Introduction
Carcinosarcoma, formerly called spindle-cell,
pseudosarcomatous or sarcomatoid carcinoma (1), is a rare biphasic tumor characterized
by a combination of malignant epithelial and mesenchymal cell
proliferations. These tumors occur in various organs, including the
upper aerodigestive tract, gastrointestinal tract, liver, bladder,
prostate, uterus, ovary and breast, and often show an aggressive
clinical course (2–4). Of these, esophageal carcinosarcoma
(ECS) is a rare malignant neoplasm that accounts for 0.5–2.8% of
all esophageal malignancies (5).
ECS is usually composed of invasive and/or in situ squamous
cell carcinoma and sarcoma-like cells (6). However, ECS is generally thought to be
derived from a single-cell clone of epithelial cells, with the
sarcoma-like cells emerging as a subclone from the carcinoma cells
through mesenchymal metaplasia (7).
Regarding patient prognosis, investigators have
suggested that ECS often presents as a polypoid lesion protruding
into the esophageal lumen, and is detected at a relatively earlier
stage than pure squamous cell carcinoma, leading to a comparatively
good prognosis (8). Other reports,
however, have indicated similar 5-year survival rates in patients
with pure esophageal squamous cell carcinoma (ESCC) and ECS
(5).
Radical esophagectomy with lymph node dissection is
currently the standard therapy for ECS patients, and systemic
adjuvant therapies may be considered in progressive cases, as for
ESCC patients. However, chemotherapies, which generally involve the
same regimen as for ESCC, are usually insufficient to control the
growth of ECS at metastatic sites (9). Although lymph node metastases occur in
~50% of ECS cases (10),
sarcoma-predominant components preferentially metastasize to
distant organs or the peritoneum, and rarely result in lymph node
metastasis (11). These sarcomatous
components at metastatic sites may define the prognosis of patients
with ECS, since unlike carcinomas, most soft tissue sarcomas are
notoriously resistant to standard chemotherapies (12).
Overexpression of receptor tyrosine kinases (RTKs)
has recently been reported in various types of malignant tumors,
and these represent attractive molecular targets for alternative
therapies using effective and safe selective inhibitors. RTKs are
key molecules in normal cellular differentiation and proliferation,
but are commonly deregulated in various types of human cancers. RTK
inhibitors have recently been reported to be effective in the
treatment of several tumor types, including breast, lung and colon
cancer, gastrointestinal stromal tumors and renal cancers (13,14).
RTKs also play an important role in ESCC, and certain RTK
inhibitors may represent useful therapeutic strategies for
esophageal cancer (15). However,
no studies have analyzed the expression of RTKs in ECS, and their
status in these tumors thus remains poorly understood.
We previously reported variable histological and
immunohistochemical phenotypes of the sarcomatous components in ECS
cases (16), suggesting that the
expression of RTKs in ECS may differ from that in ESCC. The optimal
chemotherapeutic approach for ESC might thus also differ from the
standard therapy for ESCC.
We examined for the first time the expression
patterns and genetic alterations of various RTKs in each squamous
cell and sarcomatous component of ECS, and provides some rationale
for the administration of molecular-targeted drugs for ECS.
Materials and methods
Patient characteristics and tissue
samples
This study included 20 cases of ECS as described
previously (16), and 1 additional
case, making a total of 21 patients diagnosed with ECS at Gunma
Prefectural Cancer Center, Gunma University Hospital, Niigata
University Hospital and Jichi Medical School Hospital. These
patients included 20 surgical cases and 1 autopsy case. All the
patients were males, with a mean age of 67 years (range 51–81
years). The surgical specimens were fixed with 10% formalin and
embedded in paraffin, and 3-μm sections were prepared and stained
with hematoxylin and eosin. The diagnosis of ECS was confirmed
histologically by two pathologists. Clinical information was
obtained from medical records in all cases.
Immunohistochemistry (IHC)
Formalin-fixed paraffin-embedded tissue specimens
for each patient were cut into 3-μm sections and used for IHC. The
antibodies used in this study, as well as the dilution and
antigen-retrieval method for each antibody are listed in Table I.
 | Table IAntibodies used for
immunohistochemistry. |
Table I
Antibodies used for
immunohistochemistry.
| Antibody | Clone | Source | Dilution | Antigen
retrieval |
|---|
| KIT | mAb, Y145 | Epitomics | 1:100 | MW |
| PDGFRA | pAb | Santa Cruz
Biotechnology | 1:200 | AC |
| PDGFRB | pAb | Santa Cruz
Biotechnology | 1:400 | AC |
| MET | pAb | Santa Cruz
Biotechnology | 1:200 | AC |
| EGFR | mAb, 31G7 | Invitrogen | 1:200 | Trypsin |
| HER-2 | pAb | Dako | 1:100 | MW |
| Vimentin | mAb, V9 | Dako | 1:10 | - |
| Smooth muscle
actin | mAb, 1A4 | Dako | 1:160 | - |
| Desmin | mAb, D33 | Dako | 1:50 | - |
| S100 | pAb | Dako | 1:800 | - |
| Ki-67 | mAb, MIB-1 | Dako | 1:40 | Trypsin/MW |
The cellular differentiation of the mesenchymal
component in each ECS was characterized immunohistochemically using
the following antibodies: smooth muscle actin (α-SMA) and desmin as
markers of muscle differentiation; S100 protein as a marker of
neural differentiation or chondroid differentiation and vimentin as
a marker of mesenchymal differentiation.
The expression levels of various RTKs [KIT,
platelet-derived growth factor receptor (PDGFR)A, PDGFRB, MET,
epidermal growth factor receptor (EGFR) and HER-2] were also
examined by IHC in each epithelial and mesenchymal component of the
21 ECSs.
Tissue sections were deparaffinized with xylene and
rehydrated through decreasing concentrations of alcohol. Endogenous
peroxidase activity was blocked by immersion with 0.3% hydrogen
peroxide in absolute methanol for 30 min. After antigen retrieval,
or without antigen retrieval, the primary antibody was applied and
incubated overnight at 4°C in a high-humidity chamber. EnVision+
(Dako, Glostrup, Denmark) was used with a secondary antibody for 60
min at room temperature. The slides were incubated in
3′-diaminobenzidine tetrahydrochloride solution, counterstained
with hematoxylin and mounted. Serial sections of selected tissue
samples were immunostained in the absence of the primary antibody,
as a negative control.
The expression levels of the RTKs were evaluated
separately in the epithelial and mesenchymal components in each
case. Immunoreactivity for each antibody was quantitated by scoring
the intensity of staining (0, negative; 1+, weak; 2+, moderate; 3+,
strong), and the percentage of positive cells was calculated for
each section without reference to any clinical information. IHC was
judged to be positive when ≥5% of the tumor cells were stained
moderately (2+) to strongly (3+).
Ki-67 expression was also evaluated to assess the
proportion of proliferating cells. The percentage of Ki67-positive
nuclei among 1,000 tumor cells was evaluated and defined as the
Ki67-labeling index (LI) in each epithelial and mesenchymal
component.
