Introduction
Reactive oxygen species (ROS) including hydrogen
peroxide (H2O2), superoxide anion
(O2•−) and hydroxyl radical (•OH)
regulate many essential cellular events such as gene expression,
differentiation, cell proliferation and cell death (1). A change in the redox state of tissues
and cells affects an alteration in the generation or metabolism of
ROS. They are mainly generated as by-products of mitochondrial
respiration or are specifically made by oxidases such as nicotine
adenine diphosphate (NADPH) oxidase and xanthine oxidase (XO)
(2). The principal metabolic
pathways include superoxide dismutases (SODs) [cytoplasmic (SOD1),
mitochondrial (SOD2) or extracellular (SOD3) isoforms], which
metabolize O2•− to
H2O2(3).
Further metabolism by catalase (CAT) or glutathione (GSH)
peroxidase (GPX), yields O2 and H2O (4). Especially, thioredoxin (TXN) system
consisting of TXN, TXN reductase (TXNR) and NADPH is critically
involved in maintaining cellular redox homeostasis (5). TXN as a thiol reductase is a potent
anti-oxidant and acts as a scavenger of ROS (5). Oxidative stress may be the result of
either overproduction of ROS or accumulation of it. This stress can
initiate events that lead to cell death depending on cell types
(6–8).
Arsenic trioxide (ATO; As2O3)
has long been used as therapeutic agents for some severe diseases
including leukemia in East Asia, especially China (9). Recently, ATO has been reported to cure
patients with relapsed acute promyelocytic leukemia (APL) without
severe marrow suppression (10,11).
The antiproliferative effect of ATO is not restricted to APL cells
but can also be implicated in a variety of hematological
malignancies (12,13). Furthermore, ATO may be active
against other malignancies such as solid tumor since the mechanisms
of action of ATO are mainly the induction of apoptosis and the cell
cycle arrest (14). In fact,
accumulating literature has demonstrated that ATO regulate many
biological functions of cell proliferation, differentiation and
angiogenesis as well as apoptosis in the solid tumor cells of renal
(15), head and neck (16), ovarian (17), prostate (17), hepatoma (18), bladder (19), colon (20), lung (21), breast (22), cervical (23) and gastric cancer cells (24). ATO as a mitochondrial poison can
induce the failure of the mitochondrial transmembrane potential
(MMP; Δ Ψm) and, as such, it generates high amounts of
ROS (14,25,26).
ATO also stimulates ROS generation via the activation of NADPH
oxidase (27) or the inhibition of
GPX and TXNR (28,29). These phenomena can trigger the
apoptosis of target cells. In addition, it has been reported that
the intracellular GSH content has a decisive effect on ATO-induced
apoptosis (26,30–32).
Furthermore, a combination of ATO and L-buthionine sulfoximine
(BSO; an inhibitor of GSH synthesis) induces synergistic
cytotoxicity in several cancer cells of renal cell carcinoma
(30), bladder cancer (19), leukemia (32), lung cancer (33), hepatocellular carcinoma (34) and solid tumors (35).
Lung cancer is a main cause of cancer death in
developed countries. Various novel remedial strategies including
new drug development are currently under consideration due to
intrinsic or acquired resistant and toxicity of conventional drugs
(36). Studies of the molecular
mechanisms of cytotoxic drug action have shed light on the
treatment of lung cancer. It has been reported that ATO alone or
its combination with other agents inhibits the growth of lung
cancer cells (33,37,38).
We also reported that ATO induces apoptosis in Calu-6 lung cancer
cells via GSH depletion (39).
However, little is known about the toxicological effects of ATO on
normal primary lung cells. Because we observed that ATO induces the
growth inhibition and death in human pulmonary fibroblast (HPF)
cells (40), in the present study
we investigated the effects of N-acetyl cysteine (NAC) and vitamin
C (well-known antioxidants) or L-buthionine sulfoximine [BSO; an
inhibitor of GSH synthesis (41)]
on ATO-treated HPF cells in relation to cell growth, cell death,
ROS and GSH levels. Furthermore, we examined the effects of
antioxidant-related siRNAs on cell death and ROS levels in
ATO-treated HPF cells.
Materials and methods
Cell culture
The human pulmonary fibroblast (HPF) cells from
PromoCell GmbH (Heidelberg, Germany) were maintained in humidified
incubator containing 5% CO2 at 37°C. HPF cells were
cultured in RPMI-1640 supplemented with 10% fetal bovine serum
(FBS) and 1% penicillin-streptomycin (Gibco-BRL, Grand Island, NY).
HPF cells were used between passages 4 and 8.
Reagents
ATO was purchased from Sigma-Aldrich Chemicals (St.
Louis, MO) and was dissolved in 1.65 M NaOH at 1×10−1 M
as a stock solution. NAC and BSO were obtained from Sigma-Aldrich
Chemicals. NAC was dissolved in the buffer [20 mM HEPES (pH 7.0)].
BSO was dissolved in water. Vitamin C purchased from Riedel-de Haen
(Hannover, Germany) was also dissolved in water. Cells were
pretreated with 2 mM NAC or 10 μM BSO or 0.4 mM vitamin C for 1 h
prior to ATO treatment.
Detection of intracellular ROS
levels
Intracellular ROS were detected by a fluorescent
probe dye, 2′,7′-Dichlorodihydrofluorescein diacetate
(H2DCFDA, Ex/Em = 495 nm/529 nm; Invitrogen Molecular
Probes, Eugene, OR) as previously described (42). H2DCFDA is poorly
selective for superoxide anion radical
(O2•−). In contrast, dihydroethidium (DHE,
Ex/Em = 518 nm/605 nm; Invitrogen Molecular Probes) is a
fluorogenic probe that is highly selective for
O2•− among ROS as previously described
(42). Mitochondrial
O2•− level was detected using MitoSOX™ Red
mitochondrial O2•− indicator (Ex/Em=510
nm/580 nm; Invitrogen Molecular Probes) as previously described
(42). In brief, 1×106
cells in 60-mm culture dish (Nunc, Roskilde, Denmark) were
incubated with the indicated doses of ATO in the presence or
absence of NAC, BSO, vitamin C or antioxidant-related siRNA duplex
for the indicated times. Cells were then washed in PBS and
incubated with 20 μM H2DCFDA, 20 μM DHE or 5 μM
MitoSOXTM Red at 37°C for 30 min. DCF, DHE and
MitoSOXTM Red fluorescences were detected using a
FACStar flow cytometer (Becton-Dickinson, Franklin Lakes, NJ). ROS
and O2•− levels were expressed as mean
fluorescence intensity (MFI), which was calculated by CellQuest
software (Becton-Dickinson).
Detection of the intracellular
glutathione (GSH)
Cellular GSH levels were analyzed using a
5-chloromethylfluorescein diacetate dye (CMFDA, Ex/Em = 522 nm/595
nm; Invitrogen Molecular Probes) as previously described (42). In brief, 1×106 cells in
60-mm culture dish (Nunc) were incubated with the indicated doses
of ATO in the presence or absence of NAC, BSO or vitamin C for the
indicated times. Cells were then washed with PBS and incubated with
5 μM CMFDA at 37°C for 30 min. CMF fluorescence intensity was
determined using a FACStar flow cytometer (Becton-Dickinson).
Negative CMF staining (GSH depleted) cells were expressed as the
percent of (-) CMF cells.
Cell growth assay
The effect of drugs on HPF cell growth was
determined by measuring
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT;
Sigma-Aldrich Chemicals Co.) dye absorbance as previously described
(12). In brief, 5×103
cells per well were seeded in 96-well microtiter plates (Nunc).
After exposure to 50 ATO μM with or without NAC, BSO or vitamin C
for 24 h, 20 μl of MTT solution [2 mg/ml in phosphate-buffered
saline (PBS)] were added to each well of the 96-well plates. The
plates were incubated for an additional 3 h at 37°C. Media in wells
were withdrawn by pipetting, and 200 μl of DMSO was added to each
well to solubilize the formazan crystals. Optical density was
measured at 570 nm using a microplate reader (SpectraMAX 340,
Molecular Devices, Sunnyvale, CA).
