Introduction
Breast cancer is the most frequently diagnosed
cancer in women. Triple-negative breast cancer (TNBC) is a distinct
subtype lacking the expression of receptors for estrogen (ER),
progesterone (PR) and HER2/neu. Systemic treatment options are
limited to chemotherapy. TNBC accounts for 10–17% of all breast
cancers and is burdened by a poor prognosis (1–3). Thus,
the development of novel therapeutic strategies is of paramount
importance. Growth hormone-releasing hormone (GHRH) is a peptide
hormone found in the hypothalamus (4), which stimulates growth hormone
secretion from the pituitary gland and thus IGF-1 secretion by the
liver. Various antagonists of GHRH were synthesized in our
laboratory including JMR-132. GHRH antagonists suppress the release
of GH from the pituitary resulting in an inhibition of IGF-I
production by the liver. However, recent studies have shown an
ectopic expression of GHRH and its receptors in various benign
tissues (5–7), but more importantly in various types
of cancers suggesting that GHRH acts as a local tumor growth factor
via an autocrine/paracrine loop in these tissues (8–20).
Several preclinical in vitro studies where
the hypothalamic/pituitary/hepatic axis is clearly excluded,
underlined this hypothesis of an autocrine/paracrine function.
Additional studies demonstrated the existence of several splice
variants of the pituitary GHRH receptor (pGHRH-R) of which splice
variant 1 (SV 1) is a fully functional receptor (21–24).
GHRH receptors and its splice variants are present
in surgical specimens of breast cancers. Our group demonstrated
recently, that the pGHRH-R is expressed in 25% and the SV1 receptor
in 70% of TNBC tissue samples. Additionally, 80% of these samples
expressed GHRH, suggesting the presence of an autocrine/paracrine
growth stimulatory loop (25). In
HCC1806 human TNBC cells, MAP-kinases ERK-1/2 were activated by
treatment with GHRH, and GHRH stimulated proliferation of
MDA-MB-231 human breast cancer cell lines through activation of the
MAP-kinase phosphorylation pathway via Ras/Raf/MEK/ERK (26,27).
Taxanes such as paclitaxel and docetaxel have been
observed to impinge upon several signaling pathways to bring about
cell cycle arrest and apoptosis. Some of the most common changes
after treatment are Bcl-2 phosphorylation and the activation of
mitotic spindle assembly checkpoint (28,29).
This class of substances is now well established as one of the most
efficacious for the treatment of early and metastatic breast cancer
of all molecular subtypes.
We previously demonstrated that combined treatment
with GHRH antagonist JMR-132 and docetaxel led to a potentiation of
the antiproliferative effect of both compounds in human MX-1 breast
cancers xenografted into nude mice (30).
To further explore this novel promising treatment
approach, the combination of a GHRH antagonist and a taxane was
investigated in in vitro and in vivo models of TNBC,
given the dismal prognosis of this subgroup of breast cancers.
Materials and methods
Peptides and chemicals
The GHRH antagonist JMR-132 was synthesized in our
laboratory. For daily injection, JMR-132 was dissolved in 0.1%
dimethyl sulfoxide (DMSO) in 10% aqueous propylene glycol solution
(vehicle solution). For in vitro experiments, JMR-132 was
dissolved in DMSO and diluted further with PBS to the used
concentration not exceeding 0.1% DMSO. Docetaxel (DOC) ready-to-use
vials from Sanofi-Aventis were used for the experiments and diluted
according to the manufacturer’s instructions.
Cell lines and animals
The triple negative breast cancer cell line
MDA-MB-231 was obtained from the American Type Culture Collection
(Manassas, VA, USA). Cells were cultured in Dulbecco’s modified
Eagle’s medium (DMEM) supplemented with 10% fetal bovine serum
(FBS) and penicillin/streptomycin at 37°C and 5% CO2
atmosphere.
Female athymic nude mice (Ncr nu/nu) (5–6
weeks old) were obtained from the National Cancer Institute (NCI,
Bethesda, MD, USA). The animals were housed in sterile cages under
laminar flow hoods in a temperature-controlled room with a 12-h
light/12-h dark schedule. They were fed autoclaved chow and water
ad libitum. The Institutional Animal Care and Use Committee
reviewed the protocols of the animal experiments and granted full
approval.
