Introduction
DNA methylation and related modulation of gene
expression contribute to the development of malignancies (1,2).
Specifically, methylation of CpG dinucleotides in promoter regions
has been associated with transcriptional silencing of tumor
suppressor genes, suggesting DNA methylation as a target for novel
therapeutics (3). 5-Azacytidine and
5-Aza-2′-deoxycytidine (5-Aza-CdR) belong to a class of cytosine
analogues, which are developed as inhibitors of DNA methylation.
Both analogues have been shown to have significant cytotoxic and
antineoplastic activities in many experimental tumors (3,4).
5-Aza-CdR, however, is reported to be non-carcinogenic. It
incorporates into only DNA but not RNA or proteins (5). Additionally, considerable evidence
shows that 5-Aza-CdR has more potent therapeutic effects than
5-Azacytidine in cell culture and animal models of human
cancers.
5-Azacytidine is the only DNA methyltransferase
(DNMT) inhibitor (MTI) approved by the Food and Drug Administration
for treating myelodysplastic syndromes and chronic myelomonocytic
leukemia (6). Recently, several
clinical trials of 5-Aza-CdR have been reported, including a phase
II study of 5-Aza-CdR in patients with metastatic prostate cancer
and a phase III study of 5-Aza-CdR (Decitabine) in patients with
myelodysplasia (7). Clinical trials
evaluating 5-Aza-CdR as a cancer chemotherapeutic have shown
promise for the treatment of leukemia, but less effective against
solid tumors.
5-Aza-CdR binds DNMT in an irreversible, covalent
manner, thus sequestering DNMT enzyme activity and preventing
maintenance of the DNA methylation state (8). Consequently, silenced genes are
demethylated and are re-expressed. Two major non-mutually exclusive
mechanisms of its tumor cytotoxicity have been proposed. One states
that 5-Aza-CdR demethylates cellular DNA, with reactivation of
silenced genes; and the other states that 5-Aza-CdR induces DNA
damage due to the formation of irreversible, covalent enzyme-DNA
adducts (9). However, the relative
contribution of gene reactivation and enzyme-DNA adduct formation
to the efficacy and toxicity of 5-Aza-CdR in vivo remains
unresolved.
As one of the major causes of cancer death, gastric
cancer remains threatening around the world, and most patients in
advanced stages need chemotherapy. To date, however, the effects of
5-Aza-CdR and mechanisms against gastric cancer have not been
completely characterized. Here, we show that 5-Aza-CdR is cytotoxic
against human gastric cancer BGC-823 cell growth. Mechanistic
studies demonstrated that 5-Aza-CdR induces DNA damage and causes
apoptosis of gastric cancer cells. These responses were mediated
mainly through upregulation of DNMT and partially through
demethylation of runt-related transcription factor 3 (RUNX3), which
was independent of p53 status and its ability to activate
p21Waf1/Cip1 expression. Taken together, these studies
provide the preclinical rationale for the clinical evaluation of
5-Aza-CdR to improve patient outcome in gastric cancer.
Materials and methods
Cells and treatments
Human lung cancer cell line A549 and the gastric
cancer cell line BGC-823 were obtained from the China Center for
Type Culture Collection (CCTCC). Both cell lines were grown in
Dulbecco’s modified Eagle’s medium (DMEM) containing 10% fetal
bovine serum at 37°C in a humidified atmosphere with 5%
CO2. For treatment with 5-Aza-CdR (Sigma), cells were
exposed to a single dose of 0.01–100 μM of drug. 5-Aza-CdR was
dissolved in phosphate-buffered saline (PBS) and fresh medium
containing 5-Aza-CdR was added every 24 h.
MTT assay
Cell proliferation was measured using the MTT assay.
Cells were plated in triplicate at 1×103 cells/well in
96-well plates, cultured as previously described, and treated with
different concentrations of 5-Aza-CdR or z-VAD-fmk (Beyotime
Institute of Biotechnology) for indicated times respectively.
Twenty microliters of 5 mg/ml of MTT (Amresco) was then added into
each well and the cells were cultured at 37°C for an additional 4–6
h. The resulting formazan crystals were solubilized by the addition
of 150 μl of DMSO to each well. The optical density (OD) level
under 570 nm was measured and the percentage of cell viability was
calculated using the following formula: percentage of cell
viability = (absorbanceexperimental well − absorbance
blank)/(absorbanceuntreated control well −
absorbance blank) × 100%.
Flow cytometric analysis of DNA
content
Cells were seeded into 6-well plates at a density of
4 to 5×105 cells/well. After 72 h of treatment with 0.5,
1 and 5 μM 5-Aza-CdR, cells were washed with PBS, permeabilized
with 70% ethanol overnight. The next day, ethanol was removed and
cells were incubated for 15–20 min at 37°C with 1 ml PI solution
(0.1% Triton X-100, 50 μg PI and 200 μg RNase A). Distribution of
cell cycle phases with different DNA contents was determined using
a flow cytometer (Beckman Coulter, Miami, FL, USA).
Annexin V staining
Cells (5 to 7×105) were seeded into
6-well plates and treated with 0.5, 1 and 5 μM of 5-Aza-CdR for 48
h. The cells were then trypsinized and washed in PBS. Annexin V
staining was then performed following the manufacturer’s
instructions of the Annexin V Staining kit (Multi Sciences Biotech
Co., Ltd).
Measurement of caspase-3 activity
Caspase-3 activity in the cell lysate was measured
using a colorimetric kit according to the manufacturer’s
instructions (Beyotime Institute of Biotechnology). The absorbance
was determined at 405 nm and activity of caspase-3 was determined
by calculating the ratio of OD at 405 nm of drug treated cells to
untreated cells.
Comet assay for detecting DNA strand
breaks
The Comet assay was performed as previously
described (10). In brief, slides
were cleaned with acid and scrapped with 40 μl of 0.6% agarose.
