Introduction
ARHI, first described in 1999 by Yu et al
using differential display PCR (1),
is a maternally imprinted tumor-suppressor gene located on
chromosome 1p31 that encodes a 26-kDa small GTP-binding protein
sharing 54–62% amino acid homology with Ras and Rap (2,3).
Rather than acting as a proto-oncogene similar to ras and rap, ARHI
is responsible for inducing apoptosis and inhibiting cancer cell
growth and motility (4,5). It has been reported that ARHI is
expressed consistently in primary ovarian and breast epithelial
cells but is dramatically downregulated in the majority of ovarian,
breast, and even pancreatic and hepatocellular cancer cells
(6). Subsequently, loss of
heterozygosity (LOH) of ARHI was found in ~40% of ovarian cancer
cells. Multiple factors appear to contribute to the abnormal gene
conversion (2). Although the
definite molecular pathogenesis of LOH has not been elucidated,
oxidative stress is suggested to be associated with the
LOH-mediated carcinogenesis of ovarian cancer (7). On the other hand, promoter
methylation-induced ARHI silencing is also critical to
downregulated expression of ARHI.
Three potential CpG islands in the ARHI gene of ~300
bp each have been identified by genomic structure analysis. Two of
them (CpG islands I and II) are located within the promoter and
adjacent exon 1 of the ARHI gene, while CpG island III is located
in the protein-encoding region of exon 2 (8). Accumulating evidence indicates that
the promoter methylation status is closely related to reduced ARHI
expression and malignant proliferation of ovarian cancer. Feng
et al(9) reported that ARHI
CpG islands I and II are hypermethylated in 31 and 12% of ovarian
cancers, respectively. However, how ARHI is methylated and
regulated requires further investigation.
STAT3, signal transducer and activator of
transcription 3, a well-known transcription activator for many
genes, has been reported to inhibit gene expression. The importance
of phosphorylation in regulating STAT3 functions has been the
subject of intense scrutiny; at the same time, acetylation of STAT3
in mediating cancer progression is gradually surfacing and has
attracted increased attention (10–12).
STAT3 has been shown to increase methylation of CpG islands in
certain tumor-suppressor genes through regulation of the expression
and interaction with DNA methyltransferase 1 (DNMT1), which plays
key roles in maintenance of the methylation status as well as
inhibition of tumor-suppressor genes through aberrant CpG island
methylation. Thus, dysregulation of STAT3 could relate, in part, to
loss of function of critical tumor-suppressor genes, which may
exaggerate the malignant proliferation and invasiveness of ovarian
cancer cells.
To date, ovarian cancer still ranks third in
incidence and first in mortality among all gynecological
malignancies worldwide, of which epithelial ovarian cancer (EOC),
including endometrioid adenocarcinoma (EAC), clear cell carcinoma
(CCC) and other types, is the leading cause of cancer-related death
in women, with a 5-year survival rate less than 20% (13,14).
Therefore, understanding the underlying mechanisms of cellular
carcinogenesis and apoptosis regulation are of paramount
importance. Our study was carried out to investigate the specific
functions of ARHI and its methylation in ovarian cancer cell
proliferation. Furthermore, we examined the possible role of
acetylated STAT3 in modulating the expression of ARHI and its
methylation, which may be an important therapeutic strategy for
treating ovarian cancer.
Materials and methods
Cell lines, tissue samples and
regents
The human ovarian cancer cell lines (SKOV3 and
HO-8910) were obtained from the American Type Cell Culture
Collection (ATCC, Manassas, VA, USA), and early passages of normal
human ovarian epithelial cell lines (NOE095 and HOSEpiC) were
purchased from ScienCell Research Laboratories (San Diego, CA, USA)
and deemed free of mycoplasma and bacterial contaminants. The SKOV3
and HO-8910 cells were maintained in RPMI-1640 medium supplemented
with 10% heat-inactivated FBS at 37°C with 5% CO2, while
the NOE095 and HOSEpiC cells were cultivated in Ovarian Epithelial
Cell Medium (OEpiCM) obtained from ScienCell Research Laboratories
under the same conditions. Furthermore, an HO-8910 cell line
harboring the endogenous STAT3 K685R mutation was established as
previously described (15) using a
homologous recombination-mediated knock-in strategy.
Twenty pairs of matched ovarian cancers and normal
tissues from the same patients (mean age 38±7 years) were obtained
from the Department of Gynecology and Obstetrics, at General
Hospital of PLA, between January 2011 and April 2011. All tissues
and brushings were fresh-frozen and stored at −80°C. The present
study was approved and monitored by the Ethics Committee of the
General Hospital of PLA.
5-Aza-2′-deoxycytidine (5-Aza), resveratrol and
trichostatin were purchased from Sigma-Aldrich, and administered to
the cell cultures at 0.5–1 μM, 500 ng/ml and 10 μM,
respectively.
Immunohistochemical staining
Paraffin-embedded normal ovarian epithelium tissues
and ovarian cancer tissues were sectioned and mounted on
polylysine-coated slides, dewaxed in xylene and rehydrated using a
graded ethanol series, before being washed three times with
phosphate-buffered saline (PBS). After initial deparaffinization,
endogenous peroxidase activity was blocked using 0.3% hydrogen
peroxide, and the sections were steamed in 1X Diva Decloaker
(Biocare Medical, Concord, CA, USA) for 1 h to restore latent
epitopes. The slides were then incubated with an anti-ARHI antibody
(C-12) obtained from Santa Cruz Biotechnology, Inc. (Santa Cruz,
CA, USA) at a final concentration of 15 μg/ml for 3 h at 37°C.
