Introduction
Selective and specific induction of apoptotic cell
death in cancer cells has been increasingly recognized as a
promising therapeutic approach for the treatment of many types of
cancers. The tumor necrosis factor (TNF)-related apoptosis-inducing
ligand (TRAIL), a member of the TNF ligand superfamily, is
considered as a promising anticancer agent in cancer therapy due to
its ability to induce apoptosis in a variety of tumor cell types
while having no significant side-effects on normal cells (1,2).
Therefore, the discovery of agents that induce TRAIL and promote
TRAIL-mediated apoptotic cell death has received considerable
attention. TRAIL induces apoptosis in cancer cells via the death
receptor pathway using a mechanism similar to that of TNF (2–4).
Although many cancer cells express functional TRAIL receptors, such
as death receptor 4 (DR4) and DR5, resistance to TRAIL is common
due to a decreased level or mutation in DR4 and DR5 or the loss of
distal signaling cascades (4–6). For
these reasons, TRAIL is not recommended for use as a single agent;
therefore, chemotherapeutic agents that can sensitize cells to
TRAIL-mediated apoptosis are urgently needed.
The polyadenylation inhibitor cordycepin
(3′-deoxyadenosine) is an active component of the caterpillar
fungus Cordyceps militaris. Due to the absence of oxygen in
the 3′ position of its ribose moiety, incorporation of cordycepin
during RNA synthesis results in termination of chain elongation
(7,8). This activity has been well described
in vitro with purified RNA polymerases and poly(A)
polymerases from a number of organisms, including yeasts and
mammals (9,10). Cordycepin has anticancer activities
including induction of apoptosis, DNA double-strand break activity,
and cell cycle arrest in cancer cells (11–15).
This compound also inhibits metastasis and angiogenesis (16–19).
Despite these observations, the molecular mechanisms underlying the
anticancer effects of cordycepin on TRAIL-mediated apoptosis have
not been fully elucidated.
In the present study, we investigated the mechanisms
involved in cordycepin-induced apoptosis in TRAIL-resistant Hep3B
human hepatocellular carcinoma cells. Our results indicate that a
combined treatment with subtoxic concentrations of cordycepin and
TRAIL dramatically induced Hep3B cell death through activation of
caspase and loss of mitochondrial membrane potential (MMP) via
inhibition of c-Jun N-terminal kinase (JNK) signaling. Therefore, a
combined treatment with cordycepin and TRAIL may synergistically
stimulate and accelerate the JNK-mediated apoptotic signaling
pathway.
Materials and methods
Regents and antibodies
Cordycepin (MW, 251.2; product no. C3394) from C.
militaris, 4′,6-diamidino-2-phenylindole (DAPI),
N-acetyl-L-cysteine,
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT),
5,5′,6,6′-tetra- chloro-1,1′,3,3′
tetraethylbenzimidazolylcarbocyanine iodide (JC-1) and propidium
iodide (PI) were obtained from Sigma-Aldrich Chemical Co. (St.
Louis, MO, USA). Caspase activity assay kits were obtained from
R&D Systems (Minneapolis, MN, USA). An enhanced
chemiluminescence (ECL) kit and RNeasy kit were purchased from
Amersham Corp. (Arlington Heights, IL, USA) and Qiagen (La Jolla,
CA, USA), respectively. RPMI-1640 medium and fetal bovine serum
(FBS) were purchased from Invitrogen Life Technologies (Carlsbad,
CA, USA) and Gibco-BRL (Gaithersburg, MD, USA), respectively. All
other chemicals were purchased from Sigma-Aldrich. Antibodies
against cIAP-1, cIAP-2, XIAP, Bcl-2, Bcl-xL, Bax, Bid,
poly(ADP-ribose) polymerase (PARP), β-catenin, capase-3, -8 and -9
were purchased form Santa Cruz Biotechnology Inc. (Santa Cruz, CA,
USA). Antibodies against extracellular signal-regulated kinase
(ERK), phospho (p)-ERK, p38 mitogen-activated protein kinase
(MAPK), p-p38 MAPK, JNK, p-JNK, Akt and p-Akt were purchased from
BD Biosciences (San Jose, CA, USA). Antibody against actin was from
Sigma-Aldrich. Peroxidase-labeled donkey anti-rabbit and sheep
anti-mouse immunoglobulin were purchased from Amersham Corp.
Cell culture and MTT assay
Hep3B human hepatocellular carcinoma cells were
obtained from the American Type Culture Collection (Manassas, VA,
USA). Cells were cultured at 37ºC in a 5% CO2 humidified
incubator, and maintained in RPMI-1640 culture medium containing
10% heat-inactivated FBS. For the cell viability assay, cells were
grown to 70% confluence and treated with the indicated
concentrations of cordycepin, TRAIL, or a combined treatment
(cordycepin plus TRAIL). After treatment, MTT working solution was
added to 6-well culture plates and incubated continuously at 37ºC
for 2 h. The culture supernatant was removed from the wells, and
DMSO was added to dissolve the formazan crystals. The absorbance of
each well was measured at 540 nm with an enzyme-linked
immunosorbent assay (ELISA) reader (Molecular Devices, Sunnyvale,
CA, USA).
Nuclear staining
After treatment with cordycepin and TRAIL alone or
together for the indicated times, the cells were harvested, washed
with phosphate-buffered saline (PBS) and fixed with 3.7%
paraformaldehyde in PBS for 10 min at room temperature. Fixed cells
were washed with PBS, and stained with 2.5 μg/ml DAPI solution for
10 min at room temperature. The cells were washed 2 more times with
PBS and analyzed via a fluorescence microscope (Carl Zeiss,
Oberkochen, Germany).
Flow cytometric analysis for assessment
of the sub-G1 phase population
The cells were washed twice with cold PBS and then
centrifuged. The resulting pellet was fixed in 75% (v/v) ethanol
for 1 h at 4ºC. The cells were washed once with PBS and resuspended
in cold PI solution (50 μg/ml) containing RNase A (0.1 mg/ml) in
PBS for 30 min in the dark. Flow cytometric analyses were performed
using FACSCalibur (Becton-Dickinson, San Jose, CA, USA). Forward
light scatter characteristics were used to exclude cell debris from
the analysis. The sub-G1 population was calculated to estimate the
apoptotic cell population.
Determination of caspase activity
Caspase activities were determined by colorimetric
assays using caspase-3, -8 and -9 activation kits according to the
manufacturer’s protocol. The kits utilize synthetic tetrapeptides
labeled with p-nitroaniline. Briefly, the cells were lysed in the
supplied lysis buffer. The supernatants were collected and
incubated with the supplied reaction buffer containing
dithiothreitol (DTT) and substrates at 37ºC. Caspase activity was
determined by measuring changes in absorbance at 405 nm using an
ELISA reader.