Annexin V/PI staining for cell death
detection
Apoptosis was determined by staining cells with
Annexin V-fluorescein isothiocyanate (FITC, Ex/Em = 488 nm/519 nm;
Invitrogen Molecular Probes) and propidium iodide (PI, Ex/Em = 488
nm/617 nm; Sigma-Aldrich Chemicals). In brief, 1×106
cells in 60-mm culture dish (Nunc) were incubated with 50 μM ATO in
the presence or absence of NAC, BSO, vitamin C or
antioxidant-related siRNA duplex for 24 h. Cells were washed twice
with cold PBS and then resuspended in 500 μl of binding buffer (10
mM HEPES/NaOH pH 7.4, 140 mM NaCl, 2.5 mM CaCl2) at a
concentration of 1×106 cells/ml. Annexin V-FITC (5 μl)
and PI (1 μg/ml) were then added to these cells, which were
analyzed with a FACStar flow cytometer (Becton-Dickinson).
Measurement of MMP (Δ Ψm)
MMP (Δ Ψm) levels were measured using a
rhodamine 123 fluorescent dye (Ex/Em = 485 nm/535 nm; Sigma-Aldrich
Chemical) as previously described (43). In brief, 1×106 cells in
60-mm culture dish (Nunc) were incubated with 50 μM ATO in the
presence or absence of NAC, BSO or vitamin C for 24 h. Cells were
washed twice with PBS and incubated with the rhodamine 123 (0.1
μg/ml) at 37°C for 30 min. Rhodamine 123 staining intensity was
determined by flow cytometry (Becton-Dickinson). An absence of
rhodamine 123 from cells indicated the loss of MMP (Δ
Ψm) in HPF cells.
Western blot analysis
The changes of antioxidant-related protein in
ATO-treated cells were determined by western blotting as previously
described (12). In brief,
1×106 cells in 60-mm culture dish (Nunc) were incubated
with 50 μM ATO in the presence or absence of 2 mM NAC for 24 h. The
cells were then washed in PBS and suspended in five volumes of
lysis buffer (20 mM HEPES, pH 7.9, 20% glycerol, 200 mM KCl, 0.5 mM
EDTA, 0.5% NP40, 0.5 mM DTT, 1% protease inhibitor cocktail).
Supernatant protein concentrations were determined using the
Bradford method. Samples containing 10 μg total protein were
resolved by 12.5% SDS-PAGE gels, transferred to Immobilon-P PVDF
membranes (Millipore, Billerica, MA) by electroblotting and then
probed with anti-SOD1, anti-SOD2, anti-TXN, anti-TXNR1 and
anti-β-actin antibodies (Santa Cruz Biotechnology, Santa Cruz, CA).
Membranes were incubated with horseradish peroxidase-conjugated
secondary antibodies. Blots were developed using an ECL kit
(Amersham, Arlington Heights, IL).
Transfection of cells with
antioxidant-related siRNAs
Gene silencing of SOD1, SOD2, CAT, GPX and TXN was
performed as previously described (44). A non-specific control siRNA duplex
[5′-CCUACGCCACCAAUUUCGU(dTdT)-3′], SOD1 siRNA duplex
[5′-GAAAACACGGUGGGCCAAA(dTdT)-3′], SOD2 siRNA duplex
[5′-CUGGGAGAAUGUAACUGAA (dTdT)-3′], CAT siRNA duplex
[5′-CACUGAUUUCACAACAGAU(dTdT)-3′], GPX siRNA duplex
[5′-CAAGCUCAUCACCUGGUCU(dTdT)-3′] and TXN siRNA duplex
[5′-GCAUGCCAACAUUCCAGUU(dTdT)-3′] were purchased from the Bioneer
Corp. (Daejeon, South Korea). In brief, 2.5×105 cells in
6-well plates (Nunc) were incubated in RPMI-1640 supplemented with
10% FBS. The next day, cells (~30–40% confluence) in each well were
transfected with the control or each siRNA duplex [80 pmol in
Opti-MEM (Gibco-BRL)] using Lipofectamine 2000, according to the
manufacturer’s instructions (Invitrogen, Brandford, CT). Two days
later, cells were treated with or without 50 μM ATO for additional
24 h. The transfected cells were collected and used for the
measurement of Annexin V-FITC/PI staining cells and ROS levels.
Statistical analysis
The results represent the mean of at least three
independent experiments (mean ± SD). The data were analyzed using
InStat software (GraphPad Prism 4, San Diego, CA). The Student’s
t-test or one-way analysis of variance (ANOVA) with post hoc
analysis using Tukey’s multiple comparison test was used for
parametric data. Statistical significance was defined as
P<0.05.
Results
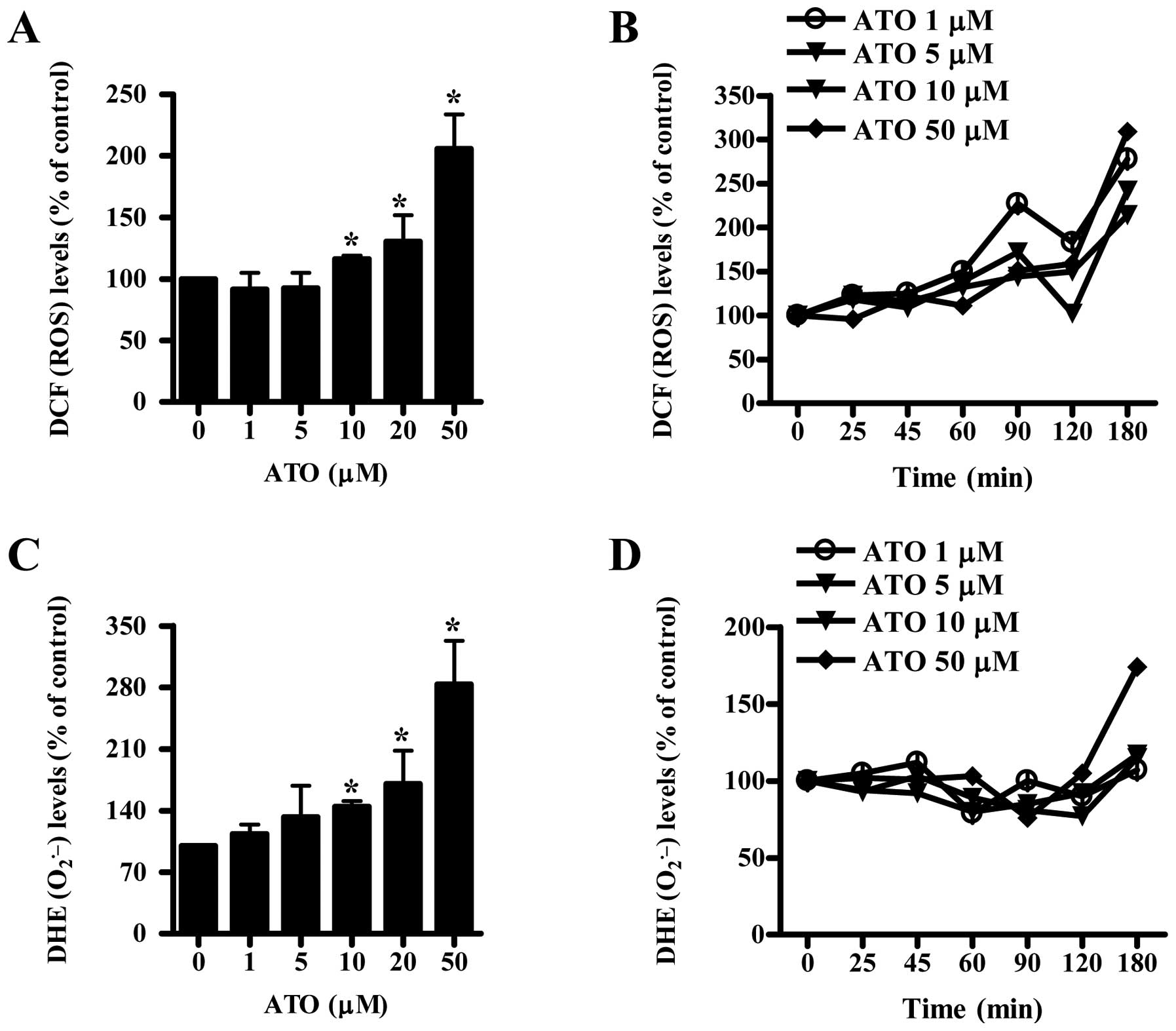
ATO changes ROS levels in HPF cells
We used doses of 1–50 μM ATO to assess ROS levels in
HPF cells. Treatment with 1 or 5 μM ATO increased the growth of HPF
cells at 24 h whereas 10–50 μM ATO inhibited the growth
(unpublished data). As shown in Fig.
