Introduction
The receptor for advanced glycation end products
(RAGE) is a type I transmembrane receptor belonging to the
immunoglobulin superfamily. The receptor is involved in the
pathogenesis of a broad range of inflammatory, degenerative and
hyperproliferative diseases (1,2). It
binds to diverse ligands, including advanced glycation end products
(AGEs) (3), high-mobility group box
1 (HMGB1) (4,5) and S100 family proteins (6). Binding of ligands to RAGE results in
the activation of multiple intracellular signaling pathways,
eventually leading to apoptosis, hyperproliferation, production of
inflammatory cytokines and increased motility (2,5,7).
Our previous study demonstrated that the cytoplasmic
domain of RAGE is phosphorylated at Ser391 by PKCζ upon the binding
of ligands (12). TIRAP and MyD88
are recruited to the phosphorylated RAGE and transduce a signal to
downstream effector molecules, such as NF-κB, Akt, p38 and JNK1.
This finding may assist in elucidating the mechanisms of the
various RAGE-mediated cellular processes.
Hudson et al(8) reported that diaphanous-1 (Dia-1) also
functions as an adaptor for the RAGE cytoplasmic domain. This
protein plays a major role in the control of the RAGE-mediated
activation of the small guanine nucleotide triphosphatases
(GTPases), Rac1 and Cdc42. In this pathway, the effector small
GTPases are responsible for the formation of lamellipodia and
filopodia, in turn facilitating cellular migration (9,10).
However, it remains unclear how the ligand-bound RAGE leads to the
activation of Rac-1 and Cdc42. For the activation of the small
GTPases, involvement of a guanine nucleotide exchange factor (GEF)
is essential, since GEFs catalyze the replacement of GDP with free
cytoplasmic GTP to generate active GTPases. There have been no
reports demonstrating that Dia-1 has GEF activity and a GEF-like
domain is not present in Dia-1.
In the present study, we aimed to identify a
possible GEF(s) that is involved in the RAGE-Rac1/Cdc42 signaling
axis. Our efforts resulted in the identification of an atypical
DOCK180-related GEF, dedicator of cytokinesis protein 7 (DOCK7).
DOCK7 bound to the cytoplasmic domain of RAGE and the
downregulation of DOCK7 resulted in marked interference with signal
transduction from RAGE to Cdc42, indicating the involvement of
DOCK7 in the RAGE-Cdc42 signaling axis in human glioblastoma
cells.
Materials and methods
Cell lines
The following human cancer cell lines were used:
HEK293T (embryonic kidney cell line stably expressing the SV40
large T antigen; Riken BioResource Center, Tsukuba, Japan), SH-SY5Y
(neuroblastoma; ATCC, Manassas, VA), U-87MG (glioblastoma; ATCC),
PK-8 (pancreatic carcinoma; Cell Resource Center for Biomedical
Research, Tohoku University, Sendai, Japan), HepG2 (hepatocellular
carcinoma; ATCC), Hep3B (hepatocellular carcinoma; ATCC), HeLa
(cervix adenocarcinoma; ATCC), PC-3 (prostate adenocarcinoma;
ATCC), LNCaP (prostate carcinoma; ATCC), DU145 (prostate carcinoma;
ATCC), A431 (skin epidermoid carcinoma; ATCC), HCT116 (colorectal
carcinoma; ATCC), DLD-1 (colorectal adenocarcinoma; ATCC), KPK1
(renal clear cell carcinoma; Clonetics, San Diego, CA), Caki-1
(renal clear cell carcinoma; ATCC), Caki-2 (renal clear cell
carcinoma; ATCC) and MCF7 (mammary gland adenocarcinoma; ATCC).
These cell lines were cultivated in D/F medium (Invitrogen,
Carlsbad, CA) supplemented with 10% FBS.
Liquid chromatography coupled with
electrospray tandem mass spectrometry (LC-MS/MS)
Immunoprecipitated proteins were identified using a
shotgun-type protein identification approach as previously
described (11). Briefly, we
composed a nano-flow liquid chromatography-mass spectrometry
platform consisting of a Nanospace SI-2 high-performance liquid
chromatography modified to adjust flow rate (Shiseido Co., Tokyo,
Japan), a nano-electrospray ionization source modified according to
Washburn et al(12) and an
LTQ-Orbitrap hybrid mass spectrometer (Thermo Fisher Scientific,
San Jose, CA) to obtain tandem mass spectra of tryptic peptides.
The resulting tandem mass spectrometry spectra were analyzed using
the Sequest algorithm against a non-redundant human protein
database (NCBI, Feb 2007) for putative protein identification.
Plasmids
We prepared two mammalian expression vectors. The
CMV promoter-intron (CMVi) from the phCMV-FSR™ vector (Genlantis,
San Diego, CA) was inserted into the Promoterless pDNR-1r vector
(Clontech-Takara, Mountain View, CA) and this was named pDNR-CMVi.
The CMVi with a part of the HTLV type 1 LTR (RU5′) was integrated
into the pIDTSmart vector (Integrated Device Technology, San Jose,
CA) and this was named pIDT-CMViR. Both vectors efficiently express
short cargo cDNAs.
Human cDNAs encoding constitutively active types of
HRAS (G12V), RHOA (G14V), RAC1 (G12V) and CDC42 (G12V) were
conjugated with green fluorescent protein (GFP) at the N-terminal
side and inserted into the pDNR-CMVi vector.
Human cDNAs encoding wild-type (WT) cytoplasmic
domain (364–404 aa) lacking a dominant-negative (DN) form and a
cytoplasmic domain (Cyt: 364–404 aa) of RAGE were inserted into the
pIDT-CMViR vector (13). WT, DN and
Cyt of RAGE were designed to be expressed as C-terminal
Myc-HA-Flag-6His-tagged forms. Human cDNA encoding S100B was also
tagged with C-terminal Myc-6His and inserted into the pIDT-CMViR
vector.