Cell suspension (20 μl) and 80 μl of 1.1% low-melting agarose were
mixed before adding to the first gel layer. The gel layer was
covered with a coverslip immediately, and then was kept at 4°C for
15 min to allow it to solidify. After gently removing the
coverslip, the slides were immersed in fresh prepared cold lysis
solution (2.5 M NaCl, 100 mM Na2EDTA, 10 mM Tris, pH
10.0) with 1% Triton X-100 and 10% DMSO for at least 1 h at 4°C.
After electrophoresis in fresh solution (1 mM Na2EDTA,
300 mM NaOH, pH 13.0) for 30 min, the slides were placed in Tris
buffer (0.4 M Tris, pH 7.5) for 15 min twice. The slides were then
stained with 40 μl of 0.1 mg/ml propidium iodide (PI). One hundred
randomly selected cells were counted per slide. The images were
captured and scored for each sample using an Image Analysis
software system (IMI version 1.0). The standard of assessing DNA
single strand breaks was based on the percentage of cells with tail
and tail length (a distance from DNA head to the end of DNA tail)
by visual estimation.
RNA preparation and RT-PCR
Total RNA was isolated using TRIzol reagent
(Invitrogen) and 1 μg of RNA was used as a template for the
synthesis of cDNA using the RevertAid™ first strand cDNA synthesis
kit (Fermentas) according to the manufacturer’s instructions.
Polymerase chain reaction (PCR) analysis was performed in a final
volume of 25 μl using the PCR Master Mix (Fermentas). Primer
sequences and annealing temperatures are indicated in Table I. PCR products were separated on
1.5% agarose gels, stained with ethidium bromide and
photographed.
 | Table IPrimers and conditions used for
RT-PCR. |
Table I
Primers and conditions used for
RT-PCR.
| Primers | Sequences | Temperature (°C) | Product length
(bp) |
|---|
| GAPDH | F:
5′-ACGGATTTGGTCGTATTGGG-3′
R: 5′-TCCTGGAAGATGGTGATGGG-3′ | 56 | 211 |
| RUNX3 | F:
5′-GAAAAGGCGTAAGGGAACTC-3′
R: 5′-ACCTGGGACCAGCTATAACC-3′ | 56 | 395 |
|
p21Waf1/Cip1 | F:
5′-GTGAGCGATGGAACTTCGACT-3′
R: 5′-CGAGGCACAAGG GTACAAGAC-3′ | 56 | 229 |
| p53 | F:
5′-GTCTACCTCCCGCCATAA-3′
R: 5′-CATCTCCCAAACATCCCT-3′ | 55 | 316 |
| DNMT1 | F:
5′-CGCTGTATCTAGCAAGGGTCA-3′
R: 5′-TCGAATCTCGCGTAGTCTTG-3′ | 56 | 313 |
| DNMT3a | F:
5′-ACCACAGAGGCGGAAATACC-3′
R: 5′-GTCTCCCTGCTGCTAACTGG-3′ | 56 | 543 |
| DNMT3b | F:
5′-CAGGAGACCTACCCTCCACA-3′
R: 5′-TTACGTCGTGGCTCCAGTTA-3′ | 56 | 436 |
Western blotting
After cells were cultured with the indicated
concentrations of 5-Aza-CdR for the specified times, cells were
lysed using RIPA buffer containing protease inhibitors. After
normalization for total protein content (50 μg/lane), samples were
subjected to 15% SDS-PAGE and then transferred to nitrocellulose
membranes (Bio-Rad Laboratories). After blocking with 5% non-fat
dry milk and 0.1% Tween-20 in Tris-buffered saline, membranes were
incubated with mouse anti-p21Waf1/Cip1, p53 (Santa Cruz
Biotechnology), goat anti-RUNX3 (R&D Systems), rabbit
anti-caspase-3, β-tubulin (Santa Cruz Biotechnology). After
extensive rinsing with TBST buffer, blots were incubated with
HRP-conjugated anti-rabbit, anti-mouse or anti-goat secondary
antibodies (Pierce) and developed with the use of an enhanced
chemiluminescence system (Millipore) and captured on
light-sensitive imaging film (Kodak, Tokyo, Japan).
Statistical analysis
All experiments were repeated in triplicate. Data
are presented as means ± SD. Levels of significance for comparisons
between samples were determined using the Student’s t-test
distribution. Statistical analyses among more than three samples
were performed by ANOVA with Tukey’s honest significant difference
post hoc test applied to significant main effects or interactions
(SPSS 11.5 for Windows).
