Introduction
Hepatocellular carcinoma (HCC) represents the 6th
most common cancer and the 3rd leading cause of cancer death
globally (1). More than 80% of HCC
occurs in developing countries, particularly Southeast Asia and
Africa. However, the incidence of HCC has recently been increasing
in western countries. In the United States, HCC incidence rates
increased by more than 2-fold between 1976 to 2002 and are expected
to double over the next 2 decades (1,2).
Although surgical resection, orthotopic liver transplantation and
radiofrequency ablation form the cornerstone of curative treatment
of HCC, patients should be treated within a multidisciplinary
setting. HCC is generally regarded as not being sensitive to
systemic chemotherapy. Other strategies such as transarterial
chemoembolization (TACE) or sorafenib alone or combined
radiotherapy provide only modest survival benefits. Due to low
liver tolerance to radiation therapy, the role of radiotherapy in
the management of HCC has traditionally been ignored (3). With developments of imaging
techniques, conformal radiotherapy has allowed the delivery of a
high dose of radiation to a precise tumor volume while sparing the
surrounding normal liver parenchyma. However, survival after
radiotherapy remains limited as a result of the high frequency of
hepatic recurrences (4–6). Therefore, it is imperative to discover
new therapeutics to improve survival in HCC patients.
Metformin (N′,N′-dimethylbiguanide) is the most
widely prescribed oral hypoglycemic agent. It is believed to exert
its effect by reducing hepatic glucose production and by increasing
insulin sensitivity and metabolism in peripheral tissues. Metformin
has been successfully used in a wide variety of indications, such
as in polycystic ovarian syndrome (PCOS) (7) and in the management of metabolic
syndrome (8). Currently, increasing
attention has been paid to metformin for its potential ability to
suppress cancer cell growth. This has been demonstrated by multiple
in vitro as well as in vivo studies (9,10) and
by its use in several randomized clinical trials as an adjuvant to
conventional chemotherapeutic agents (11,12).
Although the mechanism of metformin anticancer action is not fully
understood, there is substantial evidence indicating that metformin
treatment activates AMP-activated protein kinase (AMPK), inhibits
mTOR-dependent translation initiation (13–15)
and affects cancer cell metabolism (16). Our previous study indicated that
metformin exhibits protective effects on the metabolic disorder
caused by a high-carbohydrate high-fat diet, which may shed light
on its translational role in its antitumorigenic potential
(17). Recently, epidemiological
evidence has suggested that the anti-diabetic drug, metformin,
reduces HCC risk and lowers the rates of cancer mortality among
diabetics (18) without adverse
side-effects. Metformin rarely causes lactic acidosis (19). However, little is known about the
possible cytotoxic effects of metformin combined with radiotherapy
in human hepatoma carcinoma.
In this study, we assessed the in vitro
effect of the combination of metformin and ionizing radiation (IR)
on hepatoma cell survival and explored the underlying mechanisms.
Our data may contribute to the understanding of the mechanisms of
action of the conventional drug and highlight potential
implications of the combination of metformin and radiotherapy as an
anticanc treatment.
Materials and methods
Chemicals and reagents
Metformin was purchased from Sigma Chemical Co. and
dissolved in culture medium. RPMI-1640 medium, fetal bovine serum
(FBS), 100 U/ml penicillin and 100 μg/ml streptomycin were obtained
from Gibco. Antibodies used for western blot analyses were provided
by Cell Signaling Technology, Inc. (Danvers, MA, USA).
Cell lines and treatment
Two human hepatoma cell lines (HepG2 and Bel-7402
cells with high and low invasive ability, respectively) were
cultured in RPMI-1640 medium supplemented with 10% FBS, 100 U/ml
penicillin and 100 μg/ml streptomycin, at 37°C and 5%
CO2. The cells were a kind gift from Professor X. Guo
(Sun Yat Sen University, Guangzhou, China). To evaluate the effect
of the combination of metformin and IR, the cells were
pre-incubated with 10 μM metformin for 1 h and then exposed to
various doses of radiation (0–5 Gy) using a Varian CL21EX
accelerator (Varian Medical Systems, Palo Alto, CA, USA) with the
source-skin-distance technique. The depth was set at 2 cm to the
bottom of the 6-well plate.
Clonogenic assay
Cells were seeded in 6-well plates in triplicates at
a density of 1,000 cells/well in 2 ml of medium containing 10% FBS.
After 24 h, the cells were treated with metformin (10 μM) followed
by IR (1, 2 and 5 Gy, separately) and grown for 2 weeks. The cell
clones were fixed and stained for 15 min with a solution containing
0.5% crystal violet and 25% methanol, followed by 3 rinses with tap
water to remove the excess dye. Colonies containing >50 cells
were counted.
Cell cycle analysis
Cells (1×106) were seeded in 6-well
plates in triplicates. After 24 h, cells were treated with
metformin (10 μM) for 1 h followed by IR (2 Gy). After 36 h, the
cells were harvested by trypsinization and fixed with 70% ethanol.
The cells were stained for total DNA content with a solution
containing 50 μg/ml propidium iodide and 100 μg/ml RNase I in PBS
for 30 min at 37°C. The cell cycle distribution was then analyzed
in a FACSCalibur cytometer (BD Bioscience, Mountain View, CA,
USA).
Determination of mitochondrial complex I
and glycolytic activity
Complex I activity was determined using the Dipstick
Assay kit (MitoSciences, Eugene, OR, USA) according to the
manufacturer’s instructions. The cellular glycolytic activity was
evaluated by measuring the activity of the key enzyme, lactate
dehydrogenase (LDH) and lactate production. LDH activity was
monitored spectrophotometrically by measuring the increase in NADH
at 340 nm produced in the reaction of lactate to pyruvate as
described previously (20). Lactate
concentration in the culture medium was assessed with a
chromatometric kit obtained from Sigma.
