Introduction
Wilms’ tumor (WT) is a heterogeneous neoplasia
characterized by several genetic and epigenetic abnormalities,
involving tumor suppressor genes, oncogenes and genes related to
the Wnt signaling pathway. The incidence of WT is 1/10,000 and
bilateral presentation is observed in 10% of affected individuals.
In approximately 1–2% of WT, recurrence occurs in the family
(1).
The WT1 gene is an essential regulator of
kidney development, critical to the survival and subsequent
differentiation of kidney cells (2). The WT1 somatic, biallelic
inactivation is seen in 5–10% of sporadic WT (1). The WT1 protein contains an
amino-terminal transactivator and a carboxyl-terminal DNA-binding
domain consisting of four zinc fingers. Alternative WT1
splicing results in four different isoforms of the protein, and the
most abundant isoform (+KTS) is generated by insertion of amino
acids lysine, threonine and serine (KTS), coded by exons 9 and 10
(3–6). Exon 9 represents an important target
for germline mutations associated with Denys-Drash syndrome (DDS),
and specific constitutional point mutations affect the properties
of WT1 to bind with EGR1 (early growth response 1) consensus
sequence (7).
Mutations in exon 3 of the CTNNB1 gene, which
encodes β-catenin, were initially observed in 15% of tumor samples
from WT patients (8). The β-catenin
N-terminal region contains consensus phosphorylation sites for the
serine/threonine kinase GSK-3β (glycogen synthase kinase 3β)
protein, whose function is to phosphorylate β-catenin at multiple
sites (Ser33, Ser37, Thr41 and Ser45). In the absence of signs of
growth and differentiation, this phosphorylation results in
β-catenin degradation mediated by ubiquitin (9,10).
β-catenin stabilization in the nucleus activates the Wnt signaling
pathway mediated by β-catenin/TCF (11), leading to the deregulation of
β-catenin signaling, which is critical for the development of
various malignancies, including WT (8).
Rivera et al(2) identified another gene, WTX,
found inactivated in one third of the studied WT samples. The WTX
protein forms a complex with β-catenin, APC, and other proteins to
negatively regulate the Wnt/β-catenin signaling pathway, leading to
the degradation of β-catenin (12).
Rivera et al(13) showed
that WTX shuttles between the cytoplasm and the nucleus, and
mediates the binding of WT1, modulating its activity. Mutations in
WTX and WT1 were initially thought to be mutually
exclusive, while most mutations observed in CTNNB1 coincided
with WT1 mutations (2).
In the present study, we screened germline and
somatic mutations in the WT1 exons 8, 9 and 10, the
WTX coding region and the CTNNB1 exon 3 in 43 WT
patients.
Materials and methods
Patients
This study involved 43 patients with documented WT.
All tumor samples were collected following neoadjuvant
chemotherapy. This study was approved by the local ethics committee
and the parents or tutors of all participant patients signed an
informed consent.
DNA extraction
DNA extraction from peripheral blood and fresh tumor
samples followed procedures established by Miller et
al(14) and Sambrook et
al(15).
Sequencing of WT1, WTX and CTNNB1
Blood and fresh tumor DNA samples were screened for
WT1, WTX and CTNNB1 mutations with previously
reported primers (2). PCR reactions
contained 5 pmol of forward and reverse primers (Prodimol), 1 μm
dNTPs (Life Technologies), 0.9 mM MgCl2, 10 mM Tris-HCl
(pH 8.0), 25 μM KCl, 1 U Taq DNA polymerase (Life Technologies) and
100 ng of DNA in a 25 μl final volume. PCR conditions for
WT1 exons 8, 9 and 10 assay consisted of 94°C for 5 min, 35
cycles at 94°C for 30 sec, 58°C for 30 sec and 72°C for 30 sec and
72°C for 7 min. PCR conditions for WTX exon 2 assay
consisted of 94°C for 5 min, 35 ‘step down’ cycles at 94°C for 30
sec, 66°C (decreasing 0.3°C per cycle) for 30 sec and 72°C for 30
sec and 72°C for 7 min. Finally, CTNNB1 exon 3 amplification
conditions were 94°C for 5 min, 35 ‘step down’ cycles at 94°C for
30 sec, 60°C (decreasing 0.2°C per cycle) for 30 sec, 72°C for 30
sec and 72°C for 7 min. PCR products were purified using the GFX™
PCR DNA and Gel Band Purification kit (GE Healthcare) and were
subjected to nucleotide sequencing using BigDye v3.1 (Life
Technologies). DNA samples and reference sequences (NG_009272 for
WT1; NG_021345 for WTX; and NG_013302.1 for
CTNNB1) (16) were aligned
and compared to identify homozygous and heterozygous nucleotide
positions using ChromasPro v. 1.41 and MEGA 5 software.