Thorough washing in 0.01 mol/l PBS was then performed. The slides
were incubated for 30 min with each biotin-labeled secondary
antibody, and then with streptavidin/peroxidase. Subsequently,
after administration of diaminobenzidine substrate, the tissue
sections were lightly counterstained with hematoxylin for ~40 sec
and then fixed with neutral balata for observation.
Real-time RT-PCR
RT-PCR was performed to investigate the expression
of ARHI. Total RNA of all normal ovarian epithelial cells (NOE095
and HOSEpiC) and cancer cells (SKOV3 and HO-8910) as well as normal
and cancerous tissues were extracted and purified using an RNeasy
Mini kit according to the manufacturer’s instructions (Qiagen,
Santa Clarita, CA, USA). After assessing RNA purity, 5 μg of RNA
was used as the template for PCR. ARHI primers (sense
5′-TCTGCCCGCCCTGCTTAT-3′ and antisense 5′-TTGCCGTCGCCACTCTTG-3′)
were used with GAPDH primers (sense 5′-GCCAAAAGGGTCATCATCTC-3′ and
antisense 5′-GTAGAGGCAGGGATGATGTTC-3′) as the internal control.
Amplification cycles consisted of 94°C for 3 min, then 33 cycles at
94°C for 1 min, 58°C for 1 min, and 72°C for 1.5 min, followed by
72°C for 15 min. Aliquots of the PCR products were electrophoresed
on 1.5% agarose gels, and PCR fragments were visualized by ethidium
bromide staining.
Methylation analysis of the ARHI CpG
islands
Pyro-sequencing analysis performed as previously
described (16) was used to measure
the ARHI promoter CpG I and II methylation status in all cell
lines. Briefly, genomic DNA was extracted from the cell lines using
the QIAamp DNA Mini kit (Qiagen) and treated with bisulfite to
convert all the unmethylated cytosines to uracils while conserving
all the methylated cytosines. After treatment, 1-μl aliquots were
amplified by PCR. The pyrosequencing primers were as follows:
CpGI-F, 5′-GTAAGGGAGAAAGAAGTTAGA-3′ and CpGI-R (50-Biotin),
5′-biotin-TA CTATCCTAACAAAACCCTC-3′; CpGII-F,
5′-GTTGGGTTAGTTTTTTATAGTTGGTT-3′ and CpGII-R (50-Biotin),
5′-biotin-AACCAAACAACCTAAAAAACAAATAC-3′.
The biotinylated PCR products were purified and
subjected to pyrosequencing using the PSQHS 96A Pyrosequencing
System and Pyro Gold reagents (PSQ 96MA; Biotage, Charlottesville,
VA, USA). A methylation density cut-off point of 15% was considered
significant.
Western blotting
Expression of Ac-STAT3 in the HOSEpiC and HO-8910
cell lines was analyzed by western blotting using commercially
available antibodies from Santa Cruz Biotechnology. The cell
lysates were subjected to sodium dodecyl sulfate (SDS)-PAGE and
subsequently transferred to a PVDF membrane. Blots were visualized
using Amersham western blot detection reagent (GE Healthcare).
Cell proliferation assay
The cells were seeded in 96-well plates
(5×104/well) and incubated with 5-Aza (0.5–1 μM) for
three days. The culture medium was changed daily to maintain the
desired concentration of drugs. The cell proliferation assay was
performed using the CyQuant® NF Cell Assay kit
(Invitrogen, Carlsbad, CA, USA) following the manufacturer’s
instructions. Cell viability was measured at a wavelength of 490
nm.
Chromatin immunoprecipitation
ChIP assays were performed as previously described
(17) with minor modifications.
Briefly, chromatin samples isolated from HO-8910 cells and the
STAT3 mutant were sonicated to shear the DNA to an average length
of 200–500 bp. Samples were centrifuged and resuspended in dilution
buffer, then incubated overnight, at 4°C with primary antibody
against STAT3 or DNMT1 (3 mg/ml). Protein agarose beads were added
to isolate the immune complexes. After being washed from the
agarose beads, the DNA-protein crosslinks in the immunopreciptated
chromatin complexes were reversed by heating at 65°C overnight, and
the DNA was eluted. The ARHI DNA in the immunoprecipitates was then
detected by PCR using gene-specific primers. Similarly, input was
prepared by treating aliquots of chromatin with proteinase K,
heated at 65°C for 6 h for decrosslinking and the resulting DNA was
subjected to RT-PCR for amplification. The primers for ARHI
amplification were described above. The PCR products were analyzed
by electrophoresis on 2% agarose gels. The signals were normalized
as the amount of ARHI DNA in the immunoprecipitate/amount of DNA
input.
Immunoprecipitation
Immunoprecipitation assays were performed as
previously described (18). Control
HO-8910 cells and the mutant STAT3 cells (1×106) were
incubated for 24 h and then treated with lysis buffer. For
immunoprecipitation, 3 mg of anti-Stat3 or anti-NMT1 antibody was
added to 2 mg of protein lysate. After overnight incubation at 4°C,
protein-G beads were added and incubated for 1.5 h at 4°C. After
washing with wash buffer, the immunoprecipitated proteins were
eluted for western blotting.
Statistical analysis
Data are expressed as means ± SD. All statistical
analyses were carried out using SPSS 13.0 (SPSS Inc., Chicago, IL,
USA). Statistical comparisons were performed by one-way analysis of
variance followed by the Student-Newman-Keuls test. P<0.05 was
considered to indicate a statistically significant result.