RNA extraction and reverse
transcription-polymerase chain reaction (RT-PCR)
Total RNA was prepared using an RNeasy kit and
primed with random hexamers to synthesize complementary DNA using
AMV reverse transcriptase (Amersham Corp.) according to the
manufacturer’s instructions. PCR was carried out in a Mastercycler
(Eppendorf, Hamburg, Germany) with the indicated primers.
Conditions for the PCR reactions were, 1× (94ºC for 3 min); 35×
(94ºC for 45 sec, 58ºC for 45 sec and 72ºC for 1 min); and 1× (72ºC
for 10 min). Amplification products obtained by PCR were
electrophoretically separated on 1% agarose gels and visualized by
ethidium bromide staining.
Protein extraction and western
blotting
The cells were harvested and lysed with lysis buffer
(20 mM sucrose, 1 mM EDTA, 20 μM Tris-Cl, pH 7.2, 1 mM DTT, 10 mM
KCl, 1.5 mM MgCl2, and 5 μg/ml aprotinin) for 30 min.
Protein concentration was measured using a Bio-Rad protein assay
(Bio-Rad Laboratories, Hercules, CA, USA) according to the
manufacturer’s instructions. For western blot analysis, an equal
amount of protein was subjected to electrophoresis on
SDS-polyacrylamide gels and was transferred by electroblotting to a
nitrocellulose membrane (Schleicher & Schuell, Keene, NH, USA).
The blots were probed with the desired antibodies for 1 h,
incubated with the diluted enzyme-linked secondary antibody, and
visualized with an ECL solution according to the recommended
procedure.
Measurement of MMP (ΔΨm)
The MMP of intact cells was measured by DNA flow
cytometry with the lipophilic cation JC-1. JC-1 is a rationmetric,
dual-emission fluorescent dye that is internalized and concentrated
by respiring mitochondria and reflects changes in MMP in living
cells. There are two excitation wavelengths. At low values of MMP,
JC-1 remains a monomer (FL-1, green fluorescence; 527 nm) whereas
it forms aggregates at high MMP (FL-2, red fluorescence; 590 nm)
according to the recommended procedure (20). Cells were trypsinized, the cell
pellets were resuspended in PBS, and incubated with 10 μM JC-1 for
30 min at 37ºC. The cells were subsequently washed once with cold
PBS, suspended and analyzed by flow cytometry.
Statistical analysis
All data are presented as means ± standard
deviation. Significant differences among the groups were determined
using the unpaired Student’s t-test. A p-value <0.05 was
considered to indicate a statistically significant result. All
values were obtained from at least 2 or 3 independent
experiments.
Results
Induction of apoptosis by co-treatment
with cordycepin and TRAIL in Hep3B cells
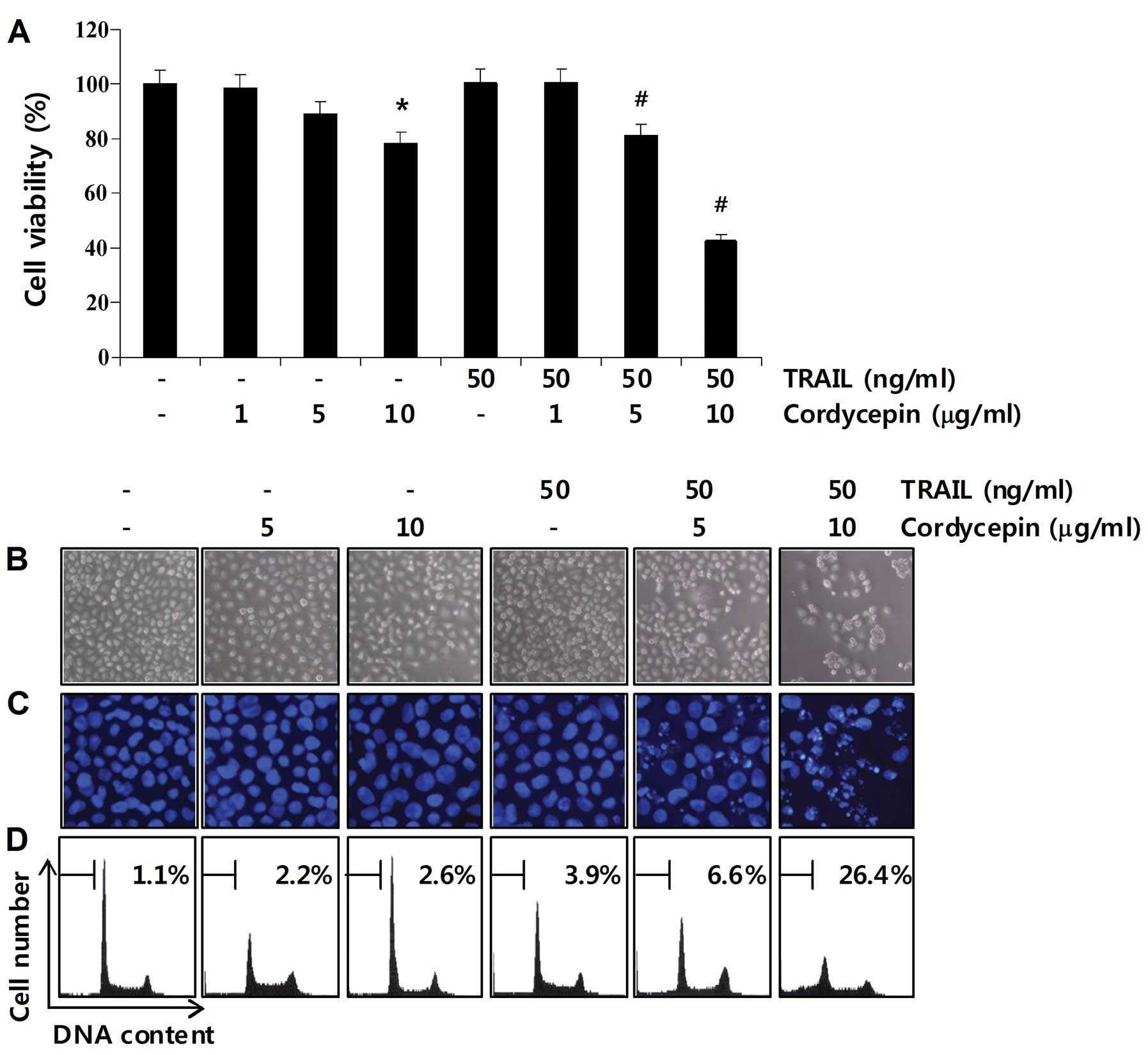
We first assessed whether cordycepin sensitizes
Hep3B cells to TRAIL-mediated apoptosis. Treatment with 50 ng/ml
TRAIL resulted in negligible growth inhibition of Hep3B cells at 24
h (Fig. 1A), suggesting that this
cell type is resistant to TRAIL-induced apoptosis. We next examined
the cytotoxic effects of cordycepin alone or in combination with
TRAIL in Hep3B cells. The concentrations (1–10 μg/ml) of cordycepin
alone used in this study did not significantly induce morphological
changes or inhibit growth. However, cell viability was
significantly inhibited by the co-treatment with cordycepin and
TRAIL (Fig. 1A) indicating that the
combination of cordycepin and TRAIL substantially induces cell
death. To investigate whether decreased cell viability by the
combined treatment was due to apoptotic signaling, morphological
changes and sub-G1 phase populations of Hep3B cells were determined
using DAPI staining and flow cytometric analysis, respectively. As
shown in Fig. 1B and C, a
significant number of cells co-treated with cordycepin and TRAIL
were observed with apoptotic shrinkage, chromatin condensation,
loss of nuclear construction and formation of apoptotic bodies,
whereas these features were not observed in control cells or cells
treated with cordycepin or TRAIL alone. Additionally, treatment of
Hep3B cells with a combination of cordycepin and TRAIL
significantly increased the accumulation of sub-G1 phase cells,
whereas treatment with cordycepin or TRAIL alone did not (Fig. 1D). These results showed that the
combined treatment of cordycepin and TRAIL sensitized
TRAIL-resistant Hep3B cells to apoptosis.