Measurement of cellular adenosine
triphosphate (ATP) concentration
An ATP assay kit was purchased from the Beyotime
Institute of Biotechnology (Haimen, China). The process was
performed according to the manufacturer’s instructions. Harvested
cultured cells were lysed with a lysis buffer, followed by
centrifugation at 12,000 × g for 5 min at 4°C. Finally, the level
of ATP was determined by mixing 100 μl of the supernatant with 100
μl luciferase reagent, which catalyzed the light production from
ATP and luciferin. The emitted light was linearly associated with
the ATP concentration and measured using a luminometer (Promega,
Madison, WI, USA).
Comet assay
The comet assay was performed under alkaline
conditions as previously described (21,22),
with minor modifications. Briefly, HepG2 and Bel-7402 cell
suspension was separately mixed with low-melting agarose at
1×104 cells/ml at 37°C and evenly pipetted into the
microscope slides pre-coated with 250 μl of 0.6% normal
high-melting agarose. The slides were maintained on ice for 10 min
to solidify. The remaining cells were exposed to metformin (10 μM)
for 1 h followed by IR (2 Gy). After 1 h, the treated cells were
washed with ice-cold PBS and spread on the slides as described
above. The slides were then immersed in chilled lysis solution (2.5
M NaCl, 100 mM Na2 EDTA, 10 mM Tris-HCl, 1% Triton X-100
and 10% DMSO, pH 10.0) at 4°C for 2 h in the dark. Thereafter, the
slides were rinsed in freshly prepared and chilled electrophoresis
buffer (1 mM Na2 EDTA, 300 mM NaOH and pH >13.0) for
20 min to allow DNA unwinding. Electrophoresis was then performed
at 22 V, 300 mA at 4°C for 20 min. The slides were washed with a
neutralizing buffer (0.4 M Tris-HCl and pH 7.5) 3 times and then
stained with 30 μl ethidium bromide (5 μg/ml) for 15 min. Images of
the comets were captured under a fluorescence microscope (Leica
CTR6000) at ×100 magnification. For each sample, a minimum of 50
comets was obtained and the olive tail moment [tail DNA (%)× (tail
mean - head mean)] was analyzed using the Comet Assay Software
Project (CASP).
Western blot analysis
Cell lysates were prepared in a buffer containing
0.5 mM Tris HCl (pH 7.0), 0.1% β-mercaptoethanol, 0.5 mM EDTA (pH
7.0), 0.5 mM EGTA (pH 7.0), 2 mM leupeptin, 1 mM PMSF, 2.5 mg/ml
aprotinin, 1 mM DTT and 0.5% Triton X-100. After protein
quantification using the Bradford assay, 25 μg of proteins were
