Introduction
Chronic inflammation increases the risk of cancer
development and progression (1). In
the tumor microenvironment, intracellular molecules are released
from tumor cells damaged by immune cells. Some of these molecules
are inflammatory mediators referred to as damage-associated
molecular pattern molecules (DAMPs) (2). DAMPs play critical roles in triggering
immune responses and activating repair mechanisms to produce both
antitumor and protumor effects (2).
In tumor progression, the inflammatory response is triggered by
interactions between intracellular molecules and Toll-like
receptors (TLRs) expressed on tumor cells or local immune cells.
These interactions promote neoplastic cell growth and have been
proposed as initiating factors in ~15% of human tumors (3). In addition to passive release from
necrotic cells and pulsatile release from apoptotic cells, DAMPs
can be secreted in response to feed forward signals from identical
or other DAMPs (4). This mechanism
is complex and has yet to be fully elucidated.
Heat shock protein (HSP) 70 is a well-characterized
DAMP involved in chronic inflammation (5). Stress-inducible HSP70 functions as a
cytoprotective protein when cells are exposed to stressful stimuli.
However, stress-inducible HSP70 can also be passively released from
necrotic cells (6) as well as
actively released when tumor cells suffer from exogenous stress
(7). In the tumor microenvironment,
extracellular HSP70 can bind to TLR2 and TLR4 expressed by tumor
cells and causes immune tolerance, cancer progression and
propagation of the tumor microenvironment (6,7).
However, HSP70 also participates in preventing apoptosis through
autophagy and can act as a prosurvival mechanism (8). Indeed, HSP70 augmented autophagy
through c-Jun N-terminal kinase (JNK) phosphorylation and Beclin-1
upregulation (8). Several studies
have demonstrated that inhibition of the autophagy regulator
Beclin-1 by RNAi in tumor cells inhibited stress induced autophagy
and high-mobility group protein B1 (HMGB1) release (8). Among different signaling pathways
induced by HSP70, the promotion of tumor growth by activation of
the NF-κB cascade has been identified in several tumor cells
(9). Thus, HSP70 can stimulate both
the NF-κB and JNK-Beclin-1/HMGB1 pathways in tumor cells. However,
whether communication between the two signaling pathways is
involved in the increased malignant potential of tumor cells
remains unknown.
HMGB1 is widely expressed by numerous tumor cells
and can be secreted or released upon necrotic cell death (2,10).
HMGB1 expression is high in migrating growth cones and malignant
cells (2). HMGB1 binds tissue-type
plasminogen activator and plasminogen, promoting plasmin production
and tissue invasion (2,11). Secreted HMGB1 mediates responses to
infection and injury by binding with high affinity to several
receptors including receptor for advanced glycation end products
(RAGE), TLR2 and TLR4, thereby promoting tumor invasion and
metastasis (12,13).
The pathogenic role of HMGB1 secretion in patients
undergoing cancer treatment remains largely unexplored (14). HMGB1 release may initiate immune
responses against tumor cells in patients undergoing
chemotherapy-induced necrosis (15). In the present study, we demonstrated
that HMGB1 release is a critical regulator of the response to
various forms of metabolic stress. HSP70 production by DAMP
stimulation resulted in NF-κB activation in tumor cells via TLR
signaling accompanied by increased tumor cell proliferation.
Extracellular HSP70 also activated JNK/Beclin-1 pathways in tumor
cells and induced the release of HMGB1 from tumor cells, resulting
in a second phase of NF-κB activation and a long-lasting tumor
promoting effect, reinforcing and amplifying DAMP-induced signaling
(a positive feedback loop). These findings suggest that this
feedback signaling pathway may play an important role in the
initiation and progression of cancer.
Materials and methods
Animals and cells
BALB/c mice (6–8 weeks old) were purchased from the
Center of Medical Experimental Animals of Hubei Province (Wuhan,
China). Mice were maintained in the accredited animal facility of
Tongji Medical College, and used for studies approved by the Animal
Care and Use Committee of Tongji Medical College. Murine H22, a
human HepG2 hepatocarcinoma cell line, was purchased from China
Center for Type Culture Collection (CCTCC, Wuhan, China) and
cultured according to CCTCC guidelines.
Reagents and plasmids
Resveratrol (3,4′,5-trihydroxy-trans-stilbene) was
purchased from Sigma-Aldrich (St. Louis, MO, USA).
6-Amino-4-(4-phenoxyphenylethylamino) quinazoline (QNZ) was
purchased from Merck4Biosciences (Calbiochem, Darmstadt, Germany).
Recombinant human HSP70 and HMGB1 were prepared from engineered
bacteria carrying an expression plasmid kindly provided by Dr
Richard Morimoto and Ji-Zhong Cheng (The University of Texas
Medical Branch), purified and tested for endotoxin as previously
described (16). The content of the
endotoxin was <0.006 EU/ml for concentrations of recombinant
HSP70 up to 50 μg/ml. Eukaryotic expression vectors psTLR2 and
psTLR4 carrying cDNAs encoding the signal peptide and extracellular
domain of murine TLR2 and TLR4 were constructed by the insertion of
cDNA into plasmid pcDNA3.1 (Invitrogen, Carlsbad, CA, USA) in our
laboratory. Antibodies were purchased from Cell Signaling
Technology (Beverly, MA, USA) and Santa Cruz Biotechnology, Inc.
(Santa Cruz, CA, USA).
Construction of short interfering RNA
(shRNA)-expressing H22 tumor cell line
To downregulate HMGB1, RAGE or Beclin-1 in tumor
cells, cells were transduced with HMGB1shRNA(h), RAGE-shRNA(h) or
Beclin-1 shRNA(h) lentiviral particles or control shRNA lentiviral
particles (Santa Cruz Biotechnology, Inc.), according to the
manufacturer’s protocol.