1A, ROS (DCF) levels were not altered in 1 or 5 μM ATO-treated
HPF cells at 24 h but were increased in 10–50 μM ATO-treated HPF
cells. All the tested doses of ATO generally increased ROS (DCF)
levels from the early time of 25 min and the gradual increases
lasted for the tested times (25–80 min) although there was a
transient decrease in ROS levels at 120 min (Fig. 1B). Intracellular
O2•− (DHE) level was increased in 1–50 μM
ATO-treated HPF cells in a dose-dependent manner (Fig. 1C). However,
O2•− levels in these cells were not clearly
augmented at the early time of 25 or 45 min and the levels were
instead gradually decreased until 120 min (Fig. 1D). At 180 min, ATO seemed to
increase O2•− level in HPF cells and 50 μM
ATO showed a strong increase in this level (Fig. D).
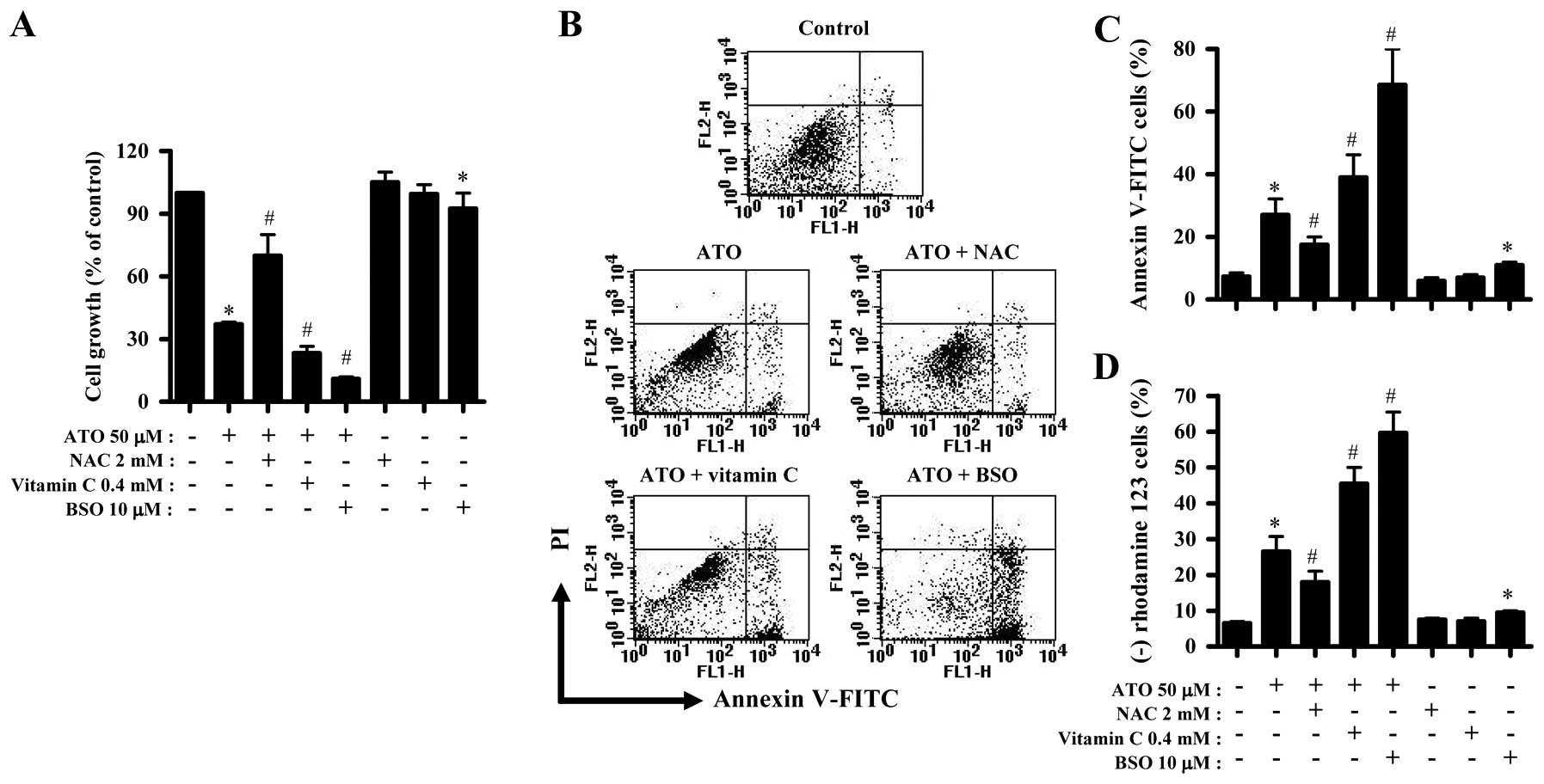
NAC, vitamin C or BSO influences the
growth inhibition and death of ATO-treated HPF cells
We examined the effect of NAC, vitamin C and BSO on
the growth and death of ATO-treated HPF cells. For this experiment,
50 μM ATO was used as a suitable dose to differentiate the levels
of cell growth inhibition and death. NAC significantly prevented
the growth inhibition by ATO whereas vitamin C and BSO enhanced the
growth inhibition (Fig. 2A). BSO
alone inhibited HPF cell growth (Fig.
2A). In relation to cell death, ATO induced cell death in HPF
cells at 24 h, as evidenced by Annexin V staining cells (Fig. 2B and C). NAC significantly rescued
HPF cells from ATO insult whereas vitamin C and BSO increased the
cell death by ATO, and BSO alone also induced cell death in HPF
control cells (Fig. 2B and C).
Apoptosis is closely related to the collapse of MMP
(Δ Ψm) (45). As
expected, loss of MMP (Δ Ψm) was observed in ATO-treated
HPF cells (Fig. 2D). Similar to the
results of Annexin V staining cells, NAC attenuated the loss of MMP
(Δ Ψm) in ATO-treated HPF cells whereas vitamin C and
BSO enhanced the loss in these cells (Fig. 2D). BSO alone induced MMP (Δ
Ψm) loss in HPF control cells as well (Fig. 2D).