separated by SDS-PAGE and transferred onto nitrocellulose membranes
(Amersham Life Sciences, Piscataway, NJ, USA). The membranes were
blocked using PBS (pH 7.4) containing 5% not-fat milk for 1 h,
probed with primary antibody phospho-H2AX (Ser139)
[anti-phosphorylated histone H2AX (γ-H2AX)] overnight at 4°C. The
membranes were then washed with PBST (PBS + 0.1% Tween-20) and
incubated with a HRP-conjugated secondary antibody (Santa Cruz
Biotechnology, Inc., Santa Cruz, CA, USA) for 1 h. Immunoreactive
proteins were detected using an enhanced chemiluminescence
detection reagent from BestBio Co. (Shanghai, China). The membranes
were stripped and reprobed with anti-H2AX and anti-β-actin
antibodies.
Statistical analysis
Data analyses were carried out using SPSS V17.0 and
GraphPad Prism 4 software with the Student’s t-test. The results
are expressed as the means ± SEM or SD. A difference was considered
statistically significant when P<0.05.
Results
Effect of metformin plus IR on cancer
cell viability
To determine the effects of metformin in combination
with IR on hepatoma cell viability, we observed the ability of the
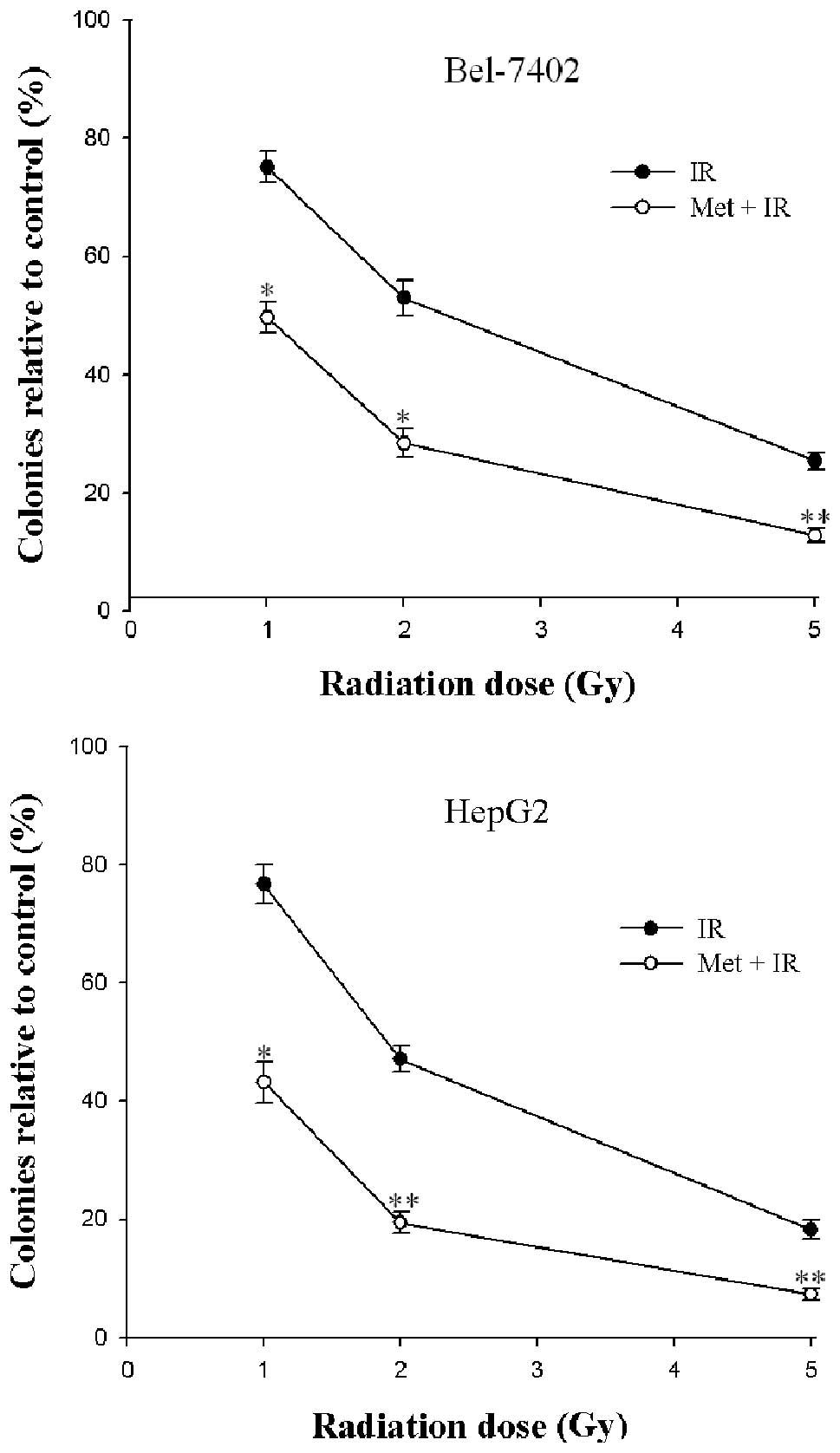
2 cell lines to form colonies on 6-well plates. IR caused a
dose-dependent inhibition of colony formation in Bel-7402 and HepG2
cells (Fig. 1). A statistically
significant enhancement of the radiation response was found in the
2 cell lines after treatment with metformin (P<0.05) (Fig. 1). These results demonstrated that
the inhibitory effect caused by the combination of metformin and IR
on cell survival was more effective than IR alone. Moreover,
metformin combined with 5 Gy X-ray radiation was found to provide
the best results with respect to the enhancement of cytotoxicity as
opposed to a lower irradiation dose. As regards clinical
radiotherapy, the dose of 2 Gy was selected to fulfill the
following experiment.
Combination of metformin and IR blocks
the cell cycle in the G2/M phase
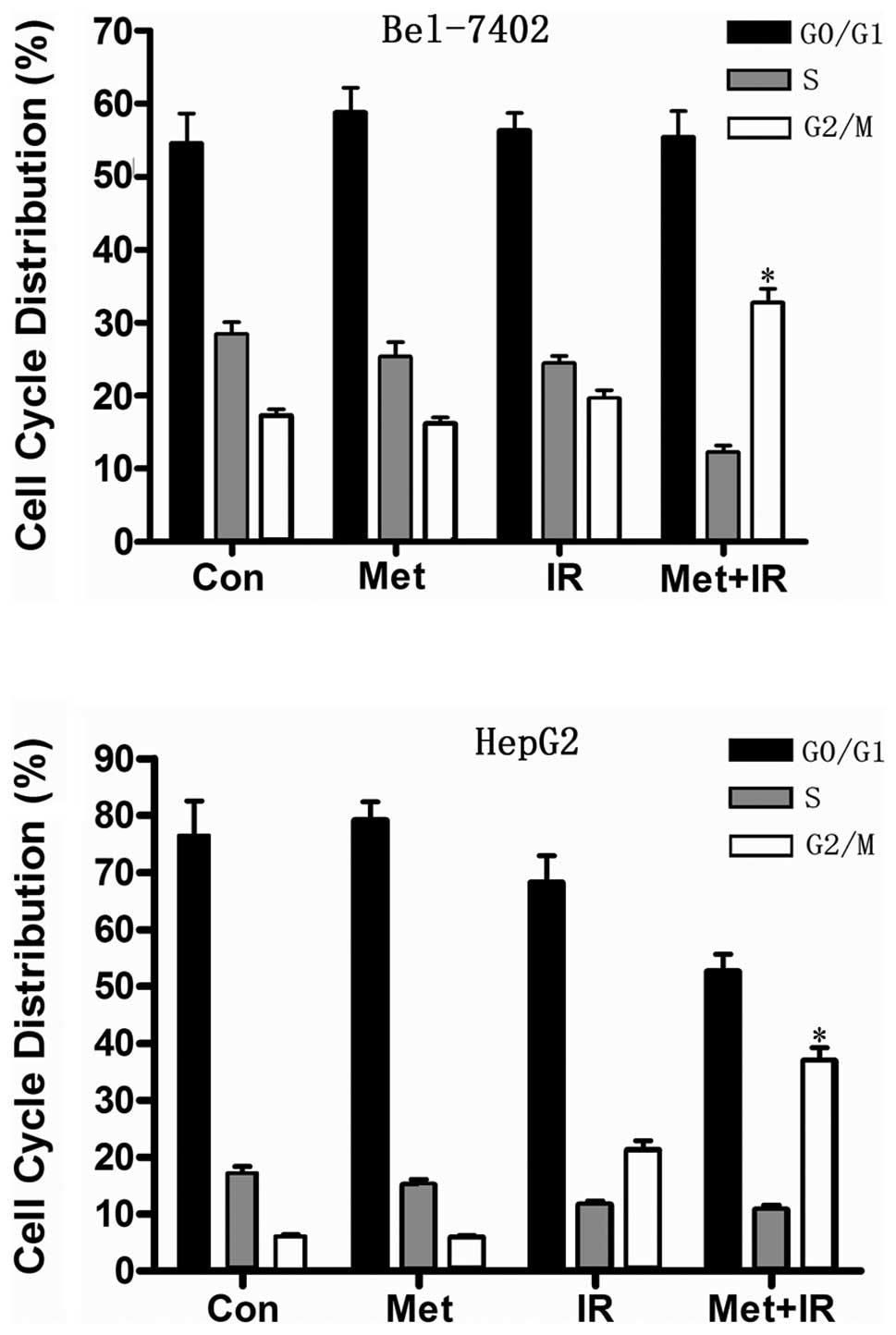
To determine whether the decrease in cell viability
induced by the combination of metformin and IR is associated with
cell cycle arrest, we analyzed the cell cycle phase distribution in
the 2 hepatoma cell lines. Bel-7402 and HepG2 cells treated for 1 h
in the presence of metformin were irradiated with 2 Gy X-rays and
subjected to flow cytometric analysis 36 h later. Metformin
treatment resulted in a minor accumulation of hepatoma cells in the
G0/G1 phase (Fig.