Transient transfection of the plasmids into cultured
cells was performed using FuGENE HD (Promega Biosciences, San Luis
Obispo, CA) for HEK293T, U-87MG and PC3 cells and
TransIT-keratinocyte (Mirus Bio LLC, Madison, WI) for MCF7
cells.
Western blot analysis and
co-immunoprecipitation
Western blot analysis was performed under
conventional conditions. Antibodies used were as follows: mouse
anti-HA tag (clone 6E2), rabbit anti-human RAGE (Santa Cruz
Biotechnology, Santa Cruz, CA), rabbit anti-human DOCK7
(Sigma-Aldrich, St. Louis, MO) and mouse anti-human tubulin
antibodies (Sigma-Aldrich). The secondary antibodies were
horseradish peroxidase-conjugated anti-mouse or anti-rabbit IgG
antibody (Cell Signaling Technology, Beverly, MA). Positive signals
were detected by a chemiluminescence system (ECL Plus; GE
Healthcare Bio-Sciences, Piscataway, NJ).
Rabbit anti-human RAGE antibody was biotinylated
using a Biotin Labeling kit-SH (Dojindo Molecular Technologies,
Rockville, MD) to recover antibody-free RAGE after
immunoprecipitation using streptavidin-agarose (13). Monoclonal anti-HA (clone
HA-7)-tagged agarose (Sigma-Aldrich) and streptavidin agarose
(Invitrogen) were used for the co-immunoprecipitation experiments.
Monoclonal anti-His tag (clone 2D8) agarose (MBL, Nagoya, Japan)
was used to purify recombinant human S100B protein expressed in the
HEK293T cells.
The GTP-bound form of Cdc42 was determined using a
Rac/Cdc42 Activation Assay kit (Millipore, Billerica, MA).
siRNA
Human RAGE (siRAGE: siGENOME SMARTpool
M-003625-02-0005), human DOCK7 (siDOCK7: siGENOME SMART pool
M-031725-01-0005) and the negative control (siCont: siGENOME
non-targeting siRNA pool #1, D-001210-01) siRNAs were purchased
from Thermo Scientific Dharmacon (Lafayette, CO). The siRNAs (100
nM) were transfected using Lipofectamine RNAiMax reagent
(Invitrogen).
Migration assay
Migration of human glioblastoma U-87MG cells was
assayed using a 24-well disposable chemotaxis system (cell culture
inserts, 8-μm pore size; BD Falcon, Franklin Lakes, NJ). The lower
wells of the chamber were loaded with DMEM supplemented with 10%
FBS. Cells (5,000) were placed in the upper wells under a
serum-free condition. After incubation for 8 h, cells on the lower
surface of the filter were counted after staining with hematoxylin
and eosin (H&E) (n=4).
Immunocytochemistry
To visualize overexpressed RAGE, fixed cells were
treated with rabbit anti-HA antibody (MBL) at RT for 1 h and were
further treated with Alexa 594-conjugated goat anti-rabbit IgG
antibody (Molecular Probes/Invitrogen, Eugene, OR) under the same
conditions as those previously reported (14).
Statistical analysis
Data are expressed as the means ± SD. We employed
simple pair-wise comparison with the Student’s t-test (two-tailed
distribution with two-sample equal variance). P<0.05 was
considered to indicate a statistically significant difference.
Results
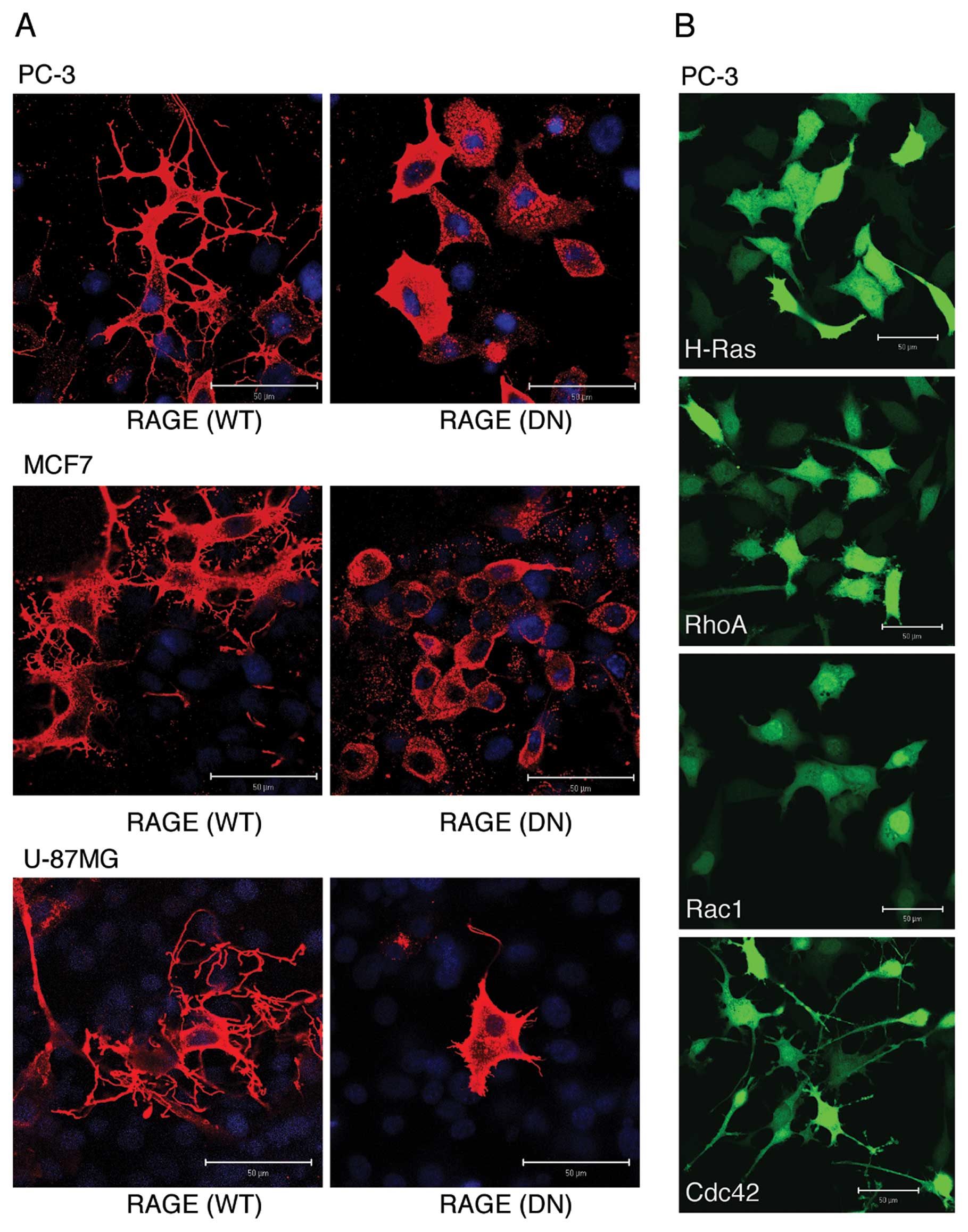
Overexpression of RAGE induces the
formation of dendritic pseudopodia in cancer cells
It has been shown that upon ligand binding RAGE
activates Rac1 and Cdc42 (8),
eventually leading to the induction of lamellipodial and filopodial
formation, respectively (15,16).