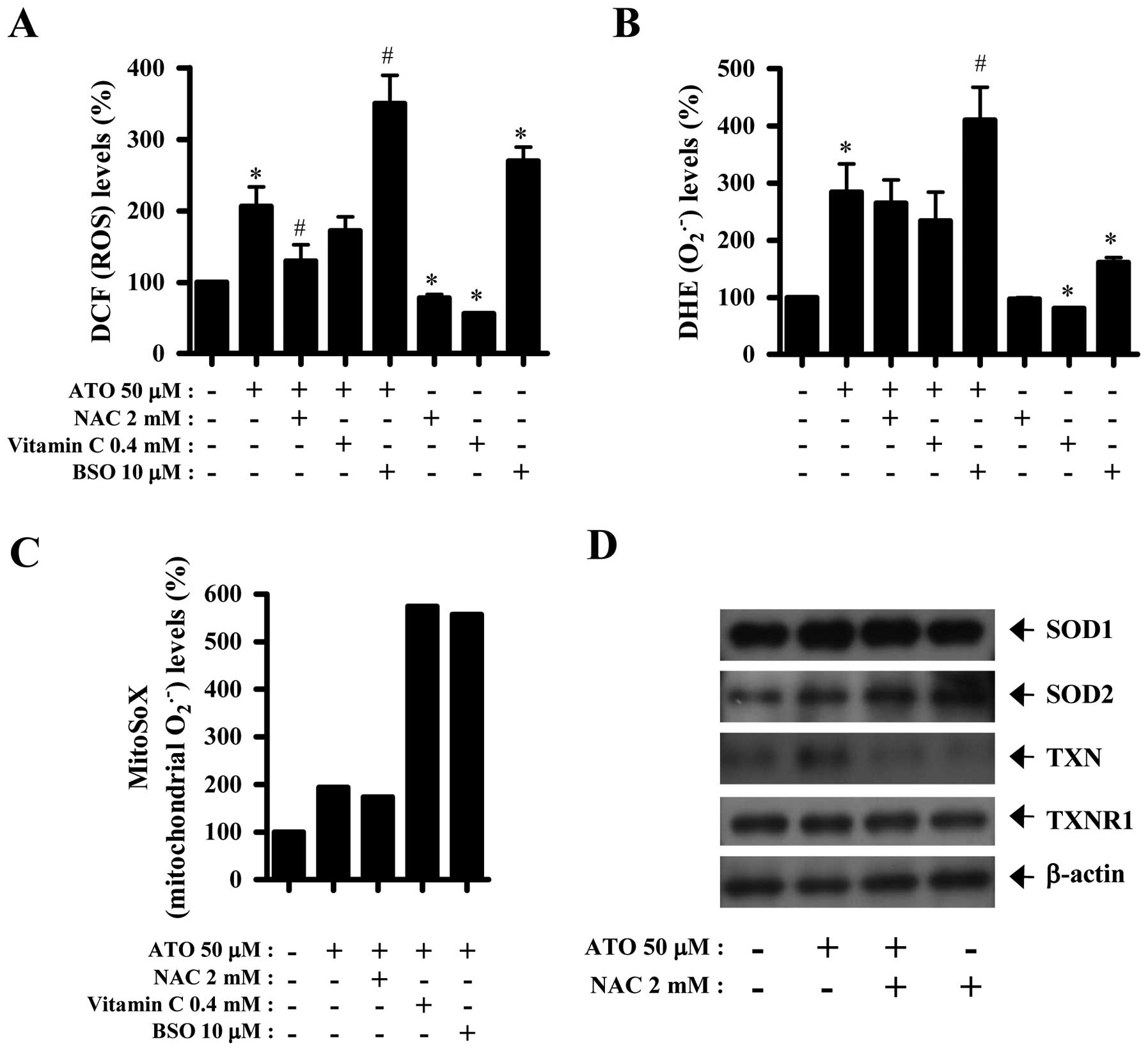
NAC, vitamin C or BSO affects ROS and
antioxidant-protein levels in ATO-treated HPF cells
Next, ROS and antioxidant-protein levels in 50 μM
ATO-treated HPF cells were assessed in the presence or absence of
NAC, vitamin C or BSO. As shown in Fig.
3A, ROS (DCF) level in ATO-treated HPF cells was significantly
decreased by NAC and was also slightly attenuated by vitamin C.
Both NAC and vitamin C decreased basal ROS (DCF) levels in HPF
control cells (Fig. 3A). In
contrast, BSO strongly increased ROS (DCF) levels in ATO-treated or
-untreated HPF cells (Fig. 3A).
Both NAC and vitamin C seemed to decrease
O2•− level in ATO-treated and -untreated HPF
cells (Fig. 3B). However, BSO
significantly increased O2•− levels in
ATO-treated or -untreated HPF cells (Fig. 3B). Furthermore, MitoSOX Red
fluorescence levels, which specifically indicate
O2•− levels in the mitochondria, were
strongly increased in 50 μM ATO-treated HPF cells at 24 h (Fig. 3C). While NAC seemed to decrease
mitochondrial O2•− level in ATO-treated HPF
cells, both vitamin C and BSO strongly enhanced the level (Fig. 3C). The expression of SOD1 was not
changed by ATO and/or NAC whereas that of SOD2 was increased by
both agents (Fig. 3D). In addition,
ATO clearly upregulated the expression of TXN in HPF cells, which
expression was completely downregulated by NAC (Fig. 3D). Neither ATO nor NAC strongly
affected the expression of TXNR1 (Fig.
3D).
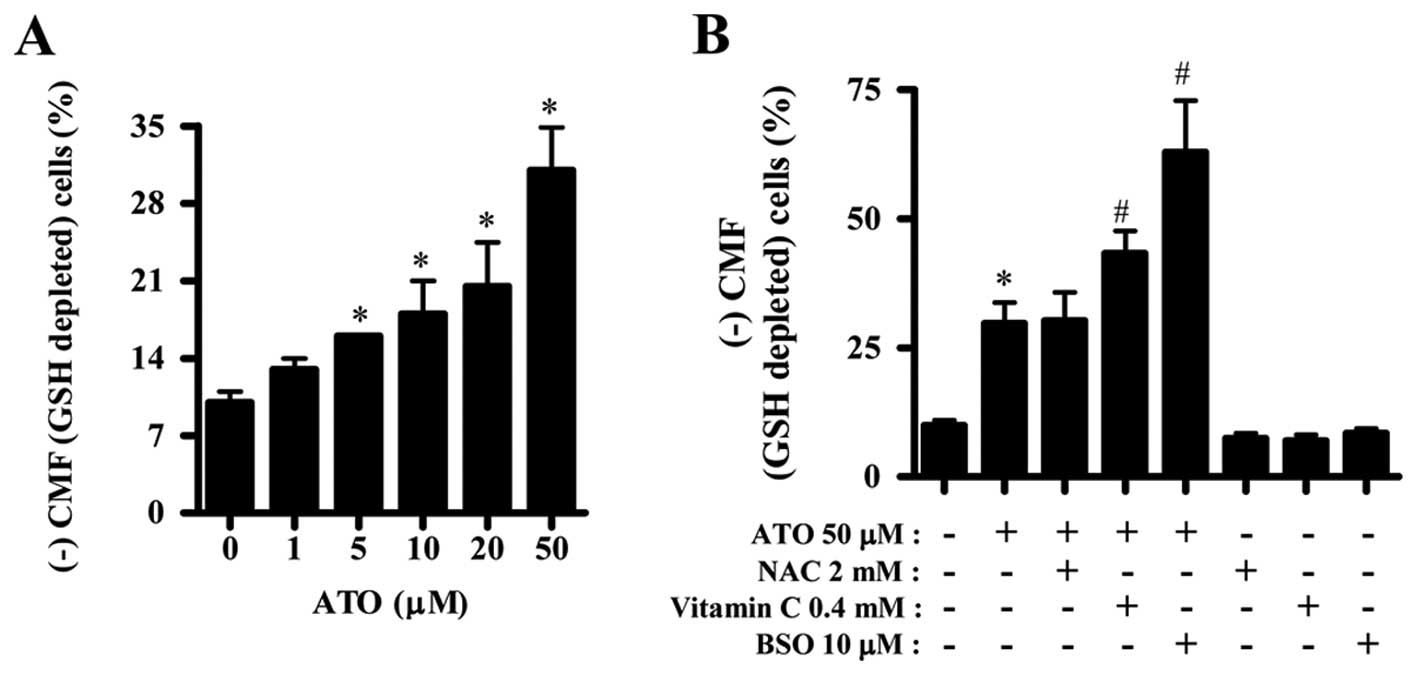
ATO and/or NAC, vitamin C or BSO affects
GSH levels in HPF cells
Next, we assessed the changes of GSH levels by ATO
in the presence or absence of NAC, vitamin C or BSO. ATO increased
GSH depleted cell number in HPF cells in a dose-dependent manner
and even low dose of 1 or 5 μM ATO induced GSH depletion (Fig. 4A). NAC did not affect GSH depleted
cell number in ATO-treated HPF cells but vitamin C and BSO
significantly increased the number in these cells (Fig. 4B). NAC, vitamin C or BSO alone did
not significantly affect the percent of GSH depletion in HPF
control cells (Fig. 4B).