Statistical analysis
Kaplan-Meier curves were used to estimate 60-month
survival rates and overall survival. The Mann-Whitney U test was
used to compare age at diagnosis.
Results
Our sample consisted of 43 unrelated patients, 18
females and 25 males, diagnosed with WT. Bilateral disease was
diagnosed in nine patients. WT mean age at diagnosis was 43 months
for the whole sample (ranging from 4 to 137 months) and 32 months
for bilateral cases only. Five patients also presented major
phenotypic abnormalities: one with Beckwith-Wiedemann syndrome, one
with hemihypertrophy, two with non-syndromic macrosomia and one
with Denys-Drash syndrome. Table I
shows clinical, histopathological and molecular data of all
patients.
 | Table IWT1, WTX and
CTNNB1 mutations, histopathology of the tumors, and patient
clinical data. |
Table I
WT1, WTX and
CTNNB1 mutations, histopathology of the tumors, and patient
clinical data.
| Patients | Gender | Laterality | Histopathology | Dx age | WT1 exons 8,
9, 10 | WTX exon
2 | CTNNB1 exon
3 | Phenotype |
|---|
| 7 | F | U | Tri | 44 | rs16754 (B) | | |
Hemihypertrophy |
| 8 | M | B | ILNR | 6 | rs16754 (B) | | | |
| 9 | F | U | Tri | 48 | rs16754 (B) | | | |
| 10 | F | U | Tri | 10 | rs2234593 (B) | | | |
| 11 | M | U | Bl | 136 | rs16754 | rs34677493 +
rs61730681 | | |
| 13 | F | U | Ep | 62 | rs2234593 | | | |
| 14 | M | U | Ep | 59 | rs16754 | | | |
| 16 | M | B | DA | 29 | rs16754 | | | |
| 18 | M | U | FA | 36 | | R560W | | |
| 19 | F | U | Tri | 48 | | rs61730681
(LOH) | | |
| 21 | F | U | Bl | 4 | | rs61730681 (T,
het) | | |
| 22 | F | U | Tri | 28 | | | T41A (T) | Macrosomia |
| 23 | M | U | FA | 57 | rs16754 | rs61730681 | | |
| 24 | F | U | Tri | 13 | | rs150075206 +
rs61730681 (het) | | |
| 25 | M | U | Tri. | 42 | rs16754 | | | |
| 27 | M | B | Tri, PLNR | 61 | | | T41A (T) | |
| 28 | M | U | Tri | 67 | rs16754 (B) | | | |
| 32 | F | U | Bl | 7 | rs16754 (T) | | | |
| 34 | F | U | Tri | 35 | | rs61730681
(LOH) | | |
| 35 | M | U | Tri | 47 | rs16754 +
rs2234593 | | | Beckwith-Wiedemann