Effects of cordycepin and TRAIL
co-treatment on the expression of Bcl-2 and IAP family members and
MMP values
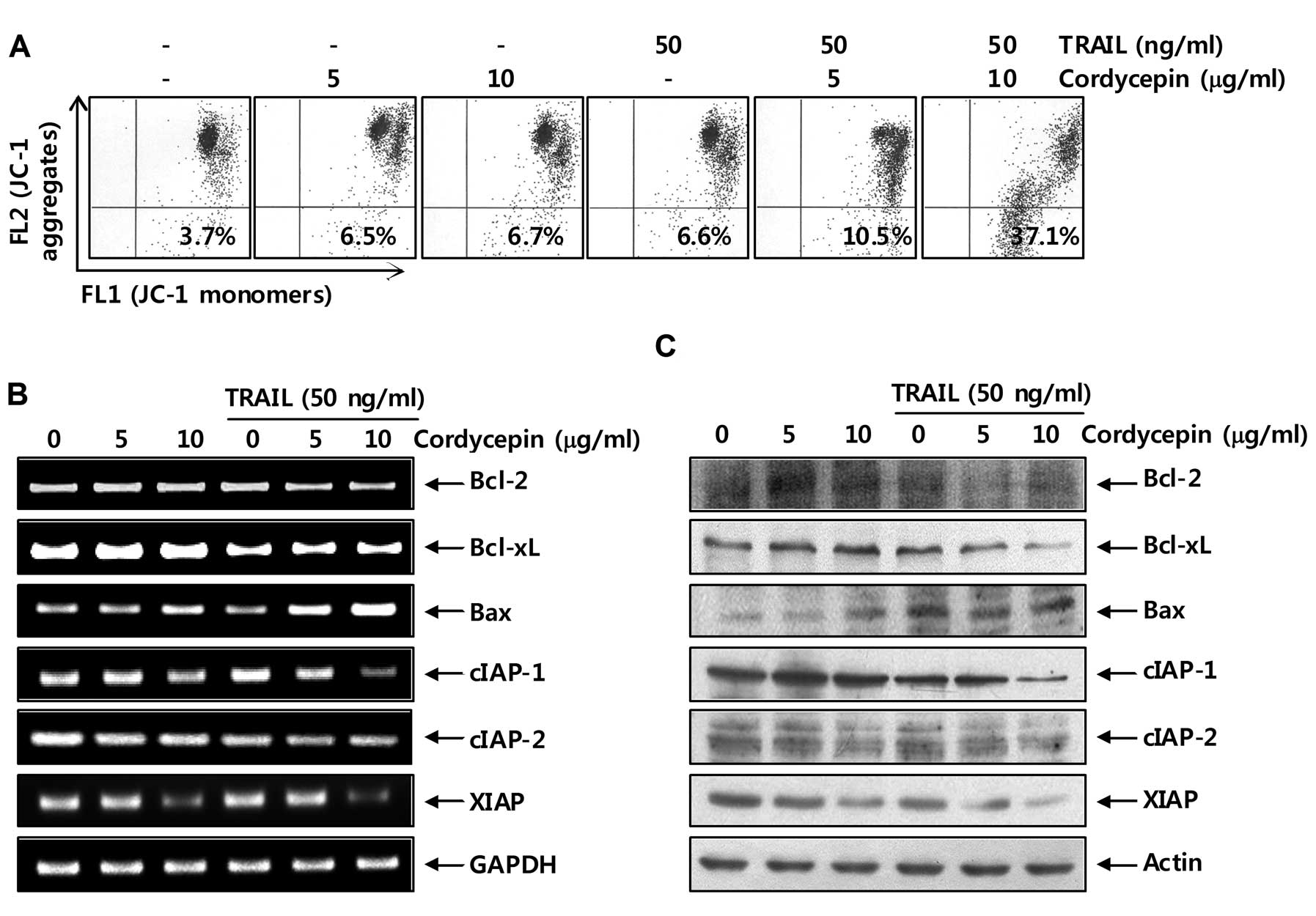
Mitochondria appear to play a central role in
apoptosis. Thus, they have been a major focus of recent studies.
The early events that occur during apoptotic cell death are
mitochondrial depolarization and loss of pro-apoptotic factors from
the mitochondrial inter-membrane space (21,22).
As shown in Fig. 2A, treatment of
cells with cordycepin or TRAIL alone induced a slight loss of MMP
in Hep3B cells; however, combined treatment with cordycepin and
TRAIL caused a significant concentration-dependent induction of MMP
loss. In particular, the anti-apoptotic Bcl-2 family molecules such
as Bcl-2 and Bcl-xL, and IAP family members protect some tumor cell
lines from TRAIL-induced apoptosis (22–24);
therefore, we investigated the expression levels of members
belonging to the Bcl-2 and IAP family. As shown in Fig. 2B and C, the mRNA and protein levels
of anti-apoptotic Bcl-2, Bcl-xL, cIAP-1, cIAP-2 and XIAP were
reduced in response to treatment with cordycepin plus TRAIL,
whereas the levels of pro-apoptotic Bax increased markedly.