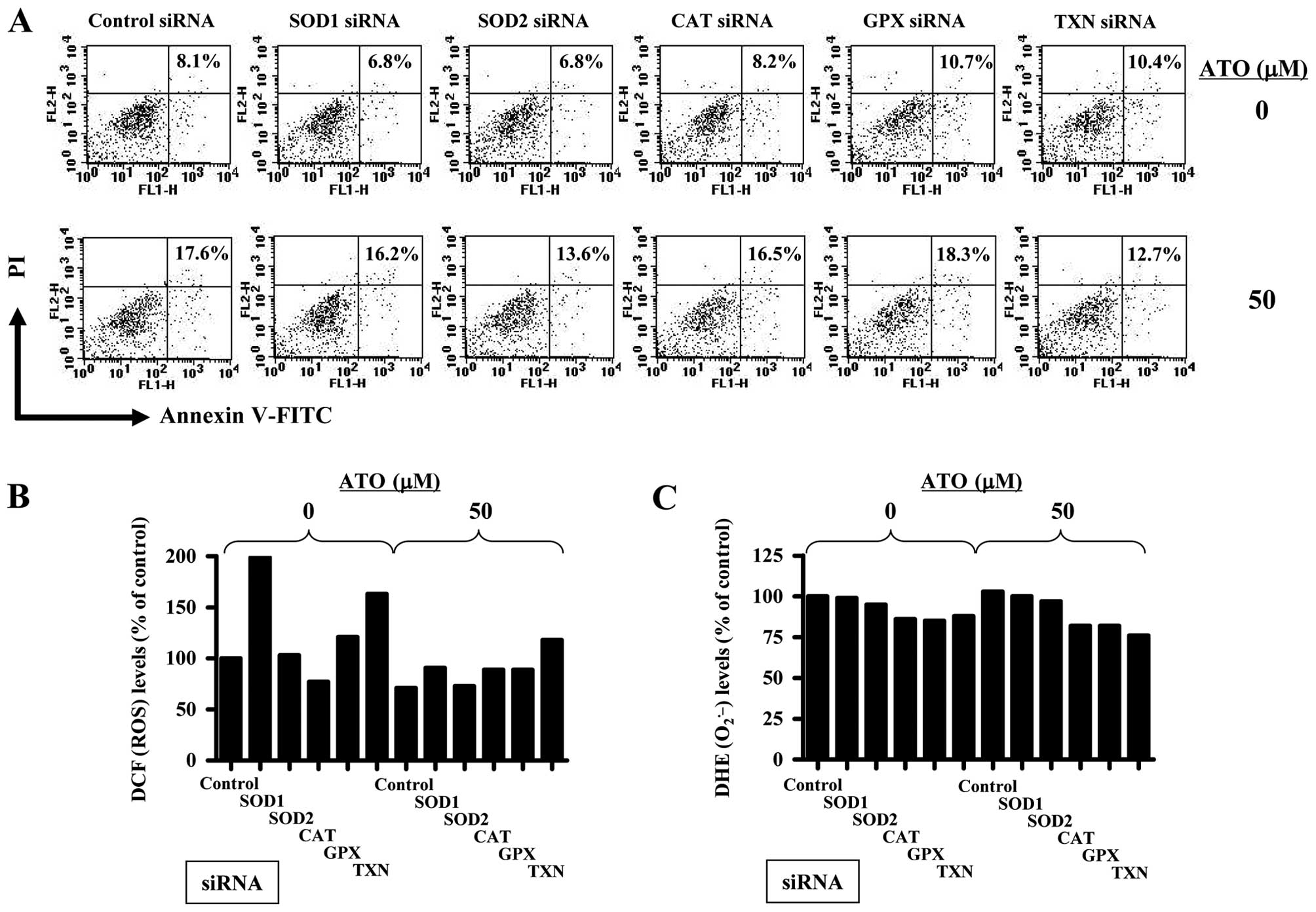
Antioxidant-related siRNAs affect cell
death, ROS and GSH depletion levels in ATO-treated HPF cells
Furthermore, it was determined whether antioxidant
(SOD1, SOD2, CAT, GPX or TXN)-related siRNAs changed cell death,
ROS and GSH depletion levels in ATO-treated HPF cells. As shown in
Fig. 5, 50 μM ATO increased the
proportion of Annexin V-stained cells about 9% compared with that
in control siRNA treated HPF cells. Probably, the siRNA knockdown
system with Lipofectamine 2000 attenuated the biological activity
of ATO. The siRNAs of antioxidant-related proteins did not
significantly alter Annexin V-stained cell number in HPF control
cells for 72 h (Fig. 5A).
Administration of SOD2 or TXN siRNA attenuated cell death in
ATO-treated HPF cells, whereas SOD1, CAT or GPX siRNA did not
(Fig. 5A). SOD1, GPX or TXN siRNA
increased ROS (DCF) level in HPF control cells whwewasCAT siRNA
decreased the level for 72 h (Fig.
5B). CAT, GPX or TXN siRNA seemed to decrease
O2•− level in HPF control cells (Fig. 5C). Treatment with 50 μM ATO in this
siRNA knockdown system did not increase ROS levels including
O2•− in control siRNA treated HPF cells
(Fig. 5B and C). Relatively, SOD1,
CAT, GPX or TXN siRNA upregulated the ROS (DCF) level in
ATO-treated HPF cells (Fig. 5B).
CAT, GPX or TXN siRNA reduced O2•− level in
ATO-treated HPF cells (Fig. 5C).
None of the antioxidant-related siRNAs affected GSH depleted cell
number in HPF control cells for 72 h, and 50 μM ATO did not clearly
increase the number in control siRNA treated HPF cells at 24 h
(data not shown). The tested siRNA did not strongly change GSH
depleted cell number in ATO-treated HPF cells (data not shown).
Discussion
ATO can disturb the natural oxidation and reduction
equilibrium in cells via changing a variety of redox enzymes
(14,27) and influencing MMP (Δ Ψm)
(14,25,26).
The increased intracellular ROS is observed in ATO-treated cervical
cancer cells (46), APL cells
(47), hepatocellular carcinoma
HepG2 (26) and glioblastoma A172
cells (48). These results suggest
that ATO-induced cell death is related to ROS accumulation.
Therefore, in the present study we focused on the molecular
mechanism of ATO-induced HPF cell death in relation to ROS and
GSH.
ROS level (as determined by DCF) was increased in
HPF cells treated with 10–50 μM ATO and O2•−
level (as determined by DHE) was also increased by all the tested
doses of 1–50 μM ATO at 24 h. ATO also ROS (DCF) levels from the
early time phases but did not increase O2•−
levels at these times. There was a transient decrease in ROS levels
at 120 min. However, an increase in O2•−
level was observed from 120 min, which level was strongly increased
at 180 min in 50 μM ATO-treated HPF cells. Because ATO increases
ROS levels including O2•− via a variety of
redox enzymes (14,27) as well as causing mitochondrial
dysfunction (14,25,26),
it is possible that ATO increased ROS (DCF) levels in HPF cells via
affecting redox enzymes until 90 min and then ATO increased ROS
levels including O2•− via damaging
mitochondria as well as changing the activities of redox enzymes
from 120 min. In addition, the increased O2•−
levels in ATO-treated HPF cells at 24 h seemed to result from the
enhanced production of O2•− itself rather
than the reduction of SOD activity since mitochondrial
O2•− level in HPF cells were increased by ATO
and the expression SOD1 or 2 was not downregulated by ATO.