We, therefore, examined which morphological changes were mainly
induced by RAGE. Overexpression of full-length WT RAGE, but not
that of cytoplasmic domain-deleted DN type of RAGE, induced
dramatic morphological changes in all human cancer cell lines
examined (Fig. 1A). The most
typically observed morphological change was the presence of highly
branched filopodia-like protrusions (dendritic pseudopodia). When
various small GTPases were overexpressed in PC-3 cells, Cdc42
induced a morphological change most similar to that induced by WT
RAGE when compared to Ras, Rho and Rac1 (Fig. 1B), suggesting that the formation of
dendritic pseudopodia is due to the activation of the RAGE-Cdc42
signaling axis.
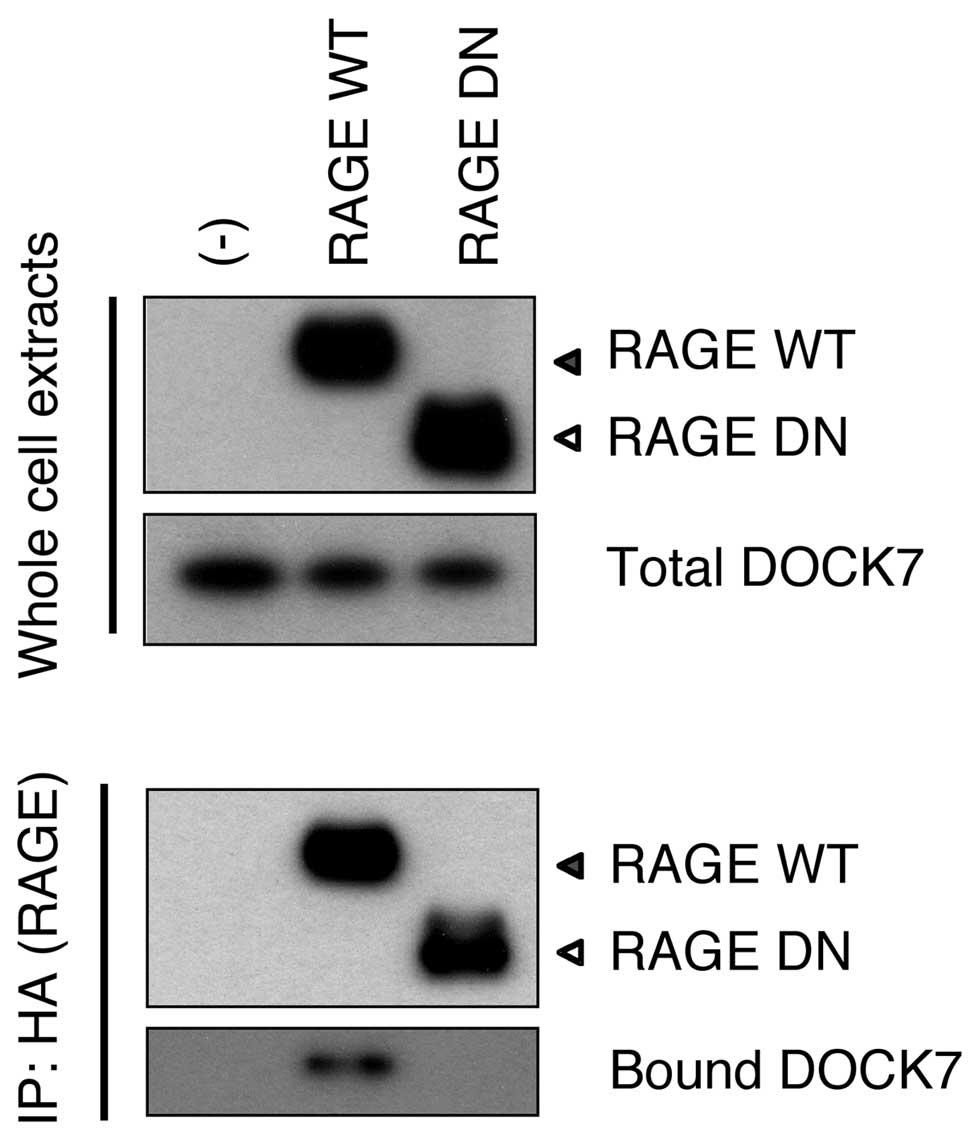
DOCK7 binds to the RAGE cytoplasmic
domain
We aimed to identify a candidate protein(s) involved
in mediating a signal from RAGE to Cdc42. We screened proteins that
bound to the overexpressed RAGE cytoplasmic domain in HEK293T
cells, employing immunoprecipitation and affinity purification
followed by mass spectrometry. Among the proteins identified by
mass spectrometry, Ras-GTPase-activating protein SH3 domain-binding
proteins (G3BP) and DOCK7 were noted as promising. However, neither
cloned and overexpressed G3BP nor endogenous G3BP were noted to
bind to Cyt RAGE in transfected cells (data not shown). Endogenous
DOCK7, the only GEF discovered in the analysis, was confirmed to
bind to WT RAGE but not to DN RAGE in HEK293T cells (Fig. 2). This indicated that RAGE interacts
with DOCK7 via its cytoplasmic domain.