Results
5-Aza-CdR inhibits BGC-823 cell growth by
induction of apoptosis
Human lung cancer A549 cells were used as a control
since it is well-established that 5-Aza-CdR potentially overcomes
growth advantages (11). Human
BGC-823 cells and A549 cells were exposed to 5-Aza-CdR at different
concentrations for 72 h. The cell viability was determined by the
MTT assay. A dose-dependent inhibition of cell proliferation was
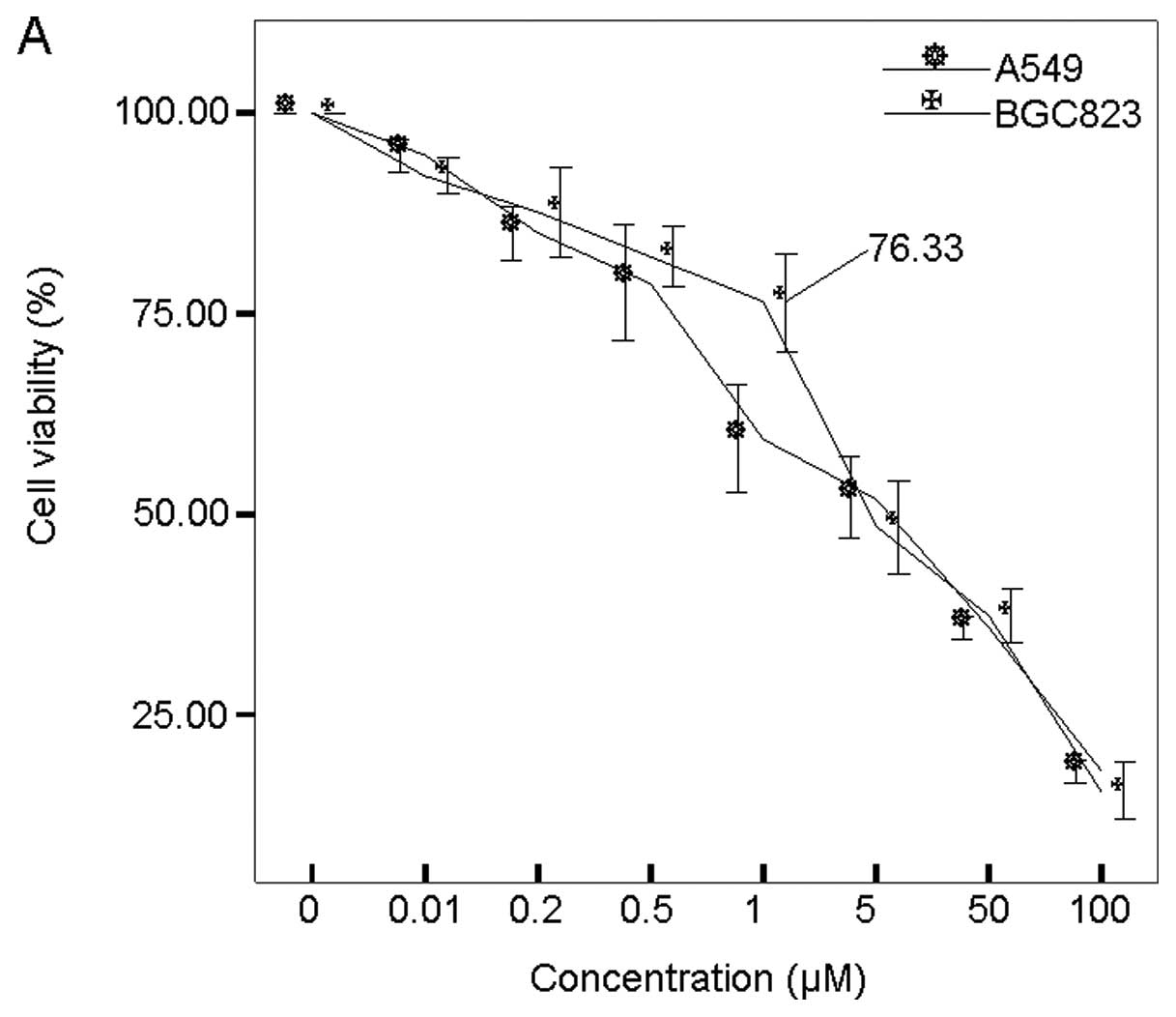
observed in both BGC-823 and A549 cells. As shown in Fig. 1A, BGC cell viability was 76.23%
after 1 μM 5-Aza-CdR treatment, while its viability was only about
25% after 100 μM 5-Aza-CdR treatment.
Thus, 1 μM 5-Aza-CdR treatment resulted in cell
growth inhibition in both cell lines. Therefore, to determine the
duration effect of 5-Aza-CdR on cell viability, BGC-823 cells were
treated with 1 μM of 5-Aza-CdR for different durations. As
expected, we observed not only a dose-dependent growth suppression,
but also a duration-dependent growth suppression in BGC-823 cells
(Fig. 1B). BGC-823 cell viability
was 90, 82, 70 and 38% after 24, 48, 72 and 96 h of treatment,
respectively.
To dissect the mechanism of the anti-proliferative
effects of 5-Aza-CdR in BGC-823 cells, we determined whether
inhibition in cell viability was associated with specific cell
cycle arrest. Exponentially growing BGC-823 cells were treated with
0.5, 1 or 5 μM 5-Aza-CdR for 72 h. The cells were harvested for
flow cytometric analysis of DNA content by PI staining as described
in Materials and methods. The results showed that BGC-823 cell
cycles were not affected by the presence of 5-Aza-CdR (Fig. 1C).
Next, we aimed to examine whether 5-Aza-CdR could
efficiently trigger apoptosis, which led to cytotoxicity against
BGC-823 cells. BGC-823 cells were treated with 0.5, 1 and 5 μM of
5-Aza-CdR for 48 h. The cells were subjected to apoptosis analysis
using Annexin V staining flow cytometry assay as described in
Materials and methods. As shown in Fig.
1D, the data revealed that 5-Aza-CdR treatment increased the
proportion of cells positive for Annexin V from 0.7% in pretreated
cells to 15.5, 32.7 and 49.6% after 5-Aza-CdR treatment at 0.5, 1
and 5 μM, respectively. The results strongly suggested that
apoptosis rather than necrosis is the mechanism of 5-Aza-CdR
induced growth inhibition in BGC-823 cells.
