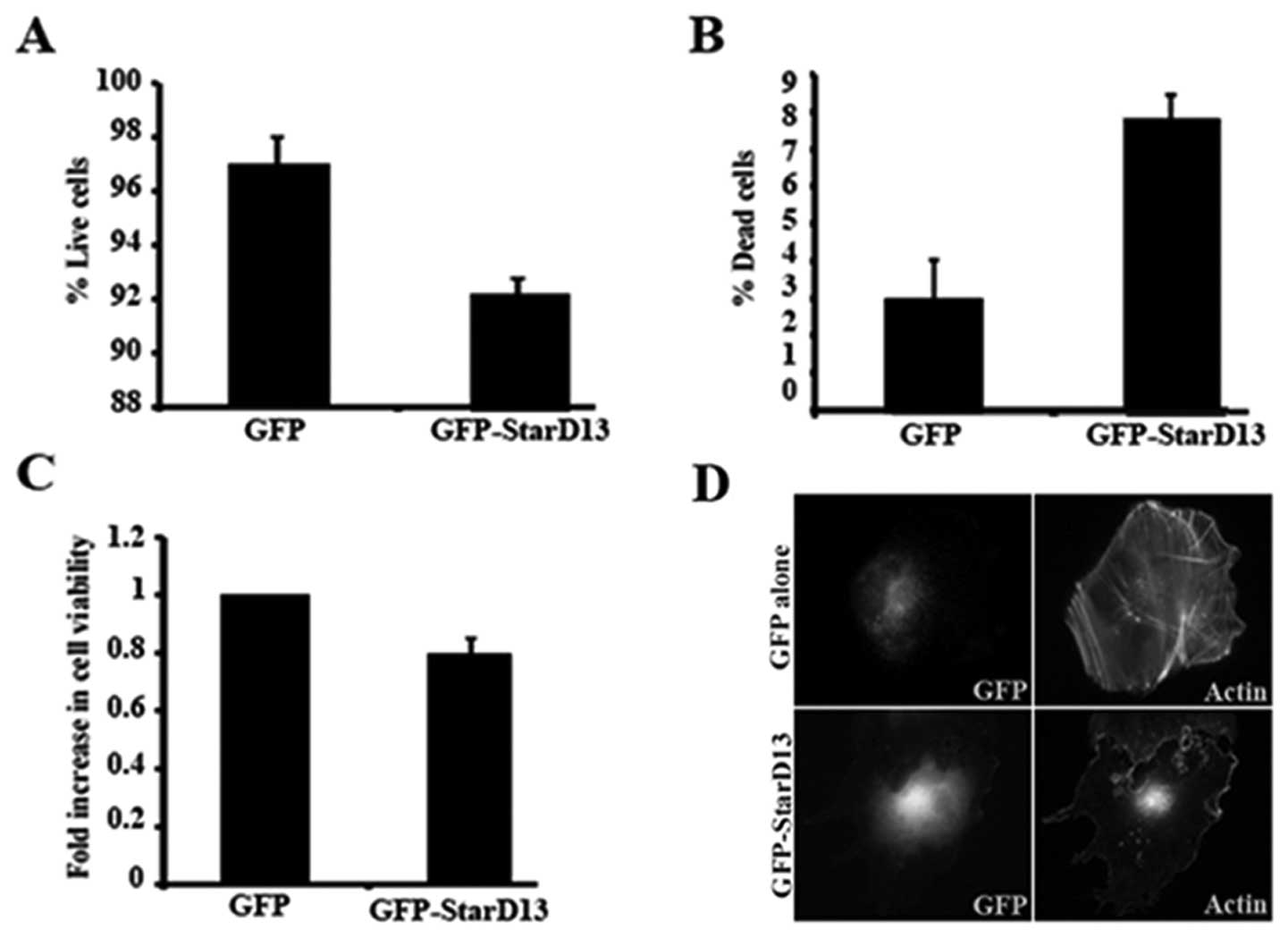
StarD13 overexpression reduces cell
viability
In order to determine the role of StarD13 in cell
viability, we transfected T98 cells with GFP-StarD13 and studied
the effect of this overexpression on cell viability using two
methods: the trypan blue exclusion method and the cell
proliferation reagent (WST-1).