Mutational analysis of c-kit, PDGFRA,
c-met and EGFR genes
Mutational analysis of previously reported hotspots
for each RTK gene was performed for all cases that were positive
for each RTK by IHC. Mutation analysis was performed as previously
described (17). Genomic DNA was
extracted from formalin-fixed, paraffin-embedded tumor tissues. The
epithelial and mesenchymal components were dissected and subjected
to proteinase K treatment in an extraction buffer (10 mmol/l Tris
HCl, pH 8.0; 1 mmol/l EDTA; and 1% Tween-20) and incubated
overnight at 62°C. Exons 9, 11, 13 and 17 of c-kit, exons 12
and 18 of PDGFRA, exon 14 of c-met and exons 19–21 of
EGFR, which were identified as mutational hot spots in
previous reports, were amplified by polymerase chain reaction. The
forward and reverse oligonucleotide primers used in this study are
listed in Table II. Nested PCR
amplification was carried out for the EGFR gene. Each of the
amplified fragments was purified from a polyacrylamide gel, and
direct sequencing was carried out using a BigDye Terminator v.3.1
cycle sequencing kit (Applied Biosystems, Foster City, CA, USA) and
an ABI PRISM 3130 DNA Sequencer (Applied Biosystems). All
sequencing reactions were carried out in forward and reverse
directions.
| Table IIOligonucleotide primers used for
direct sequence analysis. |
Table II
Oligonucleotide primers used for
direct sequence analysis.
| Gene and exons | Sequence | Fragment size
(bp) | Annealing
temperature (°C) |
|---|
| c-kit |
| Exon 9 | Forward
5′-ATGCTCTGCTTCTGTACTGCC-3′ | 238 | 55 |
| Reverse
5′-CAGAGCCTAAACATCCCCTTA-3′ | | |
| Exon 11 | Forward
5′-CCAGAGTGCTCTAATGACTG-3′ | 236 | 53 |
| Reverse
5′-ACCCAAAAAGGTGACATGGA-3′ | | |
| Exon 13 | Forward
5′-CATCAGTTTGCCAGTTGTGC-3′ | 174 | 55 |
| Reverse
5′-ACACGGCTTTACCTCCAAATG-3′ | | |
| Exon 17 | Forward
5′-TGTATTCACAGAGACTTGGC-3′ | 218 | 55 |
| Reverse
5′-GGATTTACATTATGAAAGTCACAGG-3′ | | |
| PDGFRA |
| Exon 12 | Forward
5′-TCCAGTCACTGTGCTGCTTC-3′ | 260 | 56 |
| Reverse
5′-GCAAGGGAAAAGGGAGTCTT-3′ | | |
| Exon 18 | Forward
5′-ACCATGGATCAGCCAGTCTT-3′ | 255 | 56 |
| Reverse
5′-AAGTGTGGGAGGATGAGCCTG-3′ | | |
| c-met |
| Exon 14 | Forward
5′-TTCTGGGCACTGGGTCAAAGT-3′ | 281 | 58 |
| Reverse
5′-AATGTCACAACCCACTGAGGT-3′ | | |
| EGFR |
| Exon 19 | Forward
5′-GCAATATCAGCCTTAGGTGCGGCTC-3′ | 372 | 58 |
| Reverse
5′-CATAGAAAGTGAACATTTAGGATGTG-3′ | | |
| Exon 19 (nested
PCR) | Forward
5′-CCTTAGGTGCGGCTCCACAGC-3′ | 349 | 58 |
| Reverse
5′-CATTTAGGATGTGGAGATGAGC-3′ | | |
| Exon 20 | Forward
5′-CCATGAGTACGTATTTTGAAACTC-3′ | 408 | 58 |
| Reverse
5′-CATATCCCCATGGCAAACTCTTGC-3′ | | |
| Exon 20 (nested
PCR) | Forward
5′-GAAACTCAAGATCGCATTCATGC-3′ | 379 | 58 |
| Reverse
5′-GCAAACTCTTGCTATCCCAGGAG-3′ | | |
| Exon 21 | Forward
5′-CTAACGTTCGCCAGCCATAAGTCC-3′ | 415 | 58 |
| Reverse
5′-GCTGCGAGCTCACCCAGAATGTCTGG-3′ | | |
| Exon 21 (nested
PCR) | Forward
5′-CAGCCATAAGTCCTCGACGTGG-3′ | 374 | 58 |
| Reverse
5′-CATCCTCCCCTGCATGTGTTAAAC-3′ | | |
Fluorescence in situ hybridization
(FISH)
FISH analysis was performed for all tumors
positively stained with the RTK antibodies to define the status of
the c-kit, PDGFRA, c-met and EGFR
genes. The following DNA probe mixtures were used: c-kit
(BAC clone RP11-586A2 SpectrumGreen)/CEP4 (BAC clone RP11-217B22
SpectrumOrange), PDGFRA (BAC clone RP11-231C18
SpectrumGreen)/CEP4 (BAC clone RP11-217B22 SpectrumOrange),
c-met (BAC clone RP11-163C9 SpectrumOrange)/CEP7 (BAC clone
RP11-90C3 SpectrumGreen) (Chromosome Science Labo, Sapporo, Japan)
and EGFR (LSI EGFR SpectrumOrange)/CEP7 (SpectrumGreen)
(Vysis; Abbott Laboratories, Downers Grove, IL, USA).
Representative areas of the tissue sections that
showed positive immunostaining for each RTK were selected and
trimmed for FISH. Formalin-fixed paraffin-embedded tissue was
prepared in serial 6-μm sections. After dewaxing in xylene and
dehydration in 100% ethanol, sections were immersed in 0.2 N HCl
for 20 min and incubated in 1 M NaSCN pretreatment solution (Vysis)
for 30 min at 80°C. Sections were digested with protease solution
(Vysis) for 60 min at 37°C and fixed with 10% formalin for 10 min,
denatured at 72°C for 5 min in 70% formamide/2X standard saline
citrate (SSC), and dehydrated through a series of graded ethanols.
A volume of 10 μl of the denatured DNA probe mixture was applied to
the hybridization area and covered with a glass coverslip. After
microwaving for 60 min at 40°C, the slides were hybridized at 37°C
for 48 h. Sections were washed in post-hybridization wash solution
(2X SSC, 0.3% NP-40) at 73°C for 2 min and counterstained with
4,6-diamidino-2-phenylindole (DAPI).
The signals were counted in at least 50 nuclei/slide
under ×1,000 magnification in each selected area, and the target
gene/CEP ratio was calculated. The cytogenetic patterns were
classified according to the criteria of Cappuzzo et
al(18): high polysomy (≥4
copies in ≥40% cells) and gene amplification (defined by presence
of tight gene clusters, a gene/chromosome ratio ≥2, or ≥15
copies/cell in ≥10% of analyzed cells) were considered as
FISH-positive. Disomy (≤2 copies in >90% of cells); trisomy (≤2
copies in ≥40% of cells, ≥4 copies in <10% of cells) and low
polysomy (≥4 copies in 10–40% of cells) were considered as
FISH-negative.