Reverse transcription and RT-PCR
Total RNA was isolated and DNase-treated using the
Macherey-Nagel NucleoSpin® kit according to the
manufacturer’s instructions (Macherey-Nagel, Düren, Germany). The
yield and the quality of RNA samples were determined
spectrophotometrically using 260 nm, and 260/280 and 260/230 nm
ratio. Two micrograms of RNA from the sample was reverse
transcribed into cDNA by QuantiTect reverse transcription kit
(Qiagen, Hilden, Germany) in a final volume of 40 μl. We evaluated
the mRNA expression of human pituitary GHRH receptor (pGHRH-R) and
SV1 of GHRH-R as previously reported (31). The probe and primers for pGHRH-R and
sense and antisense specific primers for SV1 were described
previously (23).
All real-time PCR reactions were performed using the
iCycler iQ™ Real-Time PCR detection system (Bio-Rad, Hercules, CA,
USA). All thermal cycling conditions were described previously
(18). All samples were run in
triplicate, and each well of the PCR reaction contained 25 μl as a
final volume including 2 μl of cDNA, 200 nM of the gene-specific
primers and 400 nM of probes. iQ™ Supermix was used in the PCR
reactions for pGHRH-R and iQ™ SYBR-Green Supermix (both from
Bio-Rad) for SV1. The efficiencies of all primers (Invitrogen Life
Technologies, Carlsbad, CA, USA) and the probe (Integrated DNA
Technologies, Coralville, IA, USA) were tested prior to the
experiments and were efficient in the range of 95–105%. Normal
human pituitary was used as the positive control, and human β-actin
as the housekeeping gene. Negative samples were run in each
reaction consisting of no-RNA in the reverse transcriptase reaction
and no-cDNA in the PCR reaction.
Cell viability assays
For cell viability studies, 5,000 cells/well were
seeded in a 96-well plate in 100 μl medium. After 24 h, the culture
medium was replaced by JMR-132 at different concentrations (0.1, 1,
10 μM) and in other studies by JMR-132 (5 μM), DOC (1.28 or 0.635
nM) or the combination of JMR-132 and DOC at different
concentrations. The cells were then incubated for 72 h in a
humidified atmosphere at 37°C. The effect of the agents on cell
proliferation was evaluated using the MTS assay (CellTiter 96
Aqueous One Solution cell proliferation assay; Promega Corporation,
Madison, WI, USA) according to the manufacturer’s instructions.
Absorbance was measured at 550 nm using an enzyme-linked
immunosorbent assay (ELISA) plate reader. All experiments were
performed in hexaplicates and repeated 3 times.
In vivo experiments
For the animal experiments, 10×106
MDA-MB-231 cells/mouse were injected into the flanks of 4 female
nude mice. The resulting tumors were harvested and minced into
~3-mm3 pieces and transplanted into both flanks of each
female nude mouse using a mini trocar. The experiment was initiated
when the MDA-MB-231 tumors had reached a volume of ~110
mm3. Mice were divided into 4 experimental groups:
control (animals n=10; tumors n=15), JMR-132 (10 μg/day) (animals
n=8; tumors n=10), DOC (10 mg/kg on day 1 and 5, i.p.) (animals
n=6; tumors n=10), and a combination of DOC (10 mg/kg on day 1 and
5, i.p.) and JMR-132 (10 μg/day) (animals n=8, tumors n=13). The
weight of the animals was measured weekly.
Tumor volume (length × width × height × 0.5236) was
measured weekly, and tumor doubling time was calculated at the end
of the study using the formula: Study duration/(log final tumor
volume - log initial tumor volume)/log2.
At the end of the experiment, mice were sacrificed
under anesthesia, tumors were excised, snap frozen at −70°C for
further experiments and a full necropsy was performed.
Human cancer PathwayFinder PCR array
Quantitative mRNA expression analysis of 84 genes
representative of the major biological pathways involved in
transformation and tumorigenesis was performed using the Human
Cancer PathwayFinder™ RT2 Profiler™ PCR array
(PAHS-033A; Qiagen, Valencia, CA, USA). RNA was extracted and
DNAse-treated using the Macherey-Nagel NucleoSpin kit from 6
representative tumors grown in the nude mice (3 tumors from the
control and 3 tumors from the animals treated with JMR-132). The
yield and the quality of RNA samples, synthesis of cDNA and
real-time RT-PCT arrays were performed as previously described
(32). Real-time PCR reactions were
performed using the iCycler iQ™ Real-Time PCR detection system. All
genes represented by the array showed a single peak on the melting
curve characteristic to the specific products. Data analysis of
gene expression was performed as previously described (9). Briefly, using Excel-based PCR array
data analysis software provided by the manufacturer, fold-changes
in gene expression were calculated using the ΔΔCt method, and 5
stably expressed housekeeping genes (B2M, HPRT1, RPL13A, GAPDH and
ACTB) were used for normalization of the results.