Collectively, these results indicate that downregulation of Bcl-2
and Bcl-xL, and IAP family member expression and increased loss of
MMP are associated with cordycepin-mediated sensitization of Hep3B
cells to TRAIL-mediated apoptosis.
Effects of co-treatment with cordycepin
and TRAIL on caspase activation
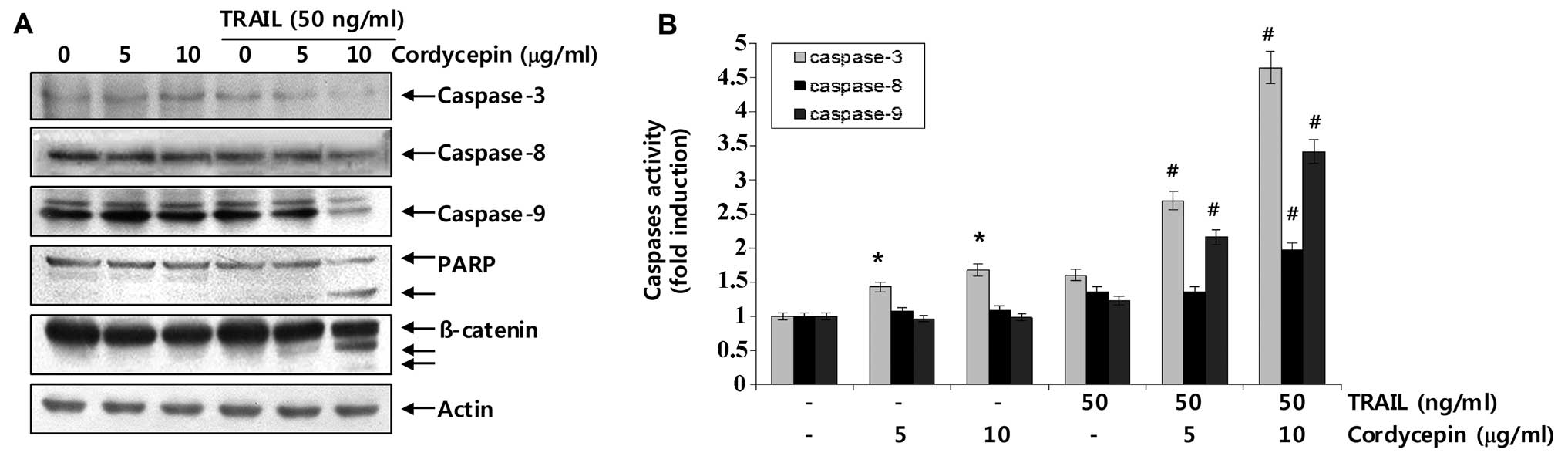
Caspases act as important mediators of apoptosis and
contribute to the overall apoptotic morphology by cleaving various
cellular substrates (24,25). Therefore, we next examined whether
caspases were actually activated during cordycepin plus
TRAIL-induced cell death in Hep3B cells. As shown in Fig. 3A, western blot analysis revealed
that treatment with 5 or 10 μg/ml cordycepin and 50 ng/ml TRAIL
alone for 24 h did not significantly decrease proteolytic
processing of caspase-3, -8 and -9; however, combined treatment
with cordycepin and TRAIL significantly decreased their levels.
Furthermore, we found that cleavage of key death substrates
indicates activation of effector caspases such as PARP and
β-catenin (activated-caspase-3 substrates) (26,27).
As a result, combined treatment with cordycepin and TRAIL
significantly induced cleavage of PARP and β-catenin in Hep3B
cells, whereas treatment with cordycepin or TRAIL alone did not.
Additionally, cell lysates containing equal amounts of total
protein from cells were assayed to assess in vitro caspase
activity. As shown in Fig. 3B, a
combined treatment with cordycepin and TRAIL significantly
increased caspase-3, -8, and -9 activities in Hep3B cells. These
results indicate that co-treatment induces apoptotic death in Hep3B
cells, at least in part, through a caspase-dependent pathway.
Effects of mitogen-activated protein
kinases (MAPKs) on cordycepin plus TRAIL-induced apoptosis
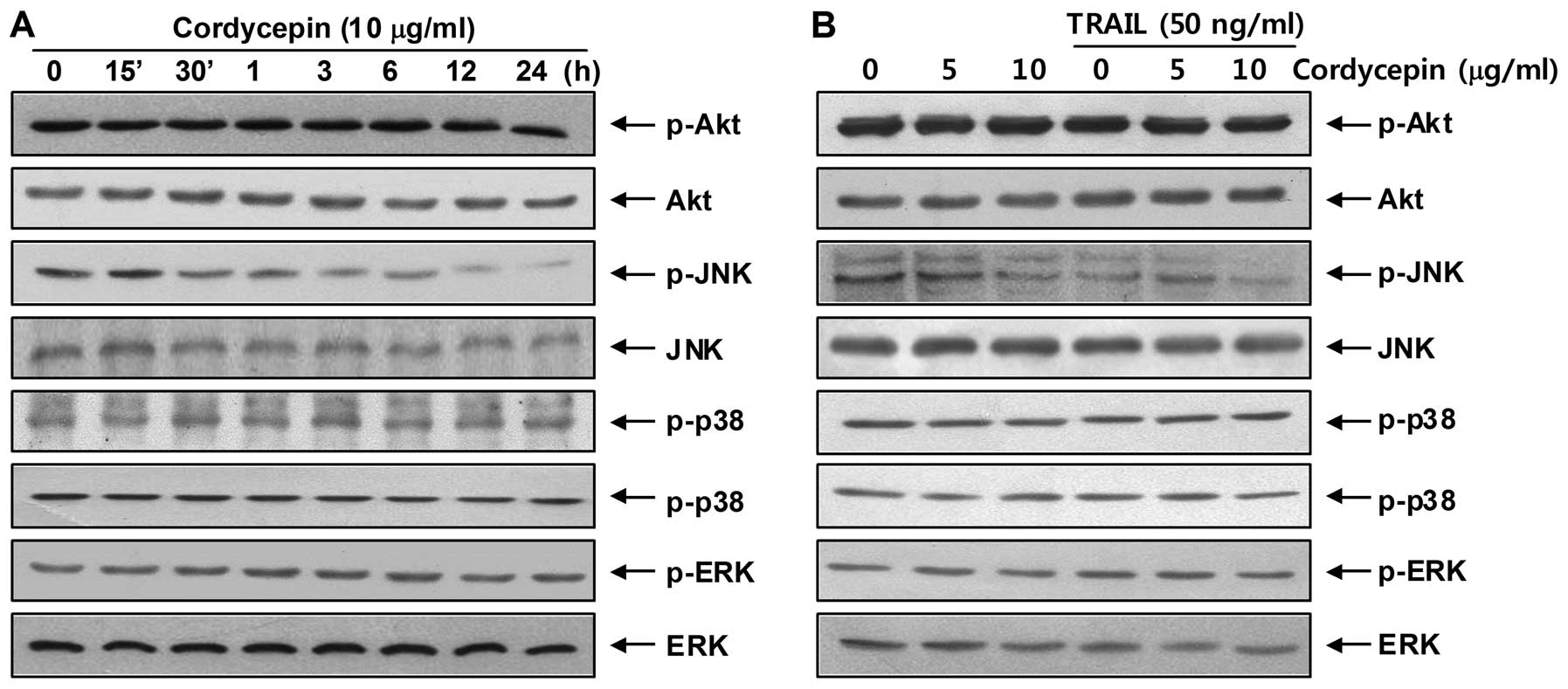
Recent studies have revealed that phosphoinositide
3-kinase (PI3K)/Akt and the MAPK signaling pathways are important
regulators during apoptotic cell regulation as a TRAIL sensitizing
signal pathway (28,29). To investigate the roles of Akt and
MAPKs in mediating the observed apoptotic response, western blot
analysis was used to assess the change in Akt, JNK, ERK, and p38
MAPK phosphorylation. As shown in Fig.
