Introduction
Cancer of the uterine cervix is the second most
common cause of gynecologic cancer mortality worldwide, and it is
reported that cervical cancer affected 493,243 women worldwide in
2002 (1). It remains a health
threat with estimated incidence and mortality rates of 12,710 and
4,290 in 2011, respectively, in the United States (2). Cervical cancer is now considered a
preventable disease (3), but it is
important to note that cervical cancer affects young women at a
higher incidence. Treatment paradigms in the primary management of
cervical cancer are well established. Early stage patients are
treated surgically and women with locally advanced disease are
managed with concomitant cisplatin chemoradiotherapy. However, the
prognosis of patients with metastatic, recurrent, or persistent
cervical cancer remains poor with a 1-year survival rate between
15–20% (4). In addition,
chemotherapy has not led to major improvements in clinical outcome
and is associated with high rates of severe toxicities. Advanced
cervical cancer is associated with significant morbidities such as
renal failure, complex fistulas and painful bone metastases.
Therefore, improvement of systemic chemotherapy is crucial and new
regimens should be further developed.
Persistent infection with an oncogenic-type human
papillomavirus (HPV) is thought to be a prerequisite for the
development of cervical cancer. The two HPV oncogenes, E6 and E7,
are required for efficient immortalization of primary epithelial
keratinocytes. The E6 proteins form a complex with p53 (5,6), and
subsequent disruption of multiple functions of p53 is an important
step in cervical carcinogenesis (7). E6 also affects the function of tumor
suppressors involved in apoptosis, cell cycle regulation, and
tissue polarity, including two human homologues of
Drosophila neoplastic tumor suppressors, hDlg and hScrib
(8,9).
Recent evidence indicates that rapidly proliferating
cells, particularly cancer cells, have a greater requirement for
proteasomal activity and a greater sensitivity to the proteasome
inhibitor compared to normal cells (10). Bortezomib, a proteasome inhibitor,
has been approved by the United States Food and Drug Administration
for the treatment of multiple myeloma, and bortezomib has shown
in vitro and in vivo activity against solid tumors,
including prostate, pancreatic and colon cancer (11). In gynecologic cancers, several
investigators have reported implicative roles of bortezomib for the
treatment of human cervical (12–16)
and ovarian cancer (17).
In this study, we report that sequential or
concomitant use of bortezomib with cisplatin markedly induces
apoptosis and inhibition of growth in cultured cervical cancer
cells and xenograft. Bortezomib may have pre-clinical activity in
cisplatin-resistant tumors and may have synergic activity when
combined with cisplatin in HeLa cells. These effects may have an
important clinical implication to maximize the stabilization of
tumor suppressors (18).
Materials and methods
Chemicals and antibodies
Bortezomib (VELCADE, formerly known as PS-341) was
kindly provided by Millennium Pharmaceuticals (Cambridge, MA, USA).
Cisplatin, carboplatin and paclitaxel were from Bristol-Myers
Squibb (Princeton, NJ, USA). Bortezomib, cisplatin, carboplatin and
paclitaxel were dissolved in dimethyl sulfoxide and the final
concentration of dimethyl sulfoxide never exceeded 0.05%.
Anti-hScrib, anti-pRb (Ser 795), anti-p53 and
anti-p21 were purchased from Santa Cruz Biotechnology (Santa Cruz,
CA, USA). Rabbit polyclonal antibody was anti-Noxa (AnaSpec, Inc.,
San Jose, CA, USA). Mouse monoclonal antibody was anti-α-Tubulin
(Calbiochem, EMD Biosciences, Inc., La Jolla, CA, USA). Alexa Fluor
488-conjugated donkey anti-mouse IgG (A-21202) and Alexa Fluor
555-conjugated goat anti-rabbit IgG (A-21428) were purchased from
Invitrogen (Carlsbad, CA, USA).
Cell culture
HeLa (CCL-2) and CaSki (HB-8307) uterine cervical
cancer cell lines were purchased from the American Type Culture
Collection (Manassas, VA, USA) and grown in DMEM supplemented with
10% fetal bovine serum.
Sequential and simultaneous treatment
regimens
To determine the effect of sequence difference on
cellular response, cells were treated with bortezomib (100 nM),
carboplatin (250 μM), paclitaxel (10 μM) and cisplatin (500 μM).
HeLa and CaSki cells were seeded and allowed to adhere for 24 h.
For the sequential treatment, after 12 h of initial treatment, the
medium was changed to fresh medium containing the other treatment.
For the simultaneous treatment, after an initial 12 h in medium,
cells were treated with fresh medium containing the same drugs. The
control cell medium was changed at similar time points. After the
second 12-h treatment, the cells were harvested for flow cytometric
analysis or western blotting. Therefore, all groups received the
same duration of exposure to each agent and the assays were
performed at the same point following the final treatment.
Cell viability test
Viability of HeLa and CaSki cells was examined using
the CellTiter 96 Aqueous One Solution Cell Proliferation Assay kit
(Promega Corp., Madison, WI, USA), as previously described
(19).
Pulse chase analysis of p53 and
hScrib
The culture medium of HeLa cells was replaced with
Met/Cys-free DMEM for 2 h and pulsed with 20 μCi/ml of EasyTag™
EXPRESS35S (Perkin-Elmer Life Sciences, Boston, MA, USA)
for 1.5 h. The 35S-labeled protein was chased with or
without bortezomib (100 nM). Cells were harvested at the indicated
time point, lysed and electrophoresed.