The overexpression was apparent looking at the GFP
channel and through the effect of overexpressing GFP-StaD13 on
stress fiber formation (due to Rho inhibition) as revealed by
rhodamine phalloidin staining (Fig.
3D). This is compared to cells transfected with GFP alone
(Fig. 3D, upper panels) where
stress fibers were not affected. The overexpression of StarD13
decreased the percentage of live cells from 97 to 92% (Fig. 3A). This was consistent with the
results of the WST-1 which showed a decrease of ~20% in cell
viability (Fig. 3C). The amount of
dead cells increased by ~2-fold (Fig.
3B).
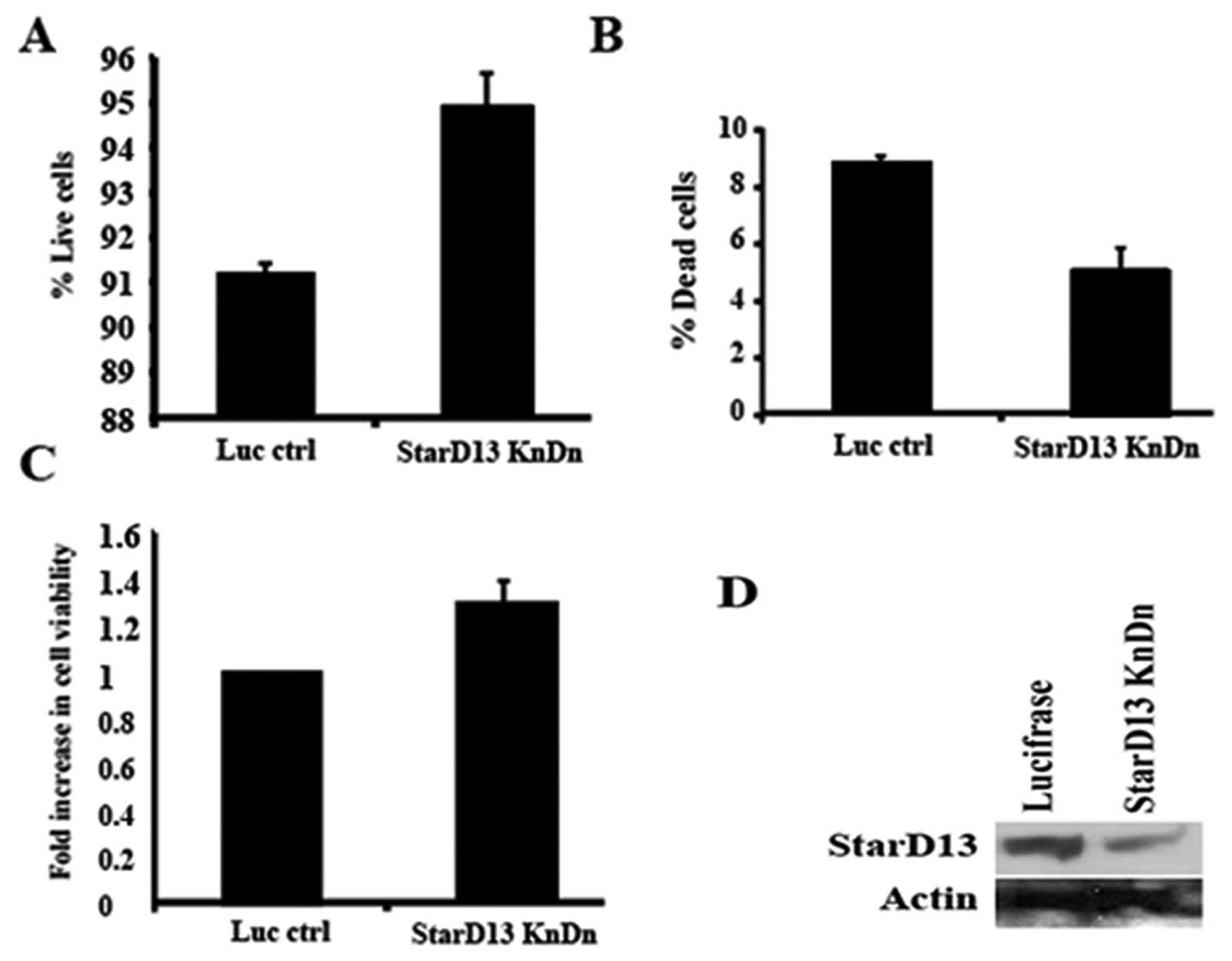
StarD13 knockdown increases cell
viability
To confirm the previous results, we knocked down
expression of StarD13 with siRNA. StarD13 expression was reduced by
50% as compared to cells transfected with control siRNA duplexes
(Fig. 4D). The percentage of live
cells in StarD13-siRNA treated cells was increased as compared to
control-siRNA treated cells (Fig.