Statistical analysis
Data are expressed as means ± SE. One-way ANOVA
followed by Bonferroni t-test or a two-tailed Student’s t-test was
used where appropriate, and significance was accepted at
p<0.05.
Results
Receptor expression for pGHRH-R and
SV1
Expression of mRNA of SV1 was detected in the
MDA-MB-231 cell line (Ct value 32.6) by real-time RT-PCR. However
the expression of the pGHRH-R could not be determined. For the
positive control (pituitary) the Ct value for pGHRH-R was 20.5 and
for SV1, 33.3.
Effect of GHRH antagonist JMR-132 and DOC
on the viability of MDA-MB-231 human breast cancer cells in
vitro
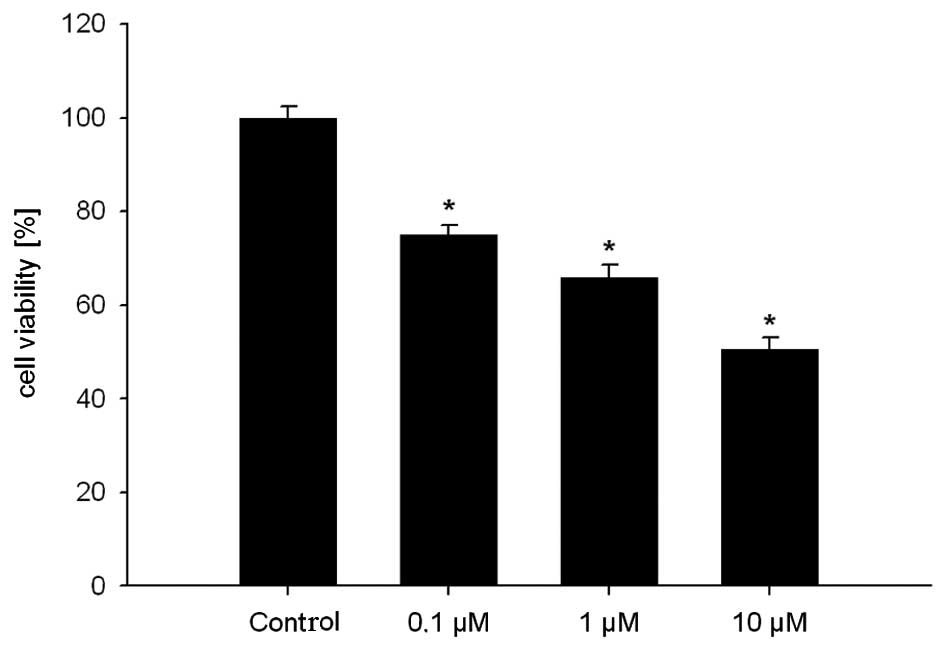
In the first experiment, we determined the effect of
GHRH antagonist JMR-132 at different concentrations on the
MDA-MB-231 cell line in the proliferation assays. A significant
(p<0.05) dose-dependent inhibition of cell viability was noted
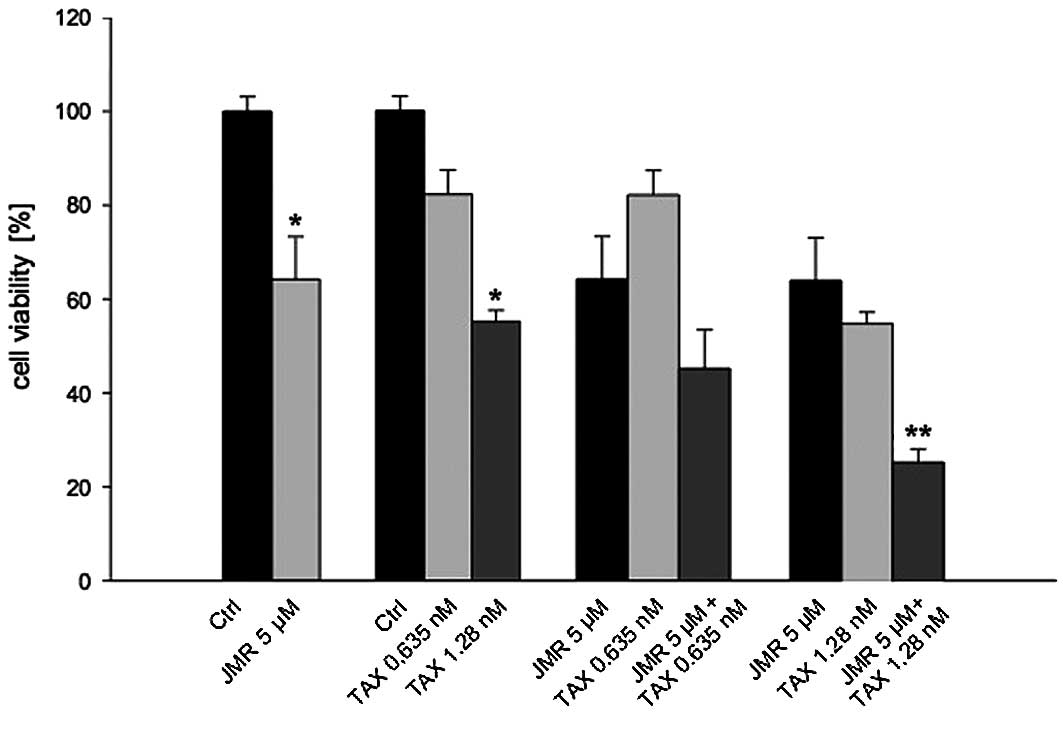
when compared to the controls. In the second experiment, we