Animal experiments and treatment
protocols
For the treatment of tumors with DTC-Ms, HSP70 or
HMGB1, BALB/c mice were inoculated with 1×105 H22 cells
by intramuscular injection into the right hind thigh. Following
inoculation, the mice of treatment groups received intramuscular
injection of DTC-Ms, HSP70 or HMGB1 (200 μg/mouse) at the
inoculation site, starting on day 1 (d1) after tumor inoculation or
d7 after tumor inoculation when the tumor was palpable, once every
2 days for 4 times. Mice from the control group received
intramuscular injection of an equal volume of PBS. Mice were
sacrificed and tumors were dissected and weighed at the indicated
times.
For treatment of tumor with sTLR2 and sTLR4, BALB/c
mice were inoculated with 1×105 H22 cells by
intramuscular injection into the right hind thigh. On d6, 8, 10,
and 12 after inoculation, the mice of the treatment groups received
an intramuscular injection (local naked DNA transfection) of 100 μg
of psTLR2 or psTLR4 at the inoculation site. The control group mice
received an equal volume of saline or an equal amount of pcDNA3.1
plasmid. In other experiments, mice received an intramuscular
injection of psTLR2, psTLR4 or psTLR2/psTLR4 at the tumor
inoculation site as described above, and also received
intramuscular injection of HSP70 (200 μg/mouse) or saline at the
inoculation site on d7, 9, 11 and 13 after tumor inoculation. Mice
were sacrificed and tumors were dissected and weighed on d15 after
inoculation.
For analysis of tumor metastasis after treatment
with HSP70 or HMGB1, H22 cells or HMGB1shRNA-transfected H22 cells
were injected into the liver of BALB/c mice (n=8/group). Mice were
treated by intravenous injection of HSP70 or HMGB1 (200 μg/mouse)
or saline once every 2 days 6 times after tumor inoculation. Mice
were sacrificed and tumors were dissected and weighed on d16 after
inoculation. The size of the main tumor nodule was measured and
satellite tumor nodes were counted.
Detection of HMGB1 in tumors
To detect extracellular HMGB1 in the tumor milieu,
BALB/c mice were inoculated with 1×105 H22 cells by
intramuscular injection into the right hind thigh. Tissues at the
inoculation site or tumor peripheral tissues were surgically
excised at the indicated time points after inoculation. Tissues
adjacent to the tumor or inoculation site were used as controls.
Interstitial molecules from the tissues were prepared by digesting
the tissue with collagenase and removing debris by centrifugation,
and were then used for detection of HMGB1 by western blotting.
HMGB1 antibody was purchased from Cell Signaling Technology
(Danvers, MA, USA).
Analysis of gene expression by reverse
transcriptase (RT)-PCR and real-time PCR
Total RNAs were isolated from cells or muscle
tissues of mice using TRIzol reagent (Invitrogen), according to the
manufacturer’s instructions. RT-PCR was used to detect mRNAs of
psTLR2 and psTLR4 as previously described (17). To detect the mRNAs of sTLR2 and
sTLR4 expressed by expression vectors, the primer sequences were:
sTLR2 sense, 5′-CCAAGCTGGCTAGCGTTTA-3′ and antisense,
5′-CAAATGTTCAAGACTGCCCA-3′; sTLR4 sense,
5′-GACCCAAGCTGGCTAGCGTTT-3′ and anti-sense,
5′-TTTGTCTCCACAGCCACCA-3′; β-actin sense, 5′-ATGGG
TCAGAAGGACTCCTATG-3′ and antisense, 5′-ATCTCC
TGCTCGAAGTCTAGAG-3′.
The quantitative real-time RT-PCR for MMP-9 and
β-actin was performed as previously described (17). MMP-9 sense,
5′-CAGATGATGGGAGAGAAGCAG-3′ and antisense,
5′-GAAGGTGAAGGGAAAGTGAC-3′; β-actin sense,
5′-ATGGGTCAGAAGGACTCCTATG-3′ and antisense,
5′-ATCTCCTGCTCGAAGTCTAGAG-3′. The results are expressed as the
expression level of the gene relative to that of the housekeeping
gene β-actin.
Western blot analysis
Following incubation of cells in the presence or
absence of HSP70 or HMGB1, tissue samples after in vivo
transfection were lysed or homogenized for western blot analysis as
previously described (17). Primary
antibodies and horseradish peroxidase-conjugated secondary
antibodies were purchased from Chemicon (Temecula, CA, USA), Santa
Cruz Biotechnology, Inc., R&D Systems (Minneapolis, MN, USA)
and Cell Signaling Technology, respectively.
MMP assay by gelatin zymography
MMP-9 assay of protein samples was performed as
previously described (17).
Briefly, proteins prepared from the tumor tissues or tumor cells of
each group were separated by 7.5% sodium dodecyl
sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) containing 1%
gelatin. Gels were incubated in MMP activation buffer containing 50
mM Tris (pH 8.0) and 10 mM CaCl2 at 37°C overnight, and
then stained with 1% Coomassie Brilliant Blue R-250 for 3 h and
destained in 10% (v/v) methanol and 5% (v/v) acetic acid. The area
and gray scale of bands were analyzed by the HPIAS-1000 analytical
system. The relative activity of MMP-9 was calculated using the
formula: (band gray scale - background gray scale) × band area.
Flow cytometric analysis
Tumor cells were stained with PE-Cy7 anti-mouse TLR2
antibody, PE anti-mouse TLR4 antibody (eBioscience, San Diego, CA,
USA), or isotype control antibody for flow cytometric analysis.
Parameters were acquired on a FACSCalibur Flow Cytometer and
analyzed with CellQuest Software (both from BD Biosciences).
Proliferation assays
H22 cells were labeled with carboxyfluorescein
succinimidyl ester (CFSE) and cultured for 3 days. Cell
proliferation was analyzed by flow cytometry. Flow cytometric
analysis acquired at least 1×104 events on a BD LSR II
Flow Cytometer. The proliferation index was calculated in the
responder population gate using the ModFit LT for Win32
Software.
Preparation of DTC-Ms
H22 cells were washed with phosphate-buffered saline
(PBS), and resuspended in PBS to a final concentration of
5×107/ml. After four-rounds of freeze-thaw cycles
followed by vortexing for 30 sec, the cells were removed by
centrifugation. The supernatant contained a mixture of DTC-Ms. The
concentration of DTC-Ms was defined by the concentration of
protein, determined using Coomassie Bradford reagent (Thermo Fisher
Scientific, Inc., Rockford, IL, USA), according to the
manufacturer’s instructions.