Furthermore, ATO induced the loss of MMP (Δ Ψm) in HPF
cells. Taken together, ATO induced growth inhibition and death in
HPF cells accompanied by an increase in ROS levels including
O2•−. These results suggest the possibility
that changes in ROS levels in ATO-treated HPF cells by NAC, vitamin
C or BSO affect cell death in these cells. Thus, we assessed ROS
and cell death levels in ATO-treated HPF cells with or without NAC,
vitamin C or BSO.
Expectedly, a well-known antioxidant NAC seemed to
attenuate ROS levels including mitochondrial
O2•− in ATO-treated or -untreated HPF cells.
It also significantly prevented cell growth inhibition, cell death
and MMP (Δ Ψm) loss in ATO-treated HPF cells.
Furthermore, NAC upregulated the expression of SOD2 protein in HPF
cells. These results are similar to other reports that NAC
attenuated cell death and ROS increase in ATO-treated cells
(26,49). In contrast, BSO showing a strong
enhancement in cell death and MMP (Δ Ψm) loss in
ATO-treated HPF cells intensified ROS level including mitochondrial
O2•− in these cells. Therefore, ATO seemed to
induce HPF cell death through the induction of ROS. BSO alone
induced cell growth inhibition, cell death and MMP (Δ
Ψm) loss in HPF control cells and strongly increased ROS
levels. Therefore, an increased ROS by BSO treatment seemed to be
tightly related to HPF cell growth inhibition and death. However,
although another antioxidant vitamin C seemed to decrease ROS
levels including O2•− in ATO-treated or
-untreated HPF cells, this agent strongly intensified mitochondrial
O2•− level, cell growth inhibition and cell
death in ATO-treated HPF cells. It was assumed that the enhancement
of HPF cell death and mitochondrial O2•−
level by co-treatment with ATO and vitamin C resulted from the
severe loss of MMP (Δ Ψm) by them. Our result is similar
to the reports that vitamin C enhances ATO-induced cytotoxicity in
multiple myeloma cells (50–52),
leukemia cells (53,54) and hepatocellular carcinoma cells
(26). Therefore, vitamin C plays a
role as a prooxidant rather than antioxidant in ATO-treated cells
including HPF cells and it can be used with ATO for treatment of
cancer including hematological malignancies.
In relation to the administration of
antioxidant-related siRNAs in ATO-treated HPF cells, 50 μM ATO
mildly increased Annexin V-stained cell number and did not increase
ROS levels including O2•− in control siRNA
treated HPF cells. SOD2 or TXN siRNAs attenuated cell death in
ATO-treated HPF cells without convincible changes in ROS levels.
Therefore, the alteration of ATO-induced HPF cell death by
antioxidant-related siRNAs seemed not to be correlated with ROS
changes. SOD2 or TXN as a potent antioxidant can stimulate cell
proliferation or may confer resistance to anti-cancer drugs
(55–57). Thus, the downregulation of SOD2 or
TXN may render cells sensitive to several cytotoxic drugs. However,
our results showed that SOD2 or TXN siRNA did not enhance HPF cell
death by ATO but instead attenuated it. In addition, ATO clearly
upregulated the expressions of SOD2 and TXN. The possibility that
SOD2 and TXN are involved in HPF cell death in respond to ATO via
an unidentified mechanism is worthy of further study. Moreover,
administration with SOD1, GPX or TXN siRNA increased ROS (DCF)
level in HPF control cells but SOD2 or CAT siRNA did not. None of
these siRNAs increased the O2•− level in HPF
control cells. Because a change in the generation or metabolism of
ROS in the cells is influenced by various prooxidant or antioxidant
enzymes as well as activities in various cellular organelles such
as mitochondria and endoplasmic reticulum, our results suggest that
the downregulation of each antioxidant protein by its corresponding
siRNA does not simply increase ROS levels in HPF cells but can
individually affect different ROS levels. Therefore, the role of
ROS level change by antioxidant-related siRNAs in ATO-treated HPF
cells need to be further studied in relation to cell death.
It has been reported that apoptotic effects are
inversely comparative to GSH content (19,34,35,58–60).
The intracellular GSH content has a decisive effect on ATO-induced
apoptosis (26,30–32).
In addition, BSO or vitamin C enhances GSH depletion in ATO-treated
cells (26,33,50,54,61).
Likewise, ATO dose-dependently increased the number of GSH-depleted
cells in HPF cells. As expected, BSO as a GSH synthesis inhibitor
increased the numbers of GSH depleted cells in ATO-treated HPF
cells. Vitamin C showing an increase in HPF cell death by ATO
augmented the numbers of GSH depleted cells in these cells.
However, NAC did not affect GSH depletion in ATO-treated HPF cells.
Although NAC is also known to be a GSH precursor, NAC did not seem
to be a GSH precursor but be an antioxidant in HPF cells. Treatment
with 10 μM BSO showing a cell death effect in HPF control cells did
not induce GSH depletion whereas the low dose of 1 or 5 μM ATO
induced GSH depletion in HPF cells without cell growth inhibition
and death. Moreover, SOD2 or TXN siRNAs did not influence GSH
depletion in ATO-treated HPF control cells. Taken together, our
results suggest that the intracellular GSH content seem to be the
decisive role on ATO-induced HPF cell death but changes of the
content are not sufficient enough to predict cell death
correctly.
In conclusion, ATO induced growth inhibition and
death of HPF cells, which were accompanied by increasing ROS level
and GSH depletion. NAC attenuated HPF cell death by ATO via
decreasing ROS levels whereas vitamin C and BSO enhanced the death
via increasing ROS and GSH depletion levels. Our present data
provide useful information for understanding the cytotoxic or
toxicological effects of ATO in normal lung cells in relation to
ROS and GSH levels.
Acknowledgements
This research was supported by the Basic Science
Research Program through the National Research Foundation of Korea
(NRF) funded by the Ministry of Education, Science and Technology
(2010-0007059).
Abbreviations:
|
HPF
|
human pulmonary fibroblast
|
|
ATO
|
arsenic trioxide
(As2O3)
|
|
ROS
|
reactive oxygen species
|
|
MMP (Δ Ψm)
|
mitochondrial membrane potential
|
|
NADPH oxidase
|
nicotine adenine diphosphate
oxidase
|
|
XO
|
xanthine oxidase
|
|
SOD
|
superoxide dismutase
|
|
CAT
|
catalase
|
|
GPX
|
GSH peroxidase
|
|
TXN
|
thioredoxin
|
|
TXNR
|
TXN reductase
|
|
FBS
|
fetal bovine serum
|
|
PI
|
propidium iodide
|
|
FITC
|
fluorescein isothiocyanate
|
|
H2DCFDA
|
2′,7′-dichlorodihydrofluorescein
diacetate
|
|
DHE
|
dihydroethidium
|
|
GSH
|
glutathione
|
|
CMFDA
|
5-chloromethylfluorescein
diacetate
|
|
MTT
|
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide
|
|
siRNA
|
small interfering RNA
|
|
NAC
|
N-acetyl cysteine
|
|
BSO
|
L-buthionine sulfoximine
|
References
|
1
|
Baran CP, Zeigler MM, Tridandapani S and
Marsh CB: The role of ROS and RNS in regulating life and death of
blood monocytes. Curr Pharm Des. 10:855–866. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Zorov DB, Juhaszova M and Sollott SJ:
Mitochondrial ROS-induced ROS release: an update and review.
Biochim Biophys Acta. 1757:509–517. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Zelko IN, Mariani TJ and Folz RJ:
Superoxide dismutase multigene family: a comparison of the CuZn-SOD
(SOD1), Mn-SOD (SOD2), and EC-SOD (SOD3) gene structures,
evolution, and expression. Free Radic Biol Med. 33:337–349. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Wilcox CS: Reactive oxygen species: roles
in blood pressure and kidney function. Curr Hypertens Rep.