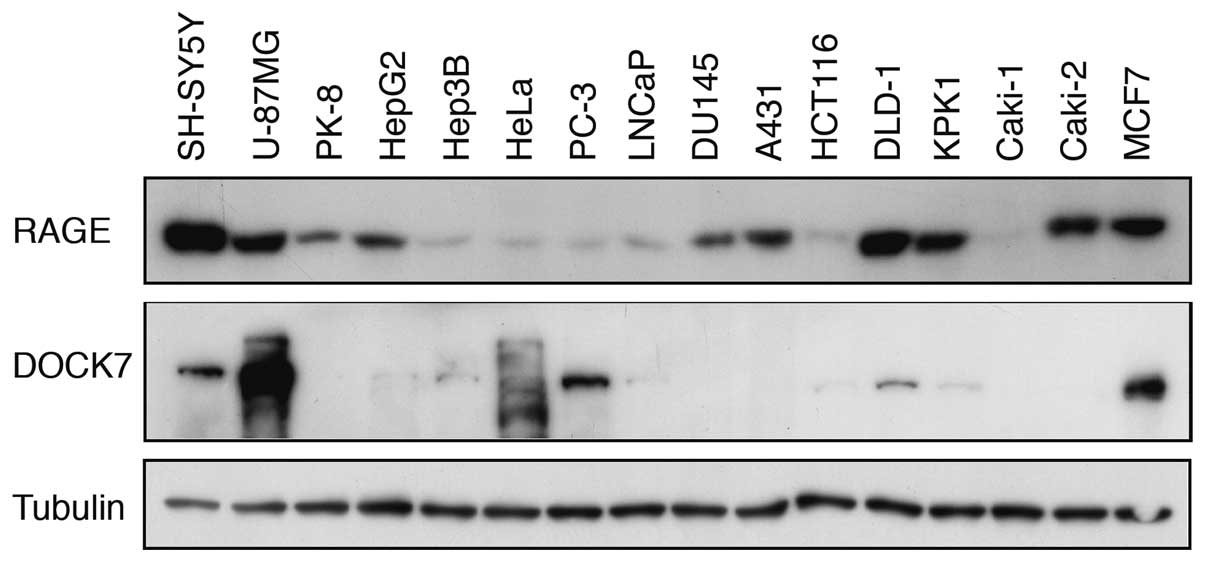
Expression of RAGE and DOCK7 in human
cancer cell lines
We next examined the expression of RAGE and DOCK7 in
various human cancer cell lines by western blot analysis. RAGE
protein was detected in all cancer cell lines examined and higher
expression levels were noted in SH-SY5Y, U-87MG, PK-8, HepG2,
DU145, A431, DLD-1, KPK1, Caki-2 and MCF7 cell lines (Fig. 3). DOCK7 protein was clearly detected
in SH-SY5Y, U-87MG, HeLa, PC-3, DLD-1 and MCF7 cell lines with the
highest expression level in U-87MG. Thus, the notion that the
induction of dendritic pseudopodia formation by overexpressed WT
RAGE (Fig. 1) is mediated by DOCK7
appeared plausible.
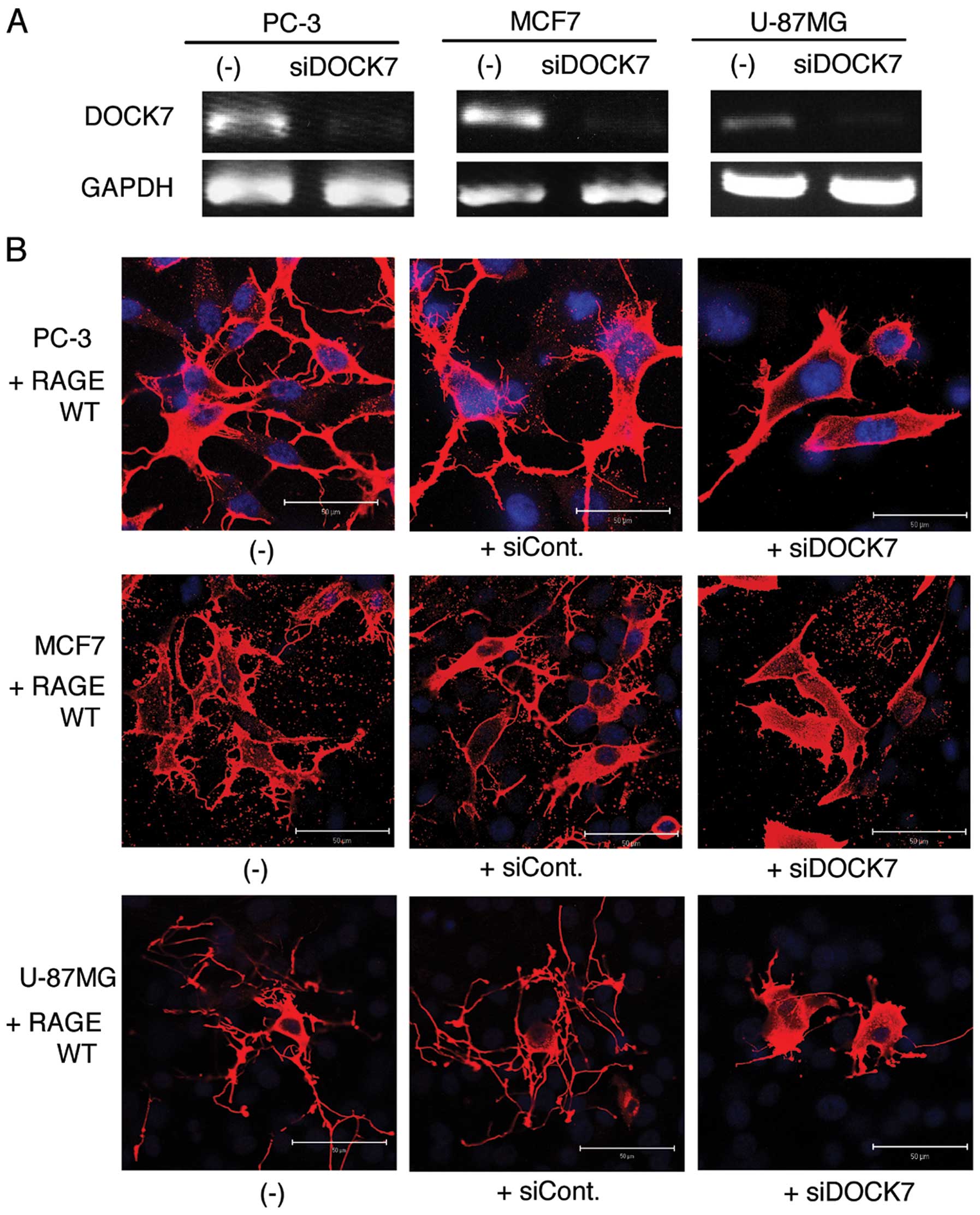
Knockdown of DOCK7 suppresses formation
of dendritic pseudopodia in RAGE-overexpressed cells
