Introduction
Granulocyte colony-stimulating factor (G-CSF)
stimulates the proliferation, survival and neutrophilic
differentiation of hematopoietic progenitor cells, making G-CSF a
secreted hematopoietic growth factor that is important for
granulopoiesis (1,2). Physiologically, G-CSF affects the
mobilization of hematopoietic stem cells, progenitors, and mature
cells, particularly neutrophils, into blood circulation (3). Healthy individuals have low levels of
G-CSF in serum ranging from <30 to 163 pg/ml. By comparison,
infection raises G-CSF serum levels to a range of 30–3199 pg/ml
(4). Elevation of G-CSF in serum
during infection increases the number of granulocytic progenitor
cells and neutrophils, and has highlighted the potential
therapeutic value of G-CSF for decades. Administration of
recombinant G-CSF has been used clinically to increase the
neutrophil count when treating chemotherapy- or
radiotherapy-induced neutropenia (5,6).
Many clinical reports have documented that
leukocytosis occurs in patients with various types of
non-hematological malignant tumors with no signs of infection, but
higher serum G-CSF produced by the tumor. The G-CSF-producing
tumors exhibit significant hyperplastic and metastatic properties
and a poor prognosis. Although the pathological significance of
G-CSF is poorly known, G-CSF has been reported to induce tumor-cell
proliferation and migration (7,8) or
induce infiltration of inflammatory cells, which leads to stromal
activation providing a neoplastic-promoting microenvironment
(7,9). Thus, G-CSF is considered a therapeutic
target.
Expression of G-CSF in monocytes and macrophages is
induced by IL-1, GM-CSF, TNF-α or LPS (5). However, there is limited knowledge
concerning the regulation of G-CSF expression in cancer cells. In
this study, we compared the expression levels of G-CSF in various
malignant cancer cells and investigated the signaling pathway that
is critical for G-CSF expression. The results showed that G-CSF
expression is highly correlated with the invasiveness of cancer
cells, and activation of extracellular signal-regulated kinase
(ERK) 2, but not ERK1, is essential for G-CSF expression.
Materials and methods
Reagents
Dulbecco’s modified Eagle’s medium (DMEM) and fetal
bovine serum (FBS) were obtained from Hyclone Laboratories (Logan,
UT, USA). TRIzol reagent and penicillin/streptomycin were obtained
from Gibco-BRL Life Technologies (Rockville, MD, USA). MMLV reverse
transcriptase was obtained from Promega Corporation (Madison, WI,
USA). Pharmacological inhibitors PD98059 and U0126 (MEK
inhibitors), LY294002 [a phosphatidylinositol 3-kinase (PI3K)
inhibitor],
1L6-hydroxymethyl-chiro-inositol-2-(R)-2-O-methyl-3-O-octadecyl-sn-glycerocarbonate
(an Akt inhibitor, referred to as Akti in the text), and
rapamycin (an mTOR inhibitor) were purchased from Calbiochem (San
Diego, CA, USA) and were dissolved in dimethyl sulfoxide (DMSO)
which was used at a maximum concentration of 0.1% in culture
medium. Antibodies against Akt, phospho-Akt, β-actin, ERK1/2,
phospho-ERK1/2, p70S6K and phospho-p70S6K were purchased from Santa
Cruz Biotechnology, Inc. (Santa Cruz, CA, USA).
Cell culture
Human oral squamous cell carcinoma (OSCC) cell lines
(HSC-3, SCC-15 and OC3), human breast cancer cell lines (MCF-7,
T47D and MDA-MB-231), and human lung adenocarcinoma cell lines (CL1
and CL1–5) (10) were used in this
study. HSC-3 cells were grown in DMEM containing 5% FBS. SCC-15,
MCF-7, T47D and MDA-MB-231 cells were grown in DMEM/F12 (1:1
mixture) supplemented with 10% FBS. OC3 cells were grown in
DMEM/KSFM (1:1 mixture) supplemented with 10% FBS. CL1 and CL1–5
cells were grown in RPMI-1640 supplemented with 5% FBS. All culture
medium contained 2 mM of glutamine and 1,000 U/ml of
penicillin-streptomycin. In these experiments, G-CSF mRNA and
protein levels were compared in untreated cells and in cells
treated with 10 μM of U0126 for 7 h, unless otherwise
specified.
RNA isolation and analysis of G-CSF
mRNA
Total cellular RNA was isolated from cells using
TRIzol reagent. RNA concentrations were estimated from the
absorbance at 260 nm. First-strand cDNA was synthesized from total
RNA using the MMLV reverse transcriptase and Oligo(dT) primer. The
mRNA levels of G-CSF and GAPDH were determined by RT-PCR, using
specific primers: 5′-CACTCTGGACAGTGC AGGAAG-3′ (F) and
5′-CGACACCTCCAGGAAGCTCTG-3′ (R) for G-CSF;
5′-AAAGGATCCACTGGCGTCTTC-3′ (F) and 5′-GAATTCGTCATGGATGACCTT-3′ (R)
for GAPDH. Quantitative real-time PCR (qPCR) was used to quantify
the level of G-CSF mRNA. The amplifications were carried out in an
ABI Prism 7300 sequence detection system (Applied Biosystems,
Foster City, CA, USA). The reaction was performed in a 20-μl
mixture containing 1× reaction buffer (SYBR-Green Master Mixture;
Applied Biosystems) and 100 nM of the primers:
5′-TCCCCATCCCATGTATTTATCT-3′ (F) and 5′-AACTCAGAAATGCAGGGAAGGA-3′
(R) for G-CSF; 5′-CATGGCCTTCCGTGTTCCTA-3′ (F) and 5′-GC
GGCACGTCAGATCCA-3′ (R) for GAPDH. The specificity was verified by
an additional dissociation curve. The interpolated number of cycles
to reach a fixed threshold above background noise (Ct) was used to
quantify the amplification.