4, treatment with cordycepin decreased only JNK
phosphorylation, whereas phosphorylation levels of other kinases,
such as Akt, p38 and ERK, did not change. The results also indicate
that cordycepin exerted a concentration-dependent effect on JNK
dephosphorylation with a fixed concentration of TRAIL. Thus, Hep3B
cells were incubated with cordycepin and TRAIL in the presence of
SP600125, a well-known JNK inhibitor, to investigate the functional
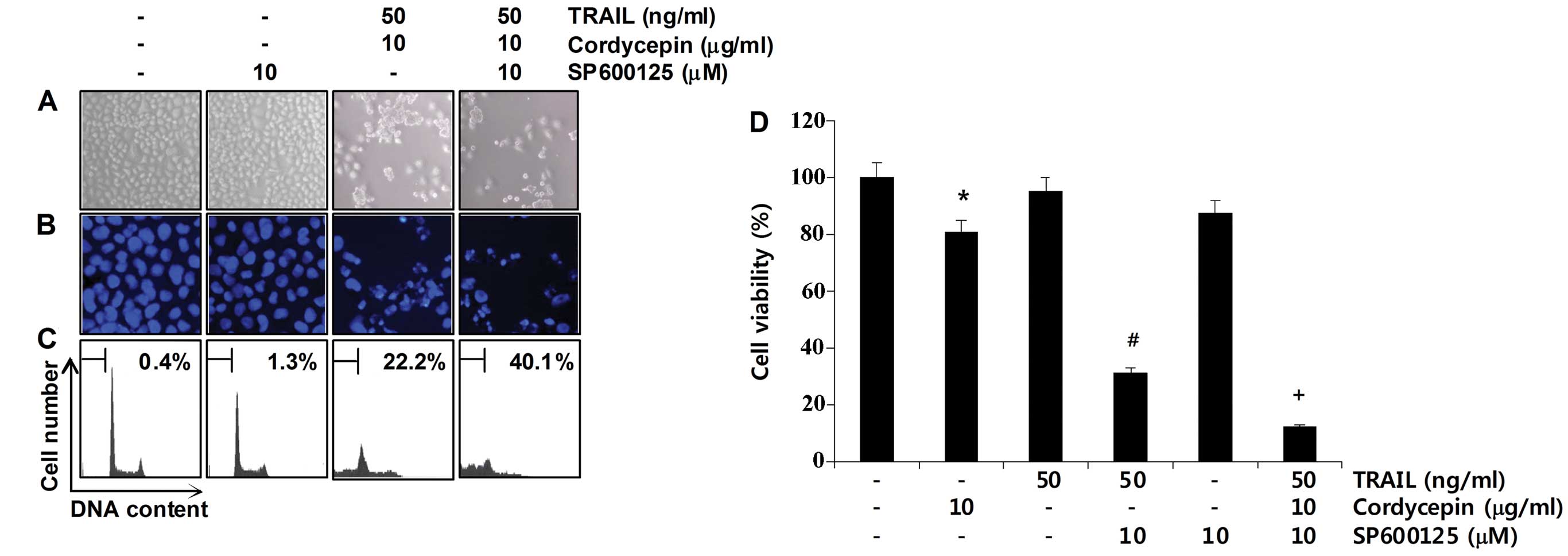
roles of these dephosphorylation events. As shown in Fig. 5A–C, pretreatment with SP600125
markedly increased the morphological changes and condensed
chromatin in the nuclei, and the number of cells with sub-G1 DNA
content. Consistent with the increase in sub-G1 DNA content,
pretreatment with SP600125 significantly increased the growth
inhibition induced by the combined treatment (Fig. 5D). These results indicate that the
combined treatment with cordycepin and TRAIL triggered the
inhibition of JNK activation; therefore, we verified the inhibition
of JNK signaling responsible for the TRAIL-sensitizing effect of
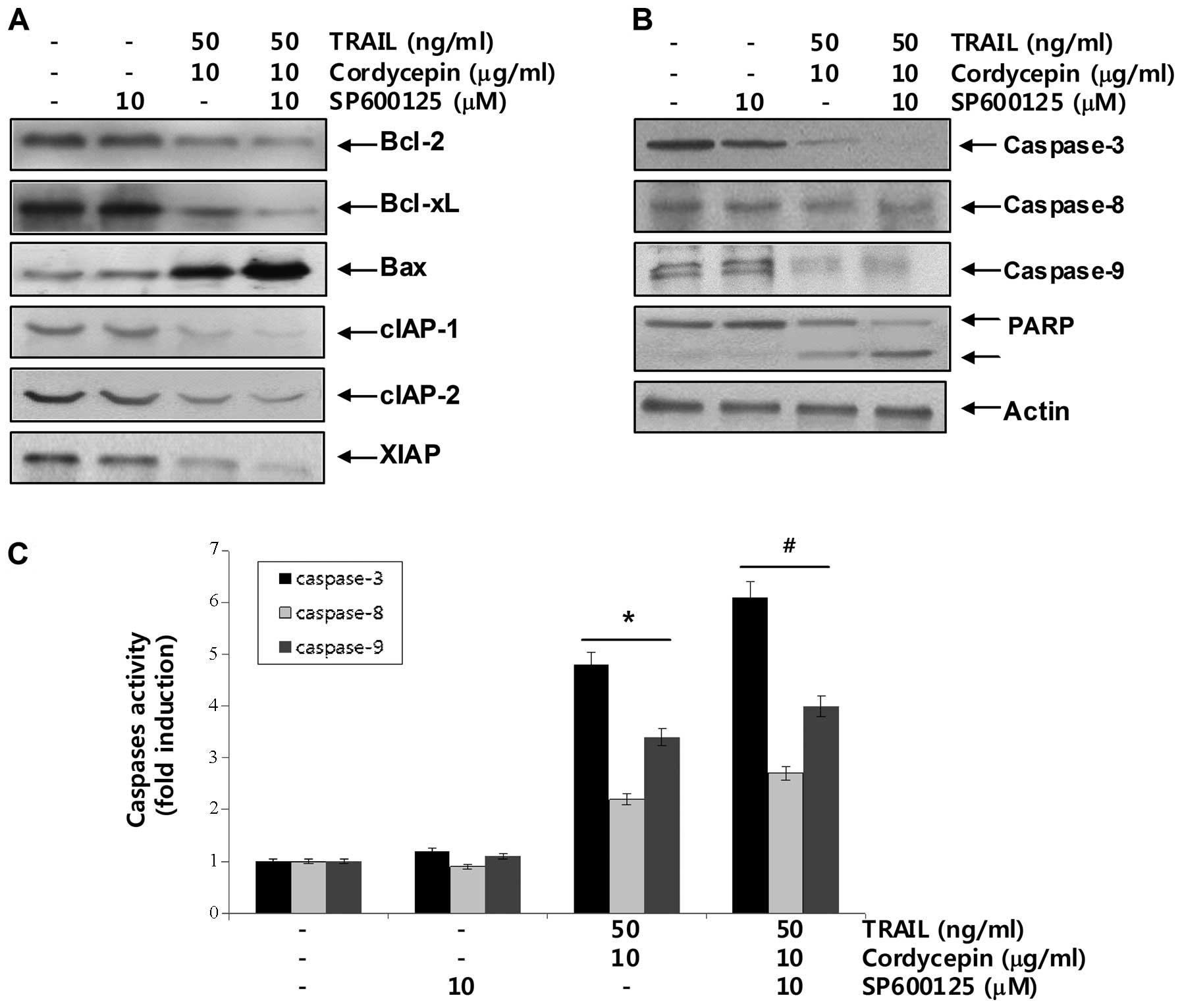
cordycepin on apoptosis in Hep3B cells. As a result, SP600125
enhanced upregulation of pro-apoptotic protein Bax levels and
downregulation of anti-apoptotic Bcl-2, Bcl-xL and IAP family
protein levels in TRAIL and cordycepin-treated Hep3B cells
(Fig. 6A). Moreover, SP600125
enhanced caspase activities, as well as PARP and β-catenin cleavage
under the same experimental conditions. Taken together, these
findings suggest that the JNK signaling pathway acts as a key
regulator of apoptosis in response to the combined cordycepin and
TRAIL treatment in Hep3B cells.
Discussion
TRAIL is a member of the TNF superfamily and is able
to trigger programmed cell death. When cells receive the cell death
signal, TRAIL binds its receptors, such as DR4 and DR5, which are
located at the cell surface, and form a death-inducing signaling
complex (DISC) that triggers activation of the caspase cascade
(1,2). As results, TRAIL initiates the
extrinsic cell death pathway through formation of DISC and
activation of caspases (2,30,31).
In addition, TRAIL is relatively non-toxic to normal cells, but it
can selectively induce apoptotic cell death in many types of
transformed or malignant cells. Thus, TRAIL is a major mediator of
acquired immune tumor surveillance and is currently being tested in
clinical trials as a novel cancer therapy. However, recent reports
have demonstrated that many cancer cells, including the
hepatocellular carcinoma Hep3B cell line (3,11),