Statistical analysis
The significance of differences was analyzed by
applying the χ2 test or Fisher’s exact test. Differences
were considered significant when the P-value was <0.05.
Results
Clinicopathological characteristics
All cases were morphologically defined as protruded
type, type 1 according to the Japanese macroscopic classification.
Four cases also had ulcerative or infiltrative lesions.
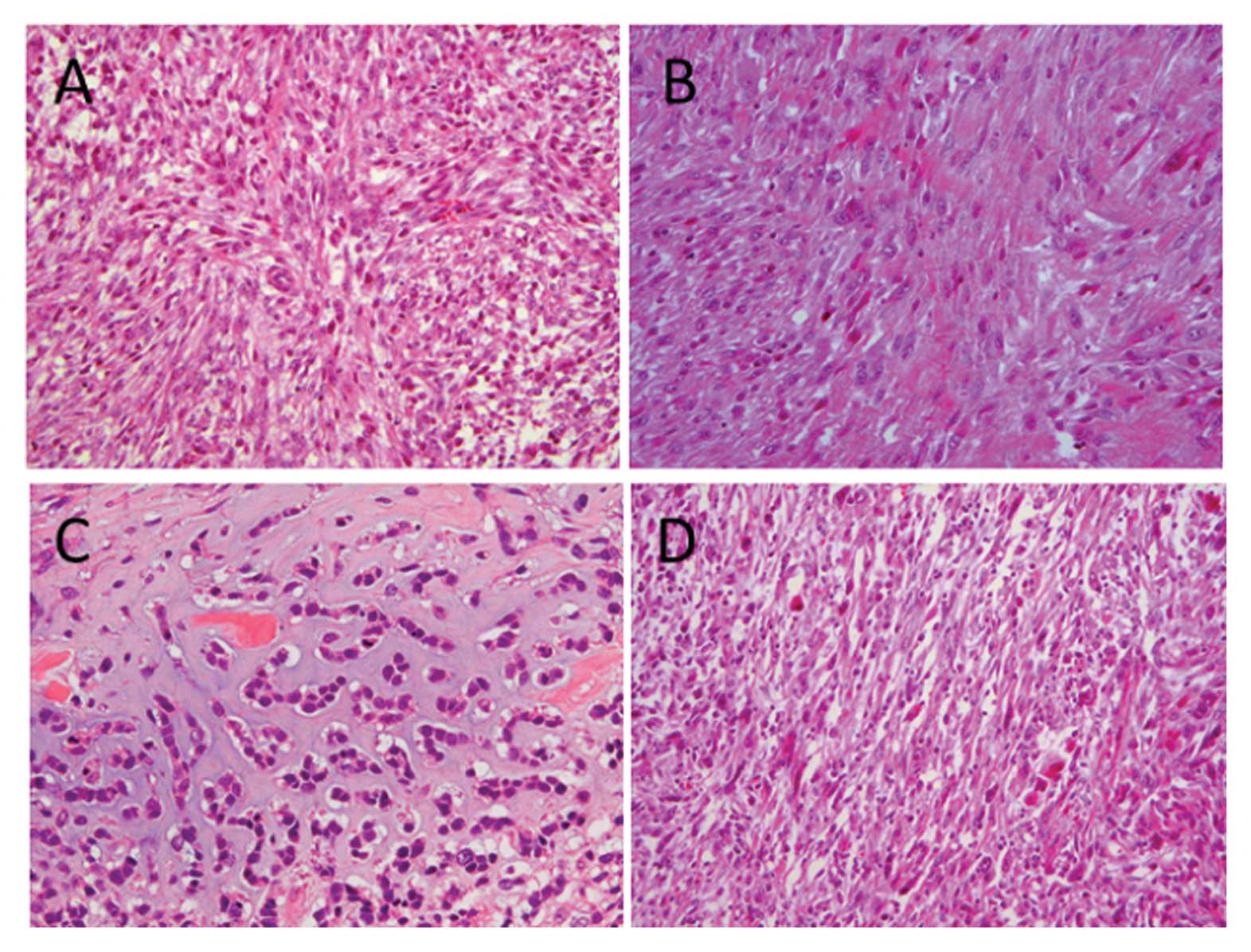
All tumors consisted microscopically of both
epithelial and mesenchymal components, although the proportions of
the components varied among the cases. The epithelial components in
all cases were squamous cell carcinoma. Histologically, the
mesenchymal components were malignant fibrous histiocytoma
(MFH)-like in 3 (Fig. 1A),
leiomyosarcoma-like in 2 (Fig. 1B)
and chondrosarcoma-like in 1 case (Fig.
1C). The remaining 15 cases were composed of pleomorphic
spindle cells (Fig. 1D).
Immunohistochemical expression of
mesenchymal markers and Ki-67
Some of the results were previously reported
(16). All histologically
classified mesenchymal components were immunohistochemically
positive for more than one mesenchymal marker. Of the 21 ECSs,
vimentin, α-SMA, desmin and S-100 were expressed in the mesenchymal
component in 19 (90.5%), 16 (76.2%), 0 and 3 cases (14.3%),
respectively. The corresponding values in the epithelial component
were 1 (4.8%), 1 (4.8%), 0 and 1 (4.8%), respectively (Table III).
| Table IIIOverexpression of receptor tyrosine
kinases and mesenchymal markers in epithelial and mesenchymal
components of the esophageal carcinosarcoma cases. |
Table III
Overexpression of receptor tyrosine
kinases and mesenchymal markers in epithelial and mesenchymal
components of the esophageal carcinosarcoma cases.