Western blot analysis
Cellular protein lysates (20 μg of protein/lane)
were separated by SDS-PAGE and transferred to a PVDF membrane,
which was blocked with 5% nonfat dried milk in phosphate-buffered
saline (PBS) overnight at 4°C and then incubated for 1 h at RT with
0.5 μg/ml of polyclonal anti-ERK, anti-phospho-ERK or anti-β-actin
Ab and for 40 min at RT with peroxidase-conjugated anti-rabbit IgG
Ab. The bound Abs were detected using an improved chemiluminescence
detection system (NEN Life Science Products, Boston, MA, USA).
Protein concentrations were determined by the Bradford method (DC
protein assay; Bio-Rad, Hercules, CA, USA).
Quantitation of G-CSF in culture
media
The concentration of human or mouse G-CSF in the
culture medium was measured with a human G-CSF Quantikine ELISA kit
(R&D Systems, Minneapolis, MN, USA) according to the
manufacturer’s instructions.
Transwell invasion assay
An in vitro cell invasion assay was carried
out using Transwell chambers (8-μm pore size; BD Biosciences,
Franklin Lakes, NJ, USA). The chambers were coated with Matrigel
(BD Biosciences). Cells were plated at a density of
5×104 cells/chamber in a 24-well plate containing
serum-free DMEM in the upper chamber and DMEM with 10% FCS in the
bottom chamber as the chemo-attractant. The cells that had invaded
into the bottom chambers were stained with crystal violet at 16 and
24 h post-seeding for breast and lung cancer cells, respectively.
Cells were counted in 4 random fields using MetaMorph software
(Molecular Devices, Sunnyvale, CA, USA). Data were averaged for
three independent experiments, each with four replicates.
Recombinant lentivirus construction and
gene transduction
The pLKO.1-shRNA plasmids encoding an shRNA with a
sequence targeting firefly luciferase or with a sequence targeting
human ERK1 (5′-CTATACCAAGTCCATCGACAT-3′) and ERK2
(5′-CAAAGTTCGAGTAGCTATCAA-3′) were obtained from the National RNAi
Core Facility at Academia Sinica, Taiwan. These plasmids and
lentiviral packaging vectors pMD.G and pCMV-ΔR8.91 were
co-introduced into HEK293T cells by calcium-phosphate transfection
method. Viruses were collected from the medium 60 h after
transfection. For knockdown experiments, HSC-3 and MDA-MB-231 cells
were infected with the collected viruses over 24 h in the presence
of Polybrene, followed by selection in medium containing puromycin
(10 μg/ml) for 7 days.
Statistical analysis
Results are shown as means ± SD. Differences between
mean values were evaluated using the Student’s t-test and were
considered significant at p<0.05.
Results
Expression of G-CSF in SCC-15 oral cancer
cells is governed by the MEK-ERK signaling pathway
To identify the pathway(s) involved in G-CSF
expression in cancer, the effects of various kinase inhibitors on
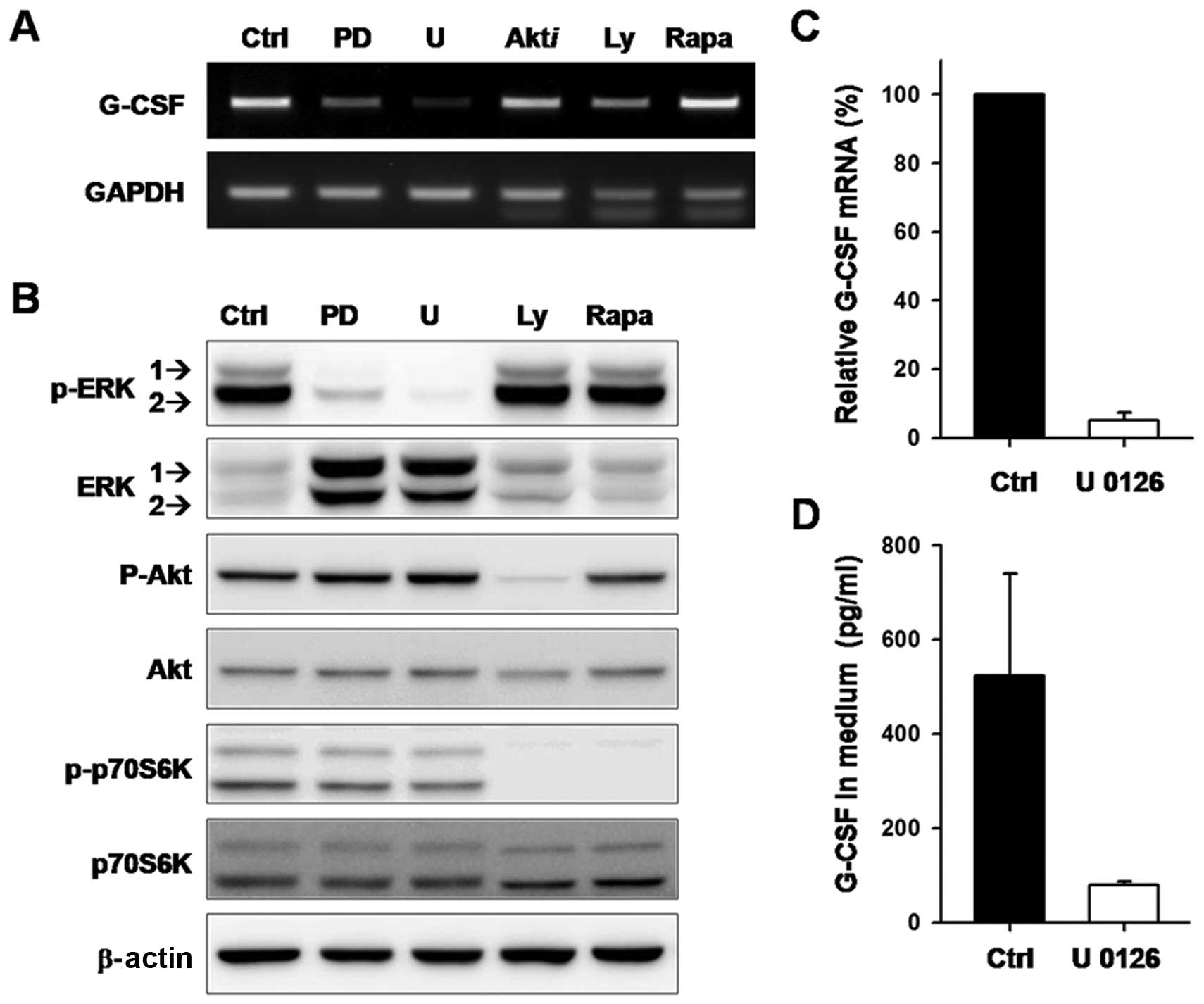
G-CSF expression were examined in SCC-15 cells. As shown in
Fig. 1A, a slight decrease in G-CSF
mRNA was detected in cells treated with inhibitors of Akt, PI3K or
mTOR when compared to the untreated and DMSO-treated control cells.