4A). These results were consistent with those of the WST-1
which showed an increase of about 30% in cell viability (Fig. 4C). The amount of dead cells
increased by ~2-fold (Fig. 4B).
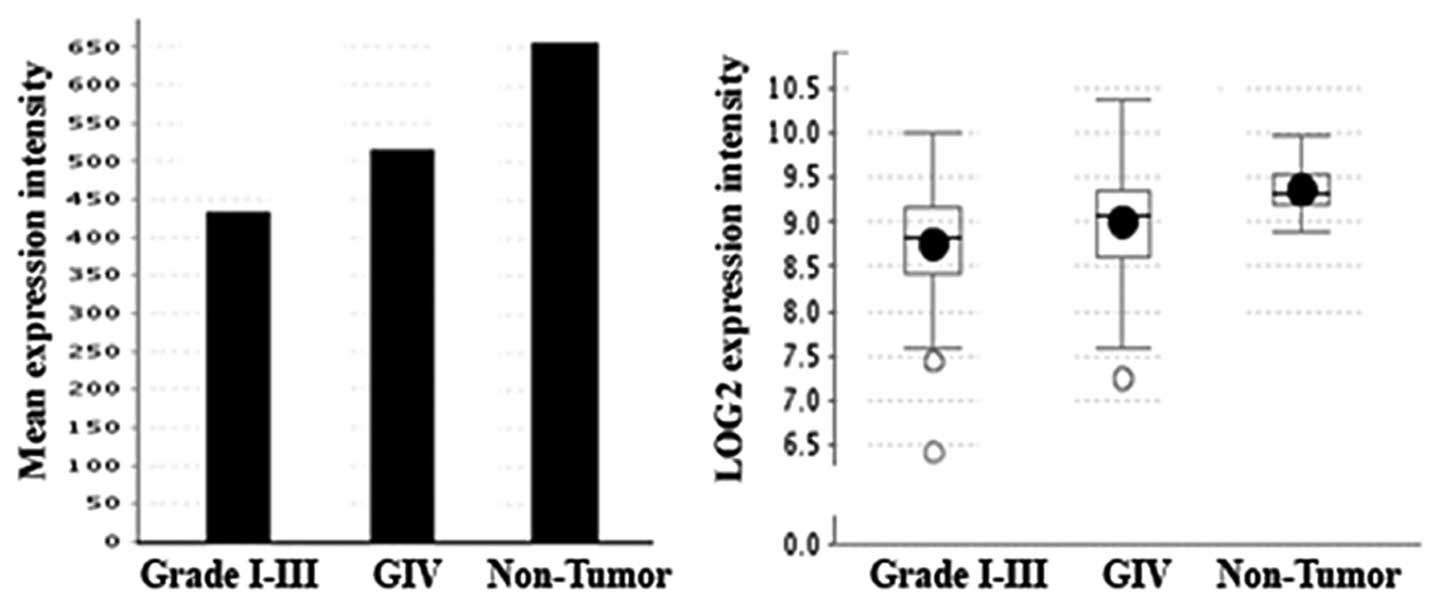
StarD13 is overexpressed in
glioblastoma
Surprisingly, our immunochemistry results suggested
StarD13 might be an oncogene and not a tumor suppressor, as the
literature suggested, since StarD13 looked to be overexpressed in
grade IV astrocytoma, as compared to grade I. However,
overexpressing and knocking down StarD13 led to a decrease and
increase in cell proliferation, respectively, as would be expected
of a tumor suppressor. In order to reconcile our results, we mined
the REMBRANDT (Repository of Molecular Brain Neoplasia Data)
database which hosts diverse types of molecular research and
clinical trials data related to brain cancers, including gliomas.