| Expression of
proteins by immunohistochemistry | |
|---|
|
| |
|---|
| Case no. | KIT | PDGFRA | PDGFRB | MET | EGFR | HER-2 | Vimentin | Mesenchymal
markers | Ki-67
LI (%) |
|---|
| 1 | E | − | − | − | + | + | − | − | - | 41.3 |
| M | − | − | − | + | − | − | + | α-SMA | 34.2 |
| 2 | E | − | − | − | + | + | − | − | - | 50.9 |
| M | − | − | − | + | + | − | + | - | 42.7 |
| 3 | E | − | − | − | + | + | − | − | - | 30.9 |
| M | − | − | − | + | − | − | + | α-SMA | 45.7 |
| 4 | E | − | − | − | − | − | − | − | - | 46.2 |
| M | − | − | − | + | + | − | + | α-SMA | 58.5 |
| 5 | E | − | − | − | + | + | + | − | - | 45.0 |
| M | − | − | − | + | + | − | + | α-SMA | 46.9 |
| 6 | E | − | − | − | + | + | − | − | - | 15.6 |
| M | − | − | − | + | + | − | + | - | 38.5 |
| 7 | E | − | − | − | − | − | − | − | - | 19.8 |
| M | − | − | − | − | + | − | + | α-SMA | 23.8 |
| 8 | E | − | − | − | − | − | − | − | - | 22.5 |
| M | + | − | − | + | + | − | + | α-SMA, S100 | 32.8 |
| 9 | E | + | − | − | + | + | − | − | S100 | 44.0 |
| M | − | − | − | − | − | − | + | α-SMA | 51.6 |
| 10 | E | − | − | − | − | + | − | − | - | 36.2 |
| M | − | − | − | + | + | − | + | α-SMA | 39.4 |
| 11 | E | − | − | − | + | + | − | − | - | 35.3 |
| M | − | − | − | + | − | − | + | α-SMA | 19.5 |
| 12 | E | − | − | − | + | + | + | − | α-SMA | 58.4 |
| M | + | − | − | + | + | − | + | α-SMA, S100 | 61.4 |
| 13 | E | − | − | − | − | − | − | − | - | 39.1 |
| M | − | − | − | − | − | − | + | - | 42.4 |
| 14 | E | − | − | − | − | − | − | − | - | 30.8 |
| M | − | + | − | + | + | − | − | α-SMA, S100 | 35.7 |
| 15 | E | − | − | − | − | − | − | − | - | 37.4 |
| M | − | + | − | − | + | − | + | - | 43.9 |
| 16 | E | − | − | − | − | − | − | − | - | 14.4 |
| M | − | − | − | + | + | − | + | α-SMA | 31.6 |
| 17 | E | − | − | − | + | + | − | − | - | 40.3 |
| M | − | − | − | − | − | − | + | - | 40.6 |
| 18 | E | − | − | − | − | + | − | − | - | 41.5 |
| M | − | − | − | − | − | − | + | α-SMA | 49.4 |
| 19 | E | − | − | − | + | + | − | − | - | 37.9 |
| M | − | − | − | − | − | − | − | α-SMA | 59.6 |
| 20 | E | − | + | − | + | − | − | + | - | 23.6 |
| M | − | − | − | − | − | − | + | α-SMA | 36.8 |
| 21 | E | − | − | − | − | + | − | − | - | 40.9 |
| M | − | − | − | − | − | − | + | α-SMA | 20.1 |
| Total | | 3 (14.3%) | 3 (14.3%) | 0 (0%) | 16 (76.2%) | 19 (90.5%) | 2 (9.5%) | 19 (90.5%) | 16 (76.2%) | |
The Ki-67 LI in the epithelial component ranged from
15.6 to 58.4%, while that in the mesenchymal component ranged from
19.5 to 61.4% (Table III). In 7
cases (case nos. 3, 4, 6, 8, 16, 19 and 20), the Ki-67 LI of the
mesenchymal component was ≥10% higher than that of the epithelial
component, whereas the Ki-67 LI of the mesenchymal component was
≥10% lower in case nos. 11 and 21. There was no significant
difference in average Ki-67 LI between the epithelial and
mesenchymal components.
Immunohistochemical expression of
receptor tyrosine kinases
The immunohistochemical expression of the various
RTKs is summarized in Table III.
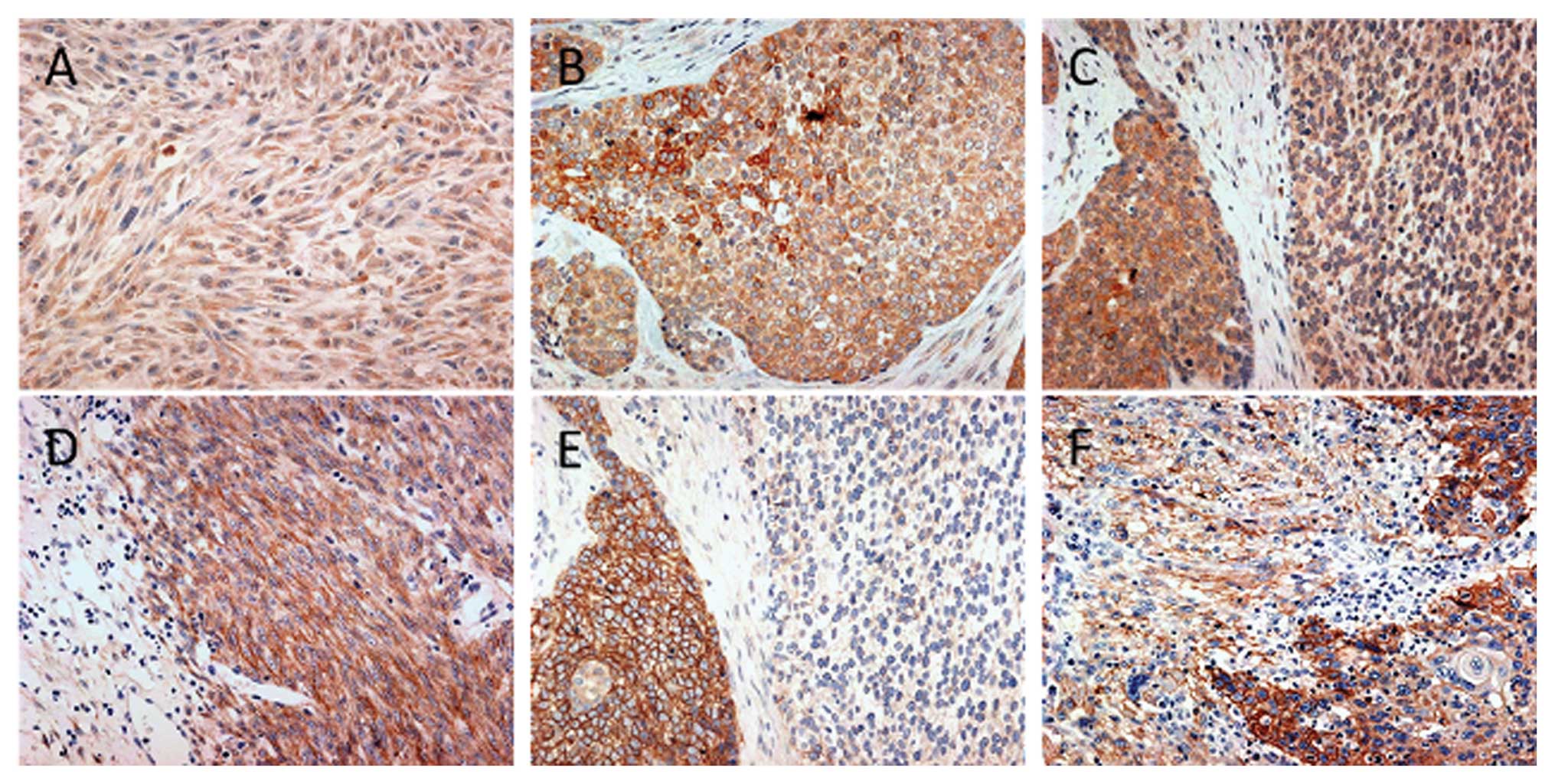
Representative IHC results for MET and EGFR are shown in Fig. 2.