However, G-CSF mRNA was nearly eliminated in cells treated with
PD98059 or U0126 (Fig. 1A). The
signaling pathways obstructed by the specific inhibitors were
verified by western blotting (Fig.
1B). A decrease in phosphorylated ERK1/2 confirmed MEK1/2
inhibition by PD98059 and U0126. Blockage of Akt and p70S6K
phosphorylation confirmed PI3K inhibition by Ly294002. Suppression
of p70S6K phosphorylation confirmed mTOR inhibition by papamycin.
Downregulation of G-CSF mRNA expression in U0126-treated cells was
validated by quantitative real-time PCR (Fig. 1C). Moreover, the amount of G-CSF
protein decreased significantly after U0126 treatment for 48 h
(Fig. 1D). These results suggest
that the expression of G-CSF in SCC-15 cells is regulated through
the MEK-ERK signaling pathway.
G-CSF mRNA levels are correlated with
ERK1/2 activation and the invasiveness of cancer cells
Since elevated G-CSF levels have been detected in
the serum of patients with aggressive tumors, this study examined
whether G-CSF expression and ERK1/2 activation are correlated with
the invasiveness of cancer cells. Results of the Transwell invasion
assay in breast and lung cancer cell lines (Fig. 2A) confirmed previous studies that
MDA-MB-231 was the most invasive cell line among the breast cancer
cell lines analyzed. In vitro invasiveness was more evident
in CL1–5 than in CL1 lung cancer cells. Among these invasive cell
lines, G-CSF expression and ERK1/2 activation were detected in the
MDA-MB-231 and CL1–5 cells, but not in the less invasive cell lines
(Fig. 2B). These results suggest
that increased G-CSF expression and ERK1/2 activation may be
associated with cancer cell aggressiveness.