The results showed StarD13 is underexpressed in tumor tissues as
compared to non-tumor tissues. However, if we compare the
expression level in GBM (glioblastoma) which is grade IV
astrocytoma to lower grade tumors, we find that the levels of
StarD13 mRNA in grade IV is higher (Fig. 5). This was consistent with our IHC
results.
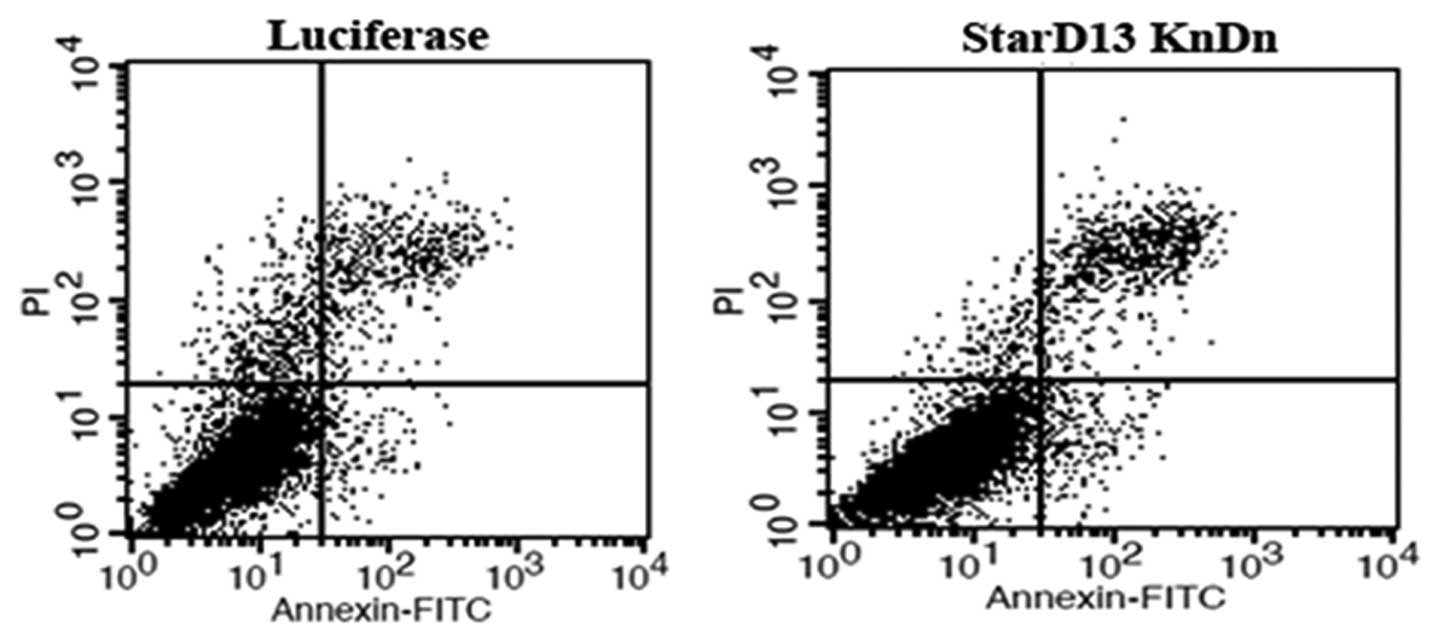
StarD13 does not affect the apoptosis
pathway
The effect of StarD13 on cell viability could be
through inducing apoptosis. To study the effect of StarD13 on
apoptosis, cells were stained with Annexin V-FITC. The fluorescence
was immediately measured by a flow cytometry. Cells, which are
early in the apoptotic process, will stain with the Annexin V-FITC
conjugate alone. Live cells will show no staining by either the
propidium iodide solution or Annexin V-FITC conjugate. Necrotic
cells will be stained by both the propidium iodide solution and
Annexin V-FITC conjugate. Our results showed that the knockdown of
StarD13 did not affect apoptosis. The percentage of apoptotic cells
was not significantly different between the controls and the
transfected cells (Fig. 6 and
Table I).