syndrome |
| 36 | M | U | Bl, DA | 32 | rs16754 (T) | | | |
| 37 | M | U | Tri | 47 | | | S45C (T) | |
| 38 | F | U | Bl/Ep, PLNR | 19 | rs16754 (T) | | | Macrosomia |
| 39 | M | U | Tri | 28 | rs16754 (B) | | | |
| 40 | M | U | Tri | 28 | | g.17896insT,
c.439insT p.157X (T) | | |
| 41 | M | U | Bl | 25 | rs16754 +
R458X | | | Proteinuria |
| 42 | M | U | St | 56 | rs16754 (B) +
rs2234593 (B) | | | |
| 43 | F | U | Tri | 12 | rs16754 +
rs2234593 | | | |
| 44 | F | B | NA | 12 | C428Y (B) | | | Denys-Drash
syndrome |
Mutation analysis of WT1 exons 8, 9 and 10
identified four sequence variants, namely, two single-nucleotide
polymorphisms (SNPs), one novel missense mutation and one nonsense
mutation. The synonymous sequence variant, SNP rs16754 (p.R369R),
located at exon 8, was the most frequent mutation, having been
observed in 17 patients. With the exception of case 7, all blood
samples were heterozygous for this SNP, and no loss of
heterozygosity (LOH) was observed in the available tumors. The only
possible case of LOH was patient 38, whose tumor sample showed this
variation in a homozygous (or hemizygous) state, but no blood
sample was available from this patient. The second most frequent
sequence variant, located at intron 9, was SNP rs2234593, observed
in five patients. In three of these patients (patients 13, 35 and
43) blood and tumor samples were studied and did not show LOH. The
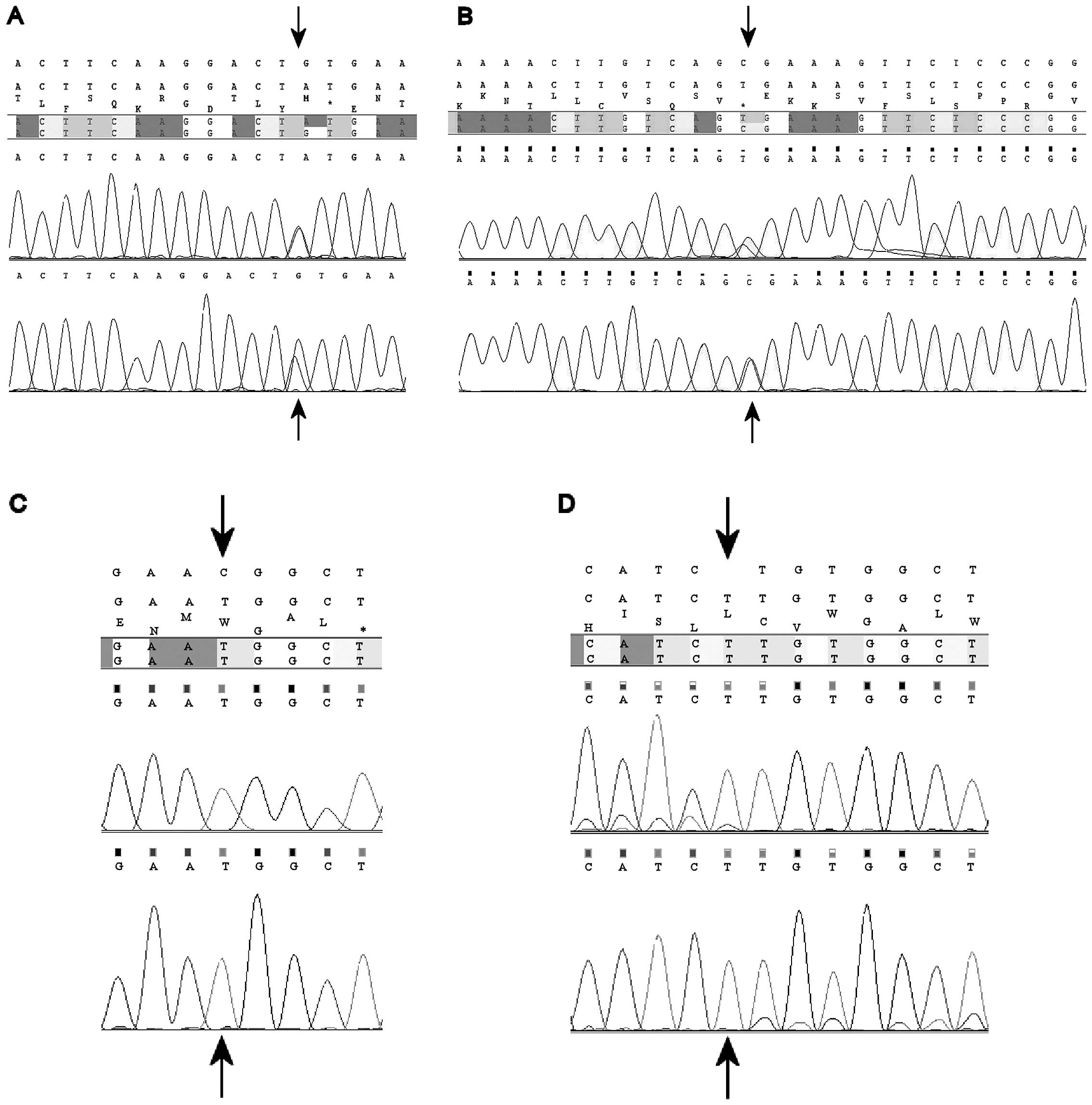
novel missense mutation, p.C428Y (g.47820G>A; c.1283G>A)
(Fig. 1A), was observed in
heterozygosis in patient 44, a female patient with bilateral WT
diagnosed at 12 months and clinical findings of Denys-Drash
syndrome. Finally, patient 41 presented a nonsense g.48510C>T
(c.1372C>T) transition, resulting in the replacement of an
arginine for a stop codon (p.R458X) (Fig. 1B). This mutation was observed in
heterozygosis in both blood and tumor samples. This male patient
developed unilateral blastematous WT diagnosed at 25 months and
proteinuria without other clinical findings of Denys-Drash
syndrome.