Results
ARHI expression is downregulated in
ovarian cancer cells
ARHI has been identified as a tumor-suppressor gene
and is of significant importance in modulating cell growth and
apoptosis (19). Consistent with
previously conducted epidemiological studies, we found that ARHI
expression was strikingly decreased in the majority of ovarian
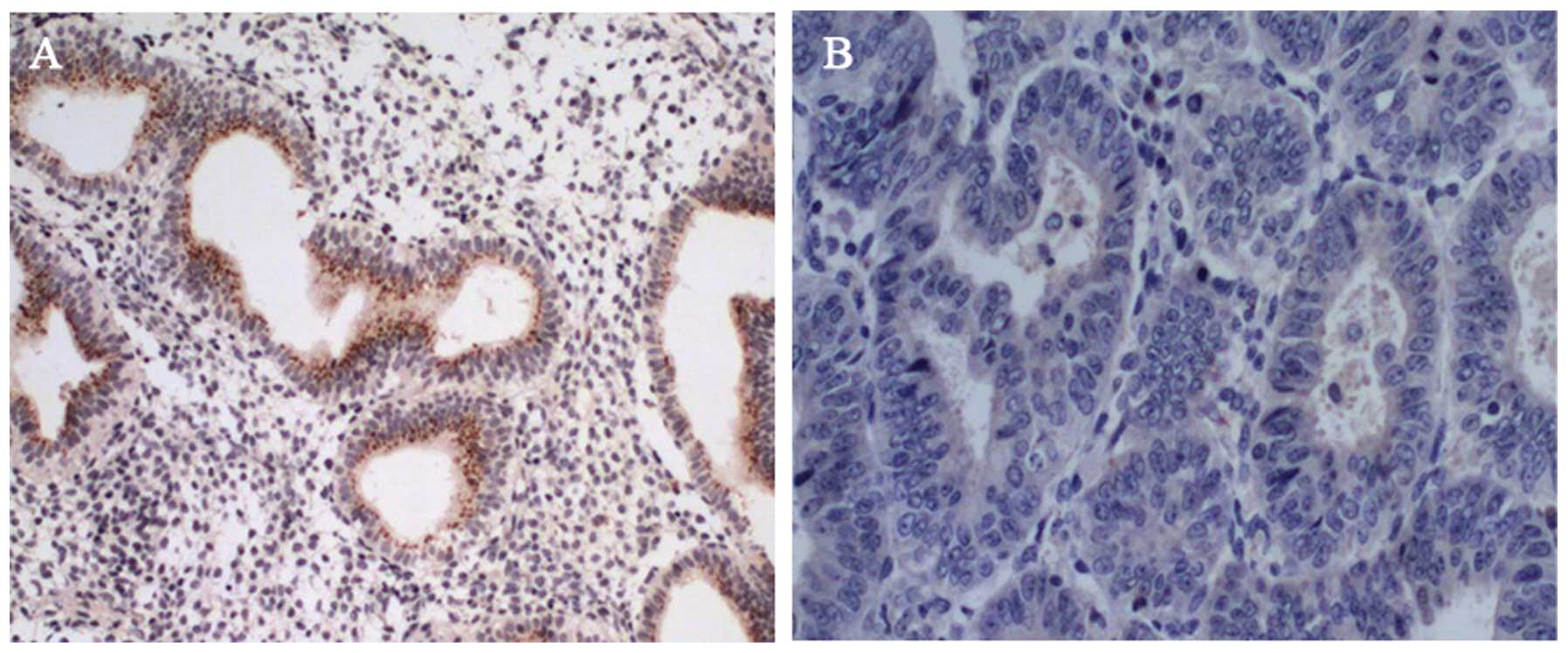
cancer tissues with loss of cell cycle control. Obvious staining
for ARHI was present in epithelial cells in normal ovarian tissue
sections (Fig. 1A) while almost no
detectable expression in cancerous tissues was noted by
immunohistochemical staining (Fig.
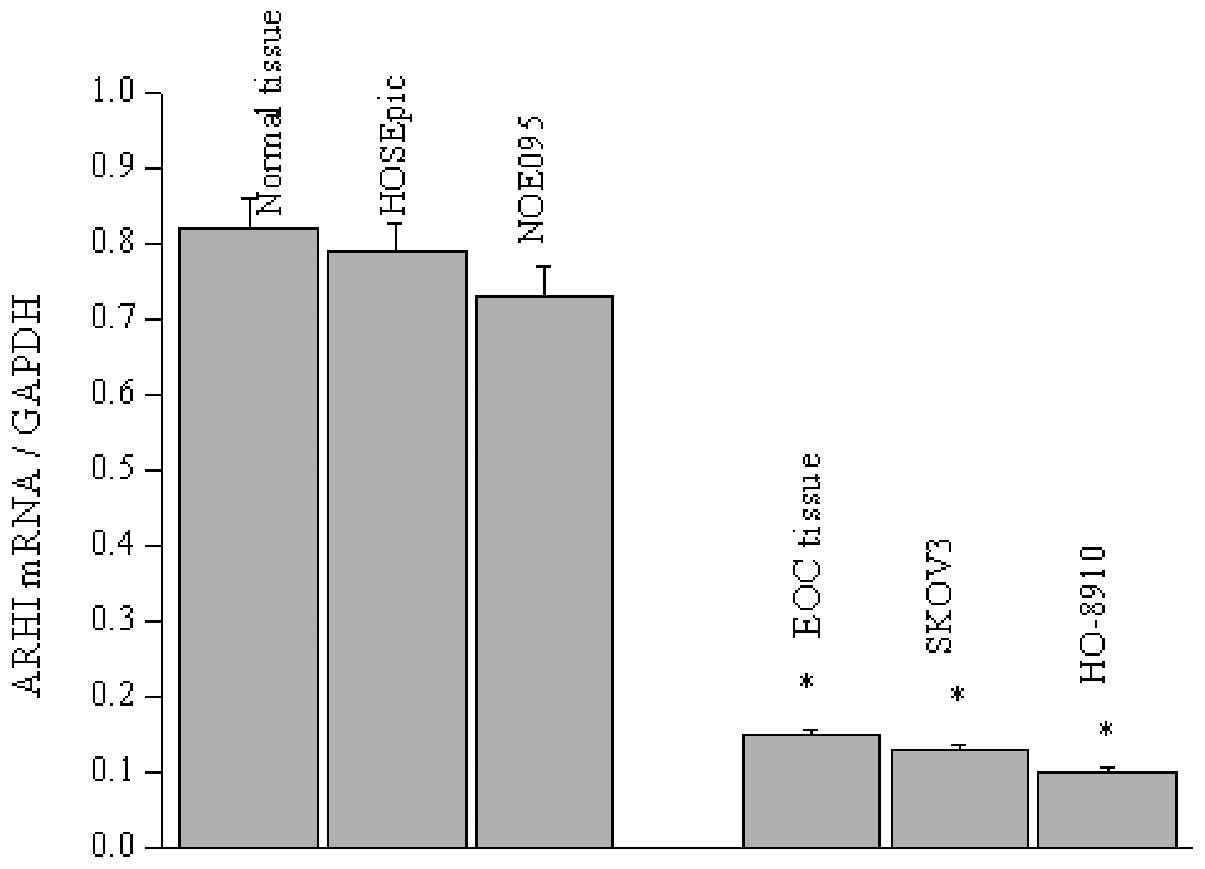
1B), which was confirmed by RT-PCR measurement. Likewise, the
ARHI mRNA level was determined in two normal and ovarian cancer
epithelial cell lines, since EOC is clearly responsibility for 90%
of ovarian neoplasias. Similar to the expression observed in the