2). Importantly, the combination of metformin and IR notably
disturbed the cell cycle progression and showed a dramatic increase
in the G2/M phase cell populations in the hepatoma cells
compared with metformin or IR treatment alone (Fig. 2), which may in part account for the
strong inhibition of cell viability induced by G2/M
arrest.
Metformin combined with IR initiates a
strong energy stress in hepatoma cells
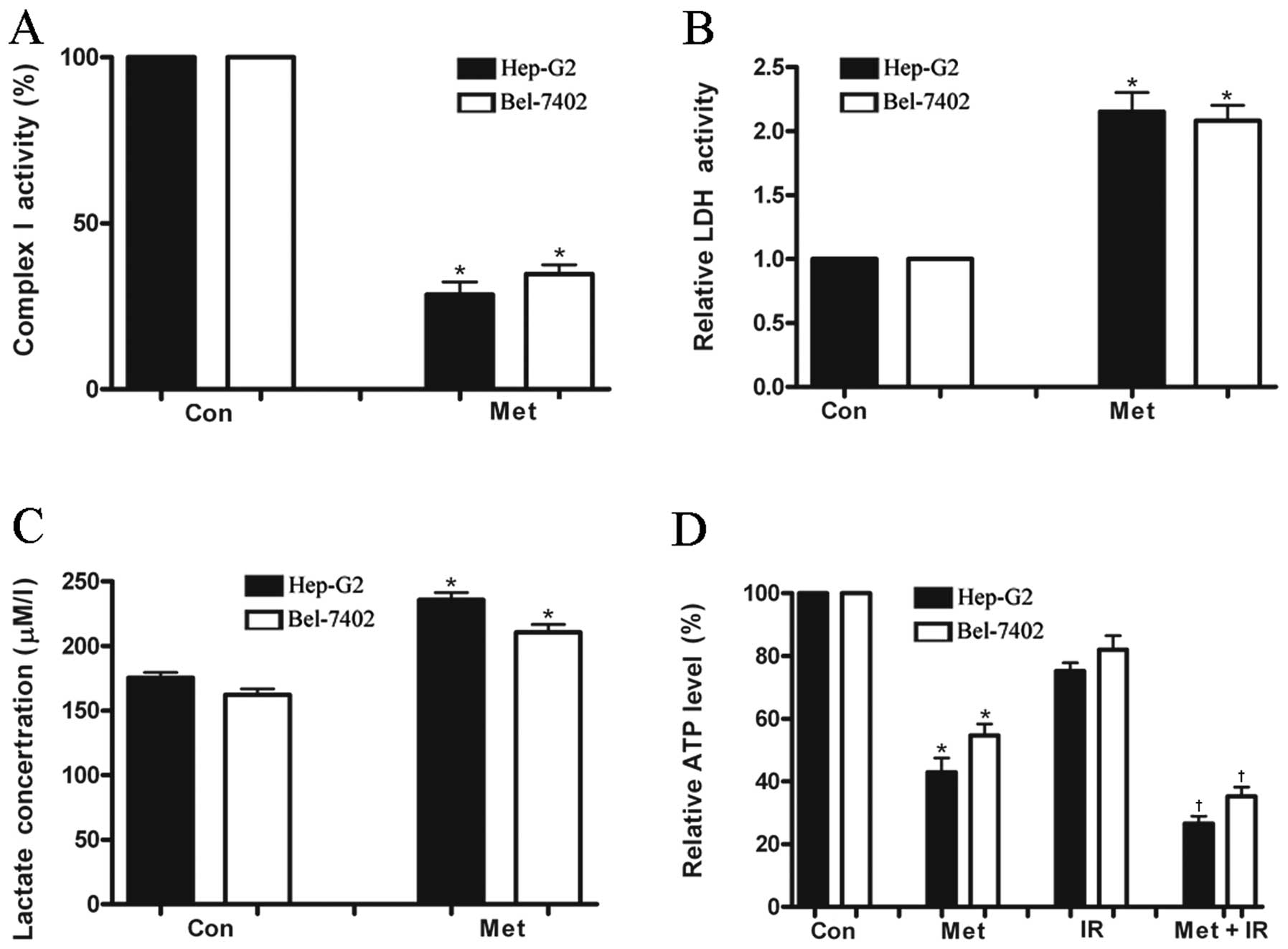
To investigate whether metformin has an effect on
hepatoma cell energy metabolism, mitochondrial complex I activity
that is involved in ATP generation was measured in a panel of 2
human malignant hepatoma cell lines. Metformin diminished
mitochondrial respiratory chain complex I activity by 72 and 65% in
HepG2 and Bel-7402 cells, respectively (Fig. 3A). As it has been known, a decrease
in oxidative phosphorylation is equivalent to a nutrient depletion
in terms of ATP supply which forces cells to improve their
bioenergetics, such as increased glycolysis and autophagy (23,16).
Hepatoma cells treated with metformin produced a significantly
greater level of LDH activity (a key enzyme involved in glycolysis
and catalyzing the conversion of pyruvate to lactate) and lactate
accumulation than the untreated cells (Fig. 3B and C) 24 h after treatment.
However, the ATP level was greatly diminished in both cell lines
after treatment with metformin (Fig.
3D). These results indicated that as a consequence of complex I
inhibition, metformin significantly enhanced the glycolytic ability
of the hepatoma cells to compensate ATP deprivation from the
mitochondrial respiratory defect. Increased glycolytic activity is
unable to produce enough compensatory ATP eventually leading to
cell death as the ATP generation by glycolysis is less efficient
than by mitochondrial oxidative phosphorylation (2 vs. 36
ATP/glucose). To evaluate the effects on cell energy metabolism
induced by IR, cellular ATP production was determined in hepatoma
cells. A small decrease in ATP levels was observed in hepatoma
cells 24 h post-irradiation, while the combination of metformin and
IR decreased ATP concentration by >70% in HepG2 cells and ~60%
in Bel-7402 cells, suggesting that metformin plus IR initiate a
strong energy stress in hepatoma cells compared with IR treatment
(Fig. 3D).