 | Table IStarD13 had no significant effect on
apoptosis. |
Table I
StarD13 had no significant effect on
apoptosis.
| Luciferase
control | StarD13 KnDn |
|---|
| Dead cells | 2.265 | 1.23 |
| Necrotic cells | 3.58 | 5.72 |
| Apoptotic cells | 1.21 | 2.42 |
| Live cells | 92.945 | 90.63 |
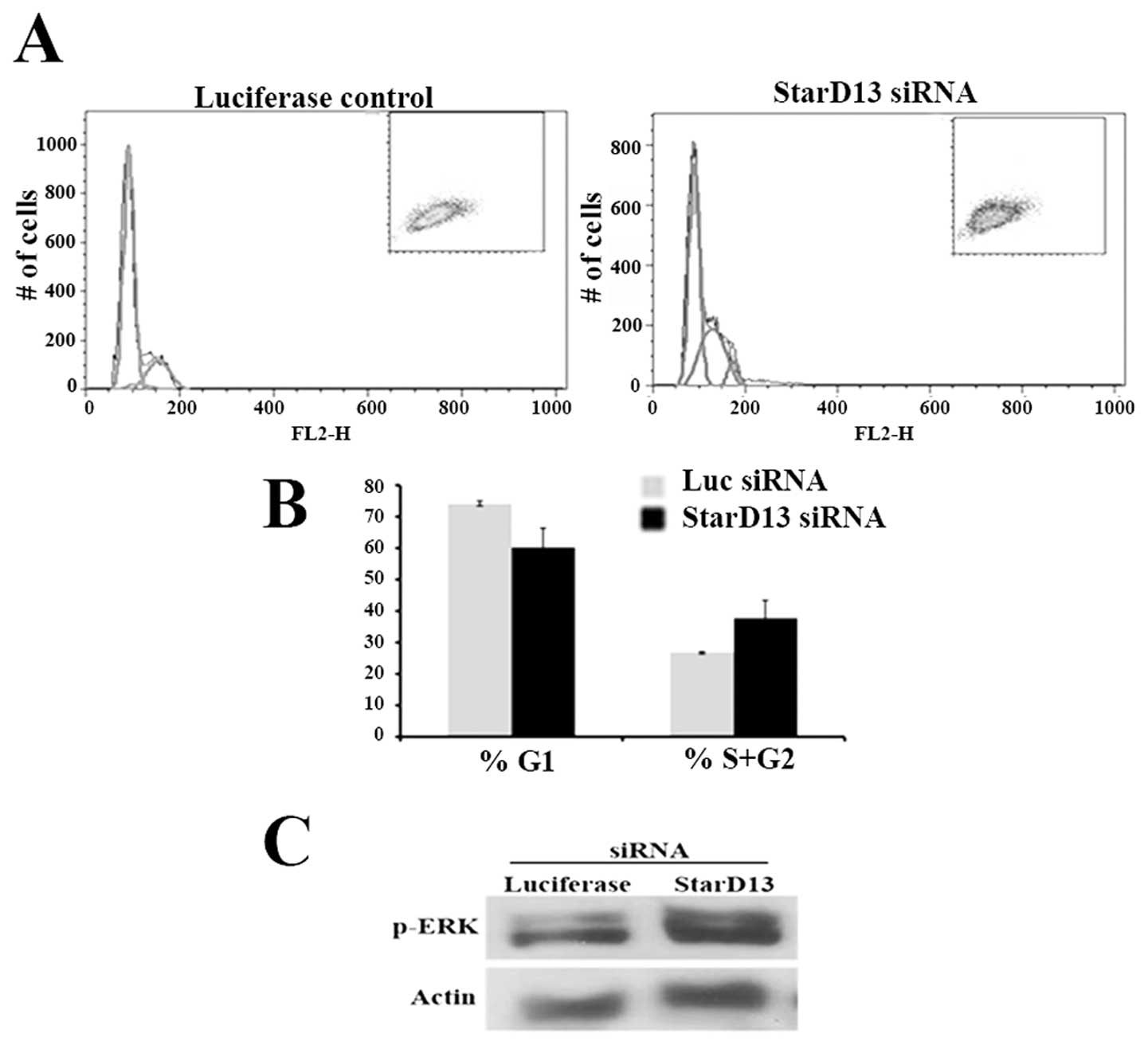
StarD13 affects the cell cycle
In order to understand the mechanism through which
StarD13 is affecting cell viability, we analyzed the effect of
StarD13 knockdown on the cell cycle. This was achieved using flow
cytometry. Our results showed an increase in the percentage of
cells in the S/G2 phase as compared to the cells in the G0 phase
(Fig. 7A and B). This showed that
knocking down StarD13 led to an increase in cycling cells, meaning
StarD13 plays the role of a tumor suppressor inhibiting the cell
cycle.