tissue sections, ARHI mRNA levels present were significantly
decreased in the SKOV3 and HO-8910 cells, with less mRNA detected
in the HO-8910 cells (Fig. 2).
Promoter methylation is associated with
ARHI expression and tumor growth
It has been reported that promoter methylation plays
vital roles in the inactivation of the ARHI gene; thus, we examined
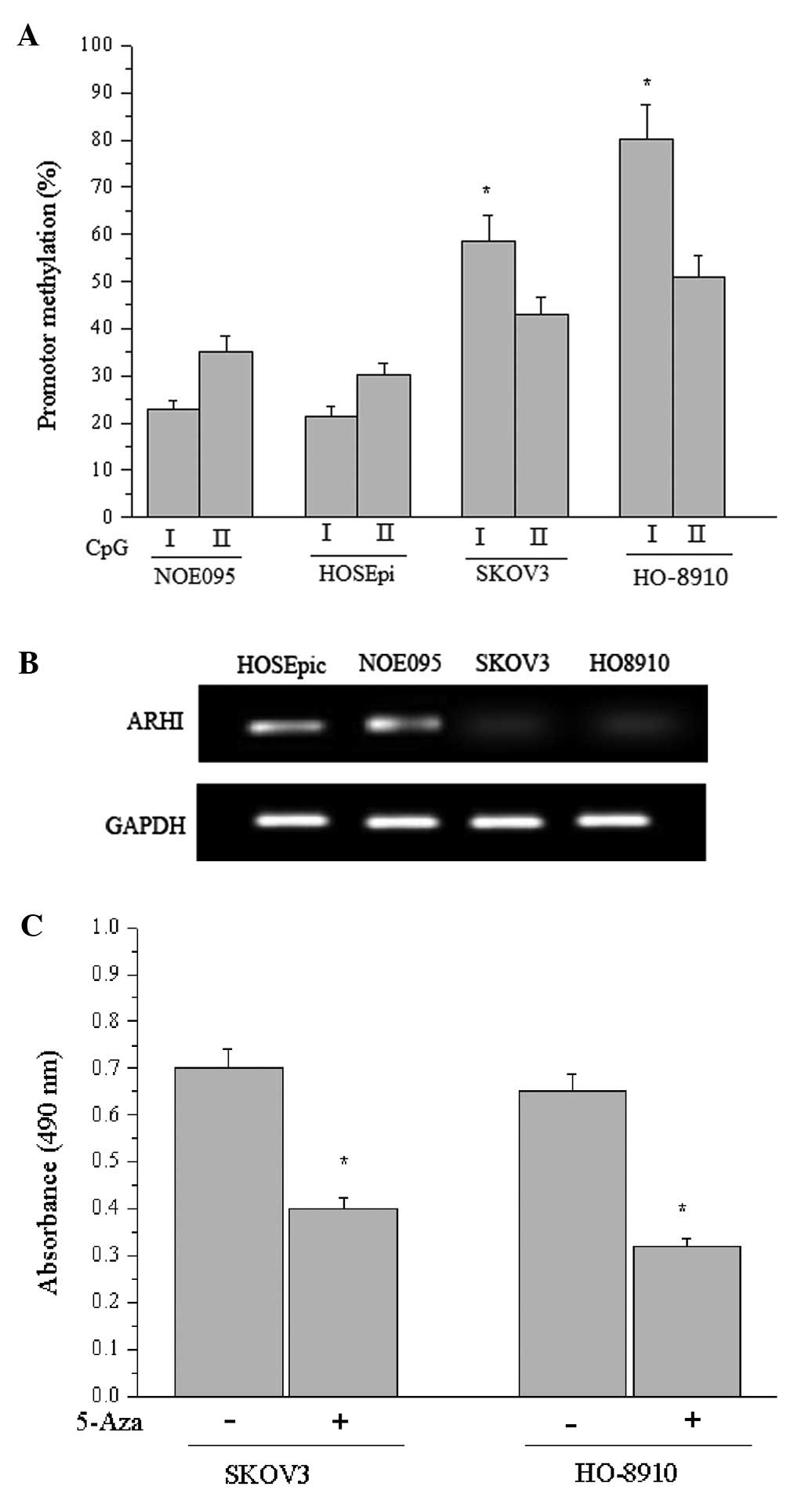
the methylation status of ARHI. As reported by Luo et
al(8), two CpG islands I and II
were located within the ARHI promoter and adjacent exon 1. As shown
in Fig. 3A, the proportions of
methylated CpG island I in NOE095 and HOSEpiC cells were 22.8±4.2%
and 21.5±3.1%, respectively, whereas they were 58.7±5.4% and
80.6±10.2% in the SKOV3 and HO-8910 cells (P<0.05), suggesting
that CpG island I in the cancer cells was partially methylated or
hypermethylated. Similarly, the density of CpG island I methylation
was increased in EOC cells compared with normal cells.