Western blotting
Cultured cells and mouse xenograft tissues were
harvested and soluble protein was extracted. The procedure of
western blotting and subsequent immunoblot was performed as
previously described (20).
Flow cytometric analysis
To determine the apoptosis rate, cells were grown in
DMEM and treated with chemotherapeutic drugs. Bortezomib,
cisplatin, carboplatin and paclitaxel treatments in 12-h increments
over a 24-h period were as described. Following treatment, cells
were harvested and stained with Annexin V-FITC and propidium iodide
(PI) according to the manufacturer’s protocol (BD Biosciences,
Bedford, MA, USA). The percentage of specific apoptosis was
analyzed on FACSCalibur and calculated by CELLQuest Pro software
(BD Biosciences).
Quantification of the synergism of
bortezomib with cisplatin in the manner of sequential or
simultaneous administration (Chou-Talalay assay)
For the Chou-Talalay assay, experiments were carried
out as previously described (21).
Dose-response curves and 50% effective dose values
(ED50) were obtained, and fixed ratios of drugs and
mutually exclusive equations used to determine combination indices
(CI) (22). The potency of the
combination was calculated with the CalcuSyn software (Biosoft,
Ferguson, MO, USA). CI<1, CI=1, and CI>1 indicate
synergistic, additive and antagonistic interactions,
respectively.
Tumor growth suppression in vivo
Athymic C.B-17/Icr-scid Jcl mice 5–7 weeks of age
(CLEA Japan, Inc., Tokyo, Japan) were maintained in an SPF facility
according to the institutional guidelines, and experiments were
conducted under an approved animal protocol of The University of
Tokyo. Subcutaneous xenograft tumors were established by the
injection of cell suspension of 1×107 HeLa and CaSki
cells. After the appropriate tumors were formed, the mice were
sacrificed. The tumors were removed, cut into 3-mm sections and
transplanted subcutaneously into other mice. Mice were randomly
assigned to one of the four treatment regimens: saline (control),
cisplatin [intraperitoneal (i.p.) injection of cisplatin at a dose
of 6 mg/kg in a volume of 0.5 ml (23)], bortezomib [i.p. injection of
bortezomib at a dose of 1 mg/kg in a volume of 0.5 ml (24)], and bortezomib followed by cisplatin
8 h later in a combined volume of 0.5 ml. Each treatment group
consisted of 6 mice. Tumors were measured and the volume of these
tumors was calculated using the formula; Volume (mm3) =
[(major axis) × (minor axis)2]/2. After 4 weeks of
treatment, the mice were sacrificed and subjected to the
analysis.
Immunofluorescence
The mouse xenograft tumors were frozen in OCT
compounds (Sakura Finetek Japan Co., Ltd., Tokyo, Japan). The
embedded tissues were cut (6 μm) and cryostat sections were
recovered and fixed with PBS containing 4% paraformaldehyde. After