evaluated the combination of JMR-132 and DOC and demonstrated a
significant (p<0.05) dose-dependent inhibition of cell viability
at different concentrations of DOC. The combination of both
substances led to an even greater inhibition of cell viability
compared to treatment with the single agents. This effect became
statistically significant at a combination dose of 5 μM JMR-132 and
1.28 nM DOC (Figs. 1 and 2).
Effect of GHRH antagonist JMR-132 and DOC
on the growth of MDA-MB-231 human breast cancer cell xenografted
tumors in nude mice
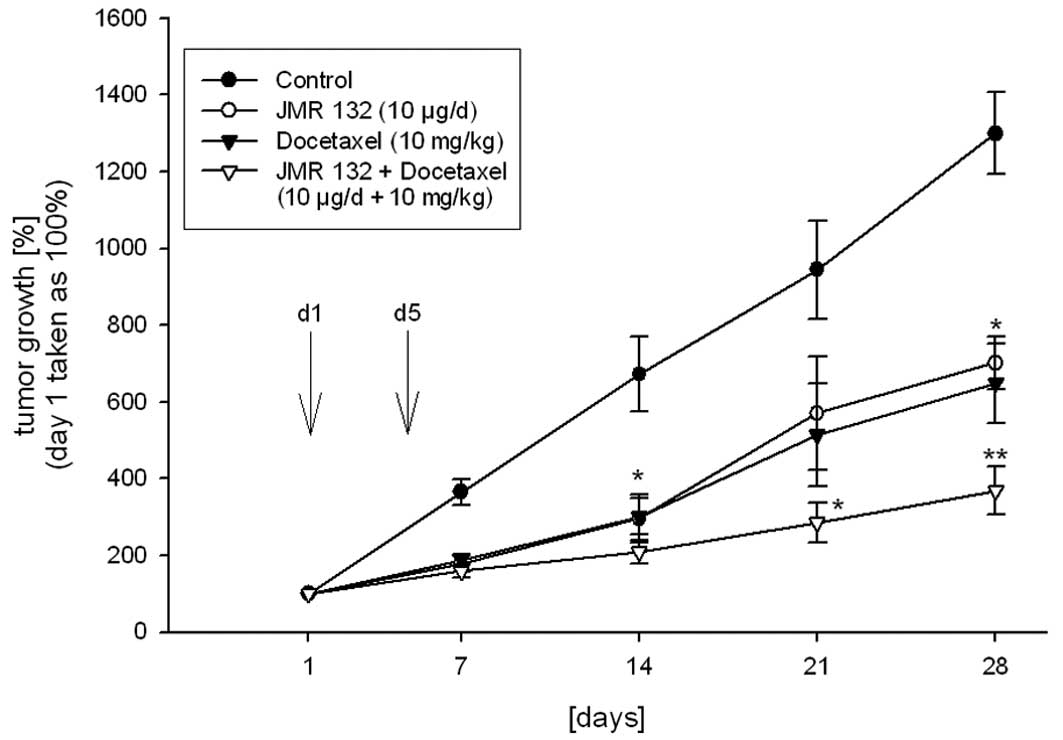
The tumor volume was significantly (p<0.05)
reduced by 46% following treatment with JMR-132 at a dose of 10
μg/day s.c., when compared to the controls at the end of the study
(Fig. 3). Additionally, JMR-132
significantly (p<0.05) increased the tumor doubling time
(Table I). When nude mice bearing
MDA-MB-231 tumors received DOC as a monotherapy, tumor growth was
inhibited by 50% (p<0.05, Fig.