Statistical analysis
The results are expressed as the mean value ±
standard deviation (SD) and are interpreted by ANOVA-repeated
measures test. Differences are considered statistically significant
when P<0.05.
Results
Damaged tumor cells promote the
proliferation of living tumor cells and increase HMGB1 expression
through initiation of HSP70
The effect of TLR2/4 signaling on the growth of
hepatocarcinoma cells was measured after inoculating
1×105 H22 cells into the hind thigh muscle and treating
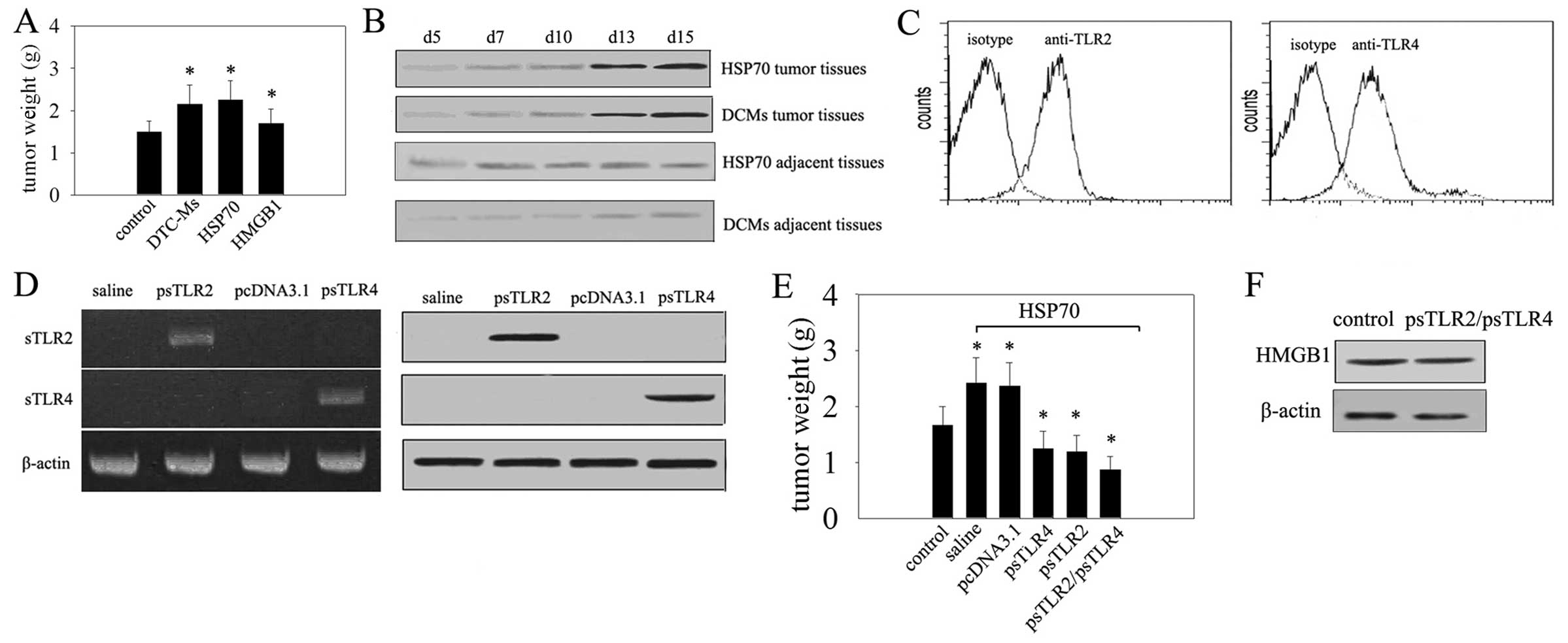
mice with DTC-Ms containing TLR2/4 ligands (18). Treatment with HSP70 and HMGB1 was
used as control. Tumor growth increased significantly after two
weeks of treatment with DTC-Ms. Notably, the effect of HSP70 on H22
cells was very similar to DTC-Ms stimulation (Fig. 1A). Moreover, at the same time
interval, a rise in HMGB1 was detected in DTC-Ms and HSP70 treated
tissues at the site of tumor cell inoculation, and increased in
local tissues with the development of the tumor (Fig. 1B). These results suggested that
HSP70 is a critical molecule(s) in DTC-Ms that can upregulate the
expression of HMGB1 and may promote tumor development. As
triggering of TLR4 and TLR2 in tumor cells promotes tumor cell
proliferation (19) and both TLR2
and TLR4 were expressed on H22 cells (Fig. 1C) we verified whether TLR2 and TLR4
mediate the increased expression of HMGB1 and tumor promoting
effects of HSP70. We expressed sTLR2 and sTLR4 in the tissue at the
site of the palpable tumor by intramuscular transfection of sTLR2
and sTLR4 expression vectors (Fig.
1D). Expression of sTLR2 and sTLR4 suppressed tumor growth
(Fig. 1E). The promoting effect of
HSP70 on tumor growth was reduced by sTLR2 or sTLR4 expression
(Fig. 1E), but the production of
HMGB1 was not significantly influenced by blockade of TLR2 and TLR4
(Fig. 1F). Thus, HSP70-inducible
expression of HMGB1 in the tumor microenvironment, independent of
both TLR2 and TLR4, may promote tumor development.