Normal esophageal epithelium adjacent to the tumor
tissue was negative for all RTKs and was scored as 0. Among the 21
ECSs, KIT overexpression was observed in 3 (14.3%) cases, including
in the mesenchymal component in 2 cases (case no. 8 and 12) and the
epithelial component in 1 case (case no. 9). PDGFRA overexpression
was detected in 3 (14.3%), including 2 in the mesenchymal component
(case nos. 14 and 15) and one in the epithelial component (case no.
20). None of the 21 tumors showed PDGFRB overexpression.
Overexpression of MET was detected in 16 of the 21 cases (76.2%).
This was limited to the mesenchymal components in 5 cases (case
nos. 4, 8, 10, 14 and 16), to the epithelial component in 4 cases
(case nos. 9, 17, 19 and 20) and occurred in both the epithelial
and mesenchymal components in 7 cases (case nos. 1, 2, 3, 5, 6, 11
and 12). EGFR overexpression was detected in 19 of 21 cases
(90.5%), restricted to the mesenchymal component in 6 cases (case
nos. 4, 7, 8, 14, 15 and 16), the epithelial component in 8 cases
(case nos. 1, 3, 9, 11, 17, 18, 19 and 21), and to both the
epithelial and mesenchymal components in 5 cases (case nos. 2, 5,
6, 10 and 12). HER-2 overexpression was observed in the epithelial
component in 2 cases (9.5%).
Among the 21 ECSs, MET and/or EGFR were co-expressed
in 15 cases (71.4%). Of these, 4 showed co-overexpression of MET
and EGFR in both the epithelial and mesenchymal components (case
nos. 2, 5, 6 and 12), 6 only in the epithelial component (case nos.
1, 3, 9, 11, 17 and 19) and 5 only in the mesenchymal component
(case nos. 4, 8, 10, 14 and 16).
There was no correlation between overexpression of
any RTK with clinicopathological factors.
Mutational analysis of c-kit, PDGFRα,
c-met and EGFR genes
No mutations were found in any of the analyzed exons
of the c-kit, PDGFRα and c-met genes.
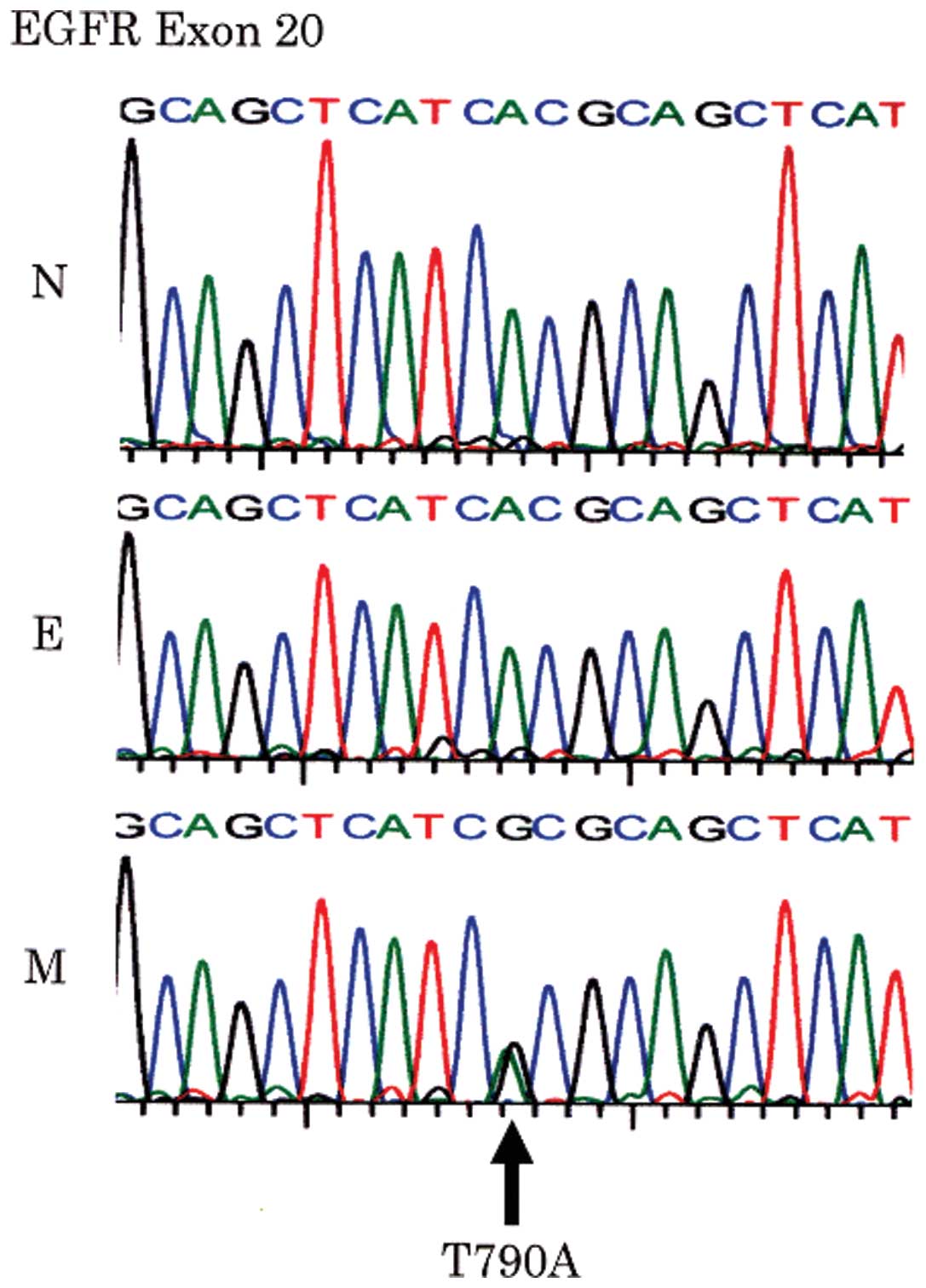
The same missense point mutation at codon 790 (ACG
to GCG) of the EGFR gene exon 20 was found in 2 of the 19
EGFR-positive ESCs (case nos. 15 and 18), resulting in substitution
of threonine by alanine (T790A). These missense mutations were only
observed in the mesenchymal components in both cases (Fig. 3). They were not detected in normal
squamous epithelium from the same patients, and were therefore
considered to be somatic mutations.