blocking, the cells were sequentially incubated with anti-p53 or
anti-hScrib antibodies and appropriate secondary antibodies. The
slides were briefly counterstained and analyzed under the
fluorescence microscope (Olympus BX50; Olympus, Tokyo, Japan).
Apoptotic cells were detected by DeadEnd™ Fluorometric TUNEL System
(Promega Corp.) in the mouse xenograft tumors.
Statistical analysis
Data represent the means ± SD or SEM from at least 3
independent experiments. Statistical analyses were performed by
one-way ANOVA with post-hoc test for multiple comparisons by using
StatView software (SAS Institute Inc., Cary, NC, USA). A P-value
<0.05 was considered to indicate statistically significant
differences.
Results
Effect of bortezomib on cellular
viability and stabilization of tumor suppressor proteins targeted
for degradation by HPV E6 and E7 in human cervical cancer cell
lines
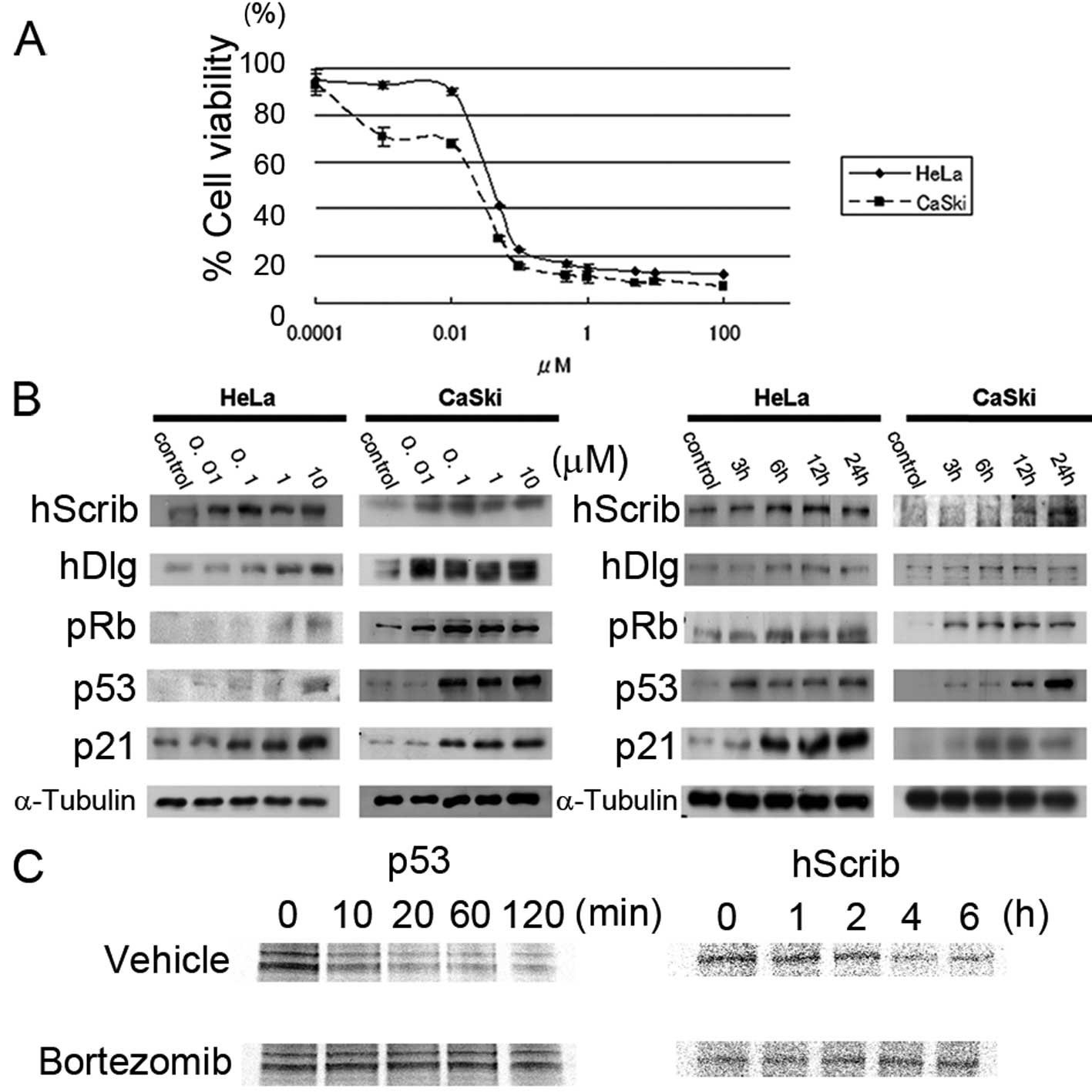
We used MTS assay to determine the effect of
bortezomib on cell viability in HeLa and CaSki cells (Fig. 1A). The approximately estimated
IC50 of bortezomib in HeLa and CaSki cells was 100 nM.
The expression of p53 increased by the exposure to bortezomib in a
dose-dependent manner (Fig. 1B left
panel) and in a time-dependent manner (Fig. 1B right panel). As a result, the
expression of p21 also increased. Elevated expression of PDZ
domain-containing scaffolding proteins (hScrib and hDlg) was shown
by the exposure to bortezomib (Fig.
1B). The expression of pRb decreases by the proteasome system
during cervical carcinogenesis (25), and the expression of pRb was
remarkably stimulated by the addition of bortezomib, particularly
in CaSki cells (Fig. 1B). These
data were translated as the antitumorigenic properties of
bortezomib and we further confirmed whether p53 and hScrib are
stabilized in the presence of bortemozib by the pulse-chase
analysis. As expected, p53 and hScrib expression remained
unaffected in the presence of bortezomib in HeLa cells (Fig. 1C).
Effect of bortezomib and chemotherapeutic
agents on cervical cancer cells
The effect of bortezomib and chemotherapeutic drugs
on the induction of apoptosis was analyzed by the flow cytometric
analysis (Fig. 2A). As a single
agent, bortezomib (lane 2) possessed a significant ability to
induce apoptosis compared to cisplatin, carboplatin, and paclitaxel
(lanes 3, 4 and 5, respectively), and the rate of apoptosis was
superior in CaSki cells. The sequential and simultaneous
combination of bortezomib with cisplatin was identified to be the
most significant treatment regimens in inducing cellular apoptosis
(Fig. 2A, lanes 6 and 9) in HeLa
cells.
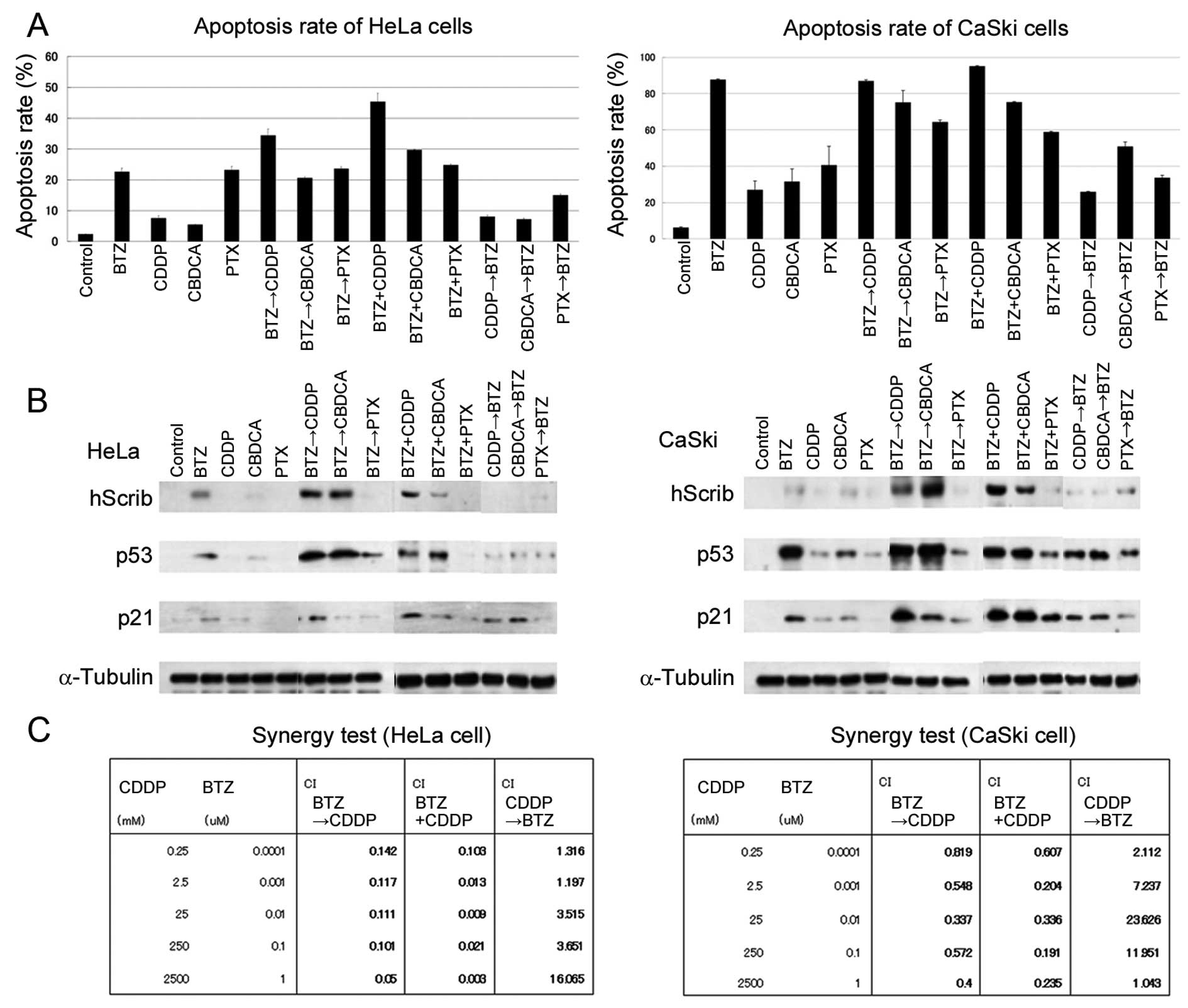 | Figure 2An analysis of bortezomib and
chemotherapeutic drugs in cervical cancer cells. (A) The rate of
apoptosis using bortezomib and chemotherapeutic drugs was measured