Treatment with 5-Aza-CdR causes DNA
damage independent of the activation of the caspase pathway in
BGC-823 cells
Since 5-Aza-CdR has been reported to incorporate
into DNA and not RNA (5), we
evaluated the effect of 5-Aza-CdR on DNA damage. BGC-823 cells were
treated with 5-Aza-CdR at 0.5, 1 and 5 μM for 72 h. DNA damage was
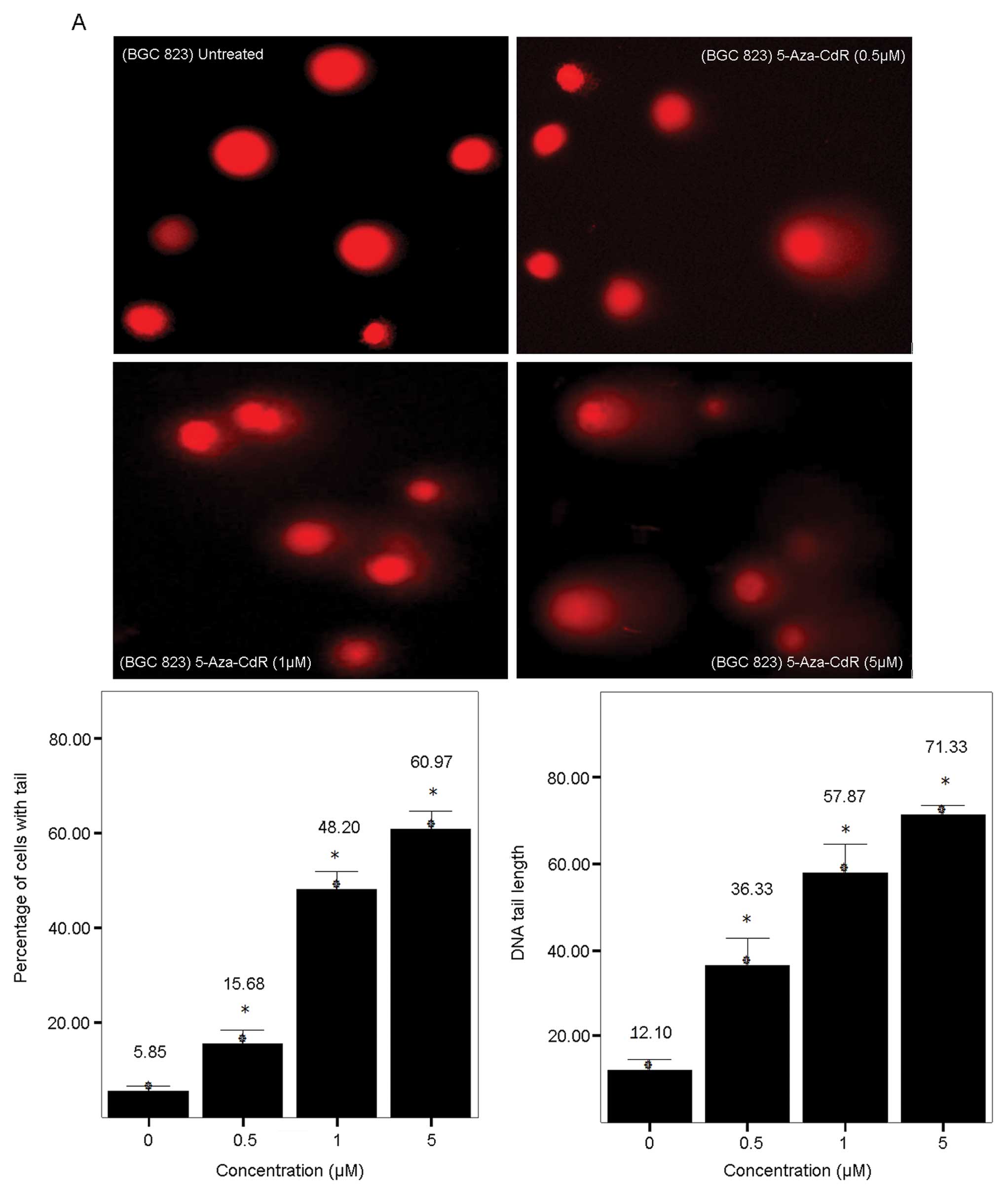
determined using the Comet assay. As shown in Fig. 2A, dose-dependent DNA damage was
observed upon 5-Aza-CdR treatment in both BGC-823 cells. Not only
more comet cells but also longer DNA tail lengths were observed
with increased dosage treatment, indicating more extensive DNA
damage at higher concentrations. As described in Materials and
methods, the image was scored for the percentage of cells with tail
and for DNA tail length. As shown in Fig. 2A, the percentage of cells with tail
increased from 5.85% in untreated cells to 15.68, 48.20 and 60.97%
in 0.5, 1 and 5 μM treated cells, respectively; while the length of
DNA tail increased from 12.10 in untreated cells to 36.33, 57.87,
and 71.33 in 0.5, 1 and 5 μM treated cells, respectively.
To analyze whether caspase-3 was responsible for the
recruitment of apoptotic pathways by 5-Aza-CdR, we performed a
colorimetric enzyme assay to examine the activity of caspase-3 and
western blotting to detect the protein level of procaspase-3. The
results showed that caspase-3 enzymatic activity was not affected
in the 5-Aza-CdR-treated cells (Fig.
2B). The protein level of procaspase-3 was not affected by
5-Aza-CdR treatment either, as monitored by western blot analysis
(Fig. 2C).
Furthermore, BGC-823 cells were pretreated with the
pan-caspase inhibitor, z-VAD-fmk (5 μM), and then treated with
various concentrations of 5-Aza-CdR for 72 h. Treatment with
z-VAD-fmk did not alter the cytotoxic effect of 5-Aza-CdR, implying
that the 5-Aza-CdR-induced apoptosis was independent of the caspase
pathway (Fig. 2D).