Status of c-kit, PDGFRα, c-met and EGFR
genes by FISH analysis
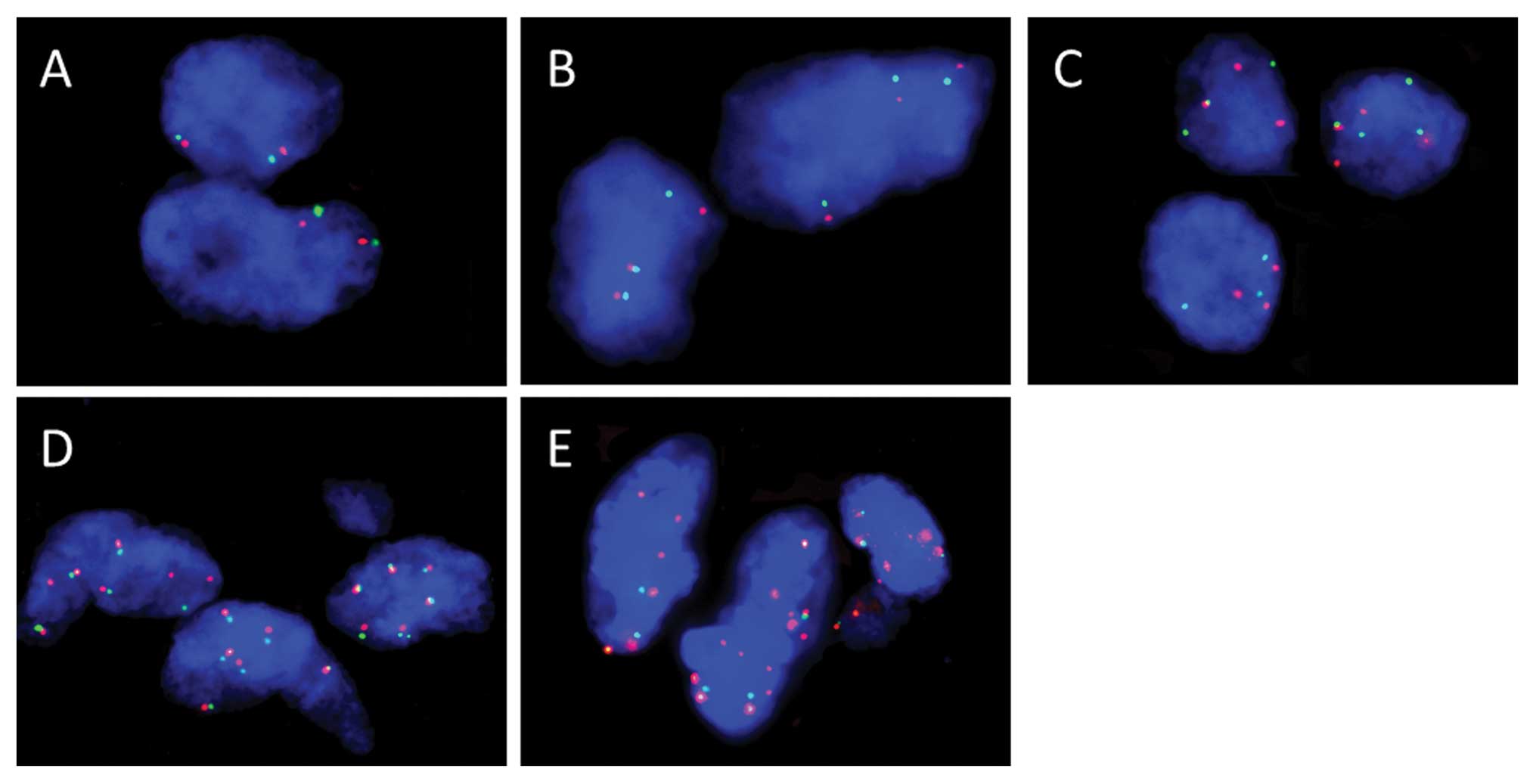
Representative results of FISH analysis of the
EGFR gene in ECS tissue samples are shown in Fig. 4. Gene status by FISH analysis is
shown in Table IV. Among the 3
cases with KIT overexpression, case no. 12 was FISH-positive (high
polysomy) and the remaining 2 cases were FISH-negative (low
polysomy in case no. 9 and disomy in case no. 8). All 3 patients
with PDGFRA overexpression were FISH-negative (trisomy in case no.
15 and disomy in case no. 14 and 20). In the 16 cases with MET
overexpression, 3 cases were FISH-positive (high polysomy in case
nos. 5, 6 and 11), and the rest were FISH-negative (low polysomy in
case nos. 9, 16, 19 and 20, trisomy in case nos. 1, 3, 12 and 17,
and disomy in case nos. 2, 4, 8, 10 and 14). Among the 19 cases
with EGFR overexpression, 10 were FISH-positive (gene amplification
in case nos. 2 and 9, and high polysomy in case nos. 3–6, 12, 16,
17 and 19), and the rest were FISH-negative (low polysomy in case
nos. 7 and 21, trisomy in case nos. 1, 10 and 11, and disomy in
case nos. 8, 14, 15 and 18).
| Table IVGene status of receptor tyrosine
kinases by fluorescence in situ hybridization. |
Table IV
Gene status of receptor tyrosine
kinases by fluorescence in situ hybridization.
| Gene status | c-kit
(n=3) | PDGFRA
(n=3) | c-met
(n=16) | EGFR
(n=19) |
|---|
| FISH-positive | Gene
amplification | 0 | 0 | 0 | 2 |
| High polysomy | 1 | 0 | 3 | 8 |
| FISH-negative | Low polysomy | 1 | 0 | 4 | 2 |
| Trisomy | 0 | 1 | 4 | 3 |
| Disomy | 1 | 2 | 5 | 4 |
There was no significant correlation between the
results of FISH and any clinicopathological factors.
Discussion
We previously demonstrated variable histological and
immunohistochemical phenotypes in the mesenchymal components of
ECSs, including MFH-like, leiomyosarcoma-like, and
chondrosarcoma-like features. The proliferative activity of tumor
cells, assessed by Ki-67 LI, also varied between cases, with the
mesenchymal component tending to show higher proliferation than the
epithelial component in each case. These results seem to be
compatible with the idea that the mesenchymal component develops by
transition from squamous cell differentiation to mesenchymal
differentiation, and plays an important role in tumor
progression.
In addition to these findings, the present study
also demonstrated that various RTKs were overexpressed in tumor
cells in ECS, with MET and EGFR especially being highly
co-expressed in most ECSs.
Overexpression of MET and/or alteration of the
c-met gene has been reported in a wide variety of tumors,
including carcinomas and sarcomas (19). The MET oncogene can be activated by
overexpression, gene rearrangements, or mutations in tumor cells,
resulting in tumor development and progression (19,20).
Previous studies have reported overexpression of MET
in up to 54% of esophageal adenocarcinomas (EAs) (21) and 92% of ESCCs (22), and expression levels are thought to
correlate with tumor development, progression, and prognosis in
patients with EA and ESCC (21–23).
However, there have been no reports of MET expression in ECS. The
present study demonstrated overexpression of MET in 76.2% of ECSs,
an intermediate percentage between EA and ESCC. Three of those
cases showed increased copy numbers of the c-met gene by
FISH analysis.