Analysis of WTX exon 2 identified five
sequence variants, two synonymous substitutions (rs61730681 and
rs150075206), a non-synonymous mutation (rs34677493), a novel
missense mutation and one frameshift mutation. SNP rs61730681
(p.Q1019Q) was observed in four female and two male patients, and
LOH was observed in two of the four female carriers. SNP
rs150075206 (p.D379D) and the non-synonymous mutation rs34677493
(p.F159L) were observed, respectively, in one female and one male
patient, in both cases in association with SNP rs61730681. The
novel missense mutation p.R560W, resulting from a C>T transition
at position g.19136 (c.1678C>T) (Fig. 1C), was identified in hemizygosis in
both blood and tumor samples of one male patient (patient 18),
whose unilateral tumor showed focal anaplasia. Mutation g.17896insT
(c.439insT), resulting in a frameshift and subsequent stop codon in
the protein (p.157X) (Fig. 1D) was
observed in the tumor sample of one male patient.
Two sequence variants in the CTNNB1 gene were
identified in three patients, in all cases in a heterozygous state
in the tumor samples. Sequence variation rs121913409 predicts the
frequently described missense mutation p.S45C and was observed in
one patient (patient 37). Variation rs121913412, predictive of
missense mutation p.T41A, was observed in two patients (patients 22
and 27).
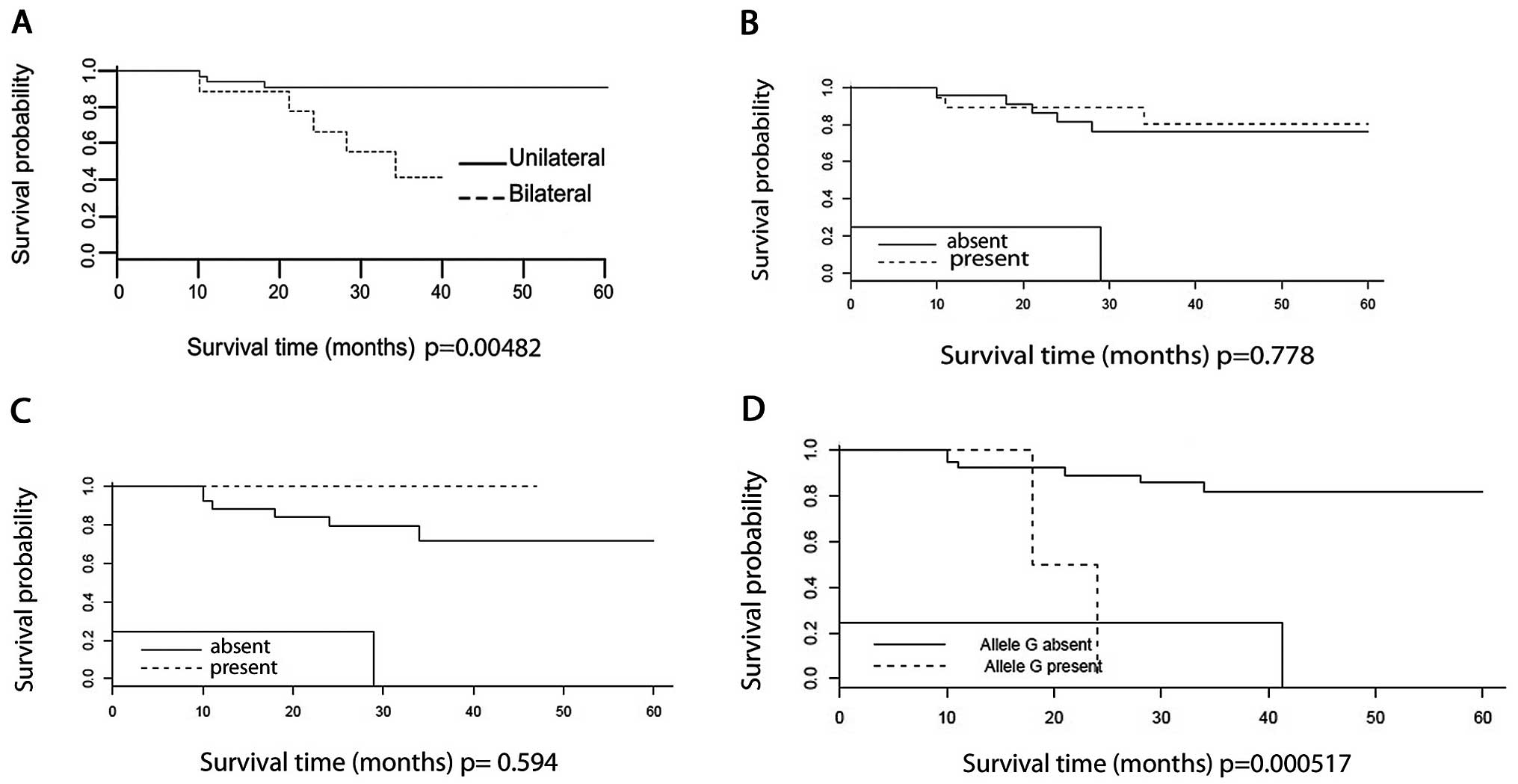
The overall 60-month survival rate for the whole
sample was approximately 80%. Overall survival of bilateral cases
was significantly lower (P=0.005) (Fig.
2A). No difference was observed when survival was analyzed
among patients with WT1 mutations (P=0.778) (Fig. 2B), or in patients with WTX
mutations (P=0.594) (Fig. 2C). On
the other hand, survival of patients 22 and 27, carriers of the
p.T41A mutation in CTNNB1, was significantly lower
(P=0.000517) than the average (Fig.
2D). Overall survival of WT1 rs16754 carriers did not differ