5-Aza-CdR has no effect on p53 and
p21Waf1/Cip1 expression
Because it has been proposed that p53 has a vital
role in growth arrest and apoptosis in response to DNA damage, we
explored the p53 expression in response to 5-Aza-CdR treatment in
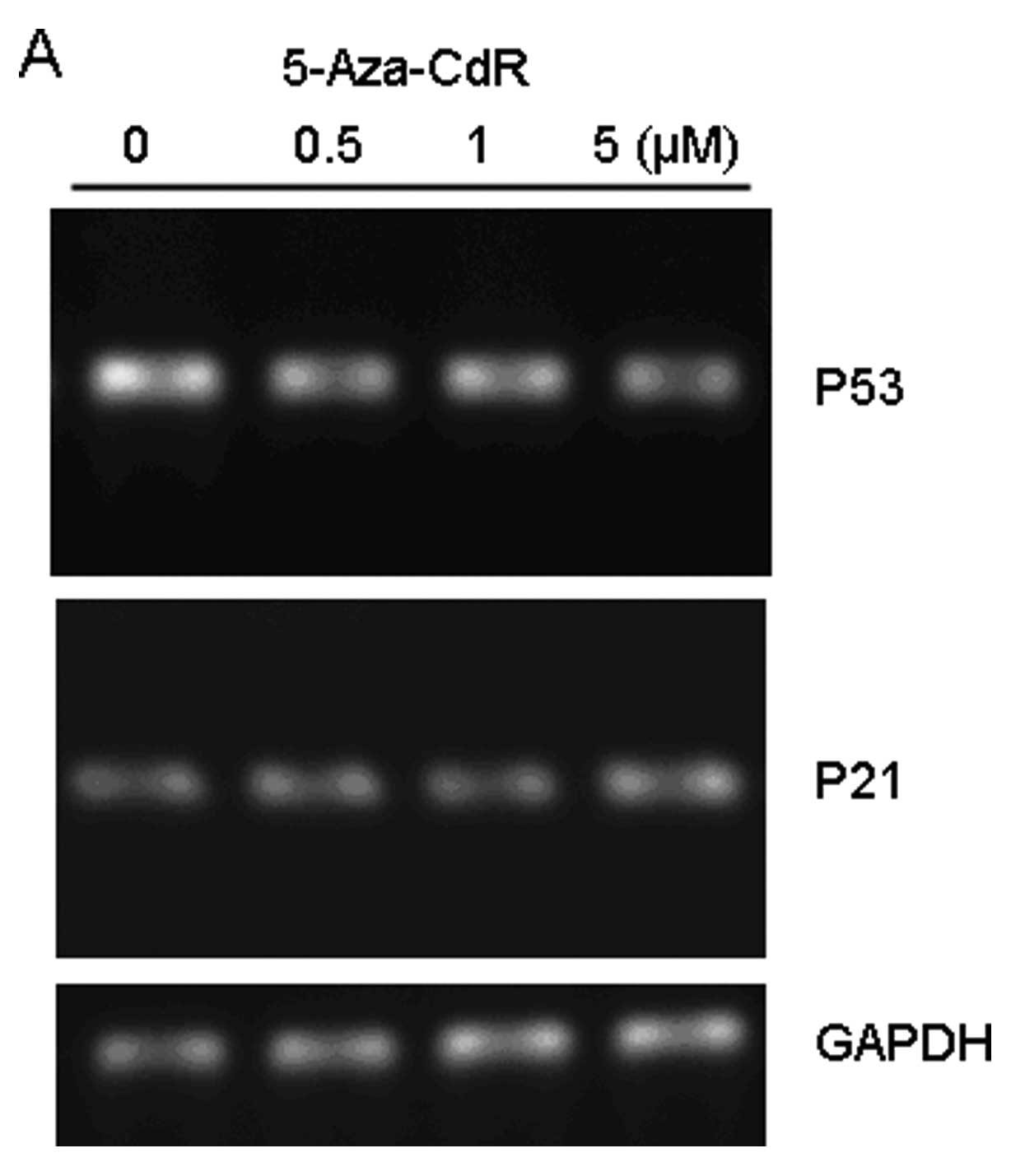
BGC-823 cells. p53 mRNA levels were determined by RT-PCR using
total-RNA isolated from BGC-823 cells treated with 0.5, 1 and 5 μM
5-Aza-CdR for 72 h. The results revealed that p53 mRNA levels were
not changed in the presence of 5-Aza-CdR (Fig. 3A). Of interest, the protein
expression of p53 was undetectable (data not shown).
It has been well established that the expression of
p21Waf1/Cip1 is regulated by p53 (12); therefore, we examined the expression
of p21Waf1/Cip1 in BGC-823 cells.
p21Waf1/Cip1 gene expression was altered in BGC-823
cells exposed to 5-Aza-CdR (Fig.
3A). As a control, 5-Aza-CdR treatment increased the protein
level of p21Waf1/Cip1 along with elevation of p53
protein levels in a dose-dependent manner in A549 cells (Fig. 3B).
5-Aza-CdR induces RUNX3, DNMT1 and DNMT3a
gene transcription
5-Aza-CdR has been reported to exert its
cytotoxicity through depleting DNA methyltransferase levels,
resulting in DNA hypomethylation (8). We thus studied the expression of
RUNX3, a 5-Aza-CdR inducible, methylation-regulated target gene.
RUNX3 promoter is highly methylated in gastric cancer cells
(13). The RUNX3 protein and mRNA
level were determined in BGC-823 cells treated with 0.5, 1 and 5 μM
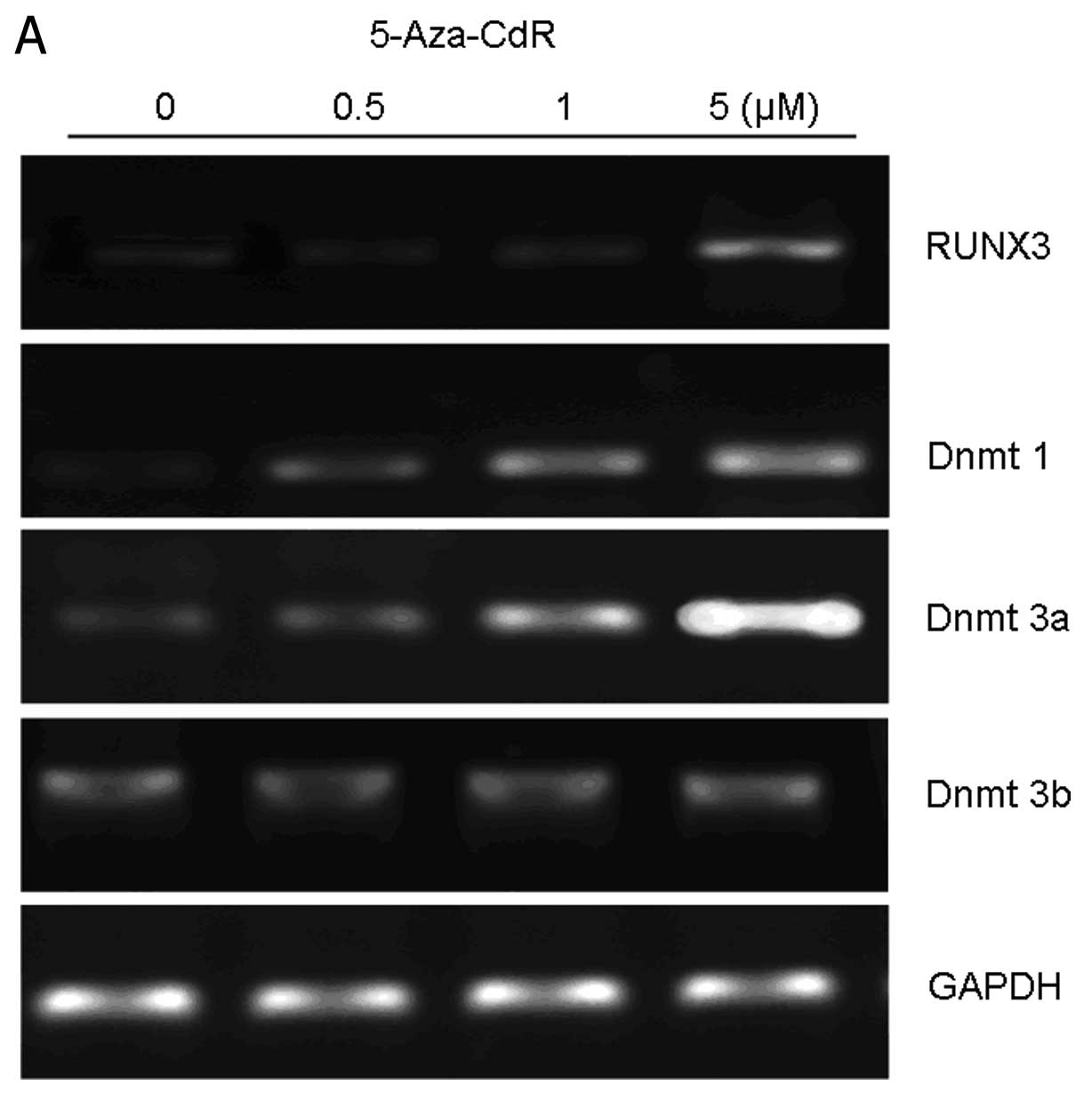
5-Aza-CdR for 72 h. As shown in Fig.
4A, it was indicated that RUNX3 mRNA levels were increased in
the presence of 5-Aza-CdR at 5 μM compared to untreated cells.
However, 0.5 and 1 μM of 5-Aza-CdR exposure did not affect the
levels of RUNX3, consistent with the finding from western blot
analysis (Fig. 4B).
Since 5-Aza-CdR is an inhibitor of DNMTs, we
analyzed the association between 5-Aza-CdR and DNMT1, DNMT3a, and
DNMT3b in BGC-823 cells with RT-PCR analysis. Upon exposure of
BGC-823 cells to 0.5 μM of 5-Aza-CdR for 72 h, the mRNA levels of
DNMT1 and DNMT3a were upregulated in a dose-dependent fashion. The
mRNA levels of DNMT3b, however, remained unchanged between
drug-treated cells and untreated cells (Fig. 4A).
Discussion
A number of studies have reported that 5-Aza-CdR
treatment decreases cell proliferation and induces apoptosis in
tumors, but the mechanisms remain unclear (14,15).
In our investigation, we observed that 5-Aza-CdR increased DNA
methyltransferease expression and demethylation of RUNX3,
independent of the activation of the caspase pathway and the
p53/p21 pathway.
From our results, we found that 5-Aza-CdR
significantly inhibited BGC-823 cell growth in a dose- and