Malignant tumor invasion is increased by
HSP70-inducible expression of HMGB1
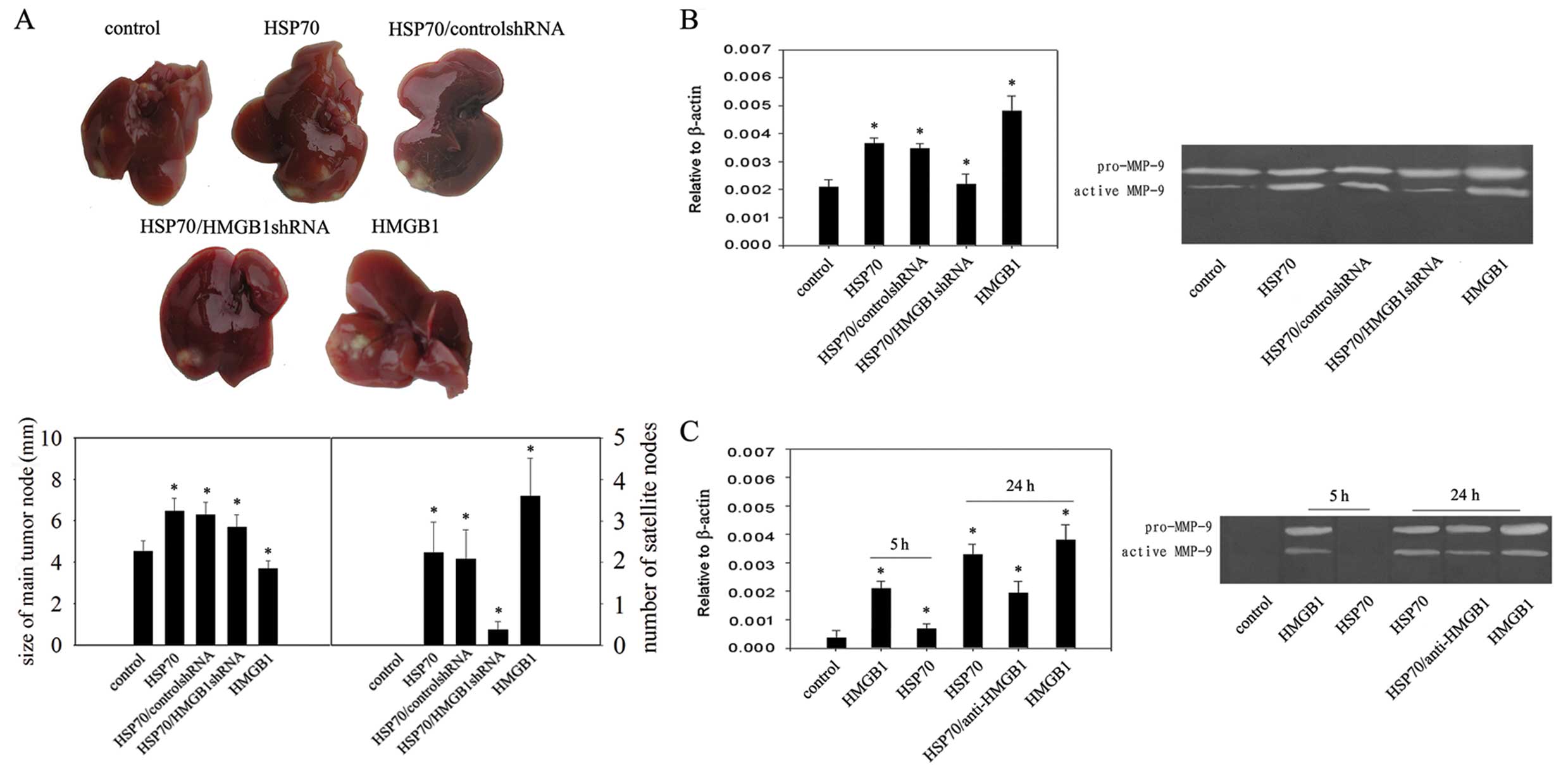
Based on the above results, we next investigated
whether HSP70 could increase H22 cell capability for malignant
invasion by inducible expression of HMGB1 in the tumor
microenvironment. In mice receiving 1×105 H22 cells in
the liver, injection of HSP70 increased the expression of HMGB1.
Production of active MMP-9 also increased significantly in the
HSP70 treatment groups (Fig. 2A),
suggesting it is a major contributor to tumor invasion (20). Both increased tumor growth and
increased satellite tumor nodes were observed in the HSP70
treatment groups (Fig. 2B), whereas
treatment of mice with HMGB1shRNA transfection suppressed tumor
growth and the formation of satellite tumor nodes (Fig. 2B). However, in the presence of
HMGB1, increased satellite tumor nodes in the liver and increased
production of active MMP-9 was observed (Fig. 2A and B), suggesting the increased
capability for malignant invasion of the tumor was not induced by
HSP70 but was due to HSP70-inducible expression of HMGB1. We used
specific antibodies to neutralize HMGB1 activity that may be
present in the HSP70 preparation to confirm the direct effect of
HMGB1 on H22 cells cultured for 5 or 24 h in the presence of HSP70
or HMGB1. The production of active MMP-9 was increased in the
presence of HMGB1 for 5 or 24 h but was unaffected by incubation in
the presence of HSP70 for 5 h (Fig.
2C). In addition, anti-HMGB1 antibody treatment significantly
reduced the increased production of active MMP-9 in the presence of
HSP70 incubated for 24 h (Fig.
2C).