3) as compared to the control group, and the tumor doubling
time was significantly prolonged compared to the controls
(p<0.05, Table I).
 | Table ITumor doubling time calculated at the
end of the study. |
Table I
Tumor doubling time calculated at the
end of the study.
| Tumor growth (%)on
day 28 | Tumor doubling times
(days) |
|---|
| Control | 1,299±106.96 | 7.90±0.47 |
| JMR-132 | 701±69.8 | 10.26±0.52b |
| Docetaxel | 649±103.26 | 12.32±1.86b |
| JMR-132 +
docetaxel | 369±62.51a | 19.52±1.02c |
Combined treatment with JMR-132 and DOC resulted in
significant tumor inhibition (p<0.001) after 14 days, and the
inhibition remained significant until the end of the experiment
when compared to the controls. A significant reduction in tumor
volume by 70% was achieved in this group (Fig. 3). The combination treatment of DOC
and JMR-132 was significantly more potent (p<0.05) with respect
to inhibition of the tumor volume when compared to the monotherapy
groups. The tumor doubling time was also significantly (p<0.05)
extended when compared to the other groups (Fig. 3 and Table I).
Toxicity
No signs of drug-induced toxicity were observed in
any of the mouse groups, as reflected by no differences in body and
organ weights between the treated groups and the control
animals.
Effect of GHRH antagonist JMR-132 on
expression of genes related to apoptosis, angiogenesis and
metastasis
The PCR array used contains 84 unique genes related
to signal transduction, cell cycle and apoptosis. All genes
represented by the array showed a single peak on the melting curve
characteristic to the specific products. Fifteen genes in the
MDA-MB-231 xenografts showed a significant change in mRNA
expression after treatment with JMR-132 compared to the controls
(Table II). Upregulated genes were
found to belong to the TNF ligand family FASLG, LTA, TNFSF10, CD70,
TNFSF8 (5.28-, 10.56-, 2.00-, 9.19-, 7.46-fold increases; p<0.05
for all). Significant transcriptional suppression of
angiogenesis-related gene ANGPT1 (2.93-fold decrease, p<0.05)
was observed. Significant alterations in transcriptional gene
expression involved in apoptosis or cell death were detected for
BCL2 (3.48-fold decrease, p<0.05), and HRK, CASP14, CASP4,
CASP5, DAPK, CIDEA (14.93-, 14.93-, 2.3-, 10.53-, 3.03-, 9.17-fold
increases, p<0.05 for all). Changes in the mRNA levels for
molecules related to invasion and metastasis were found for MMP2
(2.07-fold decrease, p<0.05) and TWIST1 (2.73-fold increase,
p<0.05).
| Table IIModulated genes with at least a 2-fold
change relative to the untreated control after treatment of
MDA-MB-231 xenografts with the GHRH antagonist JMR-132. |
Table II
Modulated genes with at least a 2-fold
change relative to the untreated control after treatment of
MDA-MB-231 xenografts with the GHRH antagonist JMR-132.
| Name of gene | Gene symbol | Accession no. | Fold changea |
|---|
| Angiopoietin 1 | ANGPT1 | NM_001146 | −2.93 |
| Antigen identified
by monoclonal antibody Ki-67 | MKI67 | NM_002417 | −2.69 |
| B-cell CLL/lymphoma
2 | BCL2 | NM_000633 | −3.48 |
| Baculoviral IAP
repeat containing 8 | BIRC8 | NM_033341 | 4.29 |
| Bcl2-like 10 | BCL2L10 | NM_020396 | 3.03 |
| Breast cancer 1,
early onset | BRCA1 | NM_007294 | −2.73 |
| Caspase 14 | CASP14 | NM_012114 | 14.93 |
| Caspase 4 | CASP4 | NM_001225 | 2.30 |
| Caspase 5 | CASP5 | NM_004347 | 10.53 |
| CD40 ligand | CD40LG | NM_000074 | 5.66 |
| CD70 molecule | CD70 | NM_001252 | 9.19 |
| Cell death-inducing
DFFA-like effector a | CIDEA | NM_001279 | 9.17 |
| Cullin 1 | CUL1 | NM_003592 | −2.18 |
| Cyclin B2 | CCNB2 | NM_004701 | −2.04 |
| Cyclin D2 | CCND2 | NM_001759 | 12.77 |
| Death-associated
protein kinase 1 | DAPK1 | NM_004938 | 3.03 |
| Fas ligand (TNF
superfamily, member 6) | FASLG | NM_000639 | 5.28 |