from the rest of the sample (P=0.561) and age at diagnosis did not
differ between carriers and non-carriers of this sequence variant
(P=0.817; data not shown).
Discussion
In the present study we screened for mutations in
selected regions of WT1, WTX and CTNNB1 in 43
WT patients.
In WT1, rs16754A>G, predictive of the
synonymous mutation p.R369R, was the most frequently observed
sequence variant (17/43 patients). No LOH was observed in 8/17
patients whose blood and tumor samples were analyzed. According to
Milani et al(17), rs16754
corresponds to a cis-acting genetic variation regulating WT1
expression levels. This SNP has been associated with better overall
survival in pediatric acute myeloid leukemia patients, and among
rs16754 carriers, an increased expression of WT1 mRNA was observed
(18). In our sample, allele
rs16754G was not associated with differential age at diagnosis
(P=0.817), or overall survival (P=0.561) among carriers.
Sequence variation WT1 rs2234593 was present
in five patients and in three of these no LOH was observed. This
intronic variant, apparently, does not alter WT1 splicing sites
(19).
The nonsense mutation p.R458X (p.R390X) was observed
in one male patient with unilateral WT. This patient also had
proteinuria and did not present genitourinary anomalies. This
mutation has been frequently described in patients with DDS, and in
at least one case of Frasier syndrome (20,21).
Royer-Pokora et al(20)
described three new cases of the WT1 p.R390X nonsense
mutation, and reviewed eight other cases from the literature
(22–25), and observed that genitourinary
anomalies were not present in 3/10 patients with p.R390X (20). Little et al(26) also described two female WT patients
with p.R390X and without genitourinary anomalies. Shibata et
al(25) studied seven cases of
WT with rhabdomyogenic components (fetal rhabdomyogenic
nephroblastoma), and found five cases with the p.R390X mutation.