by flow cytometric analysis. Single, sequential, or simultaneous
treatment regimens using bortezomib (100 nM), cisplatin (500 μM),
carboplatin (250 μM) or paclitaxel (10 μM) were applied for HeLa
and CaSki cells. Bortezomib followed by cisplatin and concomitant
bortezomib with cisplatin regimens induced higher apoptosis rates
compared with other regimens. Bars represent the means ± SD of 3
independent experiments. (B) Bortezomib stabilized the expression
of hScrib and p53 in cervical cancer cell lines in combination with
platinum agents. By contrast, cisplatin followed by bortezomib
failed to stimulate the expression of hScrib, p53 and p21. (C)
Quantification of the potency of the combination treatment. The
Chou-Talalay method was utilized. Bortezomib followed by cisplatin
and concomitant bortezomib with cisplatin regimens exhibited
synergistic effects in all the doses tested. A potent synergistic
effect was displayed with CI<0.1. By contrast, a low synergistic
or antagonistic effect was shown in the treatment using cisplatin
followed by bortezomib with CI>1. BTZ, bortezomib; CDDP,
cisplatin; CBDCA, carboplatin; PTX, paclitaxel. |
We further investigated the expression of hScrib,
p53 and p21, whether the apoptotic effects are indeed associated
with protein expressions. Cells were exposed to various
concentrations of bortezomib and/or chemotherapeutic drugs
(Fig. 2B). The elevated expression
of p53 was observed in HeLa cells treated with bortezomib followed
by cisplatin or carboplatin (lanes 6 and 7), and in CaSki cells
treated with bortezomib alone (lane 2), bortezomib followed by
cisplatin or carboplatin (lanes 6 and 7). The expression of hScrib
exhibited a similar tendency to that of p53. In terms of the
stabilization of p53 and hScrib, paclitaxel turned out to be an
unsatisfactory agent (lanes 5, 8, 11 and 14). Markedly, bortezomib
and subsequent cisplatin treatment was the most efficient regimen
to induce the expression of p21, particularly in CaSki cells (lane
6). Cisplatin followed by bortezomib treatment had an insignificant
effect on the expression of p53 and hScrib (lane 12). The apparent
recovery of pRb was also observed in HeLa cells treated by the
exposure to bortezomib followed by cisplatin (data not shown).
The synergistic effects of bortezomib and cisplatin
were analyzed using the Chou-Talalay assay. Bortezomib followed by
cisplatin and simultaneous bortezomib with cisplatin regimens were
synergistic, with CI<1. This synergy was evidently affected by
the order of addition as cisplatin followed by bortezomib showed
antagonistic interactions, with CI>1. Therefore, the synergistic
effects in HeLa cells was more pronounced compared to that in CaSki
cells and we determined to pursue the sequential effect of
bortezomib and cisplatin.