Using the DOCK7-positive cell lines, U-87MG, MCF7
and PC3, we examined the possible role of DOCK7 in the formation of
dendritic pseudopodia induced by RAGE overexpression. Application
of DOCK7 siRNA efficiently suppressed the expression of endogenous
DOCK7 (Fig. 4A). The validated
siRNA markedly inhibited the RAGE-induced formation of dendritic
pseudopodia, while the control siRNA demonstrated no effect
(Fig. 4B).
A RAGE ligand, S100B, induces the
recruitment of DOCK7 to the RAGE cytoplasmic domain and activates
Cdc42
S100B is a physiological ligand of RAGE that is
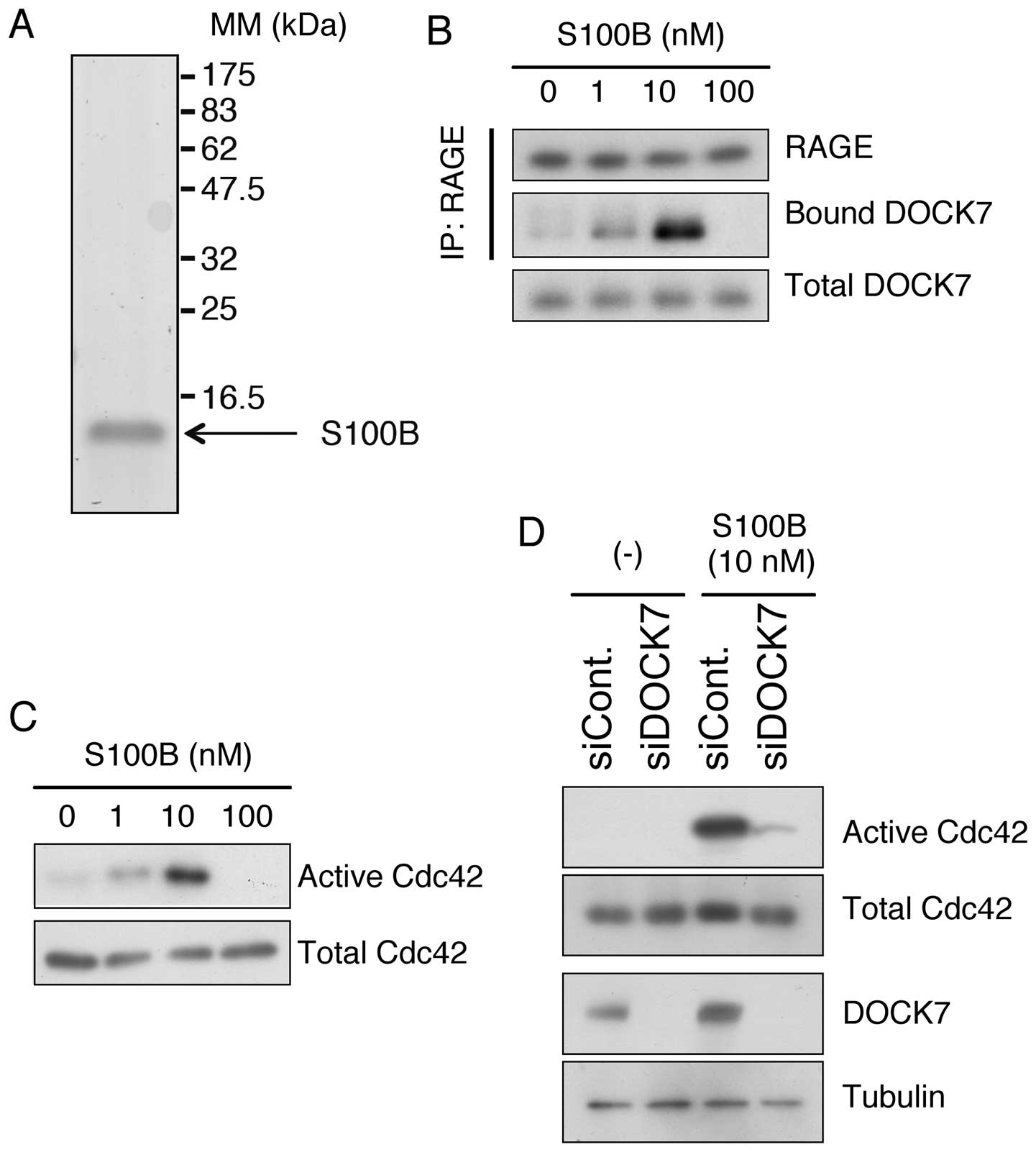
abundantly produced and secreted by glioblastoma cells (17). We prepared highly purified
recombinant human S100B using a mammalian expression system
(Fig. 5A, Materials and methods).