| Fibroblast growth
factor receptor 2 | FGFR2 | NM_000141 | 7.21 |
| Granzyme A | GZMA | NM_006144 | 3.14 |
| Harakiri, BCL2
interacting protein | HRK | NM_003806 | 14.93 |
| Integrin, β 3 | ITGB3 | NM_000212 | −3.87 |
| Interleukin 8 | IL8 | NM_000584 | 3.13 |
| Lymphotoxin α (TNF
superfamily, member 1) | LTA | NM_000595 | 10.56 |
| MAD2 mitotic arrest
deficient-like 1 (yeast) | MAD2L1 | NM_002358 | −3.08 |
| Matrix
metallopeptidase 1 | MMP1 | NM_002421 | 4.44 |
| Matrix
metallopeptidase 2 | MMP2 | NM_004530 | −2.07 |
| Platelet-derived
growth factor β polypeptide | PDGFB | NM_002608 | 2.55 |
| Serpin peptidase
inhibitor, clade B (ovalbumin), member 5 | SERPINB5 | NM_002639 | −2.56 |
| TEK tyrosine
kinase, endothelial | TEK | NM_000459 | 2.54 |
| Tumor necrosis
factor | TNF | NM_000594 | 6.28 |
| Tumor necrosis
factor | TNF | NM_000594 | 16.00 |
| Tumor necrosis
factor (ligand) superfamily, member 10 | TNFSF10 | NM_003810 | 2.00 |
| Tumor necrosis
factor (ligand) superfamily, member 8 | TNFSF8 | NM_001244 | 7.46 |
| Tumor necrosis
factor receptor superfamily, member 9 | TNFRSF9 | NM_001561 | 9.19 |
| Tumor protein
p53 | TP53 | NM_000546 | −2.19 |
| Twist homolog
1 | TWIST1 | NM_000474 | 2.73 |
Discussion
Novel therapeutic strategies for the treatment of
many types of cancers are based on agents targeting cell signaling
pathways that interacting with the cell cycle, apoptosis,
proliferation or angiogenesis. New therapeutic agents such as
trastuzumab and lapatinib have been clinically used with great
success for years.
Previously, we found that the pGHRH-R is expressed
in 25% and SV1 in 70% of TNBC tissue samples, and in 80% of these
samples GHRH is expressed (25).
Various findings suggest that GHRH acts as a growth factor in GHRH
receptor-positive malignancies, which may represent an attractive
therapeutic target for antagonists of GHRH (10–13,15–19).
Since chemotherapy is the only therapeutic option
for TNBC, GHRH antagonists could provide a novel targeted approach
for this subgroup of breast cancers.
In the present study we demonstrated the presence of
SV1 but not pGHRH-R in the human triple-negative breast cancer cell
line MDA-MB-231 and the antitumoral effect of the GHRH antagonist
JMR-132. This finding provides strong evidence for the important
role of SV1 as a functional receptor. Treatment with JMR-132
significantly inhibited cell proliferation in a dose-dependent
manner in MDA-MB-231 human breast cancers in vitro. JMR also
demonstrated antitumor activity in vivo. The in vivo
study also revealed that GHRH antagonist JMR-132 interacts with
various signal transduction pathways involved in proliferation,
apoptosis and angiogenesis. In TNBC, the MAPK and the AKT signal
transduction pathways have been found to be significantly
overactivated (33,34). These two pathways induce a variety
of cellular events, of which many are connected to proliferation
and resistance to apoptosis. Notably, targeting the GHRH signal
transduction pathway also resulted in the suppression of genes
inducing proliferation and resistance to apoptosis. Thus, targeting
the GHRH-receptor may also result in a synergistic effect in
combination with novel antitumor agents, such as AKT and ERK
inhibitors and not only with chemotherapeutic agents as
demonstrated in our study.
Docetaxel is a chemotherapeutic agent frequently
used in the treatment of breast cancers. Furthermore, taxanes have
been favorably used in combination with monoclonal antibodies and
small-molecule agents such as trastuzumab or lapatinib. Therefore,
taxane could be likely candidates for combination therapy with GHRH
antagonists. Accordingly, the present study showed that a
combination of DOC with JMR-132 potentiated the antitumor effect
in vitro and in vivo as compared to each substance
alone.