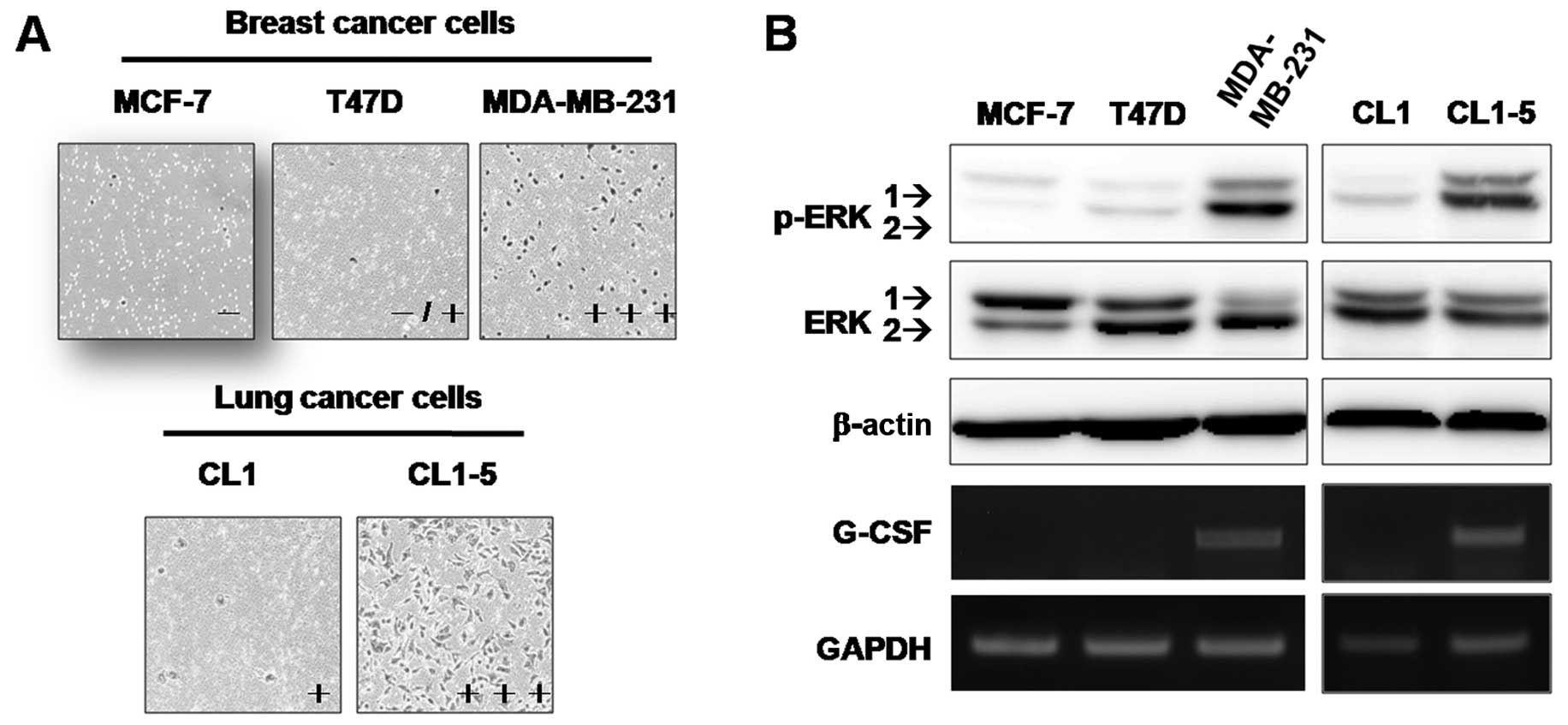 | Figure 2Relationship between expression of
granulocyte colony-stimulating factor (G-CSF), MEK activation, and
cancer cell invasiveness. (A) The invasiveness of breast and lung
cancer cells was evaluated using Matrigel-coated Transwell assay,
as described in Materials and methods. Invaded cells were counted
in 4 random fields using MetaMorph software. The number of invading
cells was the average of 3 independent experiments. Invasiveness of
each cell line was expressed as: −, <10 invasive cells; −/+,
>10 and <50 invasive cells; +, >50 and <200 invasive
cells; and +++, >300 invasive cells. (B) The levels of ERK1/2,
phospho-ERK1/2 (p-ERK1/2), and G-CSF mRNA in breast and lung cancer
cell lines were measured. Levels of β-actin protein and GAPDH mRNA
were used as internal controls for western blotting and RT-PCR,
respectively. |
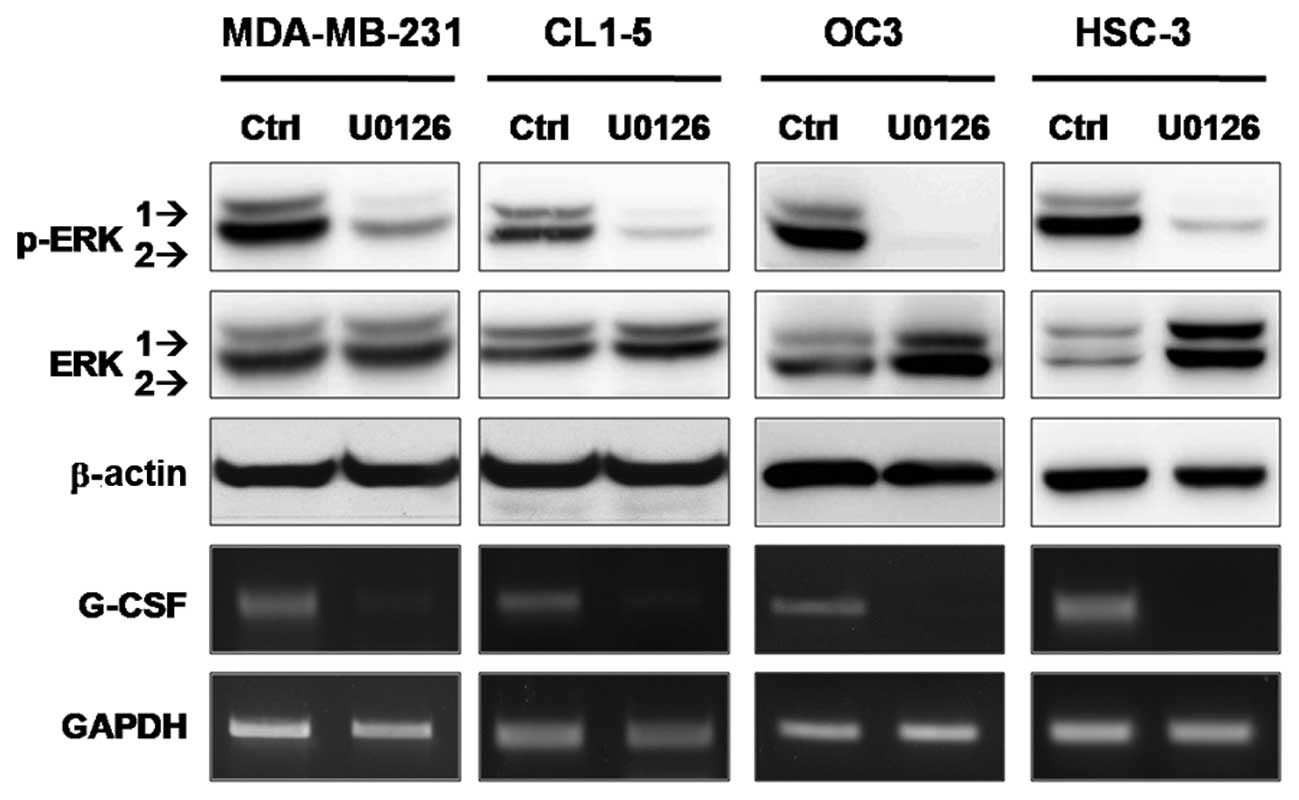
Treatment with U0126 drastically inhibits
the expression of tumor-derived G-CSF in all cancer cell lines
tested
To investigate whether the MAPK pathway is
responsible for G-CSF expression in cancer cell lines other than
SCC-15, a panel of invasive and G-CSF-producing cancer cells
(including MDA-MB-231, CL1–5, OC3 and HSC-3) were treated with
U0126. Based on the results of RT-PCR and western blot analyses
shown in Fig. 3, the expression of
G-CSF mRNA decreased dramatically (>90% inhibition determined by
RT-qPCR) when ERK1/2 phosphorylation was inhibited by U0126. These
results further emphasize the association between G-CSF expression
and ERK1/2 activation in cancer cells, suggesting that ERK1 and/or
ERK2 may affect the transcriptional regulation of G-CSF in
aggressive cancers.