Increased DNA damage induced by metformin
combined with IR
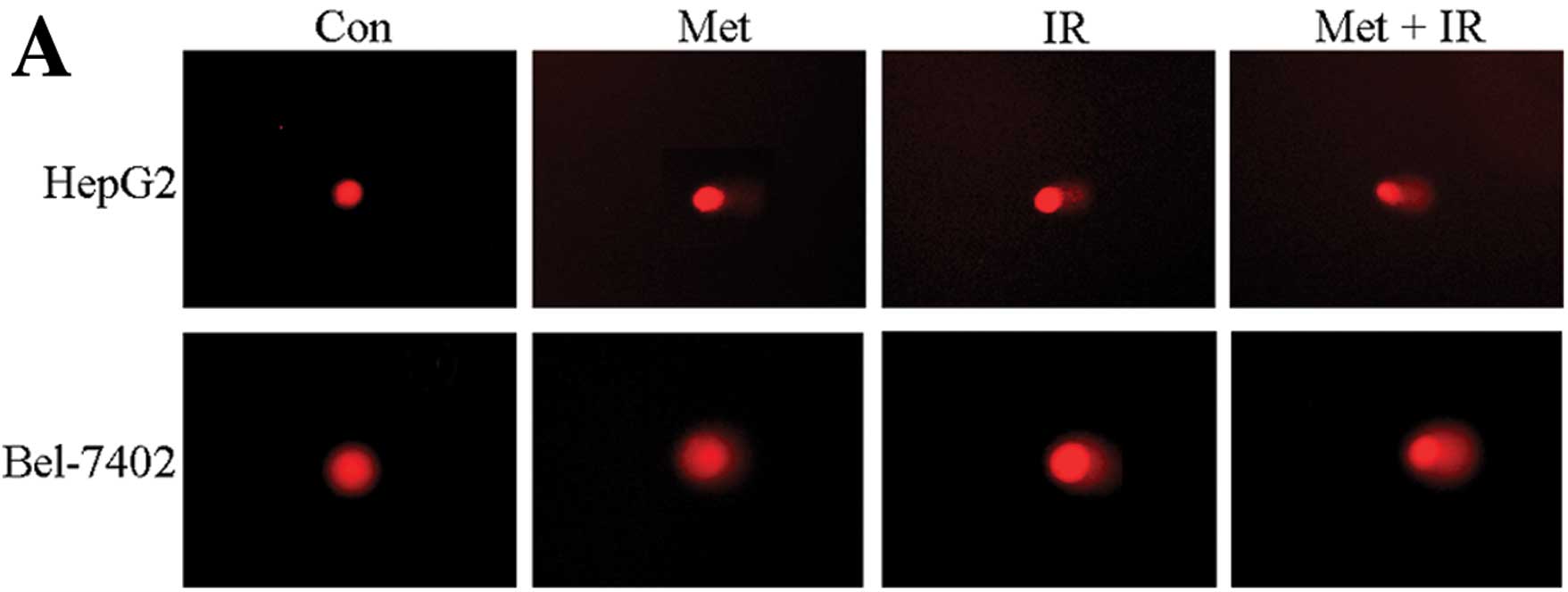
To further investigate how metformin plus IR
treatment decrease cell survival, we performed a comet assay to
ascertain DNA damage. The combination treatment significantly
increased the olive tail moment in the 2 hepatoma cell lines
compared to each individual treatment (P<0.05) (Fig. 4).
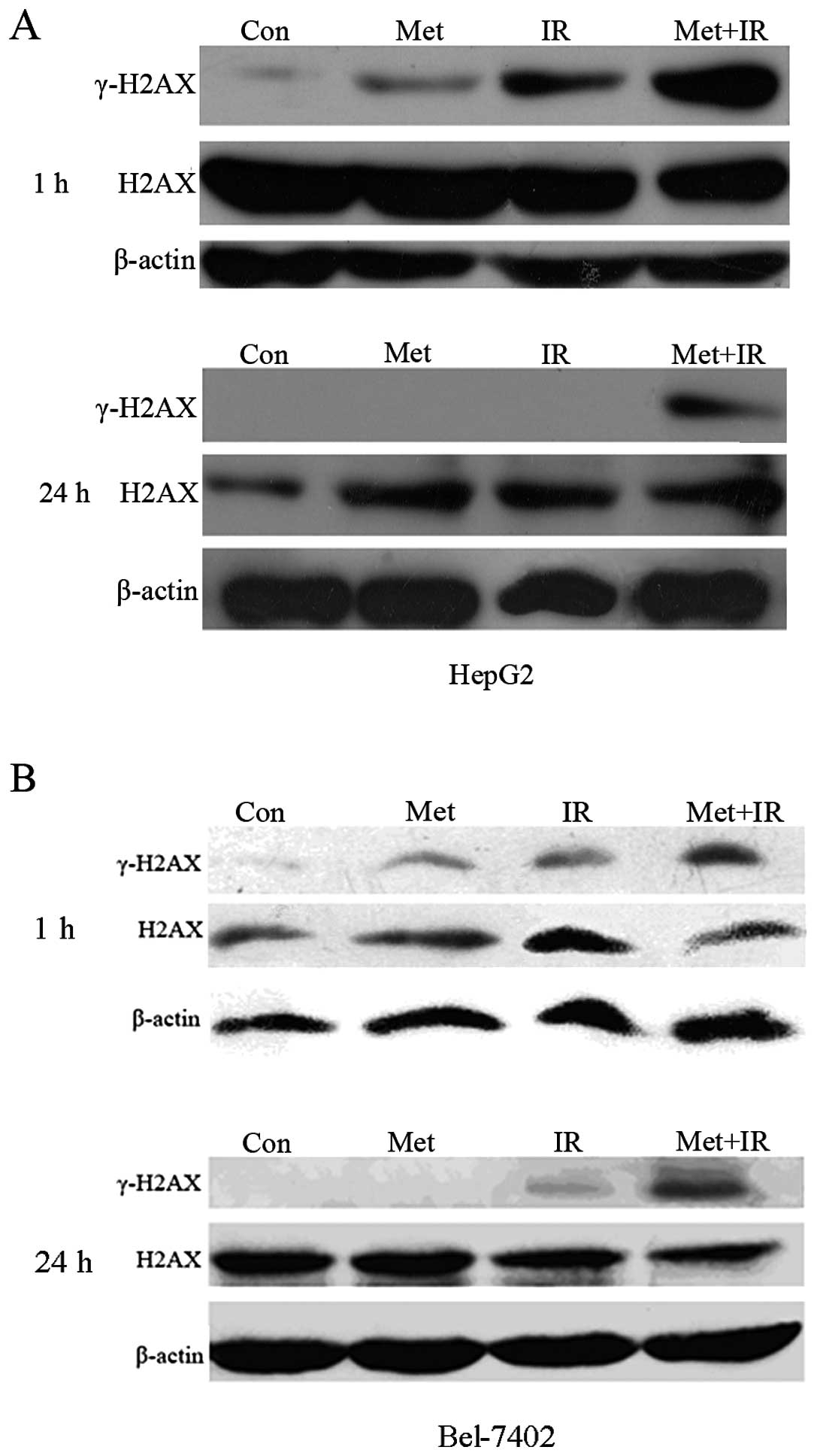
Enhanced expression of γ-H2AX protein in
response to metformin plus IR treatment
The γ-H2AX protein was infrequently expressed in the
control HepG2 cells (Fig. 5A).