In conclusion, this investigation demonstrated that
GHRH antagonists are effective for the treatment of SV1-positive
TNBC and can be combined with taxane-based chemotherapy. As in our
study and various other studies, GHRH antagonists displayed no
toxic side effects. Thus, they should be assessed in combination
with taxanes in phase I trials for the treatment of patients with
TNBC expressing the SV1 and/or pGHRH receptor.
Acknowledgements
This work was supported in part by a grant from the
American Urological Association (AUA) Foundation Research Scholars
Program and the AUA Southeastern Section (to F.G.R.). Our study was
also supported by the Medical Research Service of the Veterans
Affairs, Departments of Pathology and Medicine, Division of
Hematology/Oncology of the Miller Medical School, University of
Miami, and South Florida Veterans Affairs Foundation for Research
and Education (all to A.V.S.).
References
|
1
|
Carey LA, Dees EC, Sawyer L, et al: The
triple negative paradox: primary tumor chemosensitivity of breast
cancer subtypes. Clin Cancer Res. 13:2329–2334. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Dent R, Trudeau M, Pritchard KI, et al:
Triple-negative breast cancer: clinical features and patterns of
recurrence. Clin Cancer Res. 13:4429–4434. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Tischkowitz M, Brunet JS, Bégin LR, et al:
Use of immunohistochemical markers can refine prognosis in triple
negative breast cancer. BMC Cancer. 7:1342007. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Schally AV, Steelman SL and Bowers CY:
Effect of hypothalamic extracts on release of growth hormone in
vitro. Proc Soc Exp Biol Med. 119:208–212. 1965. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Rick FG, Schally AV, Block NL, et al:
Antagonists of growth hormone-releasing hormone (GHRH) reduce
prostate size in experimental benign prostatic hyperplasia. Proc
Natl Acad Sci USA. 108:3755–3760. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Kiaris H, Chatzistamou I, Papavassiliou AG
and Schally AV: Growth hormone-releasing hormone: not only a
neurohormone. Trends Endocrinol Metab. 22:311–317. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Rick FG, Szalontay L, Schally AV, et al:
Combining growth hormone-releasing hormone antagonist with
luteinizing hormone-releasing hormone antagonist greatly augments
benign prostatic hyperplasia shrinkage. J Urol. 187:1498–1504.
2012. View Article : Google Scholar
|
|
8
|
Bagnato A, Moretti C, Ohnishi J, Frajese G
and Catt KJ: Expression of the growth hormone-releasing hormone
gene and its peptide product in the rat ovary. Endocrinology.
130:1097–1102. 1992.PubMed/NCBI
|
|
9
|
Rick FG, Schally AV, Szalontay L, et al:
Antagonists of growth hormone-releasing hormone inhibit growth of
androgen-independent prostate cancer through inactivation of ERK
and Akt kinases. Proc Natl Acad Sci USA. 109:1655–1660. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Busto R, Schally AV, Braczkowski R, et al:
Expression of mRNA for growth hormone-releasing hormone and splice
variants of GHRH receptors in human malignant bone tumors. Regul
Pept. 108:47–53. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Busto R, Schally AV, Varga JL, et al: The
expression of growth hormone-releasing hormone (GHRH) and splice
variants of its receptor in human gastroenteropancreatic
carcinomas. Proc Natl Acad Sci USA. 99:11866–11871. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Chatzistamou I, Schally AV, Kiaris H, et
al: Immunohisto-chemical detection of GHRH and its receptor splice
variant 1 in primary human breast cancers. Eur J Endocrinol.
151:391–396. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Engel JB, Keller G, Schally AV, et al:
Inhibition of growth of experimental human endometrial cancer by an
antagonist of growth hormone-releasing hormone. J Clin Endocrinol
Metab. 90:3614–3621. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Frohman LA and Szabo M: Ectopic production
of growth hormone-releasing factor by carcinoid and pancreatic
islet tumors associated with acromegaly. Prog Clin Biol Res.
74:259–271. 1981.PubMed/NCBI
|
|
15
|
Garcia-Fernandez MO, Schally AV, Varga JL,
Groot K and Busto R: The expression of growth hormone-releasing
hormone (GHRH) and its receptor splice variants in human breast
cancer lines; the evaluation of signaling mechanisms in the
stimulation of cell proliferation. Breast Cancer Res Treat.