Additionally, the predominate change in CpG island I methylation
density may shed light on its critical function in regulating ARHI
expression. Therefore, RT-RCR was performed to determine the ARHI
transcriptional level in each cell line in response to different
methylation states, suggesting that the hypermethylated promoter
inhibited transcription of ARHI (Fig.
3B). Subsequently, we measured the effect of 5-Aza
(5-aza-2′-deoxycytidine) on the viability of SKOV3 and HO-8910
cells using a cell proliferation assay and demonstrated that
promoter methylation-mediated inactivation of the tumor-suppressor
gene ARHI accounted for the cellular malignances, while
5-Aza-induced demethylation greatly diminished the proliferation of
the cancer cells (Fig. 3C).
Elevated STAT3 acetylation results in
methylation of the ARHI gene promoter
It has been proposed that STAT3 exerts an influence
on CpG methylation, whereas the underlying mechanisms remain
elusive. The present study was carried out to investigate the
possible role of acetylated STAT3 in the methylation of CpG
islands. The ARHI gene promoter in the HO-8910 cell line exhibited
a higher methylation density than that in the SKOV3 cells;
therefore, it was selected for the following studies.
Correspondingly, the HO-8910 cell line harboring an endogenous
STAT3 mutation with K685 converted to R685 was generated via a
homologous recombination-mediated site-directed mutagenesis
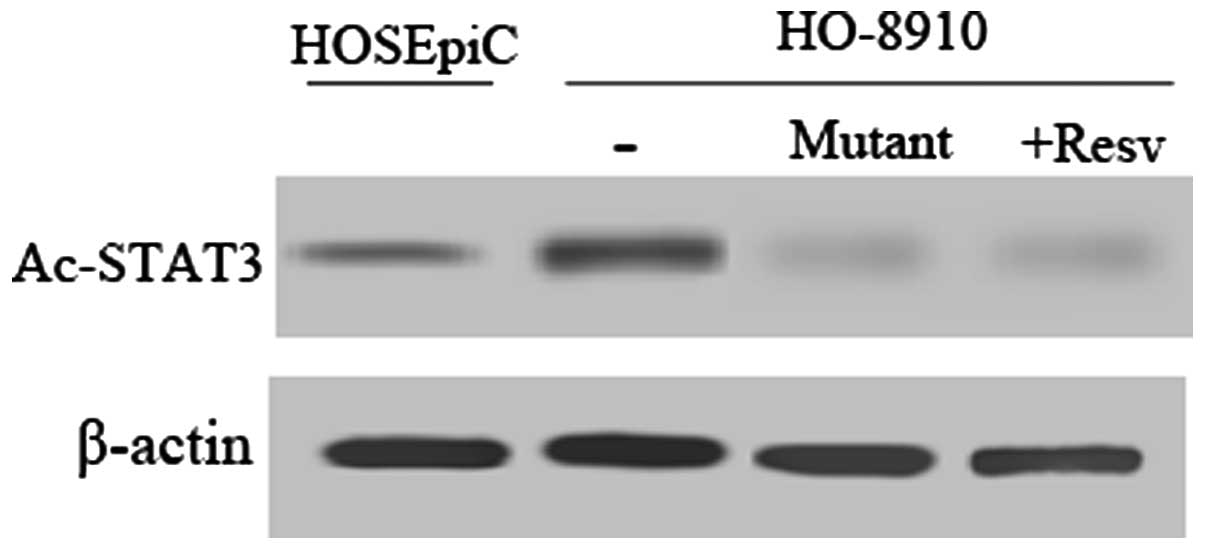
strategy as previously described (15). First, we assessed the acetylation
status of STAT3 by western blot analysis using an Ac-STAT3
monoclonal antibody. In comparison with the normal cell line
HOSEpiC, STAT3 was relatively highly acetylated in the HO-8910
cells (Fig. 4). Meanwhile, Ac-STAT3
expression decreased sharply after treatment with resveratrol, a
histone deacetylase (HDAC) activator or mutation of STAT3 K685R in
HO-8910 cells.