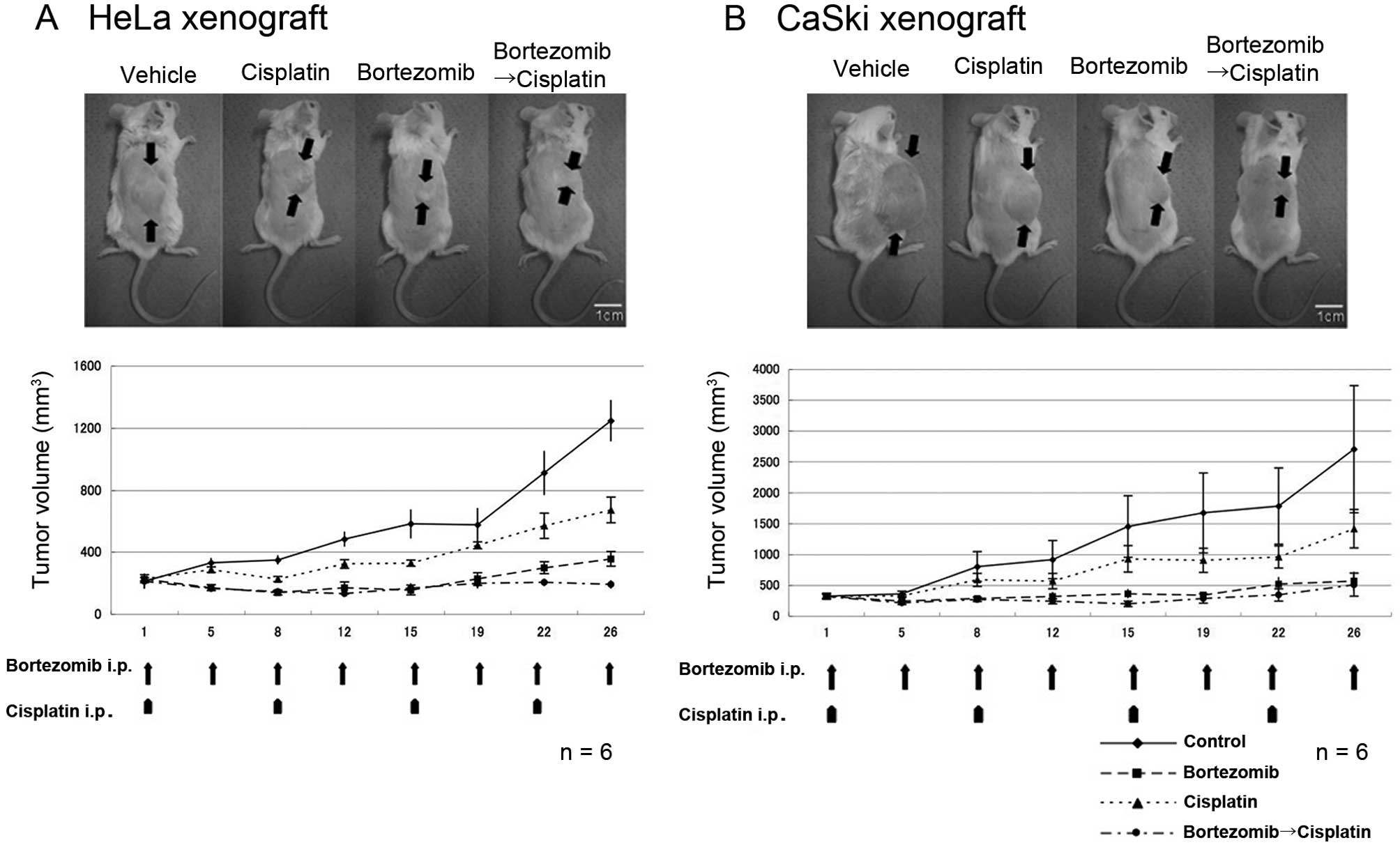
In vivo suppression of cervical cancer
growth by bortezomib in a mouse xenograft
We analyzed whether bortezomib is able to inhibit
tumor growth using a xenograft mouse model of cervical cancer
cells. Bortezomib was administered twice a week, and cisplatin once
a week according to the instructions of the suppliers. As shown in
Fig. 3, the tumor volume of the
HeLa and CaSki xenograft was significantly reduced by the injection
of bortezomib when compared with the control. The reduction rate of
the tumor growth in the bortezomib group was greater than that of
cisplatin alone and we revealed that bortezomib with cisplatin was
the most efficient treatment regimen in the HeLa xenograft. The
CaSki xenograft exhibited a pronounced sensitivity to bortezomib
alone and the effect of concomitant use was not observed as
expected from the in vitro study (Fig. 2, right panel).