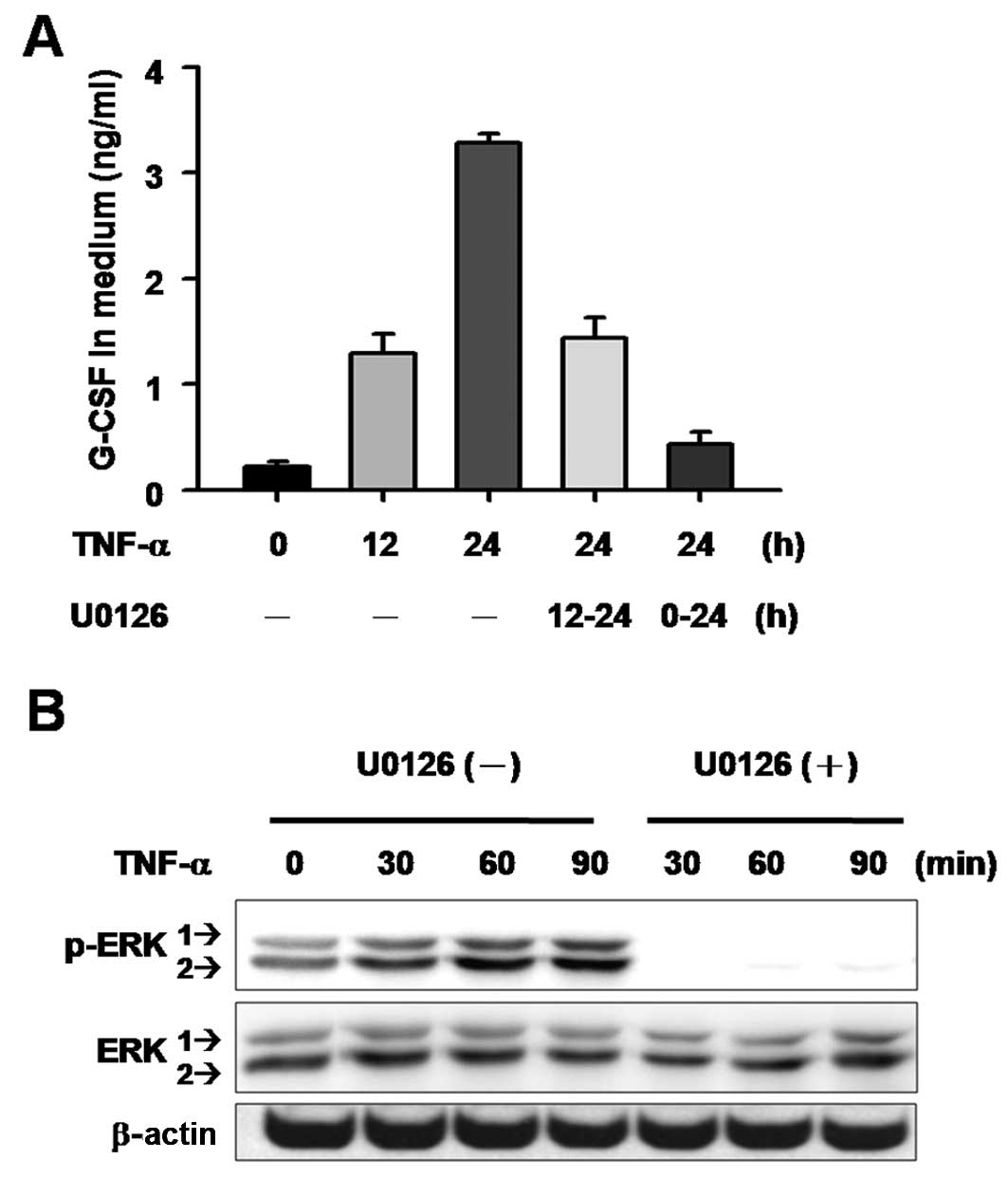
Blocking MEK activity suppresses
TNF-α-induced G-CSF expression in aggressive breast cancer
cells
Chronic inflammation with constitutive production of
TNF-α is associated with the steps in tumorigenesis in various
types of human cancers (11–13).
Koeffler et al(14) reported
that G-CSF production is stimulated by TNF-α in fibroblasts, and
the authors proposed the involvement of tumor-derived G-CSF in
TNF-α-mediated promotion of oncogenesis. The expression level of
G-CSF was relative lower in MDA-MB-231 when compared to the level
in the OC3 and HSC-3 cells. We then tested whether G-CSF production
in MDA-MB-231 cells can be further stimulated by TNF-α. Results
showed a time-dependent increase in G-CSF in culture medium after
TNF-α treatment (Fig. 4A), while
the TNF-α-induced G-CSF production was suppressed when U0126 was
added to the cells at either the start of or 12 h after TNF-α
treatment (Fig. 4A). Moreover, the
activation of ERK1/2 was analyzed using western blotting to confirm
the involvement of the MAPK pathway in TNF-α-induced G-CSF
expression. The results showed that TNF-α stimulated MEK activation
in a time-dependent manner, and phosphorylated ERK1 and ERK2 began
to increase as early as 30 min after TNF-α treatment. These effects
were completely inhibited by the presence of U0126 (Fig. 4B). These results suggest that ERK1/2
activation is involved in TNF-α-induced G-CSF production in cancer
cells.