duration-dependent manner. Regarding the mechanism underlying the
5-Aza-CdR-iduced inhibition of cell growth, we demonstrated that
induction of apoptosis resulted from DNA damage, while cell cycle
arrest and activation of the caspase pathway were not apparent.
This finding is not in line with a previous report that 5-Aza-CdR
led to a caspase-dependent induction of apoptosis (16), suggesting different apoptotic
pathways rather than caspase may be activated by 5-Aza-CdR
(17,18,25).
p53, a tumor suppressor, stands at the crossroads of
cellular responses to various stresses. Both the quantity and
activity of p53 are greatly increased in response to DNA damage.
Data from PCR and western blotting in BGC-823 cells support the
model that 5-Aza-CdR-induced cytoxicity is part of a classical
response to DNA damage through induction of the expression of p53
post-translationally rather than hypomethylation at the p53
promoter (19). However, we did not
detect any expression changes of p53 mRNA levels in
5-Aza-CdR-treated BGC-823 cells. This may be due to gene mutation
(20), as p53 protein levels were
undetectable in untreated and treated BGC-823 cells.
Actually, some investigators have indicated that the
cell lines with wild-type p53 are more sensitive to 5-Aza-CdR than
that of mutant p53 (11,21). However, Nieto et al(22) have shown that p53 null mouse
embryonic fibroblast (MEF) cells are more sensitive to 5-Aza-CdR
than p53-positive cells. In line with this report, when the null
expression of p53 was examined in BGC-823 cells, the
5-Aza-CdR-induced inhibition of cell survival was as sensitive as
that in A549 cells and triggered apoptosis in a dose-dependent
manner. Our data further showed that the cytotoxicity of 5-Aza-CdR
was independent of the p53 status in BGC-823 cells. In addition, we
observed upregulation of p21Waf1/Cip1 with increased p53
expression in A549 cells (12), in
keeping with a previous observation that p21Waf1/Cip1
functions as a downstream target of p53. Our data suggest that the
absence of cell cycle arrest and p21Waf1/Cip1 gene
expression differences between 5-Aza-CdR-treated and untreated
cells is due to the p53 mutation in BGC-823 cells.
RUNX3 is part of the runt-domain family of
transcription factors that act as master regulators of gene
expression in major developmental pathways. A more recent study
shows that RUNX3 functions as a candidate gastric cancer suppressor
gene with hypermethylation at the promoter region (13). Enhancing RUNX3 expression by
transfection or other strategies completely abrogated growth
advantages and activated apoptosis in gastric cancer (23). In line with previous reports, we
observed that 5 μM of 5-Aza-CdR increased RUNX3 expression both at
the mRNA and protein levels (24).