develops resistance to the apoptotic effects of TRAIL. In this
study, we assessed the sensitizing effects of cordycepin on
TRAIL-induced apoptosis in TRAIL-resistant hepatocellular carcinoma
Hep3B cells.
The process of apoptosis is controlled by a wide
range of cellular signals, which can be divided into extrinsic and
intrinsic pathways. Apoptosis requires caspase activity, and
caspases become active when cleaved. Adaptor proteins facilitate
the auto-cleavage of initiator caspases (e.g., caspase-8 and -9),
initiator caspases cleave effector caspases (e.g., caspase-3 and
-7), and effector caspases disrupt the cell function to elicit cell
death (21,22). Two events signal adaptor-mediated
caspase cleavage: the binding of ligands to death receptors (the
death receptor pathway), and the release of apoptotic factors such
as cytochrome c from mitochondria (the mitochondrial
pathway). Death receptors activate caspase-8, whereas cytochrome
c activates caspase-9. Caspase-3 is common to both pathways.
Many proteins modulate apoptotic signaling when apoptosis occurs,
including the Bcl-2 and IAP family proteins (22–24).
The Bcl-2 proteins such as multidomain apoptotic (Bax and Bak),
single domain apoptotic (termed BH3-only), and anti-apoptotic
(Bcl-2 and Bcl-xL) proteins damage or protect mitochondria
(32,33). In contrast, IAPs inactivate cleaved
caspases; thus, they impede the apoptotic process once it has
begun. Caspases targeted by IAPs include caspase-9 and -3 but not
caspase-8 (24,25). Our results showed that treatment
with TRAIL in combination with nontoxic concentrations of
cordycepin sensitized TRAIL-resistant Hep3B cells to TRAIL-mediated
apoptosis. Stimulation by cordycepin and TRAIL increased chromatin
condensation and the sub-G1 phase DNA content (Fig. 1). This apoptosis was associated with
downregulation of the IAP family members, such as cIAP-1, -2 and
XIAP, and anti-apoptotic Bcl-2 and Bcl-xL, and upregulation of
pro-apoptotic Bax (Fig. 2). We also
demonstrated that treatment with TRAIL and cordycepin facilitated
activation of caspase-3, -8 and -9, and concomitant degradation of
PARP and β-catenin (Fig. 3). These
data suggest that apoptosis induced by co-treatment with cordycepin
and TRAIL was caspase-dependent.
The PI3K/Akt and MAPK signaling pathways have been
associated with cellular functions as well as
TRIAL-mediated-apoptosis (28,34).
In general, many reports have described the crucial function of the
PI3K/Akt signaling pathway in survival of cancer cells, and
inhibition of the PI3K/Akt pathway sensitizes cancer cells to
TRAIL-mediated caspase-dependent apoptosis (29,30).