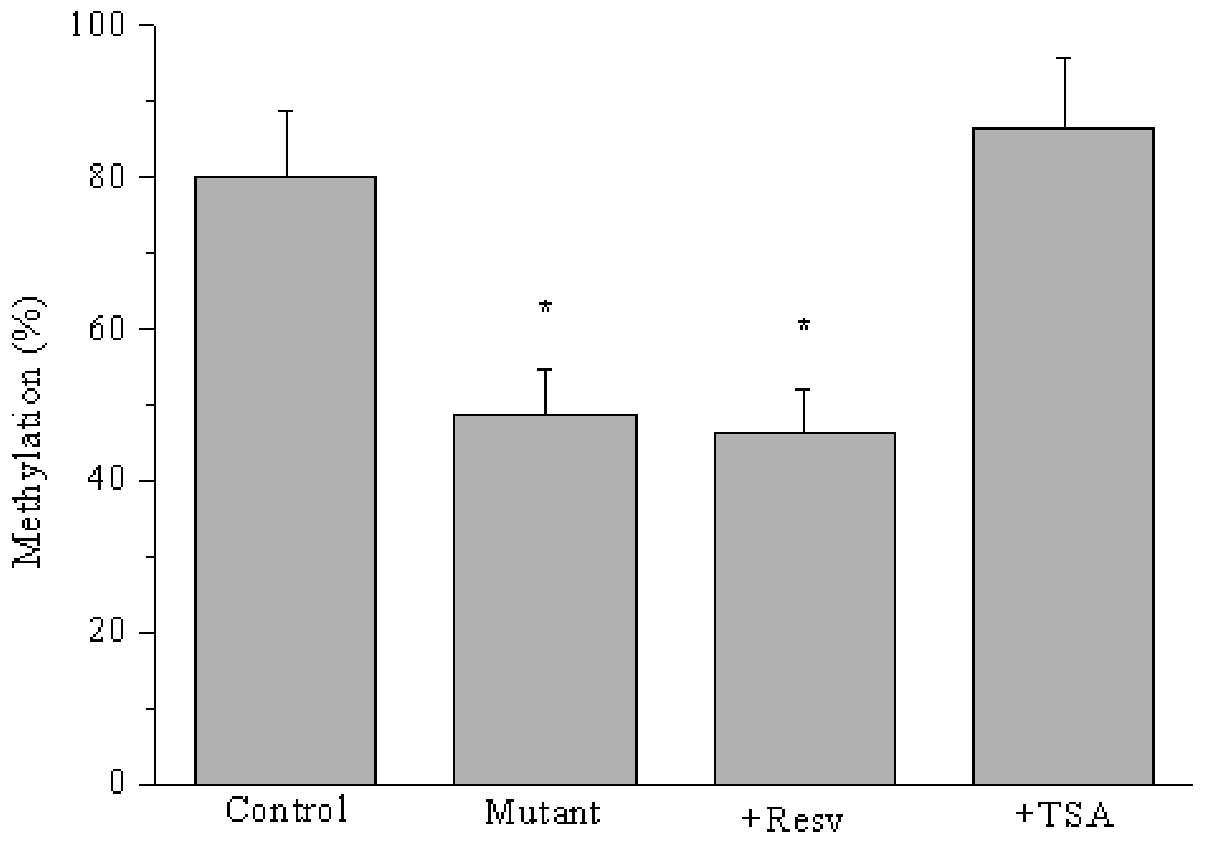
Furthermore, we determined whether CpG I island
methylation was associated with acetylated STAT3 in HO-8910 cells.
As shown in Fig. 5, ARHI promoter
methylation was reduced in response to either resveratrol or K685
mutation-mediated decreased Ac-STAT3 (P<0.05). However, CpG I
island methylation was increased by treatment with trichostatin, a
histone deacetylase inhibitor that restrains the deacetylation of
STAT3. Thus, STAT3 acetylation accelerated the methylation of the
ARHI promoter, which regulated the expression of the
tumor-suppressor gene ARHI.
STAT3 acetylation mediates STAT3-DNMT1
interaction and recruitment to the gene promoter
It has been reported that binding of inducible
transcription factors to DNA helps to recruit
chromatin-modification machinery; therefore, chromatin
immunoprecipitation was performed to determine whether DNMT1 was
attached to ARHI promoter regions in the HO-8910 cell line and the
corresponding variant STAT3 K685R cell line. Additionally,
tumor-conditioned medium prepared from U251 human glioma cells was
applied for the cultivation of HO-8910 and the variant cells to
activate STAT3 and facilitate binding to the gene promoter.