It is also noteworthy that the expression of RUNX3 was only
enhanced at 5 μM, even though 0.5 μM of 5-Aza-CdR treatment had
triggered cell apoptosis. Therefore, 5-Aza-CdR induced apoptosis
is, at least in part, due to demethylation of silenced RUNX3. In
mammals, global DNA methylation is catalyzed mainly by three DNMTs:
DNMT1, DNMT3a and DNMT3b. Recently, increased expression of DNA
methyltransferases were shown in various cancer cells (26–28).
In vitro studies indicated that 5-Aza-CdR-treated cells were
depleted of active DNA methyltransferases through sequestration of
the enzyme to azacytosine residues in DNA, resulting in genome-wide
demethylation (8). According to our
data, although 5-Aza-CdR treatment induced demethylation and
expression of RUNX3 the demethylation activity alone could not
explain the observed cytotoxicity against BGC-823 cells. We assumed
that the cellular effects of 5-Aza-CdR were a direct result of the
formation of stable adducts between DNA methyltransferases and
5-Aza-CdR-substituted genomic DNA rather than the DNA
hypomethylation. A previous study demonstrated that DNMT1 knockout
cells were more resistant to 5-Aza-CdR treatment than that of
wild-type cells (29). Also, Oka
et al(30) reported that
5-Aza-CdR may mediate DNMT3a and DNMT3b de novo DNA
methyltransferases in that DNMT3a and DNMT3b null embryonic stem
cells were highly resistant to 5-Aza-CdR when compared to that of
wild-type cells. However, our data demonstrate that the levels of
DNMT1 and DNMT3a were substantially increased rather than decreased
after 5-Aza-CdR treatment (31,32).
Our data support the hypothesis, that, cytotoxicity of 5-Aza-CdR
results from higher DNA methyltransferase levels. Higher DNA
methyltransferase levels were expected to produce more DNA
methyltransferase DNA adducts, resulting in an increased response
of BGC-823 cells to 5-Aza-CdR. Alternatively, the elevation of
DNMT1 and DNMT3a may be not a causative effect but just a side
effect of the treatment of the cells with 5-Aza-CdR. We are
planning to address this issue in a future study.
In conclusion, our results show that 5-Aza-CdR, a
cytosine analogue designed to inhibit DNMTs, inhibited gastric
cancer BGC-823 cell growth via cell apoptosis induced by DNA
damage. The upregulation of DNMT1, DNMT3a, and RUNX3 contributed to
the cytotoxicity. To our knowledge, this is the first demonstration
of p53-independent 5-Aza-CdR action on DNA methyltransferases and
demethylation.
Acknowledgements
This study was supported by a Grant-in-Aid
(302140667) from the Wuhan University.
References
|
1
|
Laird PW: Cancer epigenetics. Hum Mol
Genet. 14:65–76. 2005. View Article : Google Scholar
|
|
2
|
Robertson KD: DNA methylation and human
disease. Nat Rev Genet. 6:597–610. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Bender CM, Zingg JM and Jones PA: DNA
methylation as a target for drug design. Pharm Res. 15:175–187.
1998. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Natsume A, Wakabayashi T, Tsujimura K,
Shimato S, Ito M, Kuzushima K, Kondo Y, Sekido Y, Kawatsura H,
Narita Y and Yoshida J: The DNA demethylating agent
5-aza-2′-deoxycytidine activates NY-ESO-1 antigenicity in
orthotopic human glioma. Int J Cancer. 122:2542–2553. 2008.
|
|
5
|
Glazer RI and Knode MC:
1-beta-D-arabinosyl-5-azacytosine. Cytocidal activity and effects
on the synthesis and methylation of DNA in human colon carcinoma
cells. Mol Pharmacol. 26:381–387. 1984.PubMed/NCBI
|
|
6
|
Issa JP and Kantarjian H: Azacitidine. Nat
Rev Drug Discov Suppl. S6–S7. 2005. View
Article : Google Scholar
|
|
7
|
Kuendgen A and Lübbert M: Current status
of epigenetic treatment in myelodysplastic syndromes. Ann Hematol.
87:601–611. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Haaf T: The effects of 5-azacytidine and
5-azadeoxycytidine on chromosome structure and function:
implications for methylation-associated cellular processes.
Pharmacol Ther. 65:19–46. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Kiziltepe T, Hideshima T, Catley L, Raje
N, Yasui H, Shiraishi N, Okawa Y, Ikeda H, Vallet S, Pozzi S, et
al: 5-Azacytidine, a DNA methyltransferase inhibitor, induces
ATR-mediated DNA double-strand break responses, apoptosis, and
synergistic cytotoxicity with doxorubicin and bortezomib against
multiple myeloma cells. Mol Cancer Ther. 6:1718–1727. 2007.
View Article : Google Scholar
|
|
10
|
Wang C, Zhang Y, Liang J, Shan G, Wang Y
and Shi Q: Impacts of ascorbic acid and thiamine supplementation at
different concentrations on lead toxicity in testis. Clin Chim
Acta. 370:82–88. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Chai G, Li L, Zhou W, Wu L, Zhao Y, Wang
D, Lu S, Yu Y, Wang H, McNutt MA, et al: HDAC inhibitors act with
5-aza-2′-deoxycytidine to inhibit cell proliferation by suppressing
removal of incorporated abases in lung cancer cells. PLoS One.
3:e24452008.
|
|
12
|
Jiemjit A, Fandy TE, Carraway H, Bailey
KA, Baylin S, Herman JG and Gore SD: p21(WAF1/CIP1)
induction by 5-azacytosine nucleosides requires DNA damage.