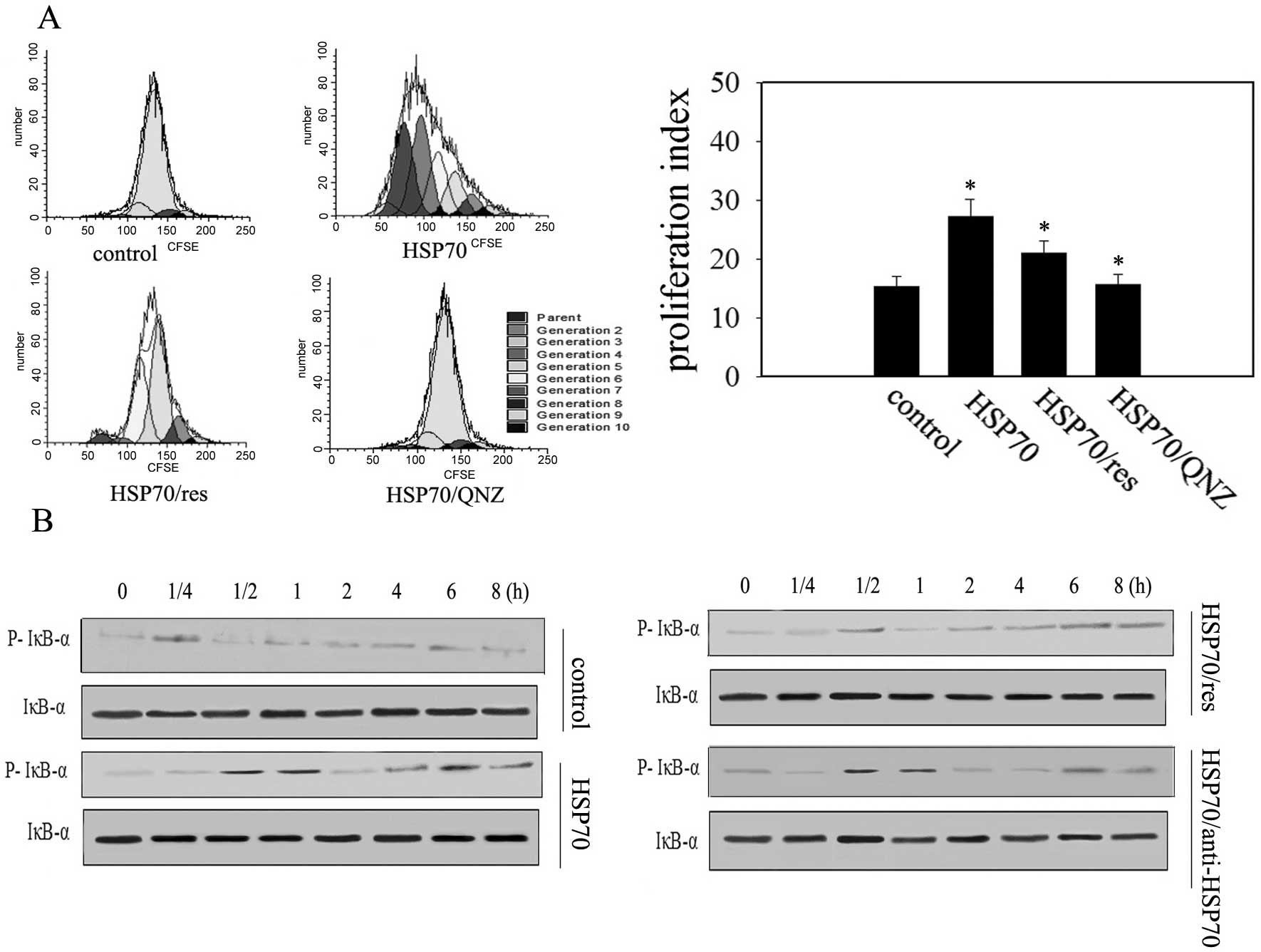
HSP70 stimulation results in different
temporal patterns of NF-κB activation in H22 cells
TLR2 and TLR4 can activate NF-κB that mediates the
effects of HSP70 (20). The
proliferation of H22 cells was promoted by administration of HSP70
(Fig. 3A). Moreover, stimulation of
H22 cells with HSP70 induced a biphasic temporal pattern of I-κB
activation, an inhibitory protein of NF-κB, and increased the
quantity of NF-κB in the cell nucleus, with a rapid increase in
phosphorylation within 30 min, followed by a second rise at ~6 h
(Fig. 3B). Preincubation of H22
cells with resveratrol, a TLR4 and TLR2 signaling inhibitor
(21), inhibited the HSP70-induced
first phase of NF-κB phosphorylation but not the second phase
(Fig. 3B), and the proliferation of
H22 cells was significantly suppressed (Fig. 3A). QNZ, an inhibitor of NF-κB,
abrogated the effect of HSP70 (Fig.
3A) suggesting that both TLR2 and TLR4 mediated the first phase
of NF-κB activation and were involved in promoting the effect of
HSP70 on tumor cell proliferation. Next, we looked at whether the
second phase of NF-κB phosphorylation may last if HSP70 was
removed. To test this, H22 cells were treated with HSP70 for 4 h
and then specific HSP70 neutralizing antibodies were administered.
I-κB activation increased at ~6 h, although not as high as that
when in the continuous presence of HSP70 (Fig. 3B). Collectively, the results suggest
that HSP70 stimulation in tumor cells may result in a previously
unrecognized activation of NF-κB but not dependent of TLR4 and TLR2
signal pathways.