When U-87MG cells were treated with different concentrations of
S100B, DOCK7 was recruited to endogenous RAGE in a dose-dependent
manner up to 10 nM, while 100 nM of S100B abolished the binding
possibly due to a cytotoxic effect (Fig. 5B). In accordance with this, Cdc42
was dose-dependently activated up to 10 nM and inactivated at 100
nM of S100B (Fig. 5C). In addition,
siRNA-mediated downregulation of DOCK7 effectively abrogated the
activation of Cdc42 caused by S100B in U-87MG cells (Fig. 5D). These results indicate that DOCK7
is an essential mediator of the RAGE-Cdc42 signaling axis.
Knockdown of DOCK7 suppresses the
migration of U-87MG cells
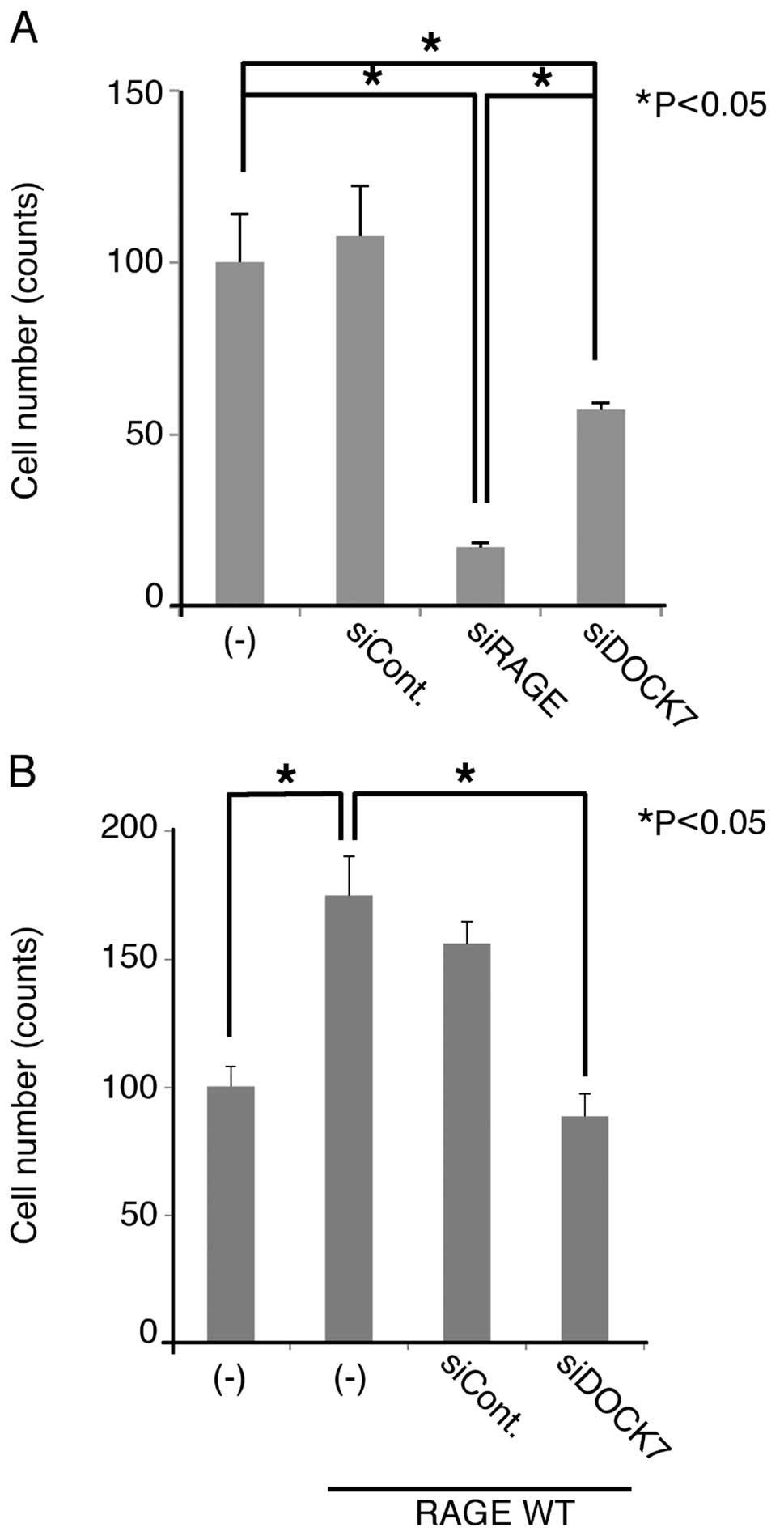
Since the RAGE-Cdc42 signaling axis is considered to
play a critical role in cellular migration (8), we examined the effect of DOCK7 siRNA
on the migration of U-87MG cells. When we treated the cells with
RAGE and DOCK7 siRNAs, cellular migration was significantly
suppressed (Fig. 6A), indicating
that endogenous RAGE and DOCK7 are involved in the migration. The
extent of suppression by the downregulation of RAGE was greater
compared to that of DOCK7 siRNA (Fig.