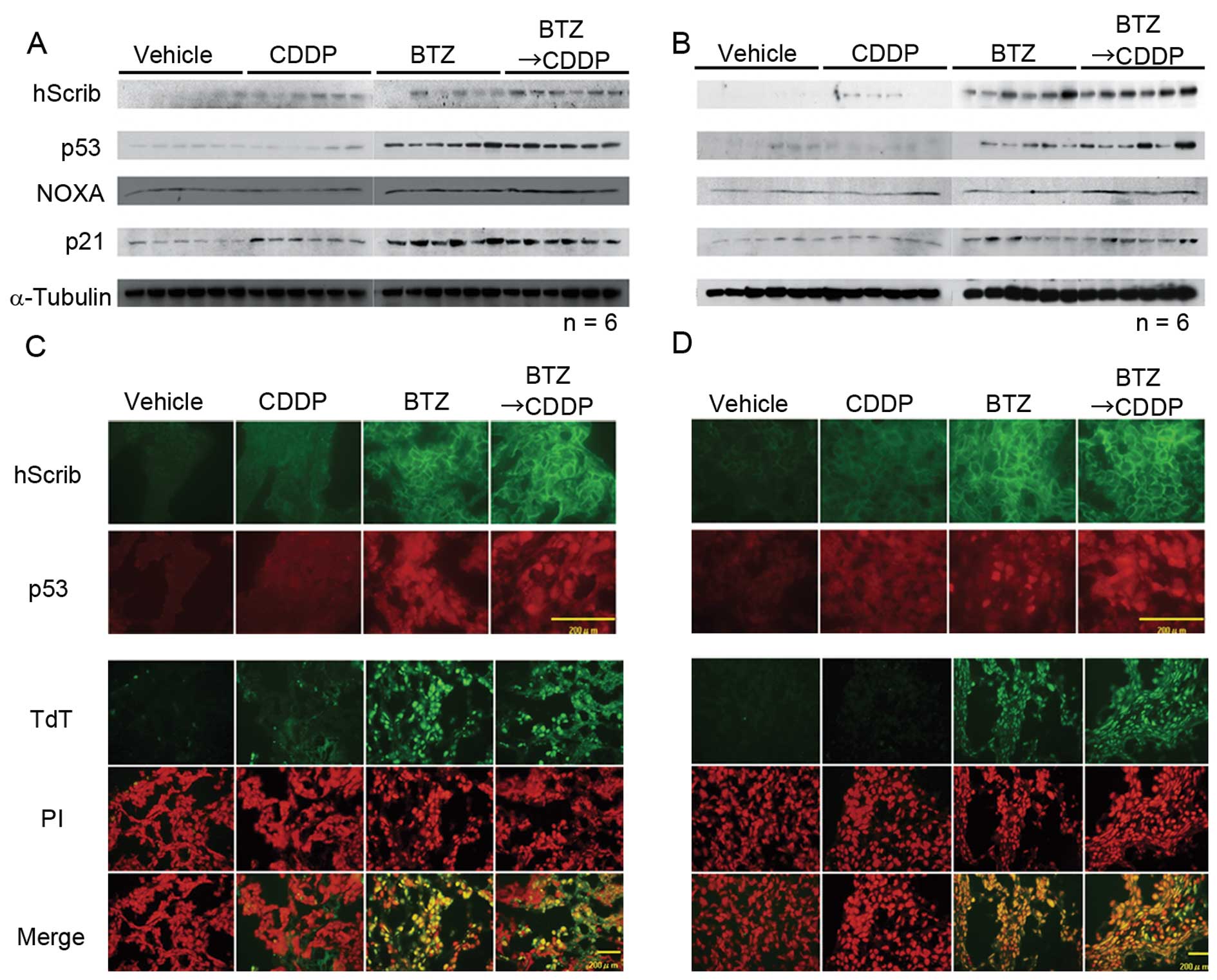
Based on the data in Fig. 2, the expression levels of p53, Noxa,
and p21 in cervical cancer xenografts were analyzed by western blot
analysis. It became evident that p53 and its downstream genes are
upregulated in xenografts treated by bortezomib (Fig. 4A and B). Our immunofluorescence
study also revealed that the expression of hScrib was recovered at
the cellular membrane in mice treated with bortezomib alone or in
combination with cisplatin (Fig. 4C and
D). The expression of p53 in the nuclei of xenograft cells also
increased as a result of treatment of bortezomib (Fig. 4C and D). HeLa and CaSki xenografts
were subjected to the detection of apoptosis using TUNEL assay and
TUNEL positive cells were observed predominantly in xenograft
tumors treated by bortezomib (Fig. 4C
and D). These data suggest the possibility that bortezomib
inhibits tumor growth in vivo through the induction of
apoptosis driven by p53 and downstream genes of p53, particularly
in HeLa cells.
Discussion
Bortezomib has been shown to be an extremely potent,
reversible and selective proteasome inhibitor (26). Several investigators have
demonstrated the ability of bortezomib to sensitize a variety of
cancer cells to the apoptotic effects of diverse chemotherapeutic
agents (12–15,17,27–29).
We confirmed that bortezomib was able to induce apoptosis in
HPV-positive cervical cancer cell lines. Cisplatin, a critical
component of therapeutic regimens in a broad range of malignancies,
has been shown to induce apoptosis in various types of cancer
cells. For the treatment of advanced or recurrent cervical cancer,
cisplatin administered every 3 weeks seems to be a reasonable
option, inducing response rates ranging from 20 to 30% and an
overall survival of 7 months (30),
but prolonged cisplatin treatment appears to have considerable
side-effects. We aimed to explore the possibility that concomitant
use of bortezomib and chemotherapeutic drugs has additive effects
to suppress ubiquitin-mediated degradation of tumor suppressors
targeted by E6 in HPV-positive cervical cancer cells both in
vitro and in vivo. As expected, bortezomib alone
increased the expression of tumor suppressors such as p53 and
hScrib in a dose- and time-dependent manner. We also observed the
elevated expression of p21 by the treatment of bortezomib since p21
is a representative downstream gene of p53. We noted that the rate
of apoptosis in cells treated with bortezomib preceded by cisplatin
was extremely lower than that in cells treated with cisplatin
preceded by bortezomib in HeLa cells. The sequential effect
(bortezomib followed by cisplatin) on the induction of apoptosis
was prominent compared with other combinations. However, this
sequential effect was less pronounced in CaSki cells both in
vitro and in vivo, and was similar to a previous report
that bortezomib did not induce a sensitization to cisplatin
treatment in SiHa cells (16). We
must take into account the fact that bortezomib alone was able to
induce significant apoptosis in CaSki cells (Fig. 2A, right panel), and this result is
in concordance with the result of the Chou-Talalay assay that
synergistic effects in HeLa cells were more pronounced compared to
those in CaSki cells (Fig. 2C). The
optimal sequence of chemotherapies with disparate mechanisms of
action has been investigated intensively. Although the mechanisms
of the sequence-specific interactions with chemotherapy remain
unclear, one possible explanation could be the effect on the
apoptotic mechanism by bortezomib. Cisplatin was shown to have a
defect in mitochondria-dependent caspase-9 activation in non-small
cell lung cancer H460 cells (29).