77:15–26. 2003. View Article : Google Scholar
|
|
16
|
Guo J, Schally AV, Zarandi M, Varga J and
Leung PC: Antiproliferative effect of growth hormone-releasing
hormone (GHRH) antagonist on ovarian cancer cells through the
EGFR-Akt pathway. Reprod Biol Endocrinol. 8:542010. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Kahán Z, Arencibia JM, Csernus VJ, et al:
Expression of growth hormone-releasing hormone (GHRH) messenger
ribonucleic acid and the presence of biologically active GHRH in
human breast, endometrial, and ovarian cancers. J Clin Endocrinol
Metab. 84:582–589. 1999.PubMed/NCBI
|
|
18
|
Schally A and Varga JL: Antagonists of
growth hormone-releasing hormone. Comb Chem High Throughput Screen.
9:163–170. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Schally AV, Comaru-Schally AM, Nagy A, et
al: Hypothalamic hormones and cancer. Front Neuroendocrinol.
22:248–291. 2001. View Article : Google Scholar
|
|
20
|
Schally AV, Varga JL and Engel JB:
Antagonists of growth-hormone-releasing hormone: an emerging new
therapy for cancer. Nat Clin Pract Endocrinol Metab. 4:33–43. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Barabutis N, Siejka A, Schally AV, Block
NL, Cai R and Varga L: Activation of mitogen-activated protein
kinases by a splice variant of GHRH receptor. J Mol Endocrinol.
44:127–134. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Barabutis N, Tsellou E, Schally AV,
Kouloheri S, Kalofoutis A and Kiaris H: Stimulation of
proliferation of MCF-7 breast cancer cells by a transfected splice
variant of growth hormone-releasing hormone receptor. Proc Natl
Acad Sci USA. 104:5575–5579. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Havt A, Schally AV, Halmos G, et al: The
expression of the pituitary growth hormone-releasing hormone
receptor and its splice variants in normal and neoplastic human
tissues. Proc Natl Acad Sci USA. 102:17424–17429. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Rekasi Z, Czompoly T, Schally AV and
Halmos G: Isolation and sequencing of cDNAs for splice variants of
growth hormone-releasing hormone receptors from human cancers. Proc
Natl Acad Sci USA. 97:10561–10566. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Köster F, Engel JB, Schally AV, et al:
Triple-negative breast cancers express receptors for growth
hormone-releasing hormone (GHRH) and respond to GHRH antagonists
with growth inhibition. Breast Cancer Res Treat. 116:273–279.
2009.PubMed/NCBI
|
|
26
|
Pombo CM, Zalvide J, Gaylinn BD and
Dieguez C: Growth hormone-releasing hormone stimulates
mitogen-activated protein kinase. Endocrinology. 141:2113–2119.
2000.PubMed/NCBI
|
|
27
|
Siriwardana G, Bradford A, Coy D and
Zeitler P: Autocrine/paracrine regulation of breast cancer cell
proliferation by growth hormone releasing hormone via Ras, Raf, and
mitogen-activated protein kinase. Mol Endocrinol. 20:2010–2019.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Berchem GJ, Bosseler M, Mine N and
Avalosse B: Nanomolar range docetaxel treatment sensitizes MCF-7
cells to chemotherapy induced apoptosis, induces G2M arrest and
phosphorylates bcl-2. Anticancer Res. 19:535–540. 1999.
|
|
29
|
Lavelle F, Bissery MC, Combeau C, Riou JF,
Vrignaud P and André S: Preclinical evaluation of docetaxel
(Taxotere). Semin Oncol. 22:3–16. 1995.
|
|
30
|
Buchholz S, Schally AV, Engel JB, et al:
Potentiation of mammary cancer inhibition by combination of
antagonists of growth hormone-releasing hormone with docetaxel.
Proc Natl Acad Sci USA. 104:1943–1946. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Kovács M, Schally AV, Hohla F, et al: A
correlation of endocrine and anticancer effects of some antagonists
of GHRH. Peptides. 31:1839–1846. 2010.PubMed/NCBI
|
|
32
|
Rick FG, Schally AV, Block NL, et al: LHRH
antagonist Cetrorelix reduces prostate size and gene expression of
proinflammatory cytokines and growth factors in a rat model of
benign prostatic hyperplasia. Prostate. 71:736–747. 2011.
View Article : Google Scholar
|
|
33
|
Umemura S, Yoshida S, Ohta Y, Naito K,
Osamura RY and Tokuda Y: Increased phosphorylation of Akt in
triple-negative breast cancers. Cancer Sci. 98:1889–1892. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Eralp Y, Derin D, Ozluk Y, et al: MAPK
overexpression is associated with anthracycline resistance and
increased risk for recurrence in patients with triple-negative
breast cancer. Ann Oncol. 19:669–674. 2008. View Article : Google Scholar : PubMed/NCBI
|