Corbin et al(27) observed
the p.R390X mutation in a homozygous state in a tumor sample of one
DDS patient who also presented the CTNNB1 p.T41A mutation in
a heterozygous state.
The missense WT1 p.C428Y (g.47820G>A;
c.1283G>A;) mutation described in this study was observed in
heterozygosis in a female patient with bilateral WT diagnosed at
age 12 months. This patient had genitourinary anomalies, and
developed early-onset proteinuria and end-stage renal disease.
Another missense mutation in the same protein residue, p.C428G, has
previously been reported. This mutation changes a cysteine residue
important for the coordination of the zinc atom in the zinc finger
domain (28).
Sixty-month overall survival among carriers of all
WT1 mutations in our sample did not differ from global overall
survival, a finding that was also observed by Royer-Pokora et
al(29).
Five sequence variants were detected in WTX,
two synonymous mutations (rs61730681; p.Q1019Q and rs150075206;
p.D379D), two non-synonymous mutations (rs34677493; p.F159L and
p.R560W) and one frameshift mutation. With the exception of
p.R560W, all other WTX mutations observed in our patients
had been previously identified by Rivera et al(2).
Transition c.1678C>T, predictive of undescribed
missense mutation p.R560W, was observed in one male patient with
unilateral WT and focal anaplasia. Corbin et al(27) observed another WTX missense
mutation, p.T429I, in a WT patient who also presented anaplasia.
Germline mutations in WTX were described in X-linked
dominant osteopathia striata with cranial sclerosis (OSCS)
(30), a disease not associated
with WT risk. To the best of our knowledge, the WTX p.R560W
mutation has not been previously described among WT (27, 31–37) or
OSCS (30,38) patients.
In our sample, 60-month overall survival among
carriers of all WTX mutations did not differ from global
overall survival (P=0.594), as observed by Wegert et
al(32).
CTNNB1 exon 3 sequencing showed two
previously well-known missense mutations (8,39,40) in
three patients of our sample, all in heterozygosis: p.T41A in two
patients and p.S45C in one patient. These somatic mutations remove
a major phosphorylation site for GSK-3β, leading to the
stabilization of β-catenin, and they exert a dominant effect at the
level of the β-catenin/TCF-mediated transcription; therefore, these
mutations may be associated with the development and/or survival of
WT (8,39). Notably, the two carries of the
p.T41A mutation showed a significantly lower overall survival rate
(P= 0.000517) than the rest of the sample. In spite of the small
number of p.T41A carriers in our group of patients (two
individuals), we could not find an association of this somatic
mutation with poorer survival rate among WT patients in the
literature.
Additional studies of the impact of WTX
mutations are essential to better understand the reasons why only
somatic, and not germline mutations in this gene result in WT.
Also, the interaction of the WT1, WTX and
CTNNB1 genes within the context of the Wnt signaling
pathway, seems to be critical for the development and survival of
various malignancies, including WT.
Acknowledgements
This study was supported by Conselho Nacional de
Desenvolvimento Científico (CNPq) grants 401966/2010-0,
476808/2010-3, 573806/2008-0 and Fundação de Amparo à Pesquisa do
Estado do Rio de Janeiro (FAPERJ) E26/170.026/2008.
References
|
1
|
Dome JS and Huff V: Wilms Tumor Overview.
GeneReviews. Pagon RA, Bird TD, Dolan CB and Stephans K: University
of Washington; Seattle: 2011
|
|
2
|
Rivera MN, Kim WJ, Wells J, et al: An X
chromosome gene, WTX, is commonly inactivated in Wilms tumor.
Science. 315:642–645. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Haber DA, Sohn RL, Buckler AJ, Pelletier
J, Call KM and Housman DE: Alternative splicing and genomic
structure of the Wilms tumor gene WT1. Proc Natl Acad Sci.
88:9618–9622. 1991. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Lamond AI: RNA processing. Wilms’ tumour -
the splicing connection? Curr Biol. 5:862–865. 1995.
|
|
5
|
Wells J, Rivera MN, Kim WJ, Starbuck K and
Haber DA: The predominant WT1 isoform (+KTS) encodes a DNA
binding protein targeting the planar cell polarity gene
Scribble in renal podocytes. Mol Cancer Res. 8:975–985.
2010.
|
|
6
|
Huff V: Wilms’ tumours: about tumour
suppressor genes, an oncogene and a chameleon gene. Nature.