ERK is a downstream effector of
StarD13
In order to determine the mechanism of action of
StarD13, we knocked it down and looked at the effect on p-ERK, the
extracellular signal-regulated kinase. The western blotting showed
an increase in pERK levels in siRNA-StarD13 transfected cells as
compared to control non transfected cells (Fig. 7C).
Discussion
In this study, we examined the role of StarD13 in
astrocytoma malignancy. We confirmed that StarD13 is a specific GAP
for Rho and Cdc42. IHC analysis showed that StarD13 is
overexpressed in grade IV astrocytoma compared to grade I. Mining
online databases explained this observation by StarD13 being indeed
overexpressed in grade IV astrocytoma as compared to grade I,
however StarD13 is underexpressed in astrocytoma (grades I and IV)
as compared to normal tissues. This confirmed StarD13 as a
potential tumor suppressor in astrocytoma. In order to directly
prove that, we overexpressed or knocked down StarD13 and looked at
the effect of cell viability, apoptosis and proliferation in a cell
culture model. In astrocytoma cell lines, overexpressing StarD13
led to no effect on apoptosis but to a decrease in cell viability
and cell proliferation as reflected by a decrease in cells in S and
G2 phase. Knocking down StarD13 with StarD13 siRNA led to no effect
on cell apoptosis but to an increase in cell viability and cell
proliferation. Knocking down StarD13 also showed an increase in
phosphorylated ERK.
Ching et al was the first to identify and
characterize StarD13 in hepatocellular carcinoma (HCC) (21). We wanted to determine the role of
this RhoGAP in another tumor model which is astrocytoma. Ching
et al reported the function of StarD13 as a GAP for RhoA and
Cdc42 not for Rac1 (21). Our
results demonstrated that cells overexpressing StarD13 possessed
reduced levels of RhoA and Cdc42 activation; however, the
activation of Rac1 was not affected. Therefore, similar to its role
in HCC, StarD13 has a RhoGAP activity for RhoA and Cdc42 in
astrocytoma. The IHC analysis on grades I–IV brain tissues from
patients showed StarD13 to be overexpressed in grades III and IV
astrocytoma tumors when compared to grades I and II. This was
contradictory to other studies where the StarD13 gene was found to
be down-regulated in several types of cancer including lung,
ovarian, renal, breast, uterine, gastric, colon and rectal tumors
(23). These results led us to
formulate the following hypothesis: contrary to other tumor models,
in brain astrocytomas, StarD13 seems to be an oncogene and not a
tumor suppressor. The REMBRANDT data, however, showed that the mRNA
levels of StarD13 are indeed higher in the higher grades but much
lower than the normal tissues. Hence, our IHC results were
consistent with role of StarD13 as a tumor suppressor in
astrocytoma, similar to hepatocellular carcinoma. We also showed
that StarD13 overexpression leads a decrease in the number of
viable cells, proving directly that StarD13 is a tumor
suppressor.
It could be informative to establish the
significance of the overexpression of StarD13 as the malignancy of
astrocytoma increases. A present study in our laboratory showed
that StarD13 is needed for astrocytoma cell motility. This might
explain why StarD13 is overexpressed in grade IV astrocytoma
compared to grade I. This pattern was also observed in the case of
DLC1, which although a known tumor suppressor, was found to be
needed for the motility of normal prostate cells (26). Could a protein play different or
even opposing roles during the course of carcinogenesis? This
remains to be investigated.