EGFR overexpression has been reported in 33–50% of
ESCCs (24,25) and 55% of EAs (26). Amplification of the EGFR gene
has also been reported in ~30% of ESCCs and 6–11% of EA cases
(25,27). The rate of EGFR overexpression in
ECS in the present study was much higher (19 of 21 cases, 90.5%),
and associated with amplification or high polysomy of the
EGFR gene. Furthermore, 2 of the 19 cases had the same
missense point mutation (T790A) in EGFR exon 20 restricted
to the mesenchymal component of ECS. Codon 790 in the EGFR
gene is a mutational hotspot for secondary resistance to gefitinib
in non-small cell lung cancer (28).
EGF/EGFR signaling pathways have recently been
reported to induce cancer cell epithelial-mesenchymal transition
(EMT) via STAT3-mediated TWIST gene expression (29), upregulation of Snail (30) and loss of E-cadherin and increased
invasion of cancer cells (31).
Snail-associated EMT has been reported to promote tumor
invasiveness, migration and proliferative activity in ESCC
(32). Dysregulated MET/HGF
signaling is also correlated with tumor proliferation and survival,
increased cell motility and migration, tumor invasion and
metastasis (33). MET/HGF signaling
recruits and activates c-Src, which subsequently phosphorylates
E-cadherin resulting in Numb dissociation from phosphorylated
E-cadherin, and several downstream signaling pathways participate
in the reduction of cell-cell adhesion, cell proliferation and cell
migration, i.e., EMT (34).
The present study identified a high frequency of MET
and EGFR co-expression in ECS (15 of 21 cases, 71%). A recent
experimental study indicated that mutant p53 and EGFR expression
potentiated HGF/MET signaling (35). The present and previous results
suggest that co-expression of MET and EGFR may play a key role in
mesenchymal sarcomatous metaplasia of squamous cell carcinoma
through mechanisms such as EMT in other type of carcinomas, with
subsequent tumor progression.
Regarding chemotherapy for esophageal cancer,
cisplatin/5-fluorouracil (5-FU) is the accepted standard treatment.
The effectiveness of combination chemotherapies such as
5-FU/nedaplatin (a third-generation platinum),
docetaxel/cisplatin/5-FU, and paclitaxel/cisplatin/5-FU has been
reported in recent years (36).
Furthermore, several molecular-targeted therapies have been
assessed for advanced esophageal cancer, including monoclonal
antibodies and signal transduction/tyrosine kinase inhibitors for
EGFR, HER2/neu receptor, vascular endothelial growth factor ligand,
cyclooxygenase-2 inhibitors and other novel drugs (36–39).
Of these trials, gefitinib appeared to have no activity in EA,
whereas limited activity was observed in patients with squamous
cell carcinoma (37). Phase II
trials of erlotinib reported activity in ESCC (38) and phase II trials of combinations of
EGFR-targeted monoclonal antibodies with chemotherapy, such as
FOLFIRI (leucovorin/5-FU/irinotecan) with cetuximab, are now
underway (39).
However, metastatic ECSs have been reported to
respond poorly to conventional chemotherapy and radiation, probably
due to the sarcomatous differentiation of tumor cells. New
treatment options for ECS patients, therefore, need to be
investigated. The results of this study suggest that
molecular-targeting therapies directed to MET and EGFR may be
effective in inhibiting the growth or progression of the epithelial
and/or mesenchymal components of ECS. Further investigations are
warranted to establish the rationale for the use of such
molecular-targeting therapies for this highly malignant cancer
type.
Acknowledgements
This study was supported, in part, by a Grant-in-Aid
for The Japanese Society of Strategies for Cancer Research and
Therapy. We thank Ms. Masako Saito, Mr. Toshiaki Hikino and Mr.
Futoshi Hara (Department of Diagnostic Pathology, Gunma University
Graduate School of Medicine, Maebashi, Japan) for their excellent
technical assistance with IHC and mutation analysis and Dr Tadashi
Hasegawa and Ms. Tomomi Inoue (Department of Surgical Pathology,
Sapporo Medical University School of Medicine, Sapporo, Japan) for
help with the FISH procedures.
References
|
1
|
Gabbert HE, Shimoda T, Hainault P,
Nakamura Y, Field JK and Inoue H: Squamous cell carcinoma of the
esophagus. Tumors of the Digestive System. Hamilton SR and Aaltonen
LA: IARC Press; Lyon, France: pp. 11–19. 2000
|
|
2
|
Perez N, Castillo M, Santos Y, et al:
Carcinosarcoma of the prostate: two cases with distinctive
morphologic and immunohistochemical findings. Virchows Arch.
446:511–516. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Cimbaluk D, Rotmensch J, Scudiere J, Gown
A and Bitterman P: Uterine carcinosarcoma: immunohistochemical
studies on tissue microarrays with focus on potential therapeutic
targets. Gynecol Oncol. 105:138–144. 2007. View Article : Google Scholar
|
|
4
|
Esses KM, Hagmaier RM, Blanchard SA,
Lazarchick JJ and Riker AI: Carcinosarcoma of the breast: two case
reports and review of the literature. Cases J. 2:152009. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Iyomasa S, Kato H, Tachimori Y, Watanabe
H, Yamaguchi H and Itabashi M: Carcinosarcoma of the esophagus: a
twenty-case study. Jpn J Clin Oncol. 20:99–106. 1990.PubMed/NCBI
|
|
6
|
Kuhajda FP, Sun TT and Mendelsohn G:
Polypoid squamous carcinoma of the esophagus. A case report with
immunostaining for keratin. Am J Surg Pathol. 7:495–499. 1983.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Wang ZY, Itabashi M, Hirota T, Watanabe H
and Kato H: Immunohistochemical study of the histogenesis of
esophageal carcinosarcoma. Jpn J Clin Oncol. 22:377–386.
1992.PubMed/NCBI
|
|
8
|
Ziauddin MF, Rodriguez HE, Quiros ED,
Connolly MM and Podbielski FJ: Carcinosarcoma of the esophagus -
pattern of recurrence. Dig Surg. 18:216–218. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Iwaya T, Maesawa C, Uesugi N, et al: True
carcinosarcoma of the esophagus. Dis Esophagus. 19:48–52. 2006.
View Article : Google Scholar
|
|
10
|
Iascone C and Barreca M: Carcinosarcoma
and pseudosarcoma of the esophagus: two names, one disease -
comprehensive review of the literature. World J Surg. 23:153–157.
1999. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Kawano S, Kusunoki R, Aimi M, et al: A
case of carcinosarcoma of the esophagus treated by
chemoradiotherapy. Nippon Shokakibyo Gakkai Zasshi. 104:535–541.