6A). Overexpression of RAGE enhanced the migration of U87MG
cells, this being abrogated by DOCK7 siRNA (Fig. 6B). These results indicate that DOCK7
functions as an essential and downstream regulator of RAGE-mediated
cellular migration in U-87MG cells
Discussion
Several lines of evidence indicate that RAGE
activation upon ligand binding promotes cancer progression by
enhancing cell proliferation and migration (4). In this study, we focused on migration
enhanced by the RAGE signaling pathway. Rho family small GTPases
are widely accepted as key regulators of cellular migration via
modulation of the structural network of actin cytoskeleton and the
eventual formation of various pseudopodia. The resemblance of the
morphological change of RAGE-transfected cells with Cdc42-induced
morphology (Fig. 1) indicated that
Cdc42 rather than Rac1 may be involved in the RAGE signaling in
cancer cells.
Hudson et al(8) demonstrated that Dia-1 functions as an
adaptor protein for RAGE and transfers a signal from RAGE to Rac1
and Cdc42, eventually enhancing cellular migration. Despite the
essential role of Dia-1 in this process, it remains unclear how
Dia-1 activates Rac1 and Cdc42. It is possible that an adaptor
GEF(s) plays a role in the activation of Rac1 and Cdc42. No obvious
domains or motifs associated with guanyl-nucleotide exchange
function were observed in Dia-1. We successfully identified DOCK7
using Cyt RAGE as a bait. Since it is difficult to efficiently
express a short cDNA such as Cyt RAGE (41 aa), we modified the
mammalian expression vectors as described in Materials and methods.
It is difficult to purify full-length recombinant DOCK7 protein due
to its high molecular weight (2,140 aa). Hence, we confirmed the
binding of DOCK7 to Cyt RAGE by immunoprecipitation using the cell
extracts (Figs. 2 and 5B and D). This did not provide an answer
as to whether the binding is direct or indirect. It is possible,
therefore, that Dia-1 functions as a mediator between RAGE and
DOCK7. We observed that the downregulation of Dia-1 by siRNA
abrogated the formation of dendritic pseudopodia in
RAGE-overexpressed cancer cells (data not shown).
DOCK7 belongs to the DOCK family, which consists of
eleven GEFs (18). DOCK proteins
contain a catalytic domain termed the DOCK homology region (DHR)-2
(19). Although the molecular
structures of DOCK proteins are similar, the small GTPases,
including Rac and Cdc42, are regulated by specific DOCK proteins
(18). DOCK180, DOCK2 and DOCK3 are
Rac-specific GEFs. DOCK4 and DOCK5 are structurally deduced GEFs
for Rac. DOCK6, DOCK7 and DOCK8 are GEFs for Rac and Cdc42. DOCK9,
DOCK10 and DOCK11 are Cdc42-specific GEFs. It is also known that
each DOCK protein is differentially expressed in different cell
types (18). We, therefore,
examined DOCK7 expression in various cancer cell lines. DOCK7 was
detected in a number of cancer cell lines with varying expression
levels. Among those, U-87MG cells demonstrated the highest level of
DOCK7 expression (Fig. 3). This may
influence the RAGE-Cdc42 signaling axis in different cancer types.
In fact, the overexpression of RAGE induced no marked morphological
change in DOCK7-deficient PK-8 cells (data not shown). Thus, we
reasonably conclude that DOCK7 binding to RAGE leads to Cdc42
activation at least in several types of cancer cells.
It should be noted that siRNA-mediated
downregulation of RAGE suppressed migration of U-87MG cells more
effectively when compared to that of DOCK7 (Fig. 6A). This may imply the involvement of
factors other than DOCK7 in the RAGE-mediated migration. We
recently demonstrated that signal transduction triggered by RAGE is
partially mediated by the adaptor proteins TIRAP and MyD88
(13). The RAGE-TIRAP/MyD88
signaling axis leads to the activation of NF-κB, resulting in the
expression of genes related to the inflammatory response, including
TNF-α and IL-6. Several studies have shown that the MyD88 signaling
pathway plays a crucial role in neutrophil migration (20,21).
Dia-1 may mediate a signal from RAGE to downstream mediators other
than DOCK7. Thus, the RAGE-induced cellular migration may be
controlled by more complicated mechanisms involving not only DOCK7
but also MyD88, Dia-1 and yet unidentified molecules.
NF-κB has been shown to be a principal mediator for
RAGE signaling in various biological contexts such as inflammation
and stress responses (22).
Recently, accumulating evidence indicates that NF-κB is critically
involved in epithelial-mesenchymal transition (EMT), a complex
reprogramming process of epithelial cells that plays an
indispensable role in cancer invasion and metastasis. Therefore, it
is possible that NF-κB-mediated EMT and DOCK7-mediated migration
coordinately regulate cancer invasion and metastasis in response to
RAGE activation in inflammatory microenvironments.
Acknowledgements
The present study was supported in part by grants
from the Ministry of Health, Labor and Welfare (Research for
Intractable Diseases) (to N.H.), the Ministry of Education,
Culture, Sports, Science and Technology of Japan (Grant-in-Aid for
Scientific Research on Innovation Areas) (to M.S.), The Naito
Foundation (to M.S.), the Senshin Medical Research Foundation (to
M.S.) and the Research Foundation for Pharmaceutical Sciences (to
M.S.).