4:160–166. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Marks PA: Thioredoxin in cancer - role of
histone deacetylase inhibitors. Semin Cancer Biol. 16:436–443.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Chen TJ, Jeng JY, Lin CW, Wu CY and Chen
YC: Quercetin inhibition of ROS-dependent and -independent
apoptosis in rat glioma C6 cells. Toxicology. 223:113–126. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Dasmahapatra G, Rahmani M, Dent P and
Grant S: The tyrphostin adaphostin interacts synergistically with
proteasome inhibitors to induce apoptosis in human leukemia cells
through a reactive oxygen species (ROS)-dependent mechanism. Blood.
107:232–240. 2006. View Article : Google Scholar
|
|
8
|
Wallach-Dayan SB, Izbicki G, Cohen PY,
Gerstl-Golan R, Fine A and Breuer R: Bleomycin initiates apoptosis
of lung epithelial cells by ROS but not by Fas/FasL pathway. Am J
Physiol Lung Cell Mol Physiol. 290:L790–L796. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Waxman S and Anderson KC: History of the
development of arsenic derivatives in cancer therapy. Oncologist.
6(Suppl 2): 3–10. 2001. View Article : Google Scholar
|
|
10
|
Shen ZX, Chen GQ, Ni JH, Li XS, Xiong SM,
Qiu QY, Zhu J, Tang W, Sun GL, Yang KQ, et al: Use of arsenic
trioxide (As2O3) in the treatment of acute promyelocytic leukemia
(APL): II. Clinical efficacy and pharmacokinetics in relapsed
patients. Blood. 89:3354–3360. 1997.PubMed/NCBI
|
|
11
|
Soignet SL, Maslak P, Wang ZG, Jhanwar S,
Calleja E, Dardashti LJ, Corso D, DeBlasio A, Gabrilove J,
Scheinberg DA, et al: Complete remission after treatment of acute
promyelocytic leukemia with arsenic trioxide. N Engl J Med.
339:1341–1348. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Park WH, Seol JG, Kim ES, Hyun JM, Jung
CW, Lee CC, Kim BK and Lee YY: Arsenic trioxide-mediated growth
inhibition in MC/CAR myeloma cells via cell cycle arrest in
association with induction of cyclin-dependent kinase inhibitor,
p21, and apoptosis. Cancer Res. 60:3065–3071. 2000.
|
|
13
|
Zhang W, Ohnishi K, Shigeno K, Fujisawa S,
Naito K, Nakamura S, Takeshita K, Takeshita A and Ohno R: The
induction of apoptosis and cell cycle arrest by arsenic trioxide in
lymphoid neoplasms. Leukemia. 12:1383–1391. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Miller WH Jr, Schipper HM, Lee JS, Singer
J and Waxman S: Mechanisms of action of arsenic trioxide. Cancer
Res. 62:3893–3903. 2002.PubMed/NCBI
|
|
15
|
Hyun Park W, Hee Cho Y, Won Jung C, Oh
Park J, Kim K, Hyuck Im Y, Lee MH, Ki Kang W and Park K: Arsenic
trioxide inhibits the growth of A498 renal cell carcinoma cells via
cell cycle arrest or apoptosis. Biochem Biophys Res Commun.
300:230–235. 2003.
|
|
16
|
Seol JG, Park WH, Kim ES, Jung CW, Hyun
JM, Kim BK and Lee YY: Effect of arsenic trioxide on cell cycle
arrest in head and neck cancer cell line PCI-1. Biochem Biophys Res
Commun. 265:400–404. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Uslu R, Sanli UA, Sezgin C, Karabulut B,
Terzioglu E, Omay SB and Goker E: Arsenic trioxide-mediated
cytotoxicity and apoptosis in prostate and ovarian carcinoma cell
lines. Clin Cancer Res. 6:4957–4964. 2000.PubMed/NCBI
|
|
18
|
Oketani M, Kohara K, Tuvdendorj D,
Ishitsuka K, Komorizono Y, Ishibashi K and Arima T: Inhibition by
arsenic trioxide of human hepatoma cell growth. Cancer Lett.
183:147–153. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Pu YS, Hour TC, Chen J, Huang CY, Guan JY
and Lu SH: Cytotoxicity of arsenic trioxide to transitional
carcinoma cells. Urology. 60:346–350. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Nakagawa Y, Akao Y, Morikawa H, Hirata I,
Katsu K, Naoe T, Ohishi N and Yagi K: Arsenic trioxide-induced
apoptosis through oxidative stress in cells of colon cancer cell
lines. Life Sci. 70:2253–2269. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Li M, Cai JF and Chiu JF: Arsenic induces
oxidative stress and activates stress gene expressions in cultured
lung epithelial cells. J Cell Biochem. 87:29–38. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Baj G, Arnulfo A, Deaglio S, Mallone R,
Vigone A, De Cesaris MG, Surico N, Malavasi F and Ferrero E:
Arsenic trioxide and breast cancer: analysis of the apoptotic,
differentiative and immunomodulatory effects. Breast Cancer Res
Treat. 73:61–73. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Woo SH, Park IC, Park MJ, Lee HC, Lee SJ,
Chun YJ, Lee SH, Hong SI and Rhee CH: Arsenic trioxide induces
apoptosis through a reactive oxygen species-dependent pathway and
loss of mitochondrial membrane potential in HeLa cells. Int J
Oncol. 21:57–63. 2002.
|
|
24
|
Zhang TC, Cao EH, Li JF, Ma W and Qin JF:
Induction of apoptosis and inhibition of human gastric cancer
MGC-803 cell growth by arsenic trioxide. Eur J Cancer.
35:1258–1263. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Kim HR, Kim EJ, Yang SH, Jeong ET, Park C,
Kim SJ, Youn MJ, So HS and Park R: Combination treatment with
arsenic trioxide and sulindac augments their apoptotic potential in
lung cancer cells through activation of caspase cascade and
mitochondrial dysfunction. Int J Oncol. 28:1401–1408. 2006.
|
|
26
|
Li JJ, Tang Q, Li Y, Hu BR, Ming ZY, Fu Q,
Qian JQ and Xiang JZ: Role of oxidative stress in the apoptosis of
hepatocellular carcinoma induced by combination of arsenic trioxide
and ascorbic acid. Acta Pharmacol Sin. 27:1078–1084. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Chou WC, Jie C, Kenedy AA, Jones RJ, Trush
MA and Dang CV: Role of NADPH oxidase in arsenic-induced reactive
oxygen species formation and cytotoxicity in myeloid leukemia
cells. Proc Natl Acad Sci USA. 101:4578–4583. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Lu J, Chew EH and Holmgren A: Targeting
thioredoxin reductase is a basis for cancer therapy by arsenic
trioxide. Proc Natl Acad Sci USA. 104:12288–12293. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Chouchane S and Snow ET: In vitro effect
of arsenical compounds on glutathione-related enzymes. Chem Res
Toxicol. 14:517–522. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Wu XX, Ogawa O and Kakehi Y: Enhancement
of arsenic trioxide-induced apoptosis in renal cell carcinoma cells
by L-buthionine sulfoximine. Int J Oncol. 24:1489–1497.
2004.PubMed/NCBI
|
|
31
|
Kitamura K, Minami Y, Yamamoto K, Akao Y,
Kiyoi H, Saito H and Naoe T: Involvement of CD95-independent
caspase 8 activation in arsenic trioxide-induced apoptosis.
Leukemia. 14:1743–1750. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Dai J, Weinberg RS, Waxman S and Jing Y:
Malignant cells can be sensitized to undergo growth inhibition and
apoptosis by arsenic trioxide through modulation of the glutathione
redox system. Blood. 93:268–277. 1999.PubMed/NCBI
|
|
33
|
Han YH, Kim SZ, Kim SH and Park WH:
Induction of apoptosis in arsenic trioxide-treated lung cancer A549
cells by buthionine sulfoximine. Mol Cells. 26:158–164.
2008.PubMed/NCBI
|
|
34
|
Kito M, Akao Y, Ohishi N, Yagi K and
Nozawa Y: Arsenic trioxide-induced apoptosis and its enhancement by
buthionine sulfoximine in hepatocellular carcinoma cell lines.