Contrary to this, bortezomib efficiently induced caspase-9
activation and apoptosis by promoting a pro-apoptotic shift in the
levels of proteins involved in mitochondrial outer-membrane
permeabilization (29). Another
mechanism might be its distribution to cell cycle arrest.
Bortezomib causes G2/M arrest (31)
and cisplatin causes long-lasting blocks at the G1/S boundary with
increasing cytotoxicity (32).
Theoretically, when bortezomib is administered first, the increased
p53 may serve to enhance G1/S checkpoint function. Then G1/S arrest
by cisplatin may efficiently enhance apoptotic cell death.
We further examined whether concomitant use of
bortezomib and cisplatin can induce apoptosis in vivo.
Although the regimen was not completely identical to that of the
in vitro experiment, the concomitant use of bortezomib and
cisplatin abrogated the tumor growth of xenografts. In
bortezomib-treated tumors, p53 is apparently stabilized in the
nuclei of tumor cells, and, subsequently, p21 and Noxa are elevated
due to the increased expression of p53. TUNEL assay showed enhanced
apoptosis in xenografts treated by bortezomib alone or bortezomib
with cisplatin, but concomitant treatment showed significant
enhancement of apoptosis. Several investigators have reported the
sequence-dependent effects of bortezomib are limited (28,33).
The optimal apoptotic effect occurs with the sequence gemcitabine
followed by bortezomib in pancreatic cancer cells (33) and in lung cancer cells (28). Therefore, the effect of bortezomib
may be sensitization of cancer cells to the apoptotic effect and
may be modulating the cellular response to the chemotherapeutics.
As a result, bortezomib may enhance cell death in combination with
cisplatin. Our data provide new evidence that the schedule of
combination treatment must be considered for the treatment of
cervical cancer, and gynecologists should consider pre-clinical
data in the design of clinical trials. While it is important to
confirm sequential effects in each cancer type studied, it appears
that chemotherapy given prior to bortezomib may yield inferior
results.
In conclusion, a proteasome inhibitor, bortezomib,
induces apoptosis in HPV-positive cervical cancer cells depending
on the stabilization of tumor suppressors, particularly p53. When
bortezomib is combined with cisplatin, a higher effect of apoptosis
induction might be expected since bortezomib plus cisplatin almost
completely abolished growth of cervical cancer xenografts with the
recovery of p53 and hScrib expression. Our data suggest the
possibility that bortezomib plus cisplatin is a promising regimen
for the treatment of advanced and/or chemotherapy or radiation
therapy-resistant cervical cancer cases.
Acknowledgements
This study was supported by the Mitsui Life Social
Welfare Foundation and the grant-in-aid for Scientific Research
from the Ministry of Education, Science and Culture, Japan.
References
|
1
|
Parkin DM, Bray F, Ferlay J and Pisani P:
Global cancer statistics, 2002. CA Cancer J Clin. 55:74–108. 2005.
View Article : Google Scholar
|
|
2
|
American Cancer Society. Cancer Facts and
Figures 2011. http://www.cancer.org/Cancer/CervicalCancer.
2011
|
|
3
|
Carter JR, Ding Z and Rose BR: HPV
infection and cervical disease: a review. Aust N Z J Obstet
Gynaecol. 51:103–108. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Berek JS and Hacker NF: Practical
Gynaecologic Oncology. 4th edition. Lippincott Williams &
Wilkins; Philadelphia, PA: 2005
|
|
5
|
Nakagawa S, Watanabe S, Yoshikawa H,
Taketani Y, Yoshiike K and Kanda T: Mutational analysis of human
papillomavirus type 16 E6 protein: transforming function for human
cells and degradation of p53 in vitro. Virology. 212:535–542. 1995.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Crook T, Tidy JA and Vousden KH:
Degradation of p53 can be targeted by HPV E6 sequences distinct
from those required for p53 binding and trans-activation. Cell.
67:547–556. 1991. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Huibregtse JM and Beaudenon SL: Mechanism
of HPV E6 proteins in cellular transformation. Semin Cancer Biol.
7:317–326. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Massimi P, Gammoh N, Thomas M and Banks L:
HPV E6 specifically targets different cellular pools of its PDZ
domain-containing tumour suppressor substrates for
proteasome-mediated degradation. Oncogene. 23:8033–8039. 2004.
View Article : Google Scholar
|
|
9
|
Nakagawa S and Huibregtse JM: Human
scribble (Vartul) is targeted for ubiquitin-mediated degradation by
the high-risk papillomavirus E6 proteins and the E6AP
ubiquitin-protein ligase. Mol Cell Biol. 20:8244–8253. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Mani A and Gelmann EP: The
ubiquitin-proteasome pathway and its role in cancer. J Clin Oncol.
23:4776–4789. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Adams J and Kauffman M: Development of the
proteasome inhibitor Velcade (Bortezomib). Cancer Invest.