The effect of cell viability could be either due to
decreased proliferation or increased apoptosis. To test which
underlying mechanism was responsible for the tumor suppressor
function of StarD13, we studied the effect of StarD13 knockdown on
cell cycle using flow cytometry. In StarD13 siRNA transfected
cells, the percentage of cells at the S/G2 phase was higher than
that of the control cells. The percentage of cells at the G1 phase
was lower. These results indicated that the deleted in liver cancer
2 blocks the cells in the G1 phase inhibiting the cell cycle
progression. This was consistent with a study conducted by Leung
et al(27) which
demonstrated that a stable expression of StarD13 caused
accumulation of cells in the G1 phase leading to the inhibition of
cell growth.
In addition to the RhoGAP domain, the START domain
plays an important role by targeting the tumor suppressor DLC2 to
mitochondria, indicating a possible role of DLC2 in lipid transport
and in the regulation of the mitochondrial pathway of apoptosis and
mitochondrial membrane permeability (22). Therefore, we examined the effect of
StarD13 on apoptosis. Our results showed that StarD13 did not
induce apoptosis. This was reflected by the percentage of apoptotic
cells which was approximately similar in the StarD13 siRNA
transfected cells and the control cells.
To examine the underlying mechanism behind the tumor
suppressor role of StarD13, we investigated the level of ERK
phosphorylation after the silencing of StarD13. The results showed
an increase in the level of p-ERK in the cells transfected with
StarD13 siRNA as compared to non-transfected cells. These findings
were in accordance with the study conducted by Leung et al
which showed that StarD13 suppresses cell growth via the regulation
of the Raf1-ERK1/2-p70S6K signaling pathway (27). Since p-ERK, the extracellular
signal-regulated kinase, is involved in the regulation of cellular
growth and proliferation of several tumor types (28), we suggest that possibly our StarD13
is affecting the cell growth of tumor cells via p-ERK pathway. This
is of interest since it directly links Rho or Cdc42 to the
inhibition of a MAPK pathway.
This study confirmed the role of StarD13 as a tumor
suppressor in astrocytoma. It also uncovered a link between the Rho
GTPase pathway and the MAPK pathway. Perhaps the most interesting
finding in this study was the variation in the level of expression
of a tumor suppressor based on the grade of the tumor. This
corroborated the fact that the same protein might play different
and even opposing roles in the cell depending on the
conditions.
Acknowledgements
The authors would like to thank Dr Noha Bajjani,
from Rizk Medical Center for the tissues collected. We would also
like to thank Dr Hitoshi Yagisawa for providing constructs. This
study was supported by the Natural Science Department at the
Lebanese American University, by the University Research Council
(URC) at LAU and by the Lebanese National Center for Scientific
Research (L-NCSR) (Ref: 03-06-10).
References
|
1
|
Buckner JC, Brown PD, O'Neill BP, Meyer
FB, Wetmore CJ and Uhm JH: Central nervous system tumors. Mayo Clin
Proc. 82:1271–1286. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Salacz ME, Watson KR and Schomas DA:
Glioblastoma: Part I. Current state of affairs. Mol Med.
108:187–194. 2011.PubMed/NCBI
|
|
3
|
Salacz ME, Watson KR and Schomas DA:
Glioblastoma. Part II: future directions. Mol Med. 108:289–291.
2011.PubMed/NCBI
|
|
4
|
Louis DN, Ohgaki H, Wiestler OD and
Cavenee WK: WHO Classification of Tumours of the Central Nervous
System. World Health Organization; 2007
|
|
5
|
Wen PY and Kesari S: Malignant gliomas in
adults. N Engl J Med. 359:492–507. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
DeAngelis LM: Brain tumors. N Engl J Med.