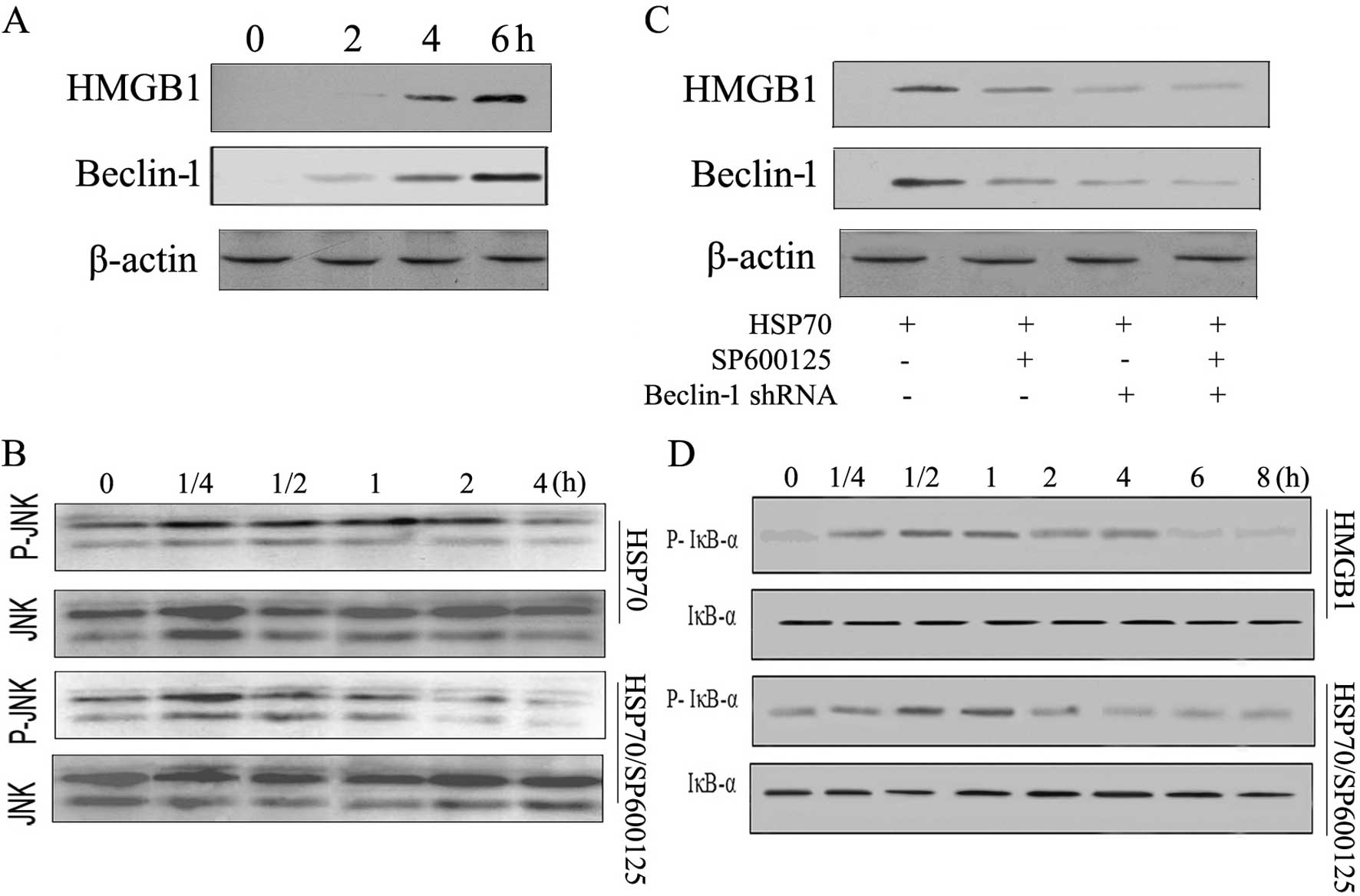
The second phase of HSP70-induced NF-κB
activation is dependent on JNK signaling and HMGB1 expression in
H22 cells
HSP70 regulates diverse signaling pathways involving
NF-κB in different types of cells (22). However, the mechanism(s) of the
second phase of NF-κB activation by HSP70 is unknown. Given that
HSP70 can induce HMGB1 release via JNK activation and Beclin-1
expression and that HMGB1 can trigger NF-κB activation (8,23), we
hypothesized that the second phase of NF-κB activation in H22 cells
is caused by Beclin-1-derived HMGB1 production induced by HSP70. To
test this hypothesis, we examined the temporal production of HMGB1
by HSP70. In H22 cells, HSP70 induced rapid JNK phosphorylation,
significant upregulation of Beclin-1 and HMGB1 at 4 h (Fig. 4A and B), which preceded the second
phase of NF-κB activation. Then, we examined the effect of
exogenously delivered HMGB1 on NF-κB signaling. Stimulation of H22
cells with HMGB1 induced rapid NF-κB phosphorylation (Fig. 4D). These results indicated that H22
cells activated NF-κB rapidly but the second phase of NF-κB
activation was not induced by HMGB1. The second phase of NF-κB
activation induced by HSP70 in H22 cells could be due to HMGB1
generation via JNK mediating Beclin-1. However, when cancer cells
were pretreated with a selective JNK inhibitor (SP600125)
HSP70-induced JNK, phosphorylation was inhibited (Fig. 4B), which prevented Beclin-1 and
HMGB1 production in H22 cells (Fig.
4C). HMGB1 production by HSP70 was also inhibited by Beclin-1
shRNA (Fig. 4C). Moreover, the JNK
inhibitor prevented the second, but not the first, phase of
HSP70-induced activation of NF-κB (Fig.
4D). From these results, we conclude that HSP70 induces the
second phase of NF-κB activation via JNK-mediated
Beclin-1-dependent HMGB1 production in H22 cells.