Treatment with 10 μM of metformin alone led to a minor increase in
γ-H2AX expression, 1 h after treatment. Moderate levels of γ-H2AX
protein were observed 1 h after irradiation with 2 Gy. More
importantly, when the HepG2 cells were treated with 10 μM of
metformin for 1 h prior to treatment with 2 Gy X-rays, the protein
expression of γ-H2AX was dramatically increased compared with
either metformin or IR treatment alone. Surprisingly, an obvious
diminution of the expression of the γ-H2AX protein in HepG2 cells
was observed 24 h after treatment with metformin or radiation
alone, showing that DNA damage, likely the presence of
double-strand breaks (DSBs), was repaired. By contrast, the
combination of metformin plus radiation significantly prolonged the
expression of γ-H2AX 24 h after treatment, with more breaks
remaining unrepaired. A similar difference was also observed when
the Bel-7402 cells were assayed for γ-H2AX (Fig. 5B). However, the total H2AX
expression levels remained almost unaltered (Fig. 5). These results revealed that
metformin enhanced tumor radiosensitivity in vitro and this
sensitization correlated with the augmentation in DNA damage and
the delayed disappearance of γ-H2AX expression, suggesting an
inhibition of the repair of constitutive DNA damage.
Discussion
HCC is a highly lethal cancer with a poor prognosis.
Radiotherapy has long been excluded from the therapeutic strategy
for HCC due to its significant toxic effects on the normal liver
parenchyma. With the development of radiation therapy equipment and
technology, radiotherapy has become one of the main non-surgical
treatments for HCC. However, increasing the dose of radiation is
unfeasible to the majority of patients with advanced HCC having
serious liver diseases, such as hepatitis and cirrhosis. Thus, more
efficient and safe agents should be developed to increase the
radiosensitivity of HCC (24).
Therefore, the present study was carried out to address the effects
of metformin in combitation with IR on human hepatoma cells and to
investigate the possible mechanisms involved.
In this study, the low doses of metformin in
combination with X-ray radiation demonstrated an obvious
enhancement of cytotoxicity in the 2 hepatoma cell lines. To the
best of our knowledge, an investigation of the radiosensitizing
potential of the combination of metformin and X-ray radiation has
not yet been reported, although metformin has recently been
reported to modify γ-ray response in lung cancer (25). Based on our results, metformin
significantly enhanced the radiation response, as the number of
colonies of hepatoma cells in the combined treatment group was
significantly lower than that of each individual treatment group,
implying that metformin may function as a potential radiation
sensitizer for cancer radiotherapy.
The results of metformin combined with X-ray
radiation are promising. However, the underlying mechanisms by
which metformin exhibits its effects and interacts with X-ray
radiation are largely unknown.
A possible explanation may be related to the changes
in DNA repair. The cellular genome is constantly exposed to
exogenous and endogenous DNA damage. DNA is regarded as the major
target of IR. IR-induced DNA damage, including base damage, sugar
damage, single-strand breaks (SSBs) and DSBs, of which DSBs are
generally accepted as the most relevant lesion for the deleterious
effect of IR. In response to DNA damage, the histone H2AX at the
break site is phosphorylated at serine 139 by DNA damage sensor
kinases, such as ataxia telangiectasia-mutated (ATM), forming
γ-H2AX. Previous biochemical studies have revealed that γ-H2AX
plays an essential role in the recruitment of other proteins to
repair the breaks. After DNA damage is repaired by homologous
recombination or non-homologous end-joining, γ-H2AX is
dephosphorylated and the cell survives (26). Additionally, γ-H2AX expression has
been shown to disappear more rapidly in radiation-resistant tumor
cells than in radiation-sensitive cells (27,28).
In the present study, the combination of metformin with X-ray
radiation remarkably induced DNA damage in the 2 hepatoma cell
lines, suggesting that radiosensitivity with metformin may result
from the augmentation of X-ray-induced DNA damage. Interestingly,
the disappearance of γ-H2AX expression was slower in the group
treated with metformin plus X-ray radiation than in the group
treated with radiation or metformin alone, indicating that the
radiosensitizing effects of metformin may be due to the impaired
repair of DNA damage, which was consistent with previous
observations that the inhibition of DNA repair was a possible cause
of the enhancement of the radiation response (29,30).