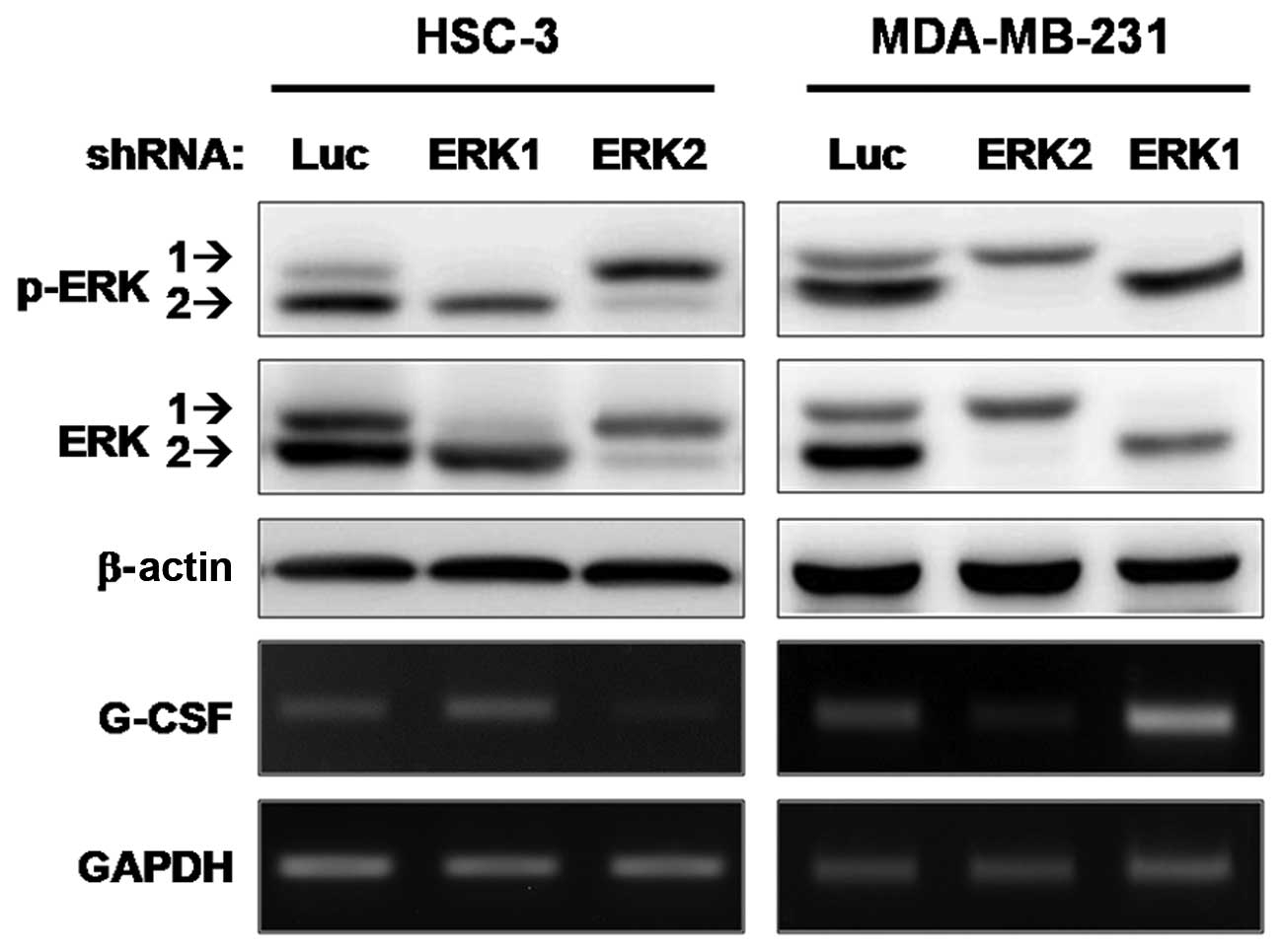
ERK2, and not ERK1, regulates G-CSF
expression in cancer cells
MEK1 and MEK2 are dual-specificity kinases that
share the consensus kinase motifs of both serine/threonine, and
tyrosine kinases. No catalytic substrates for MEK proteins have
been identified other than ERK1 and ERK2 (15). To determine whether ERK1 or ERK2
contributes to the transcriptional regulation of G-CSF, an
shRNA-knockdown experiment was performed. G-CSF mRNA was determined
in HSC-3 and MDA-MB-231 cells (which produce a high level of G-CSF
and show invasiveness in vitro) infected with a lentivirus
carrying shRNA targeting ERK1 or ERK2. Fig. 5 shows that, in both cell lines,
G-CSF expression was almost completely suppressed after shRNA
silencing of endogenous ERK2, whereas no significant changes at the
mRNA level of G-CSF occurred after ERK1 knockdown. These results
demonstrate that ERK2, but not ERK1, is the MAPK implicated in the
transcriptional regulation of G-CSF in cancer cells.
Discussion
Although recombinant G-CSF has been widely used in
cancer therapy to ameliorate neutropenia induced by anticancer
agents, the deleterious effects of tumor-derived G-CSF have been
noted in several studies including clinical case reports as well as
in vitro and in vivo experimental studies. The
detrimental activities of tumor-produced G-CSF include enhancement
of tumor growth, metastasis, and angiogenesis which also creates a
tumor-promoting microenvironment. This study found that higher
G-CSF expression and ERK activation were associated with the in
vitro invasiveness of breast and lung cancer cell lines. The
pro-inflammatory cytokine TNF-α-induced G-CSF expression in
MDA-MB-231 malignant breast cancer cells was suppressed by U0126
treatment accompanied by the blockade of ERK1/2 phosphorylation.