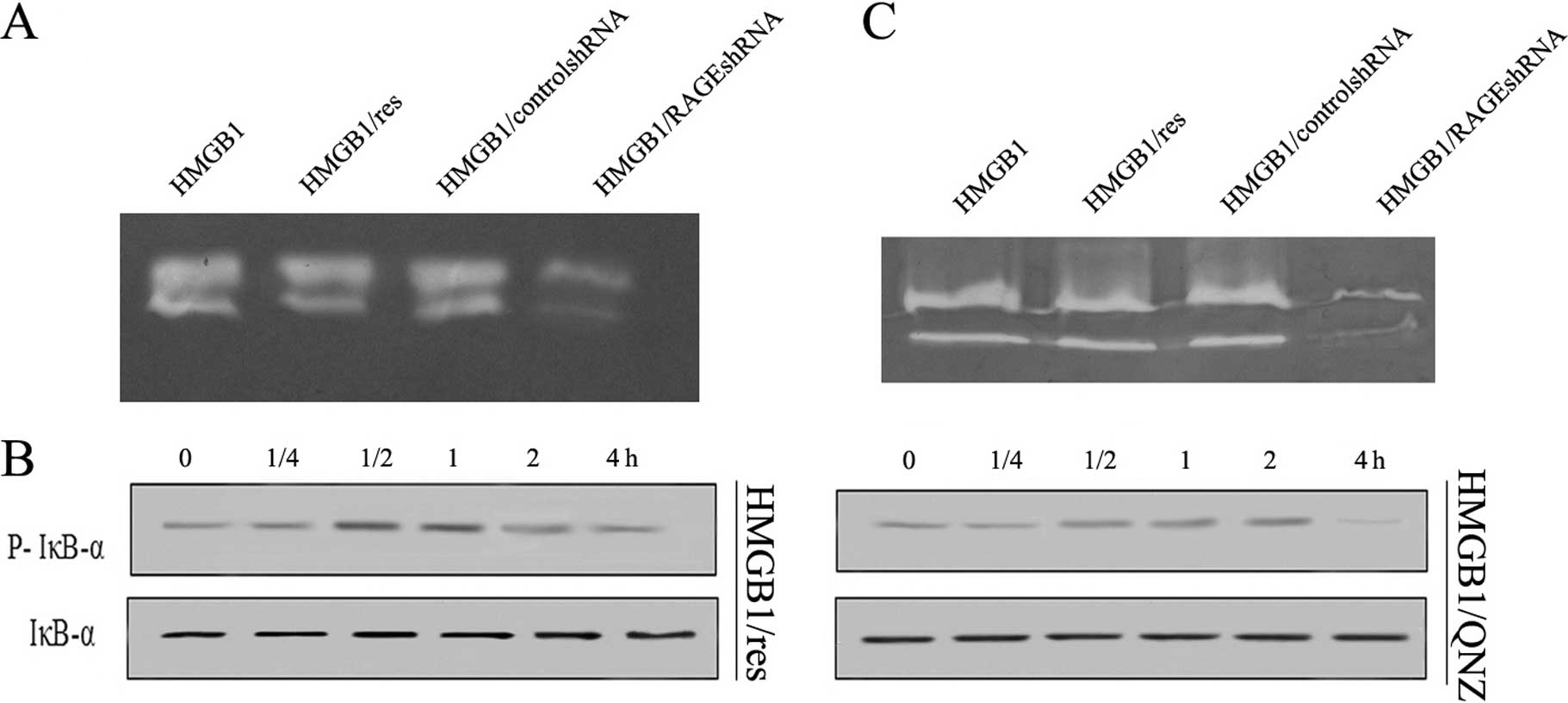
HMGB1/RAGE-mediated NF-κB phosphorylation
is involved in hepatocarcinoma cell invasion
HMGB1 mediates the response to inflammation-based
carcinogenesis by binding with high affinity to several receptors
including the RAGE and TLRs 2 and 4, thereby augmenting tumor
growth and invasion. To determine whether RAGE and/or TLR2/4
mediate HMGB1-induced increased production of active MMP-9, a
target-specific shRNA against these receptors was transfected into
tumor cells. Knockdown of RAGE in cancer cells diminished
HMGB1-induced increased production of MMP-9 (Fig. 5A). By contrast, there was no effect
on HMGB1-induced increased production of MMP-9 when resveratrol was
used to inhibit TLR2/4 (Fig. 5A).
This suggested RAGE is required for HMGB1 promotion of tumor
invasion by increased production of active MMP-9. To analyze the
pathway(s) involved in the effect of RAGE-mediated NF-κB signaling,
we stimulated H22 cells with HMGB1 in the presence of QNZ (NF-κB
inhibitor) and resveratrol (TRIF inhibitor). The effect of
RAGE-mediated NF-κB signaling was completely abolished by QNZ, but
not resveratrol (Fig. 5B),
suggesting that NF-κB was involved in RAGE signaling for MMP-9
expression in H22 cells. A human hepatocarcinoma cell line, HepG2,
was used to evaluate the effect of RAGE signaling. The human cancer
cell line showed a similar pattern of increased MMP-9 expression in
response to HMGB1 stimulation (Fig.
5C). Taken together, these results suggested that RAGE
signaling activated NF-κB to regulate the expression of MMP-9 in
hepatocarcinoma H22 cells.
Discussion
The present study demonstrated that HMGB1 secreted
from cells during the initial steps of HSP70 induction may result
in a higher invasion potential of hepatocarcinoma cells by
mediating a second phase of NF-κB activation. Stress-inducible
HSP70 is abundantly expressed in various types of tumor cells, and
is regarded as a cancer-relevant survival protein (22). However, data in the current study
showed a distinct effect of HSP70 in progressively growing tumors.
In the early stages of tumor growth, HSP70/TLR2/4 could promote H22
cell proliferation by stimulating NF-κB pathways, which is strongly
associated with tumor growth (24).
Of note, in the microenvironment of late-stage tumors, tumor cells
pretreated with HSP70 showed stronger trend to form tumors after
inoculation, and had increased metastatic potential after the
removal of HSP70. The underlying mechanisms may involve the
increased expression of HMGB1 in tumor cells and the release of
HMGB1 from tumor cells by treatment with HSP70, similar to the
effect of LPS in promoting the expression and release of HMGB1
(25). Extracellular HMGB1 has been
extensively related to tumor growth and metastasis (10) as it can activate NF-κB through
TLR2/4 and RAGE signaling (26). We
found that RAGE RNAi abolished HMGB1-induced activation of NF-κB
and induced metastasis in tumor cells. However, blocking TLR2/4 had
no effect, suggesting they are indispensable for HMGB1/RAGE in the
NF-κB pathway of hepatocarcinoma cells in response to HSP70.
Therefore, HSP70 could mediate biphasic NF-κB activation in H22
cells. In this situation, the effect of HSP70 on tumor cells in the
original microenvironment could promote invasion and metastasis of
tumor cells into a new microenvironment where HSP70 has not
accumulated.
In cancerous conditions, cells dying through
non-apoptotic pathways can release stress-inducible HSP70 into the
extracellular space. However, living tumor cells can secrete
stress-inducible HSP70 under stress conditions (7). DTC-Ms could promote survival and
proliferation of tumor cells by HSP70/TLR2/4 mediated NF-κB
activation (19,20). In addition, it was proposed that
within tumor microenvironments, stress-induced HSP70 increases JNK
phosphorylation and enhances autophagy by increasing Beclin-1
expression (8). In the current
study, we found that HSP70 enhanced HMGB1 production in H22 cells,
which constitutively expressed Beclin-1, via a JNK signaling
cascade. Furthermore, HSP70-induced HMGB1 production was prevented
completely by inhibitors of Beclin-1 and JNK. Thus, DAMPs released
during the early stages of tumor development can generate a
positive feedback loop by stimulating HMGB1 release from tumor
cells. However, our data also showed that HSP70 did not induce
Beclin-1 upregulation significantly until 4 h after stimulation,
suggesting that Beclin-1/HMGB1 is unlikely to be responsible for
the NF-κB phosphorylation observed at 30 min. Administration of a
JNK inhibitor, SP600125, inhibited the second, but not the first,
phase of NF-κB phosphorylation. These findings indicated that
Beclin-1-derived HMGB1 synthesis induced by HSP70 was mediated by
activation of the JNK signaling cascade. Activation of JNK
signaling in H22 cells resulted in HMGB1 production, which was
maximal 4 h after exposure to HSP70, and preceded the second phase
of NF-κB activation.