Among MAPKs, the JNK and p38 MAPK pathways are frequently
associated with induction of apoptosis through caspase activation,
whereas the ERK pathway is thought to deliver survival signals that
protect cells from TRAIL-mediated apoptosis (35–37).
However, increasing evidence indicates that under certain
conditions, JNK and p38 MAPK may induce anti-apoptotic,
proliferative and cell survival signals in response to a specific
stimulus, and ERK activation also results in TRAIL-mediated
apoptosis in certain cell types (38–41).
However the molecular mechanisms that link TRAIL-mediated apoptosis
to kinase activation are still not completely understood. Our
results showed that treatment with TRAIL and cordycepin selectively
inhibited phosphorylation of JNK (Fig.
4) and the JNK inhibitor SP600125 inhibited cell viability more
than co-treatment with cordycepin and TRAIL and increased the
sub-G1 population (Fig. 5).
Additionally, SP600125 enhanced modulation of the Bcl-2 family,
inhibition of the IAP family, and activation of caspases in Hep3B
cells treated with cordycepin and TRAIL (Fig. 6). These results strongly suggest
that JNK is a key regulator of apoptosis induction by cordycepin
and TRAIL in TRAIL-resistant Hep3B cells.
In summary, our results demonstrate that cordycepin
significantly enhanced apoptosis in TRAIL-resistance Hep3B cells by
inhibiting JNK signaling. Taken together, our results suggest that
JNK acts as a key regulator of apoptosis in response to combined
cordycepin and TRAIL in human hepatocellular carcinoma Hep3B cells.
These findings provide novel insight into the clinical application
of TRAIL-induced apoptosis in Hep3B cells.
Acknowledgements
This study was supported by a National Research
Foundation of Korea grant funded by the Korean government
(2012-0000897, 2008-0062611) and the Technology Development Program
for Agriculture and Forestry (610003-03-1-SU000), Ministry for
Food, Agriculture, Forestry and Fisheries, Republic of Korea.
References
|
1
|
Gruss HJ: Molecular, structural, and
biological characteristics of the tumor necrosis factor ligand
superfamily. Int J Clin Lab Res. 26:143–159. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Bonavida B, Ng CP, Jazirehi A, Schiller G
and Mizutani Y: Selectivity of TRAIL-mediated apoptosis of cancer
cells and synergy with drugs: The trail to non-toxic cancer
therapeutics (Review). Int J Oncol. 15:793–802. 1999.PubMed/NCBI
|
|
3
|
Koschny R, Ganten TM, Sykora J, Haas TL,
Sprick MR, Kolb A, Stremmel W and Walczak H: TRAIL/bortezomib
cotreatment is potentially hepatotoxic but induces cancer-specific
apoptosis within a therapeutic window. Hepatology. 45:649–658.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Secchiero P, Vaccarezza M, Gonelli A and
Zauli G: TNF-related apoptosis-inducing ligand (TRAIL): a potential
candidate for combined treatment of hematological malignancies.
Curr Pharm Des. 10:3673–3681. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Bin L, Thorburn J, Thomas LR, Clark PE,
Humphreys R and Thorburn A: Tumor-derived mutations in the TRAIL
receptor DR5 inhibit TRAIL signaling through the DR4 receptor by
competing for ligand binding. J Biol Chem. 282:28189–28194. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Lee SH, Shin MS, Kim HS, Lee HK, Park WS,
Kim SY, Lee JH, Han SY, Park JY, Oh RR, Kang CS, Kim KM, Jang JJ,
Nam SW, Lee JY and Yoo NJ: Somatic mutations of TRAIL-receptor 1
and TRAIL-receptor 2 genes in non-Hodgkin’s lymphoma. Oncogene.
20:399–403. 2001.PubMed/NCBI
|
|
7
|
Ito Y, Arita M, Adachi K, Shibata T, Sawai
H and Ohno M: Chirally selective synthesis of sugar moiety of
nucleosides by chemicoenzymatic approach: L- and D-riboses,
showdomycin, and cordycepin. Nucleic Acids Symp Ser. 10:45–48.
1981.PubMed/NCBI
|
|
8
|
Westhof E, Plach H, Cuno I and Lüdemann
HD: Proton magnetic resonance studies of 2′-,3′-, and
5′-deoxyadenosine conformations in solution. Nucleic Acids Res.
4:939–953. 1977.
|
|
9
|
Horowitz B, Goldfinger BA and Marmur J:
Effect of cordycepin triphosphate on the nuclear DNA-dependent RNA
polymerases and poly(A) polymerase from the yeast, Saccharomyces
cerevisiae. Arch Biochem Biophys. 172:143–148. 1976. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Müller WE, Weiler BE, Charubala R,
Pfleiderer W, Leserman L, Sobol RW, Suhadolnik RJ and Schröder HC:
Cordycepin analogues of 2′,5′-oligoadenylate inhibit human
immunodeficiency virus infection via inhibition of reverse
transcriptase. Biochemistry. 30:2027–2033. 1991.
|
|
11
|
Chen Y, Chen YC, Lin YT, Huang SH and Wang
SM: Cordycepin induces apoptosis of CGTH W-2 thyroid carcinoma
cells through the calcium-calpain-caspase 7-PARP pathway. J Agric
Food Chem. 58:11645–11652. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Lee HH, Park C, Jeong JW, Kim MJ, Seo MJ,
Kang BW, Park JU, Kim GY, Choi BT, Choi YH and Jeong YK: Apoptosis
induction of human prostate carcinoma cells by cordycepin through
reactive oxygen species-mediated mitochondrial death pathway. Int J
Oncol. 42:1036–1044. 2013.
|
|
13
|
Lee SJ, Moon GS, Jung KH, Kim WJ and Moon
SK: c-Jun N-terminal kinase 1 is required for cordycepin-mediated
induction of G2/M cell-cycle arrest via p21WAF1 expression in human
colon cancer cells. Food Chem Toxicol. 48:277–283. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Lee HJ, Burger P, Vogel M, Friese K and
Brüning A: The nucleoside antagonist cordycepin causes DNA double
strand breaks in breast cancer cells. Invest New Drugs.