Moreover, a similar U0126 effect on G-CSF expression was
demonstrated in other invasive cancer cells. An shRNA knockdown
approach was used to show that ERK2, but not ERK1, is the main
regulator of G-CSF transcription in cancer cells, further
demonstrating that the MEK-ERK pathway may affect the regulation of
G-CSF expression.
The MAPK pathways are activated by a wide array of
extracellular stimuli, which trigger the sequential activation of
numerous protein kinases and modulate gene expression, metabolism,
cell proliferation, apoptosis and survival. MEK1 and MEK2 are
active in ~30% of all human cancers where MAPK signaling pathways
are implicated (16). Both ERK1 and
ERK2 are downstream of MEKs and have been considered
interchangeable, since both are similar in sequence, ubiquitously
expressed in most tissues, and share upstream activators. However,
recent studies have shown that ERK1 and ERK2 are not functionally
redundant and exhibit many diverse cellular roles (17,18).
The results of this study indicate that inhibition of ERK2 is
necessary and sufficient to reduce the level of G-CSF in malignant
cancer cells. Carlson et al(19) identified 80 proteins phosphorylated
by ERK2. These ERK2 substrates are associated with diverse cellular
functions: over 10 proteins are transcription factors or
regulators. By modifying these factors, ERK2 may alter the
expression of genes such as G-CSF. Thus, the ERK2-related
transcription regulators and molecular mechanisms controlling the
expression of G-CSF in invasive cancer cells should be further
investigated. The association between activated ERK2 and elevated
G-CSF levels in cancer cells may partially explain the enhanced
neo-angiogenesis and metastasis in tumors with constitutively
activated MAPK. Hyper-activated MAPK has been reported in many
types of cancers, and several inhibitors are currently under
investigation for their potential for targeting the RAS-RAF-MEK-ERK
axis and their application as oncology drugs. The findings in this
report suggest that targeting ERK2 may be a useful strategy for
treating G-CSF-producing cancers.
The expression of G-CSF is controlled at both the
transcriptional and post-transcriptional levels. Several
transcription factors have been identified, including Oct-1, Oct-2,
C/EBPβ, and NF-κB, which bind to the G-CSF promoter and regulate
G-CSF transcription. Recently, we reported that inhibiting the
PI3K/Akt/mTOR pathway resulted in downregulation of G-CSF
expression in LPS- or LTA-induced macrophages (20,21).
Rapamycin was found to decrease Oct-2 expression and subsequently
downregulate G-CSF expression in macrophages; however, it had no
effect on G-CSF expression in SCC-15 cells (Fig. 1). The reason for this is still
unclear, although it may be due to the fact that Oct-2 is expressed
mainly in lymphoid and neuronal cells while Oct-1 is sufficient to
support G-CSF expression in SCC-15 cells. Transcription factor
C/EBPβ, also known as NF-IL6, is involved in controlling cellular
proliferation, growth and differentiation. The phosphorylation of
C/EBPβ has been shown to be regulated by MEK-ERK signaling
(22). We also observed that the
interaction of ERK2 and C/EBPβ markedly upregulated G-CSF promoter
activity (data not shown). NF-κB is constitutively activated in a
range of cancers, and has been demonstrated to promote the
development, progression and metastasis of cancers including oral,
breast and lung cancers (23–25).
Thus, inhibition of NF-κB and the related signaling pathways may
also be another attractive target for cancer therapy. In addition,
the level of G-CSF is influenced by its mRNA stability. Multiple
AU-rich elements in the 3′ untranslated region of G-CSF mRNA imply
that mRNA stability can be regulated by p38 MAPK. However, we found
that the level of G-CSF mRNA is decreased in some types of cells,
but increased in other cell type when treated with SB203580 (a
p38-specific inhibitor). Therefore, the role of p38 MAPK in
regulating G-CSF mRNA stability requires further investigation.
In summary, this study identified ERK2 as a major
regulator of tumor-derived G-CSF expression and provides evidence
to support the use of ERK2 for prognostic purposes in patients with
unusually high G-CSF levels. Therapeutic approaches targeting
downstream transcription regulators of ERK2 may offer additional
strategies for abrogating G-CSF-mediated tumor progression to
improve the efficacy of anticancer treatment.
Acknowledgements
This study was supported by grants from the National
Science Council, Taiwan (NSC97-2320-B-002-057-MY3,
99-2320-B-038-009-MY3 and NSC100-2325-B-400-009), National Health
Research Institutes, Taiwan (CA-101-PP-01), and Department of
Health, Taiwan (DOH101-TD-C-111-004).
Abbreviations:
|
ERK
|
extracellular signal-regulated
kinase
|
|
G-CSF
|
granulocyte colony-stimulating
factor
|
|
MAPK
|
mitogen-activated protein kinase
|
|
shRNA
|
short hairpin RNA
|
|
TNF-α
|
tumor necrosis factor-α
|
References
|
1
|
Welte K, Platzer E, Lu L, Gabrilove JL,
Levi E, Mertelsmann R and Moore MA: Purification and biochemical
characterization of human pluripotent hematopoietic
colony-stimulating factor. Proc Natl Acad Sci USA. 82:1526–1530.
1985. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Nicola NA and Metcalf D: Binding of the
differentiation-inducer, granulocyte-colony-stimulating factor, to
responsive but not unresponsive leukemic cell lines. Proc Natl Acad
Sci USA. 81:3765–3769. 1984. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Demetri GD and Griffin JD: Granulocyte
colony-stimulating factor and its receptor. Blood. 78:2791–2808.