Biochem Biophys Res Commun. 291:861–867. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Maeda H, Hori S, Ohizumi H, Segawa T,
Kakehi Y, Ogawa O and Kakizuka A: Effective treatment of advanced
solid tumors by the combination of arsenic trioxide and
L-buthionine-sulfoximine. Cell Death Differ. 11:737–746. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Petty RD, Nicolson MC, Kerr KM,
Collie-Duguid E and Murray GI: Gene expression profiling in
non-small cell lung cancer: from molecular mechanisms to clinical
application. Clin Cancer Res. 10:3237–3248. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Jin HO, Yoon SI, Seo SK, Lee HC, Woo SH,
Yoo DH, Lee SJ, Choe TB, An S, Kwon TJ, et al: Synergistic
induction of apoptosis by sulindac and arsenic trioxide in human
lung cancer A549 cells via reactive oxygen species-dependent
down-regulation of survivin. Biochem Pharmacol. 72:1228–1236. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Han YH, Kim SZ, Kim SH and Park WH:
Arsenic trioxide inhibits the growth of Calu-6 cells via inducing a
G2 arrest of the cell cycle and apoptosis accompanied with the
depletion of GSH. Cancer Lett. 270:40–55. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Han YH, Kim SH, Kim SZ and Park WH:
Apoptosis in arsenic trioxide-treated Calu-6 lung cells is
correlated with the depletion of GSH levels rather than the changes
of ROS levels. J Cell Biochem. 104:862–878. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Han YH, Moon HJ, You BR, Kim SZ, Kim SH
and Park WH: Effects of arsenic trioxide on cell death, reactive
oxygen species and glutathione levels in different cell types. Int
J Mol Med. 25:121–128. 2010.PubMed/NCBI
|
|
41
|
Bailey HH: L-S,R-buthionine sulfoximine:
historical development and clinical issues. Chem Biol Interact.
111–112:239–254. 1998.PubMed/NCBI
|
|
42
|
Han YH, Kim SH, Kim SZ and Park WH:
Caspase inhibitor decreases apoptosis in pyrogallol-treated lung
cancer Calu-6 cells via the prevention of GSH depletion. Int J
Oncol. 33:1099–1105. 2008.PubMed/NCBI
|
|
43
|
Han YH, Kim SZ, Kim SH and Park WH:
Arsenic trioxide inhibits growth of As4.1 juxtaglomerular cells via
cell cycle arrest and caspase-independent apoptosis. Am J Physiol
Renal Physiol. 293:F511–F520. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Elbashir SM, Harborth J, Lendeckel W,
Yalcin A, Weber K and Tuschl T: Duplexes of 21-nucleotide RNAs
mediate RNA interference in cultured mammalian cells. Nature.
411:494–498. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Yang J, Liu X, Bhalla K, Kim CN, Ibrado
AM, Cai J, Peng TI, Jones DP and Wang X: Prevention of apoptosis by
Bcl-2: release of cytochrome c from mitochondria blocked. Science.
275:1129–1132. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Kang YH, Yi MJ, Kim MJ, Park MT, Bae S,
Kang CM, Cho CK, Park IC, Park MJ, Rhee CH, et al:
Caspase-independent cell death by arsenic trioxide in human
cervical cancer cells: reactive oxygen species-mediated
poly(ADP-ribose) polymerase-1 activation signals apoptosis-inducing
factor release from mitochondria. Cancer Res. 64:8960–8967. 2004.
View Article : Google Scholar
|
|
47
|
Jing Y, Dai J, Chalmers-Redman RM, Tatton
WG and Waxman S: Arsenic trioxide selectively induces acute
promyelocytic leukemia cell apoptosis via a hydrogen
peroxide-dependent pathway. Blood. 94:2102–2111. 1999.PubMed/NCBI
|
|
48
|
Haga N, Fujita N and Tsuruo T: Involvement
of mitochondrial aggregation in arsenic trioxide (As2O3)-induced
apoptosis in human glioblastoma cells. Cancer Sci. 96:825–833.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Yedjou CG, Rogers C, Brown E and Tchounwou
PB: Differential effect of ascorbic acid and n-acetyl-L-cysteine on
arsenic trioxide-mediated oxidative stress in human leukemia
(HL-60) cells. J Biochem Mol Toxicol. 22:85–92. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Bahlis NJ, McCafferty-Grad J,
Jordan-McMurry I, Neil J, Reis I, Kharfan-Dabaja M, Eckman J,
Goodman M, Fernandez HF, Boise LH, et al: Feasibility and
correlates of arsenic trioxide combined with ascorbic acid-mediated
depletion of intracellular glutathione for the treatment of
relapsed/refractory multiple myeloma. Clin Cancer Res. 8:3658–3668.
2002.
|
|
51
|
Grad JM, Bahlis NJ, Reis I, Oshiro MM,
Dalton WS and Boise LH: Ascorbic acid enhances arsenic
trioxide-induced cytotoxicity in multiple myeloma cells. Blood.
98:805–813. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Campbell RA, Sanchez E, Steinberg JA,
Baritaki S, Gordon M, Wang C, Shalitin D, Chen H, Pang S, Bonavida
B, et al: Antimyeloma effects of arsenic trioxide are enhanced by
melphalan, bortezomib and ascorbic acid. Br J Haematol.
138:467–478. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Yedjou C, Thuisseu L, Tchounwou C, Gomes
M, Howard C and Tchounwou P: Ascorbic acid potentiation of arsenic
trioxide anticancer activity against acute promyelocytic leukemia.
Arch Drug Inf. 2:59–65. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Biswas S, Zhao X, Mone AP, Mo X, Vargo M,
Jarjoura D, Byrd JC and Muthusamy N: Arsenic trioxide and ascorbic
acid demonstrate promising activity against primary human CLL cells
in vitro. Leuk Res. 34:925–931. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Gallegos A, Gasdaska JR, Taylor CW,
Paine-Murrieta GD, Goodman D, Gasdaska PY, Berggren M, Briehl MM
and Powis G: Transfection with human thioredoxin increases cell
proliferation and a dominant-negative mutant thioredoxin reverses
the transformed phenotype of human breast cancer cells. Cancer Res.
56:5765–5770. 1996.
|
|
56
|
Kim SJ, Miyoshi Y, Taguchi T, Tamaki Y,
Nakamura H, Yodoi J, Kato K and Noguchi S: High thioredoxin
expression is associated with resistance to docetaxel in primary
breast cancer. Clin Cancer Res. 11:8425–8430. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Epperly MW, Epstein CJ, Travis EL and
Greenberger JS: Decreased pulmonary radiation resistance of
manganese superoxide dismutase (MnSOD)-deficient mice is corrected
by human manganese superoxide dismutase-Plasmid/Liposome (SOD2-PL)
intratracheal gene therapy. Radiat Res. 154:365–374. 2000.
View Article : Google Scholar
|
|
58
|
Estrela JM, Ortega A and Obrador E:
Glutathione in cancer biology and therapy. Crit Rev Clin Lab Sci.
43:143–181. 2006. View Article : Google Scholar
|
|
59
|
Higuchi Y: Glutathione depletion-induced
chromosomal DNA fragmentation associated with apoptosis and
necrosis. J Cell Mol Med. 8:455–464. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Ramos AM, Fernandez C, Amran D, Esteban D,
de Blas E, Palacios MA and Aller P: Pharmacologic inhibitors of
extracellular signal-regulated kinase (ERKs) and c-Jun
NH(2)-terminal kinase (JNK) decrease glutathione content and
sensitize human promonocytic leukemia cells to arsenic
trioxide-induced apoptosis. J Cell Physiol. 209:1006–1015. 2006.
View Article : Google Scholar
|
|
61
|
Wang T, Ma LM, Zhang HP, Wang HW, Yang LH
and Qiao ZH: The effect of arsenic trioxide (As2O3) combined with
BSO on K562/ADM cell and its mechanisms. Zhonghua Xue Ye Xue Za
Zhi. 28:438–443. 2007.(In Chinese).
|