344:114–123. 2001. View Article : Google Scholar
|
|
7
|
Karlsson R, Pedersen ED, Wang Z and
Brakebusch C: Rho GTPase function in tumorigenesis. Biochim Biophys
Acta. 1796:91–98. 2009.PubMed/NCBI
|
|
8
|
Boettner B and Van Aelst L: The role of
Rho GTPases in disease development. Gene. 286:155–174. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Ridley AJ: Rho proteins and cancer. Breast
Cancer Res Treat. 84:13–19. 2004. View Article : Google Scholar
|
|
10
|
Hornstein I, Pikarsky E, Groysman M, Amir
G, Peylan-Ramu N and Katzav S: The haematopoietic specific signal
transducer Vav1 is expressed in a subset of human neuroblastomas. J
Pathol. 199:526–533. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Wang W, Wyckoff JB, Frohlich VC, et al:
Single cell behavior in metastatic primary mammary tumors
correlated with gene expression patterns revealed by molecular
profiling. Cancer Res. 62:6278–6288. 2002.PubMed/NCBI
|
|
12
|
Kim TY, Vigil D, Der CJ and Juliano RL:
Role of DLC-1, a tumor suppressor protein with RhoGAP activity, in
regulation of the cytoskeleton and cell motility. Cancer Metastasis
Rev. 28:77–83. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Kawai K, Seike J, Iino T, et al:
START-GAP2/DLC2 is localized in focal adhesions via its N-terminal
region. Biochem Biophys Res Commun. 380:736–741. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Rittinger K, Walker PA, Eccleston JF, et
al: Crystal structure of a small G protein in complex with the
GTPase-activating protein rhoGAP. Nature. 388:693–697. 1997.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Yau TO, Leung TH, Lam S, et al: Deleted in
liver cancer 2 (DLC2) was dispensable for development and its
deficiency did not aggravate hepatocarcinogenesis. PLoS One.
4:e65662009. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Tcherkezian J and Lamarche-Vane N: Current
knowledge of the large RhoGAP family of proteins. Biol Cell.
99:67–86. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Soccio RE and Breslow JL: StAR-related
lipid transfer (START) proteins: mediators of intracellular lipid
metabolism. J Biol Chem. 278:22183–22186. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Qian X, Li G, Asmussen HK, et al:
Oncogenic inhibition by a deleted in liver cancer gene requires
cooperation between tensin binding and Rho-specific
GTPase-activating protein activities. Proc Natl Acad Sci USA.
104:9012–9017. 2007. View Article : Google Scholar
|
|
19
|
Li H, Fung KL, Jin DY, et al: Solution
structures, dynamics, and lipid-binding of the sterile alpha-motif
domain of the deleted in liver cancer 2. Proteins. 67:1154–1166.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Liao YC and Lo SH: Deleted in liver
cancer-1 (DLC-1): a tumor suppressor not just for liver. Int J
Biochem Cell Biol. 40:843–847. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Ching YP, Wong CM, Chan SF, et al: Deleted
in liver cancer (DLC) 2 encodes a RhoGAP protein with growth
suppressor function and is underexpressed in hepatocellular
carcinoma. J Biol Chem. 278:10824–10830. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Ng DC, Chan SF, Kok KH, et al:
Mitochondrial targeting of growth suppressor protein DLC2 through
the START domain. FEBS Lett. 580:191–198. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Ullmannova V and Popescu NC: Expression
profile of the tumor suppressor genes DLC-1 and DLC-2 in solid
tumors. Int J Oncol. 29:1127–1132. 2006.PubMed/NCBI
|
|
24
|
Leung TH, Ching YP, Yam JW, et al: Deleted
in liver cancer 2 (DLC2) suppresses cell transformation by means of
inhibition of RhoA activity. Proc Natl Acad Sci USA.
102:15207–15212. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Xiaorong L, Wei W, Liyuan Q and Kaiyan Y:
Underexpression of deleted in liver cancer 2 (DLC2) is associated
with overexpression of RhoA and poor prognosis in hepatocellular
carcinoma. BMC Cancer. 8:2052008. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Shih YP, Takada Y and Lo SH: Silencing of
DLC1 upregulates PAI-1 expression and reduces migration in normal
prostate cells. Mol Cancer Res. 10:34–39. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Leung LH, Wong WK, Cheng AC, et al: A new
approach to computing normal tissue complication probability of an
intensity-modulated radiotherapy treatment with stereotactic
radiotherapy boost of nasopharyngeal carcinoma: a case study. Med
Dosim. 36:138–144. 2011. View Article : Google Scholar
|
|
28
|
Zheng B, Fiumara P, Li YV, et al: MEK/ERK
pathway is aberrantly active in Hodgkin disease: a signaling
pathway shared by CD30, CD40, and RANK that regulates cell
proliferation and survival. Blood. 102:1019–1027. 2003. View Article : Google Scholar : PubMed/NCBI
|