Abbreviations:
|
RAGE
|
receptor for advanced glycation end
products
|
|
DOCK7
|
dedicator of cytokinesis protein 7
|
|
Dia-1
|
diaphanous-1
|
|
GTPase
|
guanine nucleotide triphosphatase
|
|
GEF
|
guanine nucleotide exchange factor
|
|
EMT
|
epithelial-mesenchymal transition
|
References
|
1
|
Yan SF, Ramasamy R and Schmidt AM:
Mechanisms of disease: advanced glycation end-products and their
receptor in inflammation and diabetes complications. Nat Clin Pract
Endocrinol Metab. 4:285–293. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Lin L, Park S and Lakatta EG: RAGE
signaling in inflammation and arterial aging. Front Biosci.
14:1403–1413. 2009. View
Article : Google Scholar : PubMed/NCBI
|
|
3
|
Xue J, Rai V, Singer D, Chabierski S, Xie
J, Reverdatto S, Burz DS, Schmidt AM, Hoffmann R and Shekhtman A:
Advanced glycation end product recognition by the receptor for
AGEs. Structure. 19:722–732. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Taguchi A, Blood DC, del Toro G, Canet A,
Lee DC, Qu W, Tanji N, Lu Y, Lalla E, Fu C, Hofmann MA, Kislinger
T, Ingram M, Lu A, Tanaka H, Hori O, Ogawa S, Stern DM and Schmidt
AM: Blockade of RAGE-amphoterin signalling suppresses tumour growth
and metastases. Nature. 405:354–360. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Sims GP, Rowe DC, Rietdijk ST, Herbst R
and Coyle AJ: HMGB1 and RAGE in inflammation and cancer. Annu Rev
Immunol. 28:367–388. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Leclerc E, Fritz G, Vetter SW and Heizmann
CW: Binding of S100 proteins to RAGE: an update. Biochim Biophys
Acta. 1793:993–1007. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Gebhardt C, Riehl A, Durchdewald M, Németh
J, Fürstenberger G, Müller-Decker K, Enk A, Arnold B, Bierhaus A,
Nawroth PP, Hess J and Angel P: RAGE signaling sustains
inflammation and promotes tumor development. J Exp Med.
205:275–285. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Hudson BI, Kalea AZ, Del Mar Arriero M,
Harja E, Boulanger E, D’Agati V and Schmidt AM: Interaction of the
RAGE cytoplasmic domain with diaphanous-1 is required for
ligand-stimulated cellular migration through activation of Rac1 and
Cdc42. J Biol Chem. 283:34457–34468. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Fukata M, Nakagawa M and Kaibuchi K: Roles
of Rho-family GTPases in cell polarisation and directional
migration. Curr Opin Cell Biol. 15:590–597. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Leve F and Morgado-Díaz JA: Rho GTPase
signaling in the development of colorectal cancer. J Cell Biochem.
113:2549–2559. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Motoyama A and Yates JR III:
Multidimensional LC separations in shotgun proteomics. Anal Chem.
80:7187–7193. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Washburn MP, Wolters D and Yates JR III:
Large-scale analysis of the yeast proteome by multidimensional
protein identification technology. Nat Biotechnol. 19:242–247.
2001. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Sakaguchi M, Murata H, Yamamoto K, Ono T,
Sakaguchi Y, Motoyama A, Hibino T, Kataoka K and Huh NH: TIRAP, an
adaptor protein for TLR2/4, transduces a signal from RAGE
phosphorylated upon ligand binding. PLoS One. 6:e231322011.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Sakaguchi M, Miyazaki M, Takaishi M,
Sakaguchi Y, Makino E, Kataoka N, Yamada H, Namba M and Huh NH:
S100C/A11 is a key mediator of Ca(2+)-induced growth
inhibition of human epidermal keratinocytes. J Cell Biol.
163:825–835. 2003.PubMed/NCBI
|
|
15
|
Huttunen HJ, Fages C and Rauvala H:
Receptor for advanced glycation end products (RAGE)-mediated
neurite outgrowth and activation of NF-kappaB require the
cytoplasmic domain of the receptor but different downstream
signaling pathways. J Biol Chem. 274:19919–19924. 1999. View Article : Google Scholar
|
|
16
|
Wang L, Li S and Jungalwala FB: Receptor
for advanced glycation end products (RAGE) mediates neuronal
differentiation and neurite outgrowth. J Neurosci Res.
86:1254–1266. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Davey GE, Murmann P and Heizmann CW:
Intracellular Ca2+ and Zn2+ levels regulate
the alternative cell density-dependent secretion of S100B in human
glioblastoma cells. J Biol Chem. 276:30819–30826. 2011.
|
|
18
|
Miyamoto Y and Yamauchi J: Cellular
signaling of Dock family proteins in neural function. Cell Signal.
22:175–182. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Premkumar L, Bobkov AA, Patel M,
Jaroszewski L, Bankston LA, Stec B, Vuori K, Côté JF and Liddington
RC: Structural basis of membrane targeting by the Dock180 family of
Rho family guanine exchange factors (Rho-GEFs). J Biol Chem.
285:13211–13222. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Zhelev DV and Alteraifi A: Signaling in
the motility responses of the human neutrophil. Ann Biomed Eng.
30:356–370. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Castoldi A, Braga TT, Correa-Costa M,
Aguiar CF, Bassi ÊJ, Correa-Silva R, Elias RM, Salvador F,
Moraes-Vieira PM, Cenedeze MA, Reis MA, Hiyane MI, Pacheco-Silva Á,
Gonçalves GM and Câmara NO: TLR2, TLR4 and the MYD88 signaling
pathway are crucial for neutrophil migration in acute kidney injury
induced by sepsis. PLoS One. 7:e375842012. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Li CW, Xia W, Huo L, Lim SO, Wu Y, Hsu JL,
Chao CH, Yamaguchi H, Yang NK, Ding Q, Wang Y, Lai YJ, LaBaff AM,
Wu TJ, Lin BR, Yang MH, Hortobagyi GN and Hung MC:
Epithelial-mesenchymal transition induced by TNF-α requires
NF-κB-mediated transcriptional upregulation of Twist1. Cancer Res.
72:1290–1300. 2012.
|