2007.(In Japanese).
|
|
12
|
Donato Di Paola E and Nielsen OS; EORTC
Soft Tissue and Bone Sarcoma Group. The EORTC soft tissue and bone
sarcoma group. European Organisation for Research and Treatment of
Cancer. Eur J Cancer. 38(Suppl 4): S138–S141. 2002.
|
|
13
|
Campbell L, Blackhall F and Thatcher N:
Gefitinib for the treatment of non-small-cell lung cancer. Expert
Opin Pharmacother. 11:1343–1357. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Norum J, Nieder C and Kondo M: Sunitinib,
sorafenib, temsirolims or bevacizmab in the treatment of metastatic
renal cell carcinoma: a review of health economic evaluations. J
Chemother. 22:75–82. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Ferry DR, Anderson M, Beddard K, et al: A
phase II study of gefitinib monotherapy in advanced esophageal
adenocarcinoma: evidence of gene expression, cellular, and clinical
response. Clin Cancer Res. 13:5869–5875. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Sano A, Sakurai S, Kato H, et al:
Clinicopathological and immunohistochemical characteristics of
esophageal carcinosarcoma. Anticancer Res. 29:3375–3380.
2009.PubMed/NCBI
|
|
17
|
Saito K, Sakurai S, Sano T, et al:
Aberrant methylation status of known methylation-sensitive CpG
islands in gastrointestinal stromal tumors without any correlation
to the state of c-kit and PDGFRA gene mutations and their
malignancy. Cancer Sci. 99:253–259. 2008. View Article : Google Scholar
|
|
18
|
Cappuzzo F, Hirsch FR, Rossi E, et al:
Epidermal growth factor receptor gene and protein and gefitinib
sensitivity in non-small-cell lung cancer. J Natl Cancer Inst.
97:643–655. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Eder JP, Vande Woude GF, Boerner SA and
LoRusso PM: Novel therapeutic inhibitors of the c-Met
signaling pathway in cancer. Clin Cancer Res. 15:2207–2214. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Jeffers M, Schmidt L, Nakaigawa N, et al:
Activating mutations for the met tyrosine kinase receptor in human
cancer. Proc Natl Acad Sci USA. 94:11445–11450. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Tuynman JB, Lagarde SM, Ten Kate FJ,
Richel DJ and van Lanschot JJ: Met expression is an independent
prognostic risk factor in patients with oesophageal adenocarcinoma.
Br J Cancer. 98:1102–1108. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Hu YC, Lam KY, Law S, Wong J and
Srivastava G: Profiling of differentially expressed cancer-related
genes in esophageal squamous cell carcinoma (ESCC) using human
cancer cDNA arrays: overexpression of oncogene MET correlates with
tumor differentiation in ESCC. Clin Cancer Res. 7:3519–3525.
2001.
|
|
23
|
Ren Y, Cao B, Law S, et al: Hepatocyte
growth factor promotes cancer cell migration and angiogenic factors
expression: a prognostic marker of human esophageal squamous cell
carcinomas. Clin Cancer Res. 11:6190–6197. 2005. View Article : Google Scholar
|
|
24
|
Kawaguchi Y, Kono K, Mimura K, et al:
Targeting EGFR and HER-2 with cetuximab- and trastuzumab-mediated
immunotherapy in oesophageal squamous cell carcinoma. Br J Cancer.
97:494–501. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Hanawa M, Suzuki S, Dobashi Y, et al: EGFR
protein overexpression and gene amplification in squamous cell
carcinomas of the esophagus. Int J Cancer. 118:1173–1180. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Langer R, Von Rahden BH, Nahrig J, et al:
Prognostic significance of expression patterns of c-erbB-2, p53,
p16INK4A, p27KIP1, cyclin D1 and epidermal growth factor receptor
in oesophageal adenocarcinoma: a tissue microarray study. J Clin
Pathol. 59:631–634. 2006. View Article : Google Scholar
|
|
27
|
Rygiel AM, Milano F, Ten Kate FJ, et al:
Gains and amplifications of c-myc, EGFR, and 20.q13 loci in the no
dysplasia-dysplasia-adenocarcinoma sequence of Barrett’s esophagus.
Cancer Epidemiol Biomarkers Prev. 17:1380–1385. 2008.PubMed/NCBI
|
|
28
|
Bell DW, Gore I, Okimoto RA, et al:
Inherited susceptibility to lung cancer may be associated with the
T790M drug resistance mutation in EGFR. Nat Genet. 37:1315–1316.
2005. View
Article : Google Scholar : PubMed/NCBI
|
|
29
|
Lo HW, Hsu SC, Xia W, et al: Epidermal
growth factor receptor cooperates with signal transducer and
activator of transcription 3 to induce epithelial-mesenchymal
transition in cancer cells via up-regulation of TWIST gene
expression. Cancer Res. 67:9066–9076. 2007. View Article : Google Scholar
|
|
30
|
Lee MY, Chou CY, Tang MJ and Shen MR:
Epithelial-mesenchymal transition in cervical cancer: correlation
with tumor progression, epidermal growth factor receptor
overexpression, and snail up-regulation. Clin Cancer Res.
14:4743–4750. 2008. View Article : Google Scholar
|
|
31
|
Lu Z, Ghosh S, Wang Z and Hunter T:
Downregulation of caveolin-1 function by EGF leads to the loss of
E-cadherin, increased transcriptional activity of beta-catenin, and
enhanced tumor cell invasion. Cancer Cell. 4:499–515. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Usami Y, Satake S, Nakayama F, et al:
Snail-associated epithelial-mesenchymal transition promotes
oesophageal squamous cell carcinoma motility and progression. J
Pathol. 215:330–339. 2008. View Article : Google Scholar
|
|
33
|
Ma PC, Maulik G, Christensen J and Salgia
R: c-Met: structure, functions and potential for therapeutic
inhibition. Cancer Metastasis Rev. 22:309–325. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Wang Z and Li SSC: Numb: A new player in
EMT. Cell Adh Migr. 4:176–179. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Grugan KD, Miller CG, Yao Y, et al:
Fibroblast-secreted hepatocyte growth factor plays a functional
role in esophageal squamous cell carcinoma invasion. Proc Natl Acad
Sci USA. 107:11026–11031. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Ilson DH: Esophageal cancer chemotherapy:
recent advances. Gastrointest Cancer Res. 2:85–92. 2008.
|
|
37
|
Janmaat ML, Gallegos-Ruiz MI, Rodriguez
JA, et al: Predictive factors for outcome in a phase II study of
gefitinib in second-line treatment of advanced esophageal cancer
patients. J Clin Oncol. 24:1612–1619. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Tew WP, Shah M, Schwartz G, et al: Phase
II trial of erlotinib for second line treatment in advanced
esophageal cancer. In: American Society of Clinical Oncology
Gastrointestinal Cancers Symposium; January 27–29, 2005; abs 5.
2005
|
|
39
|
Pinto C, Di Fabio F, Siena S, et al: Phase
II study of cetuximab in combination with FOLFIRI in patients with
untreated advanced gastric or gastroesophageal junction
adenocarcinoma (FOLCETUX study). Ann Oncol. 18:510–517. 2007.
View Article : Google Scholar : PubMed/NCBI
|