However, how metformin inhibits the repair process of DNA damage is
yet to be determined. According to previous studies, DNA repair is
an energy-demanding process (31,32).
If damage is excessive and the ATP concentration drops to a low
level, it may exterminate cells whose damaged DNA can not be
successfully repaired (33). The
present study provides evidence suggesting that the depletion of
ATP may be a contributing factor linked to the impairment of DNA
repair. Since pre-treatment with metformin resulted in severe ATP
reduction in the hepatoma cells (Fig.
3D), this may have impeded DNA repair, as ATP is required for
this repair. Thus, a decrease in ATP levels could lead to
persistent DNA damage or decreased cell survival to a greater
extent due to X-ray radiation. Additional investigations are
required to further elucidate the molecular processes responsible
for metformin mediated radiosensitization.
It is striking to observe a minor increase in the
expression of γ-H2AX after 1 h of treatment with 10 μM metformin
but not after 24 h. Our findings concur with the recent observation
of Halicka et al (34), who
reported that metformin treatment for 24–48 h decreased rather than
increased the level of the constitutive expression of γ-H2AX,
possibly by decreasing oxidative stress (34). Possibly the same mechanism is
involved in the reduction of γ-H2AX expression after treatment with
metformin for 1 h as compared for 24 h.
In addition, the doses of metformin used in previous
in vitro experiments including breast (35), pancreas (36), prostate (37), colon (16), lung (38) and ovary cancer cells (39), varied from 0.1 to 30 mM, which is ~5
to 1,500-fold higher than the recommended therapeutic dose (in
clinical pharmacokinetic studies, investigators have calculated 20
μM as a clinically equivalent dose in vitro) (40–42).
It is therefore quite difficult to deduce the results obtained from
in vitro studies and the potential effects of metformin in
clinical trials with a standard metformin dosage. Therefore, for
our study, the cytotoxicity of various concentrations of metformin
(0, 1, 2, 5, 10, 20 and 50 μM) was previously determined using a
clonogenic assay in hepatoma cells. We found that the appropriate
dosage of metformin for the colony formation in these cells was ~10
μM (data not shown). Thus, this dose was used in our study.
To the best of our knowledge, our study proves for
the first time that the brief exposure of hepatoma cells to
metformin at a micromolar concentration prior to IR significantly
enhances the radiation responses by inhibiting DNA repair. The data
from our study may provide the scientific foundation for further
in vivo cancer models and clinical studies as regards the
combination of metformin and radiotherapy for HCC treatment.
Acknowledgements
This study was supported by grants (A2011290) from
the Department of Health of Guangdong Province, (2012J2200032) from
the Star of Science and Technology of Guangzhou, and (20121A011163)
from Health Bureau of Guangzhou. We gratefully acknowledge Dr Y.X.
Gu and W.J. Zhang from the Central South University for their
helpful comments during the study.
References
|
1
|
El-Serag HB and Rudolph KL: Hepatocellular
carcinoma: epidemiology and molecular carcinogenesis.
Gastroenterology. 132:2557–2576. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
El-Serag HB, Davila JA, Petersen NJ and
McGlynn KA: The continuing increase in the incidence of
hepatocellular carcinoma in the United States: an update. Ann
Intern Med. 139:817–823. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Dawson LA, Normolle D, Balter JM, McGinn
CJ, Lawrence TS and Ten Haken RK: Analysis of radiation-induced
liver disease using the Lyman NTCP model. Int J Radiat Oncol Biol
Phys. 53:810–821. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Mornex F, Girard N, Beziat C, Kubas A,
Khodri M, Trepo C and Merle P: Feasibility and efficacy of
high-dose three-dimensional-conformal radiotherapy in cirrhotic
patients with small-size hepatocellular carcinoma non-eligible for
curative therapies - mature results of the French Phase II RTF-1
trial. Int J Radiat Oncol Biol Phys. 66:1152–1158. 2006. View Article : Google Scholar
|
|
5
|
Girard N and Mornex F: External
radiotherapy for hepatocellular carcinoma. Cancer Radiother.
15:49–53. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Liang SX, Zhu XD, Lu HJ, et al:
Hypofractionated three-dimensional conformal radiation therapy for
primary liver carcinoma. Cancer. 103:2181–2188. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Diamanti-Kandarakis E, Economou F,
Palimeri S and Christakou C: Metformin in polycystic ovary
syndrome. Ann NY Acad Sci. 1205:192–198. 2010. View Article : Google Scholar
|
|
8
|
Bianchi C, Penno G, Romero F, Del Prato S
and Miccoli R: Treating the metabolic syndrome. Expert Rev
Cardiovasc Ther. 5:491–506. 2007. View Article : Google Scholar
|
|
9
|
Evans JM, Donnelly LA, Emslie-Smith AM,
Alessi DR and Morris AD: Metformin and reduced risk of cancer in
diabetic patients. BMJ. 330:1304–1305. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Kourelis TV and Siegel RD: Metformin and
cancer: new applications for an old drug. Med Oncol. 29:1314–1327.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Tennant DA, Durán RV and Gottlieb E:
Targeting metabolic transformation for cancer therapy. Nat Rev
Cancer. 10:267–277. 2010. View
Article : Google Scholar : PubMed/NCBI
|
|
12
|
Ben Sahra I, Le Marchand-Brustel Y, Tanti
JF and Bost F: Metformin in cancer therapy: a new perspective for
an old antidiabetic drug? Mol Cancer Ther. 9:1092–1099. 2010.
|
|
13
|
Zakikhani M, Dowling RJ, Sonenberg N and
Pollak MN: The effects of adiponectin and metformin on prostate and
colon neoplasia involve activation of AMP-activated protein kinase.
Cancer Prev Res (Phila). 1:369–375. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Dowling RJ, Zakikhani M, Fantus IG, Pollak
M and Sonenberg N: Metformin inhibits mammalian target of
rapamycin-dependent translation initiation in breast cancer cells.
Cancer Res. 67:10804–10812. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Hardie DG: Adenosine
monophosphate-activated protein kinase: a central regulator of
metabolism with roles in diabetes, cancer, and viral infection.