22:304–311. 2004. View Article : Google Scholar
|
|
12
|
Birle DC and Hedley DW: Suppression of the
hypoxia-inducible factor-1 response in cervical carcinoma
xenografts by proteasome inhibitors. Cancer Res. 67:1735–1743.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Kamer S, Ren Q and Dicker AP: Differential
radiation sensitization of human cervical cancer cell lines by the
proteasome inhibitor velcade (bortezomib, PS-341). Arch Gynecol
Obstet. 279:41–46. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Lin Z, Bazzaro M, Wang MC, Chan KC, Peng S
and Roden RB: Combination of proteasome and HDAC inhibitors for
uterine cervical cancer treatment. Clin Cancer Res. 15:570–577.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Jiang Y, Wang Y, Su Z, et al: Synergistic
induction of apoptosis in HeLa cells by the proteasome inhibitor
bortezomib and histone deacetylase inhibitor SAHA. Mol Med Rep.
3:613–619. 2010.PubMed/NCBI
|
|
16
|
Bruning A, Vogel M, Mylonas I, Friese K
and Burges A: Bortezomib targets the caspase-like proteasome
activity in cervical cancer cells, triggering apoptosis that can be
enhanced by nelfinavir. Curr Cancer Drug Targets. 11:799–809. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Frankel A, Man S, Elliott P, Adams J and
Kerbel RS: Lack of multicellular drug resistance observed in human
ovarian and prostate carcinoma treated with the proteasome
inhibitor PS-341. Clin Cancer Res. 6:3719–3728. 2000.PubMed/NCBI
|
|
18
|
Nagasaka K, Nakagawa S, Yano T, et al:
Human homolog of Drosophila tumor suppressor Scribble
negatively regulates cell-cycle progression from G1 to S phase by
localizing at the basolateral membrane in epithelial cells. Cancer
Sci. 97:1217–1225. 2006.
|
|
19
|
Morita Y, Wada-Hiraike O, Yano T, et al:
Resveratrol promotes expression of SIRT1 and StAR in rat ovarian
granulosa cells: an implicative role of SIRT1 in the ovary. Reprod
Biol Endocrinol. 10:142012. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Wada-Hiraike O, Yano T, Nei T, et al: The
DNA mismatch repair gene hMSH2 is a potent coactivator of oestrogen
receptor alpha. Br J Cancer. 92:2286–2291. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Chou TC: Drug combination studies and
their synergy quantification using the Chou-Talalay method. Cancer
Res. 70:440–446. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Kanai R, Wakimoto H, Martuza RL and Rabkin
SD: A novel oncolytic herpes simplex virus that synergizes with
phosphoinositide 3-kinase/Akt pathway inhibitors to target
glioblastoma stem cells. Clin Cancer Res. 17:3686–3696. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Fraval HN and Roberts JJ: G1 phase Chinese
hamster V79-379A cells are inherently more sensitive to platinum
bound to their DNA than mid S phase or asynchronously treated
cells. Biochem Pharmacol. 28:1575–1580. 1979. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Nawrocki ST, Bruns CJ, Harbison MT, et al:
Effects of the proteasome inhibitor PS-341 on apoptosis and
angiogenesis in orthotopic human pancreatic tumor xenografts. Mol
Cancer Ther. 1:1243–1253. 2002.PubMed/NCBI
|
|
25
|
Munger K and Howley PM: Human
papillomavirus immortalization and transformation functions. Virus
Res. 89:213–228. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Adams J: The proteasome: structure,
function, and role in the cell. Cancer Treat Rev. 29(Suppl 1): 3–9.
2003. View Article : Google Scholar
|
|
27
|
Ling YH, Liebes L, Jiang JD, et al:
Mechanisms of proteasome inhibitor PS-341-induced G(2)-M-phase
arrest and apoptosis in human non-small cell lung cancer cell
lines. Clin Cancer Res. 9:1145–1154. 2003.PubMed/NCBI
|
|
28
|
Mortenson MM, Schlieman MG, Virudachalam S
and Bold RJ: Effects of the proteasome inhibitor bortezomib alone
and in combination with chemotherapy in the A549 non-small-cell
lung cancer cell line. Cancer Chemother Pharmacol. 54:343–353.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Voortman J, Checinska A, Giaccone G,
Rodriguez JA and Kruyt FA: Bortezomib, but not cisplatin, induces
mitochondria-dependent apoptosis accompanied by up-regulation of
noxa in the non-small cell lung cancer cell line NCI-H460. Mol
Cancer Ther. 6:1046–1053. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Tewari KS and Monk BJ: Gynecologic
oncology group trials of chemotherapy for metastatic and recurrent
cervical cancer. Curr Oncol Rep. 7:419–434. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Gregory MA and Hann SR: c-Myc proteolysis
by the ubiquitin-proteasome pathway: stabilization of c-Myc in
Burkitt’s lymphoma cells. Mol Cell Biol. 20:2423–2435.
2000.PubMed/NCBI
|
|
32
|
Jackel M and Kopf-Maier P: Influence of
cisplatin on cell-cycle progression in xenografted human head and
neck carcinomas. Cancer Chemother Pharmacol. 27:464–471. 1991.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Fahy BN, Schlieman MG, Virudachalam S and
Bold RJ: Schedule-dependent molecular effects of the proteasome
inhibitor bortezomib and gemcitabine in pancreatic cancer. J Surg
Res. 113:88–95. 2003. View Article : Google Scholar : PubMed/NCBI
|