Furthermore, the binding of DNMT1 to ARHI was also subjected to
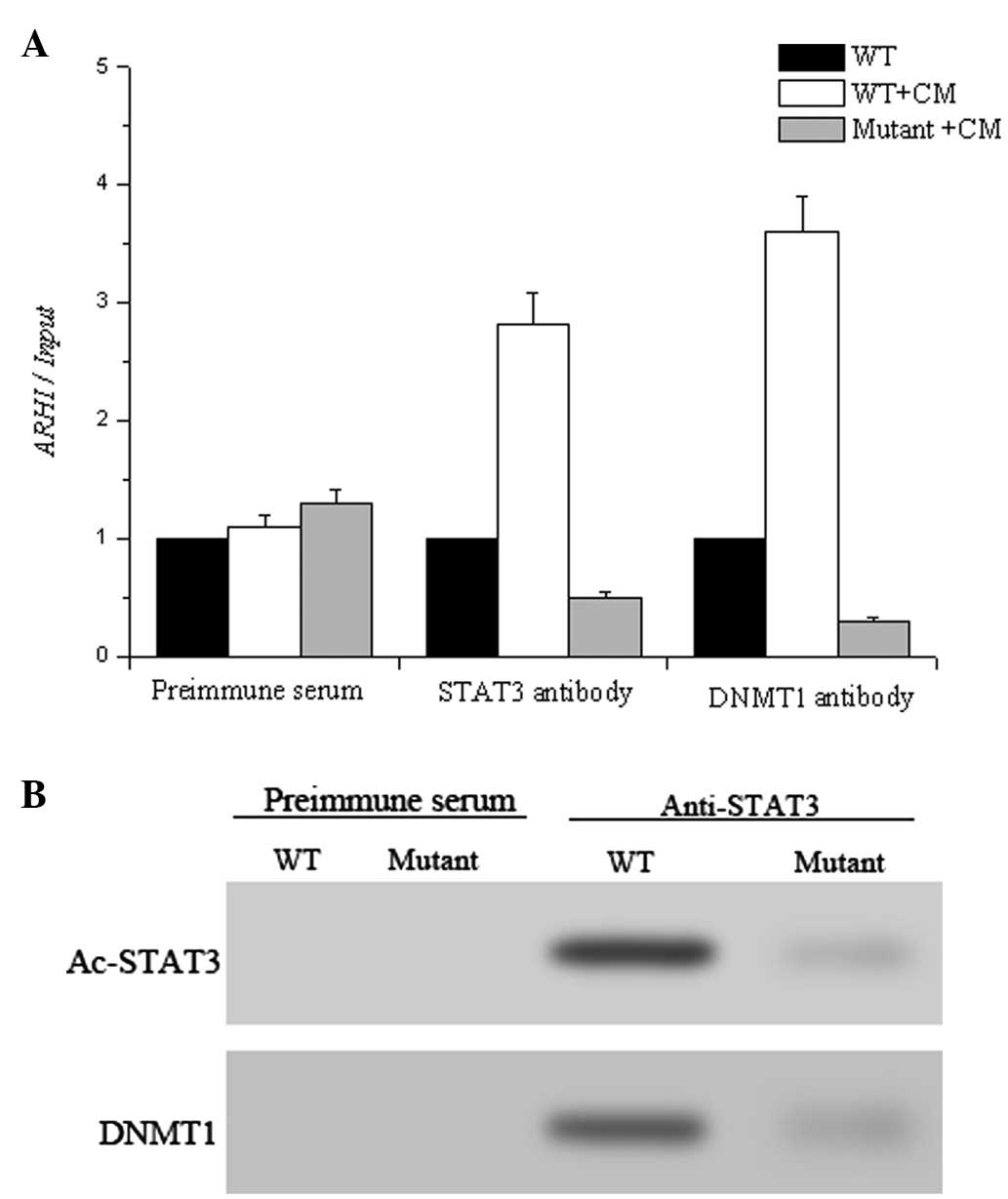
ChIP assay. As illustrated in Fig.
6A, both STAT3 and DNM1 bound to the ARHI promoter in HO-8910
cells while this binding was reduced in the STAT3 K685 mutant,
suggesting that acetylation of STAT3 regulated the attachment of
STAT3 to the promoter. Notably, the decreased Ac-STAT3 in the
mutant HO-8910 cells prevented the recruitment of DNMT1 to the ARHI
gene. We then analyzed the Ac-STAT3-DNMT1 interaction using
immunoprecipitation using anti-Ac-STAT3 antibody. Ac-STAT3 was
found to interact with DNMT1 (Fig.
6B), which is directly responsible for the modification of the
DNA.
Discussion
ARHI is a maternally imprinted tumor-suppressor gene
that is expressed in normal ovarian epithelium and is downregulated
in the majority of ovarian cancers. Accumulating evidence has shown
that decreased ARHI expression, either eliminated by LOH or
post-transcriptional modulation, is implicated in tumor
proliferation and development (20). Our study was carried out to
investigate whether methylation of the ARHI promoter gene could
mute its expression and the underlying mechanism of ARHI
methylation.
The present study also revealed that ARHI was
downregulated in EOC as measured by IHC. To the best of our
knowledge, ARHI possesses three CpG islands that are prone to
methylation. CpG I and II are located within promoter regions,
which encouraged us to consider the potential role of promoter
methylation in the regulation of ARHI expression. In comparison
with normal ovarian epithelium cells, the ARHI promoter was
partially or heavily methylated in cancer cells in concert with
reduced ARHI expression. Additionally, we demonstrated that CpG I
hypermethylation suppressed ARHI promoter activity and regulated
ARHI expression in ovarian cancer cells, which is similar to the
outcomes obtained in breast cancer cells.
DNA methylation acts in a switch-like manner and is
strongly correlated with the absence of gene expression and low
levels of additional activating epigenetic markers (21). Although the well-established role of
DNA methylation in gene silencing has been extensively
investigated, the precise mechanism of how the promoter methylation
status is modified remains elusive. It has been reported that
binding of inducible transcription factors to DNA helps recruit
chromatin-modification machinery, which is responsible for the
modification and expression of DNA (22). Furthermore, STAT3 has been shown to
increase CpG island methylation when acetylated (23). Therefore, we explored whether STAT3
was associated with the methylation-induced ARHI silencing. It was
confirmed that acetylated STAT3 bound to the ARHI promoter regions
and recruited DNMT1 to suppress ARHI expression. Simultaneously, we
found that specific inhibition of STAT3 acetylation using
resveratrol reversed the suppression of ARHI and restrained cancer
cell growth.
Taken together, our study demonstrated that the
tumor-suppressor ARHI is downregulated in ovarian epithelium cancer
cells and this downregulation can be partially ascribed to the
hypermethylation of CpG I islands. Acetylated STAT3 bound and
recruited DNMT1 to ARHI promoter regions. The activation of histone
deacetylase or inhibition of acetyltransferase may hold promise for
eliminating methylation-induced tumor-suppressor silencing, and
further prospective studies are warranted to further clarify the
modification machinery of Ac-STAT.