Cold Spring Harb Symp Quant Biol. 76:155–164. 2011. View Article : Google Scholar
|
|
16
|
Buzzai M, Jones RG, Amaravadi RK, et al:
Systemic treatment with the antidiabetic drug metformin selectively
impairs p53-deficient tumor cell growth. Cancer Res. 67:6745–6752.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Hou M, Venier N, Sugar L, et al:
Protective effect of metformin in CD1 mice placed on a high
carbohydrate-high fat diet. Biochem Biophys Res Commun.
397:537–542. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Hassan MM, Curley SA, Li D, et al:
Association of diabetes duration and diabetes treatment with the
risk of hepatocellular carcinoma. Cancer. 116:1938–1946. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Lalau JD and Race JM: Lactic acidosis in
metformin therapy. Drugs. 1:55–60. 1999. View Article : Google Scholar
|
|
20
|
Vanderlinde RE: Measurement of total
lactate dehydrogenase activity. Ann Clin Lab Sci. 15:13–31.
1985.PubMed/NCBI
|
|
21
|
van Dyk E and Pretorius PJ: DNA damage and
repair in mammalian cells exposed to p-hydroxyphenylpyruvic acid.
Biochem Biophys Res Commun. 338:815–819. 2005.PubMed/NCBI
|
|
22
|
Gu YX, Fan SS, Xiong Y, et al: Cloning and
functional characterization of TCRP1, a novel gene mediating
resistance to cisplatin in an oral squamous cell carcinoma cell
line. FEBS Lett. 585:881–887. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Ortega AD, Sánchez-Aragó M, Giner-Sánchez
D, Sánchez-Cenizo L, Willers I and Cuezva JM: Glucose avidity of
carcinomas. Cancer Lett. 276:125–135. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Rahbari NN, Mehrabi A, Mollberg NM, Müller
SA, Koch M, Büchler MW and Weitz J: Hepatocellular carcinoma:
current management and perspectives for the future. Ann Surg.
253:453–469. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Sanli T, Rashid A, Liu C, et al: Ionizing
radiation activates AMP-activated kinase (AMPK): a target for
radiosensitization of human cancer cells. Int J Radiat Oncol Biol
Phys. 78:221–229. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Kinner A, Wu W, Staudt C and Iliakis G:
Gamma-H2AX in recognition and signaling of DNA double-strand breaks
in the context of chromatin. Nucleic Acids Res. 36:5678–5694. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Judit PB, Susan HM and Peggy LO: Radiation
sensitivity, H2AX phosphorylation, and kinetics of repair of DNA
strand breaks in irradiated cervical cancer cell lines. Cancer Res.
64:7144–7149. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Klokov D, MacPhail SM, Banáth JP, Byrne JP
and Olive PL: Phosphorylated histone H2AX in relation to cell
survival in tumor cells and xenografts exposed to single and
fractionated doses of X-rays. Radiother Oncol. 80:223–229. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Raju U, Nakata E, Yang P, Newman RA, Ang
KK and Milas L: In vitro enhancement of tumor cell radiosensitivity
by a selective inhibitor of cyclooxygenase-2 enzyme: mechanistic
considerations. Int J Radiat Oncol Biol Phys. 54:886–894. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Huang G, Wang H and Yang LX: Enhancement
of radiation-induced DNA damage and inhibition of its repair by a
novel camptothecin analog. Anticancer Res. 30:937–944.
2010.PubMed/NCBI
|
|
31
|
Petermann E, Ziegler M and Oei SL:
ATP-dependent selection between single nucleotide and long patch
base excision repair. DNA Repair. 2:1101–1114. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Lammens K, Bemeleit DJ, Mockel C, et al:
The Mre11:Rad50 structure shows an ATP-dependent molecular clamp in
DNA double-strand break repair. Cell. 145:54–66. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Lyamzaev KG, Izyumov DS and Avetisyan AV:
Inhibition of mitochondrial bioenergetics: the effects on structure
of mitochondria in the cell and on apoptosis. Acta Biochim Pol.
51:553–562. 2004.PubMed/NCBI
|
|
34
|
Halicka HD, Zhao H, Li JW, Traganos1 F,
Zhang SF, Lee M and Darzynkiewicz1 Z: Genome protective effect of
metformin as revealed by reduced level of constitutive DNA damage
signaling. Aging. 3:1–11. 2011.PubMed/NCBI
|
|
35
|
Hirsch HA, Iliopoulos D, Tsichlis PN and
Struhl K: Metformin selectively targets cancer stem cells, and acts
together with chemotherapy to block tumor growth and prolong
remission. Cancer Res. 69:7507–7511. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Kisfalvi K, Eibl G, Sinnett-Smith J and
Rozengurt E: Metformin disrupts crosstalk between G protein-coupled
receptor and insulin receptor signaling systems and inhibits
pancreatic cancer growth. Cancer Res. 69:6539–6545. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Ben Sahra I, Laurent K, Loubat A, et al:
The antidiabetic drug metformin exerts an antitumoral effect in
vitro and in vivo through a decrease of cyclin D1 level. Oncogene.
27:3576–3586. 2008.PubMed/NCBI
|
|
38
|
Algire C, Zakikhani M, Blouin MJ, Shuai JH
and Pollak M: Metformin attenuates the stimulatory effect of a
high-energy diet on in vivo LLC1 carcinoma growth. Endocr Relat
Cancer. 15:833–839. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Rattan R, Giri S, Hartmann LC and Shridhar
V: Metformin attenuates ovarian cancer cell growth in an AMP-kinase
dispensable manner. J Cell Mol Med. 15:166–178. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Scheen AJ: Clinical pharmacokinetics of
metformin. Clin Pharmacokinet. 30:359–371. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Isoda K, Young JL, Zirlik A, et al:
Metformin inhibits proinflammatory responses and nuclear
factor-kappaB in human vascular wall cells. Arterioscler Thromb
Vasc Biol. 26:611–617. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Graham GG, Punt J, Arora M, et al:
Clinical pharmacokinetics of metformin. Clin Pharmacokinet.
50:81–98. 2011. View Article : Google Scholar : PubMed/NCBI
|