11:111–121. 2011.
|
|
7
|
Pelletier J, Bruening W, Kashtan CE, et
al: Germline mutations in the Wilms’ tumor suppressor gene are
associated with abnormal urogenital development in Denys-Drash
syndrome. Cell. 67:437–447. 1991.
|
|
8
|
Koesters R, Ridder R, Kopp-Schneider AK,
et al: Mutational activation of the β-catenin proto-oncogene is a
common event in the development of Wilms’ tumors. J Cancer Res.
59:3880–3882. 1999.
|
|
9
|
Maeda O, Usami N, Kondo M, et al:
Plakoglobin (γ-catenin) has TCF/LEF family - dependent
transcriptional activity in beta-catenin-deficient cell line.
Oncogene. 23:964–972. 2004.
|
|
10
|
Oloumi A, McPhee T and Dedhar S:
Regulation of E-cadherin expression and beta-catenin/Tcf
transcriptional activity by the integrin-linked kinase. Biochim
Biophys Acta. 1691:1–15. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Clevers H: Wnt/beta-catenin signaling in
development and disease. Cell. 127:469–480. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Major MB, Camp ND, Berndt JD, et al: Wilms
tumor supressor WTX negatively regulates Wnt/β-catenin signaling.
Science. 316:1043–1046. 2007.
|
|
13
|
Rivera MN, Kim WJ, Wells J, Stone A, et
al: The tumor suppressor WTX shuttles to the nucleus and modulates
WT1 activity. Proc Natl Acad Sci USA. 106:8338–8343. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Miller SA, Dykes DD and Polesky HF: A
simple salting out procedure for extracting DNA from human
nucleated cells. Nucleic Acids Res. 16:12151988. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Sambrook J, Fritsch EF and Maniatis T:
Molecular Cloning: A Laboratory Manual. 2nd edition. Cold Spring
Harbor; New York: 1989
|
|
16
|
Nacional Center for Biotechnology
Information. http://www.ncbi.nlm.nih.gov/:.
Accessed April 14, 2012
|
|
17
|
Milani L, Gupta M, Andersen M, et al:
Allelic imbalance in gene expression as a guide to cis-acting
regulatory single nucleotide polymorphisms in cancer cells. Nucleic
Acids Res. 35:e34–e43. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Ho PA, Kuhn J, Gerbing RB, et al: WT1
synonymous single nucleotide polymorphism rs16754 correlates with
higher mRNA expression and predicts significantly improved outcome
in favorable-risk pediatric acute myeloid leukemia: a report from
the children’s oncology group. J Clin Oncol. 29:704–711.
2011.PubMed/NCBI
|
|
19
|
Splice-Site Analyzer Tool. Genome
Bioinformatics group of University of California Santa Cruz:
Splice-Site analyzer tool. http://ibis.tau.ac.il/ssat/SpliceSiteFrame.htm.
Accessed January 20, 2012
|
|
20
|
Royer-Pokora B, Beier M, Henzler M, Alan
R, Schumacher V, Weirich A and Huff V: Twenty-four new cases of
WT1 germline mutations and review of the literature:
genotype/phenotype correlations for Wilms tumor development. Am J
Med Genet A. 127:249–257. 2004.
|
|
21
|
Kohsaka T, Tagawa M, Takekoshi Y,
Yanagisawa H, Tadokoro K and Yamada M: Exon 9 mutations in the
WT1 gene, without influencing KTS splice isoforms, are also
responsible for Frasier syndrome. Hum Mutat. 14:466–470.
1999.PubMed/NCBI
|
|
22
|
Little MH, Prosser J, Condie A, Smith PJ,
Van Heyningen V and Hastie ND: Zinc finger point mutations within
the WT1 gene in Wilms tumor patients. Proc Natl Acad Sci USA.
89:4791–4795. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Schumacher V, Schneider S, Figge A, et al:
Correlation of germ-line mutations and two-hit inactivation of the
WT1 gene with Wilms tumors of stromal-predominant histology.
Proc Natl Acad Sci USA. 94:3972–3977. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Maiti S, Alam R, Amos CI and Huff V:
Frequent association of β-catenin and WT1 mutations in Wilms
tumors. Cancer Res. 60:6288–6292. 2000.
|
|
25
|
Shibata R, Hashiguchi A, Sakamoto J,
Yamada T, Umezawa A and Hata J: Correlation between a specific
Wilms tumour suppressor gene (WT1) mutation and the
histological findings in Wilms tumour (WT). J Med Genet.