Acknowledgements
The present research was supported by a grant from
the Natural Science Foundation of China (no. 39830350).
References
|
1
|
Yu Y, Xu F, Peng H, et al: NOEY2 (ARHI),
an imprinted putative tumor suppressor gene in ovarian and breast
carcinomas. Proc Natl Acad Sci USA. 96:214–219. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Peng H, Xu F, Pershad R, et al: ARHI is
the center of allelic deletion on chromosome 1p31 in ovarian and
breast cancers. Int J Cancer. 86:690–694. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Luo RZ, Fang X, Marquez R, et al: ARHI is
a Ras-related small G-protein with a novel N-terminal extension
that inhibits growth of ovarian and breast cancers. Oncogene.
22:2897–2909. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Zhao X, Li J, Zhuo J and Cai L:
Reexpression of ARHI inhibits tumor growth and angiogenesis and
impairs the mTOR/VEGF pathway in hepatocellular carcinoma. Biochem
Biophys Res Commun. 403:417–421. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Zou CF, Jia L, Jin H, et al: Re-expression
of ARHI (DIRAS3) induces autophagy in breast cancer cells and
enhances the inhibitory effect of paclitaxel. BMC Cancer.
11:222011. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Yang H, Lu X, Qian J, et al: Imprinted
tumor suppressor gene ARHI induces apoptosis correlated with
changes in DNA methylation in pancreatic cancer cells. Mol Med Rep.
3:581–587. 2010.PubMed/NCBI
|
|
7
|
Kobayashi H, Kajiwara H, Kanayama S, et
al: Molecular pathogenesis of endometriosis-associated clear cell
carcinoma of the ovary (Review). Oncol Rep. 22:233–240.
2009.PubMed/NCBI
|
|
8
|
Luo RZ, Peng H, Xu F, et al: Genomic
structure and promoter characterization of an imprinted tumor
suppressor gene ARHI. Biochim Biophys Acta. 1519:216–222. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Feng W, Marquez RT, Lu Z, et al: Imprinted
tumor suppressor genes ARHI and PEG3 are the most frequently
down-regulated in human ovarian cancers by loss of heterozygosity
and promoter methylation. Cancer. 112:1489–1502. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Yuan Z, Guan Y, Chatterjee D and Chin YE:
Stat3 dimerization regulated by reversible acetylation of a single
lysine residue. Science. 307:269–273. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Wang R, Cherukuri P and Luo J: Activation
of Stat3 sequence-specific DNA binding and transcription by
p300/CREB-binding protein-mediated acetylation. J Biol Chem.
280:11528–11534. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Kimura K, Yamada T, Matsumoto M, et al:
Endoplasmic reticulum stress inhibits STAT3-dependent suppression
of hepatic gluconeogenesis via dephosphorylation and deacetylation.
Diabetes. 61:61–73. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Morgan RJ, Alvarez RD, Armstrong DK, et
al: Epithelial ovarian cancer. J Natl Compr Cancer Netw. 9:82–113.
2011.
|
|
14
|
Higashiura Y, Kajihara H, Shigetomi H and
Kobayashi H: Identification of multiple pathways involved in the
malignant transformation of endometriosis (Review). Oncol Lett.
4:3–9. 2012.PubMed/NCBI
|
|
15
|
Lee H, Zhang P, Herrmann A, et al:
Acetylated STAT3 is crucial for methylation of tumor-suppressor
gene promoters and inhibition by resveratrol results in
demethylation. Proc Natl Acad Sci USA. 109:7765–7769. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Yuan J, Luo RZ, Fujii S, et al: Aberrant
methylation and silencing of ARHI, an imprinted tumor suppressor
gene in which the function is lost in breast cancers. Cancer Res.
63:4174–4180. 2003.PubMed/NCBI
|
|
17
|
Sharda DR, Yu S, Ray M, et al: Regulation
of macrophage arginase expression and tumor growth by the Ron
receptor tyrosine kinase. J Immunol. 187:2181–2192. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Badgwell DB, Lu Z, Le K, et al: The
tumor-suppressor gene ARHI (DIRAS3) suppresses ovarian cancer cell
migration through inhibition of the Stat3 and FAK/Rho signaling
pathways. Oncogene. 31:68–79. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Bao JJ, Le XF, Wang RY, et al:
Reexpression of the tumor suppressor gene ARHI induces apoptosis in
ovarian and breast cancer cells through a caspase-independent
calpain-dependent pathway. Cancer Res. 62:7264–7272.
2002.PubMed/NCBI
|
|
20
|
Lu Z, Luo RZ, Peng H, et al:
Transcriptional and post-transcriptional down-regulation of the
imprinted tumor suppressor gene ARHI (DRAS3) in ovarian cancer.
Clin Cancer Res. 12:2404–2413. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Qidwai MT, Jamal F, Singh D and Sharma RK:
Factors modifying transcriptional regulation of signaling genes
have putative role in tumor development and progression in humans.
Med Hypotheses. 79:805–812. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Stark GR, Wang Y and Lu T: Lysine
methylation of promoter-bound transcription factors and relevance
to cancer. Cell Res. 21:375–380. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Zhang Q, Wang HY, Marzec M, Raghunath PN,
Nagasawa T and Wasik MA: STAT3- and DNA methyltransferase
1-mediated epigenetic silencing of SHP-1 tyrosine phosphatase tumor
suppressor gene in malignant T lymphocytes. Proc Natl Acad Sci USA.
102:6948–6953. 2005. View Article : Google Scholar : PubMed/NCBI
|