30:1917–1925. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Jeong JW, Jin CY, Park C, Hong SH, Kim GY,
Jeong YK, Lee JD, Yoo YH and Choi YH: Induction of apoptosis by
cordycepin via reactive oxygen species generation in human leukemia
cells. Toxicol In Vitro. 25:817–824. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Won SY and Park EH: Anti-inflammatory and
related pharmacological activities of cultured mycelia and fruiting
bodies of Cordyceps militaris. J Ethnopharmacol. 96:555–561.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Nakamura K, Konoha K, Yoshikawa N,
Yamaguchi Y, Kagota S, Shinozuka K and Kunitomo M: Effect of
cordycepin (3′-deoxyadenosine) on hematogenic lung metastatic model
mice. In Vivo. 19:137–141. 2005.
|
|
18
|
Yoshikawa N, Kunitomo M, Kagota S,
Shinozuka K and Nakamura K: Inhibitory effect of cordycepin on
hematogenic metastasis of B16–F1 mouse melanoma cells accelerated
by adenosine-5′-diphosphate. Anticancer Res. 29:3857–3860.
2009.PubMed/NCBI
|
|
19
|
Lee EJ, Kim WJ and Moon SK: Cordycepin
suppresses TNF-alpha-induced invasion, migration and matrix
metalloproteinase-9 expression in human bladder cancer cells.
Phytother Res. 24:1755–1761. 2010. View
Article : Google Scholar : PubMed/NCBI
|
|
20
|
Tak JK, Lee JH and Park JW: Resveratrol
and piperine enhance radiosensitivity of tumor cells. BMB Rep.
45:242–246. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Lemasters JJ, Nieminen AL, Qian T, Trost
LC, Elmore SP, Nishimura Y, Crowe RA, Cascio WE, Bradham CA,
Brenner DA and Herman B: The mitochondrial permeability transition
in cell death: a common mechanism in necrosis, apoptosis and
autophagy. Biochim Biophys Acta. 1366:177–196. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Fernández-Luna JL: Apoptosis regulators as
targets for cancer therapy. Clin Transl Oncol. 9:555–562. 2007.
|
|
23
|
Aggarwal BB, Bhardwaj U and Takada Y:
Regulation of TRAIL-induced apoptosis by ectopic expression of
antiapoptotic factors. Vitam Horm. 67:453–483. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Wen X, Lin ZQ, Liu B and Wei YQ:
Caspase-mediated programmed cell death pathways as potential
therapeutic targets in cancer. Cell Prolif. 45:217–224. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Patwardhan GA and Liu YY: Sphingolipids
and expression regulation of genes in cancer. Prog Lipid Res.
50:104–114. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Lazebnik YA, Kaufmann SH, Desnoyers S,
Poirier GG and Earnshaw WC: Cleavage of poly(ADP-ribose) polymerase
by a proteinase with properties like ICE. Nature. 371:346–347.
1994. View
Article : Google Scholar : PubMed/NCBI
|
|
27
|
Fukuda K: Apoptosis-associated cleavage of
β-catenin in human colon cancer and rat hepatoma cells. Int J
Biochem Cell Biol. 31:519–529. 1999.
|
|
28
|
Frese S, Pirnia F, Miescher D, Krajewski
S, Borner MM, Reed JC and Schmid RA: PG490-mediated sensitization
of lung cancer cells to Apo2L/TRAIL-induced apoptosis requires
activation of ERK2. Oncogene. 22:5427–5435. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Falschlehner C, Emmerich CH, Gerlach B and
Walczak H: TRAIL signalling: decisions between life and death. Int
J Biochem Cell Biol. 39:1462–1475. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Secchiero P, Gonelli A, Carnevale E,
Milani D, Pandolfi A, Zella D and Zauli G: TRAIL promotes the
survival and proliferation of primary human vascular endothelial
cells by activating the Akt and ERK pathways. Circulation.
107:2250–2256. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Seo OW, Kim JH, Lee KS, Lee KS, Kim JH,
Won MH, Ha KS, Kwon YG and Kim YM: Kurarinone promotes
TRAIL-induced apoptosis by inhibiting NF-κB-dependent cFLIP
expression in HeLa cells. Exp Mol Med. 44:653–664. 2012.PubMed/NCBI
|
|
32
|
Ola MS, Nawaz M and Ahsan H: Role of Bcl-2
family proteins and caspases in the regulation of apoptosis. Mol
Cell Biochem. 351:41–58. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Brenner D and Mak TW: Mitochondrial cell
death effectors. Curr Opin Cell Biol. 21:871–877. 2009. View Article : Google Scholar
|
|
34
|
Thakkar H, Chen X, Tyan F, Gim S, Robinson
H, Lee C, Pandey SK, Nwokorie C, Onwudiwe N and Srivastava RK:
Pro-survival function of Akt/protein kinase B in prostate cancer
cells. Relationship with TRAIL resistance. J Biol Chem.
276:38361–38369. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Tran SE, Holmstrom TH, Ahonen M, Kahari VM
and Eriksson JE: MAPK/ERK overrides the apoptotic signaling from
Fas, TNF, and TRAIL receptors. J Biol Chem. 276:16484–16490. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Söderström TS, Poukkula M, Holmström TH,
Heiskanen KM and Eriksson JE: Mitogen-activated protein
kinase/extracellular signal-regulated kinase signaling in activated
T cells abrogates TRAIL-induced apoptosis upstream of the
mitochondrial amplification loop and caspase-8. J Immunol.
169:2851–2860. 2002.
|
|
37
|
Jurewicz A, Matysiak M, Andrzejak S and
Selmaj K: TRAIL-induced death of human adult oligodendrocytes is
mediated by JNK pathway. Glia. 53:158–166. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Mühlenbeck F, Haas E, Schwenzer R,
Schubert G, Grell M, Smith C, Scheurich P and Wajant H: TRAIL/Apo2L
activates c-Jun NH2-terminal kinase (JNK) via
caspase-dependent and caspase-independent pathways. J Biol Chem.
273:33091–33098. 1998.PubMed/NCBI
|
|
39
|
Qu J, Zhao M, Teng Y, Zhang Y, Hou K,
Jiang Y, Yang X, Shang H, Qu X and Liu Y: Interferon-α sensitizes
human gastric cancer cells to TRAIL-induced apoptosis via
activation of the c-CBL-dependent MAPK/ERK pathway. Cancer Biol
Ther. 12:494–502. 2011.
|
|
40
|
Gupta SC, Reuter S, Phromnoi K, Park B,
Hema PS, Nair M and Aggarwal BB: Nimbolide sensitizes human colon
cancer cells to TRAIL through reactive oxygen species- and
ERK-dependent up-regulation of death receptors, p53, and Bax. J
Biol Chem. 286:1134–1146. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Phipps LE, Hino S and Muschel RJ:
Targeting cell spreading: a method of sensitizing metastatic tumor
cells to TRAIL-induced apoptosis. Mol Cancer Res. 9:249–258. 2011.
View Article : Google Scholar : PubMed/NCBI
|