39:e832002. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Little SE, Hanks SP, King-Underwood L, et
al: Frequency and heritability of WT1 mutations in
nonsyndromic Wilms’ tumor patients: a UK Children’s Cancer Study
Group Study. J Clin Oncol. 22:4140–4146. 2004.PubMed/NCBI
|
|
27
|
Corbin M, de Reyniès A, Rickman DS, et al:
WNT/β-catenin pathway activation in Wilms tumors: a unifying
mechanism with multiple entries? Genes Chromosomes Cancer.
48:816–827. 2009.
|
|
28
|
Little MH, Williamson KA, Mannens M,
Kelsey A, Gosden C, Hastie ND and van Heyningen V: Evidence that
WT1 mutations in Denys-Drash syndrome patients may act in a
dominant-negative fashion. Hum Mol Genet. 2:259–264. 1993.
|
|
29
|
Royer-Pokora B, Weirich A, Schumacher V,
et al: Clinical relevance of mutations in the Wilms tumor
suppressor 1 gene WT1 and the cadherin-associated protein β1
gene CTNNB1 for patients with Wilms tumors. Results of
long-term surveillance of 71 patients from International Society of
Pediatric Oncology Study 9/Society for Pediatric Oncology. Cancer.
113:1080–1089. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Jenkins ZA, van Kogelenberg M, Morgan T,
et al: Germline mutations in WTX cause a sclerosing skeletal
dysplasia but do not predispose to tumorigenesis. Nat Genet.
41:95–100. 2009. View
Article : Google Scholar : PubMed/NCBI
|
|
31
|
Perotti D, Gamba B, Sardella M, et al:
Functional inactivation of the WTX gene is not a frequent
event in Wilms’ tumors. Oncogene. 27:4625–4632. 2008.PubMed/NCBI
|
|
32
|
Wegert J, Wittmann S, Leuschner I,
Geissinger E, Graf N and Gessler M: WTX inactivation is a frequent,
but late event in Wilms tumors without apparent clinical impact.
Genes Chromosomes Cancer. 48:1102–1111. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Ruteshouser EC, Robinson SM and Huff V:
Wilms tumor genetics: mutations in WT1, WTX, and
CTNNB1 account for only about one-third of tumors. Genes
Chromosomes Cancer. 47:461–470. 2008.PubMed/NCBI
|
|
34
|
Fukuzawa R, Anaka MR, Weeks RJ, Morison IM
and Reeve AE: Canonical WNT signaling determines lineage
specificity in Wilms tumour. Oncogene. 28:1063–1075. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Fukuzawa R, Holman SK, Chow CW,
Savarirayan R, Reeve AE and Robertson SP: WTX mutations can occur
both early and late in the pathogenesis of Wilms tumour. J Med
Genet. 47:791–794. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Haruta M, Arai Y, Watanabe N, et al:
Different incidences of epigenetic but not genetic abnormalities
between Wilms tumors in Japanese and Caucasian children. Cancer
Sci. 103:1129–1135. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Scott RH, Murray A, Baskcomb L, Turnbull
C, et al: Stratification of Wilms tumor by genetic and epigenetic
analysis. Oncotarget. 3:327–335. 2012.PubMed/NCBI
|
|
38
|
Perdu B, Lakeman P, Mortier G, Koenig R,
Lachmeijer A and Van Hul W: Two novel WTX mutations
underscore the unpredictability of male survival in osteopathia
striata with cranial sclerosis. Clin Genet. 80:383–388. 2010.
|
|
39
|
Li CM, Kim CE, Margolin AA, Guo M, et al:
CTNNB1 mutations and overexpression of Wnt/β-catenin target genes
in WT1-mutant Wilm’s tumors. Am J Pathol. 165:1943–1953. 2004.
|
|
40
|
Uschkereit C, Perez N, de Torres C, Küff
M, Mora J and Royer-Pokora B: Different CTNNB1 mutations as
molecular genetic proof for the independent origin of four Wilms
tumours in a patient with a novel germ line WT1 mutation. J
Med Genet. 44:393–396. 2007.
|