1991.PubMed/NCBI
|
|
4
|
Kawakami M, Tsutsumi H, Kumakawa T, Abe H,
Hirai M, Kurosawa S, et al: Levels of serum granulocyte
colony-stimulating factor in patients with infections. Blood.
76:1962–1964. 1990.PubMed/NCBI
|
|
5
|
Dale DC: Colony-stimulating factors for
the management of neutropenia in cancer patients. Drugs. 62(Suppl
1): 1–15. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Carl W and Havens J: The cancer patient
with severe mucositis. Curr Rev Pain. 4:197–202. 2000. View Article : Google Scholar
|
|
7
|
Obermueller E, Vosseler S, Fusenig NE and
Mueller MM: Cooperative autocrine and paracrine functions of
granulocyte colony-stimulating factor and granulocyte-macrophage
colony-stimulating factor in the progression of skin carcinoma
cells. Cancer Res. 64:7801–7812. 2004. View Article : Google Scholar
|
|
8
|
Mueller MM, Peter W, Mappes M, Huelsen A,
Steinbauer H, Boukamp P, et al: Tumor progression of skin carcinoma
cells in vivo promoted by clonal selection, mutagenesis, and
autocrine growth regulation by granulocyte colony-stimulating
factor and granulocyte-macrophage colony-stimulating factor. Am J
Pathol. 159:1567–1579. 2001. View Article : Google Scholar
|
|
9
|
Braun B, Lange M, Oeckler R and Mueller
MM: Expression of G-CSF and GM-CSF in human meningiomas correlates
with increased tumor proliferation and vascularization. J
Neurooncol. 68:131–140. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Chu YW, Yang PC, Yang SC, Shyu YC, Hendrix
MJ, Wu R, et al: Selection of invasive and metastatic
subpopulations from a human lung adenocarcinoma cell line. Am J
Respir Cell Mol Biol. 17:353–360. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Egberts JH, Cloosters V, Noack A,
Schniewind B, Thon L, Klose S, et al: Anti-tumor necrosis factor
therapy inhibits pancreatic tumor growth and metastasis. Cancer
Res. 68:1443–1450. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Kulbe H, Thompson R, Wilson JL, Robinson
S, Hagemann T, Fatah R, et al: The inflammatory cytokine tumor
necrosis factor-α generates an autocrine tumor-promoting network in
epithelial ovarian cancer cells. Cancer Res. 67:585–592. 2007.
|
|
13
|
Stathopoulos GT, Kollintza A, Moschos C,
Psallidas I, Sherrill TP, Pitsinos EN, et al: Tumor necrosis
factor-α promotes malignant pleural effusion. Cancer Res.
67:9825–9834. 2007.
|
|
14
|
Koeffler HP, Gasson J, Ranyard J, Souza L,
Shepard M and Munker R: Recombinant human TNF alpha stimulates
production of granulocyte colony-stimulating factor. Blood.
70:55–59. 1987.PubMed/NCBI
|
|
15
|
Seger R, Ahn NG, Posada J, Munar ES,
Jensen AM, Cooper JA, et al: Purification and characterization of
mitogen-activated protein kinase activator(s) from epidermal growth
factor-stimulated A431 cells. J Biol Chem. 267:14373–14381.
1992.PubMed/NCBI
|
|
16
|
Hoshino R, Chatani Y, Yamori T, Tsuruo T,
Oka H, Yoshida O, et al: Constitutive activation of the 41-/43-kDa
mitogen-activated protein kinase signaling pathway in human tumors.
Oncogene. 18:813–822. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Vantaggiato C, Formentini I, Bondanza A,
Bonini C, Naldini L and Brambilla R: ERK1 and ERK2
mitogen-activated protein kinases affect Ras-dependent cell
signaling differentially. J Biol. 5:142006. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Yao Y, Li W, Wu J, Germann UA, Su MS,
Kuida K and Boucher DM: Extracellular signal-regulated kinase 2 is
necessary for mesoderm differentiation. Proc Natl Acad Sci USA.
100:12759–12764. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Carlson SM, Chouinard CR, Labadorf A, Lam
CJ, Schmelzle K, Fraenkel E and White FM: Large-scale discovery of
ERK2 substrates identifies ERK-mediated transcriptional regulation
by ETV3. Sci Signal. 4:rs112011. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Chou YY and Lu SC: Inhibition by rapamycin
of the lipoteichoic acid-induced granulocyte-colony stimulating
factor expression in mouse macrophages. Arch Biochem Biophys.
508:110–119. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Chou YY, Gao JI, Chang SF, Chang PY and Lu
SC: Rapamycin inhibits lipopolysaccharide induction of
granulocyte-colony stimulating factor and inducible nitric oxide
synthase expression in macrophages by reducing the levels of
octamer-binding factor-2. FEBS J. 278:85–96. 2011. View Article : Google Scholar
|
|
22
|
Armstrong DA, Phelps LN and Vincenti MP:
CCAAT enhancer binding protein-β regulates matrix
metalloproteinase-1 expression in interleukin-1β-stimulated A549
lung carcinoma cells. Mol Cancer Res. 7:1517–1524. 2009.
|
|
23
|
Coussens LM and Werb Z: Inflammation and
cancer. Nature. 420:860–867. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Karin M: Nuclear factor-κB in cancer
development and progression. Nature. 441:431–436. 2006.
|
|
25
|
Huber MA, Azoitei N, Baumann B, Grünert S,
Sommer A, Pehamberger H, et al: NF-κB is essential for
epithelial-mesenchymal transition and metastasis in a model of
breast cancer progression. J Clin Invest. 114:569–581. 2004.
|