Oncogene. 27:3615–3623. 2008.
|
|
13
|
Fujii S, Ito K, Ito Y and Ochiai A:
Enhancer of zeste homologue 2 (EZH2) down-regulates RUNX3 by
increasing histone H3 methylation. J Biol Chem. 22:17324–17332.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Fan H, Zhao ZJ, Cheng YC, Shan YF, Lu ZH,
Zhang JQ, Xie W and Fan H: Gene induction and apoptosis in human
hepatocellular carcinoma cells SMMC-7721 exposed to
5-aza-2′-deoxycytidine. Chin Med J (Engl). 120:1626–1631.
2007.PubMed/NCBI
|
|
15
|
Yang H, Hoshino K, Sanchez-Gonzalez B,
Kantarjian H and Garcia-Manero G: Antileukemia activity of the
combination of 5-aza-2′-deoxycytidine with valproic acid. Leuk Res.
29:739–748. 2005.PubMed/NCBI
|
|
16
|
Gomyo Y, Sasaki J, Branch C, Roth JA and
Mukhopadhyay T: 5-aza-2′-deoxycytidine upregulates caspase-9
expression cooperating with p53-induced apoptosis in human lung
cancer cells. Oncogene. 23:6779–6787. 2004.
|
|
17
|
Carter BZ, Kornblau SM, Tsao T, Wang RY,
Schober WD, Milella M, Sung HG, Reed JC and Andreeff M:
Caspase-independent cell death in AML: caspase inhibition in vitro
with pan-caspase inhibitors or in vivo by XIAP or Survivin does not
affect cell survival or prognosis. Blood. 102:4179–4186. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Shang D, Ito N, Kamoto T and Ogawa O:
Demethylating agent 5-Aza-2′-deoxycytidine enhances susceptibility
of renal cell carcinoma to paclitaxel. Urology. 69:1007–1012.
2007.
|
|
19
|
Wang H, Zhao Y, Li L, McNutt MA, Wu L, Lu
S, Yu Y, Zhou W, Feng J, Chai G, et al: An ATM- and Rad3-related
(ATR) signaling pathway and a phosphorylation-acetylation cascade
are involved in activation of p53/p21Waf1/Cip1 in response to
5-aza-2′-deoxycytidine treatment. J Biol Chem. 283:2564–2574.
2008.PubMed/NCBI
|
|
20
|
Shi SL, Wang YY, Liang Y and Li QF:
Effects of tachyplesin and n-sodium butyrate on proliferation and
gene expression of human gastric adenocarcinoma cell line BGC-823.
World J Gastroenterol. 12:1694–1698. 2006.PubMed/NCBI
|
|
21
|
Karpf AR, Moore BC, Ririe TO and Jones DA:
Activation of the p53 DNA damage response pathway after inhibition
of DNA methyltransferase by 5-aza-2′-deoxycytidine. Mol Pharmacol.
59:751–757. 2001.PubMed/NCBI
|
|
22
|
Nieto M, Samper E, Fraga MF, González de
Buitrago G, Esteller M and Serrano M: The absence of p53 is
critical for the induction of apoptosis by 5-aza-2′-deoxycytidine.
Oncogene. 23:735–743. 2004.PubMed/NCBI
|
|
23
|
Nagahama Y, Ishimaru M, Osaki M, Inoue T,
Maeda A, Nakada C, Moriyama M, Sato K, Oshimura M and Ito H:
Apoptotic pathway induced by transduction of RUNX3 in the human
gastric carcinoma cell line MKN-1. Cancer Sci. 99:23–30.
2008.PubMed/NCBI
|
|
24
|
Jung Y, Park J and Kim TY, Park JH, Jong
HS, Im SA, Robertson KD, Bang YJ and Kim TY: Potential advantages
of DNA methyltransferase 1 (DNMT1)-targeted inhibition for cancer
therapy. J Mol Med. 85:1137–1148. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Schmelz K, Wagner M, Dörken B and Tamm I:
5-Aza-2′-deoxycytidine induces p21WAF expression by
demethylation of p73 leading to p53-independent apoptosis in
myeloid leukemia. Int J Cancer. 114:683–695. 2005.
|
|
26
|
Zhu YM, Huang Q, Lin J, Hu Y, Chen J and
Lai MD: Expression of human DNA methyltransferase 1 in colorectal
cancer tissues and their corresponding distant normal tissues. Int
J Colorectal Dis. 22:661–666. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Park HJ, Yu E and Shim YH: DNA
methyltransferase expression and DNA hypermethylation in human
hepatocellular carcinoma. Cancer Lett. 233:271–278. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Roll JD, Rivenbark AG, Jones WD and
Coleman WB: DNMT3b overexpression contributes to a hypermethylator
phenotype in human breast cancer cell lines. Mol Cancer. 7:152008.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Jüttermann R, Li E and Jaenisch R:
Toxicity of 5-aza-2′-deoxycytidine to mammalian cells is mediated
primarily by covalent trapping of DNA methyltransferase rather than
DNA demethylation. Proc Natl Acad Sci USA. 91:11797–11801.
1994.
|
|
30
|
Oka M, Meacham AM, Hamazaki T, Rodić N,
Chang LJ and Terada N: De novo DNA methyltransferases Dnmt3a and
Dnmt3b primarily mediate the cytotoxic effect of
5-aza-2′-deoxycytidine. Oncogene. 24:3091–3099. 2005.PubMed/NCBI
|
|
31
|
Schneider-Stock R, Diab-Assef M, Rohrbeck
A, Foltzer-Jourdainne C, Boltze C, Hartig R, Schönfeld P, Roessner
A and Gali-Muhtasib H: 5-Aza-cytidine is a potent inhibitor of DNA
methyltransferase 3a and induces apoptosis in HCT-116 colon cancer
cells via Gadd45- and p53-dependent mechanisms. J Pharmacol Exp
Ther. 312:525–536. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Palii SS, Van Emburgh BO, Sankpal UT,
Brown KD and Robertson KD: DNA methylation inhibitor
5-Aza-2′-deoxycytidine induces reversible genome-wide DNA damage
that is distinctly influenced by DNA methyltransferases 1 and 3B.
Mol Cell Biol. 28:752–771. 2008.
|