NF-κB functions as a tumor promoter in
inflammation-associated cancer (3,9). The
activation of NF-κB signaling pathways is implicated in the
proliferation and metastasis of several tumors (3). Although it is known that TLR-mediated
signal transduction leads to the activation of NF-κB (3), the mechanism whereby NF-κB is
chronically activated in tumors remains to be elucidated. In the
present study, we showed that in H22 cells, HSP70, a
pathophysiological stimulus, induces biphasic NF-κB
phosphorylation; the early phase occurs ~30 min after stimulation,
involving TLR-mediated proliferation of tumor cells. Both TLR2 and
TLR4 can activate NF-κB through MyD88-dependent and TRIF-dependent
signaling pathways (27). Our data
showed that the effect of HSP70 on tumor cell proliferation was
abrogated by NF-κB inhibitor, whereas resveratrol by TRIF (27). Thus, HSP70-mediated TLR2/4 signaling
can activate NF-κB through the TRIF pathway in H22 cells. The
delayed phase occurred ~6 h after stimulation via the HMGB1-RAGE
signaling pathway, and mediated the invasion and increased
production of active MMP-9 of tumor cells. Moreover, the
involvement of HMGB1/RAGE in the NF-κB pathway has been
demonstrated in numerous studies (28), although the precise mechanism
remains unknown. We demonstrated that HMGB1/RAGE activity of the
NF-κB pathway was partly induced by IκBa degradation, since the
effect of HMGB1/RAGE was completely abrogated by QNZ.
The present results are in keeping with recent
computational analyses showing that positive feedback loops allow
cells to modulate the amplitude and duration of signaling responses
(29). Our data showed that
activation of NF-κB was indispensable for the effect of HSP70.
HSP70 induced a positive feedback loop involving Beclin-1/HMGB1
production, causing re-phosphorylation of NF-κB. This resulted in a
significant promotion of cell invasion ability of the cancer cells,
a critical aspect of tumor progression. Future studies will address
a variety of potential outcomes of this important feedback pathway,
including carcinogenic effects and effects on cell differentiation.
Thus, this signaling cascade may provide further insights into the
pathophysiology of inflammation and tumorigenesis.
Acknowledgements
This study was supported by the National Science
Foundation of Hubei province (2012FFB01906) and the ‘Important
Foundation’ of XiangYang Central Hospital (no.YY2010B05).
References
|
1
|
Sato Y, Goto Y, Narita N and Hoon DS:
Cancer cells expressing Toll-like receptors and the tumor
microenvironment. Cancer Microenviron. 2:S205–S214. 2009.
View Article : Google Scholar
|
|
2
|
Srikrishna G and Freeze HH: Endogenous
damage-associated molecular pattern molecules at the crossroads of
inflammation and cancer. Neoplasia. 11:615–628. 2009.PubMed/NCBI
|
|
3
|
Huang B, Zhao J, Shen S, et al: Listeria
monocytogenes promotes tumor growth via tumor cell toll like
receptor 2 signaling. Cancer Res. 67:4346–4352. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Korbelik M, Zhang W and Merchant S:
Involvement of damage-associated molecular patterns in tumor
response to photodynamic therapy: surface expression of
calreticulin and high-mobility group box-1 release. Cancer Immunol
Immunother. 60:1431–1437. 2011. View Article : Google Scholar
|
|
5
|
Jäättelä M: Escaping cell death: survival
proteins in cancer. Exp Cell Res. 248:30–43. 1999.PubMed/NCBI
|
|
6
|
Basu S, Binder RJ, Suto R, Anderson KM and
Srivastava PK: Necrotic but not apoptotic cell death releases heat
shock proteins, which deliver a partial maturation signal to
dendritic cells and activate the NF-kappaB pathway. Int Immunol.
12:1539–1546. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Bausero MA, Gastpar R, Multhoff G and Asea
A: Alternative mechanism by which IFN-gamma enhances tumor
recognition: active release of heat shock protein 72. J Immunol.
175:2900–2912. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Li S, Zhou Y, Fan J, Cao S, et al: Heat
shock protein 72 enhances autophagy as a protective mechanism in
lipopolysaccharide-induced peritonitis in rats. Am J Pathol.
179:2822–2834. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Pikarsky E, Porat RM, Stein I, Abramovitch
R, et al: NF-kappaB functions as a tumour promoter in
inflammation-associated cancer. Nature. 431:461–466. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Ellerman JE, Brown CK, de Vera M, et al:
Masquerader: high mobility group box-1 and cancer. Clin Cancer Res.
13:2836–2848. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Taguchi A, Blood DC, del Toro G, et al:
Blockade of RAGE-amphoterin signalling suppresses tumour growth and
metastases. Nature. 405:354–360. 2000. View
Article : Google Scholar : PubMed/NCBI
|
|
12
|
Lotze MT and Tracey KJ: High-mobility
group box 1 protein (HMGB1): nuclear weapon in the immune arsenal.
Nat Rev Immunol. 5:331–342. 2005. View
Article : Google Scholar : PubMed/NCBI
|
|
13
|
Tang D, Kang R, Cheh CW, et al: HMGB1
release and redox regulates autophagy and apoptosis in cancer
cells. Oncogene. 29:5299–5310. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Tang D, Kang R, Zeh HJ III and Lotze MT:
High-mobility group box 1 and cancer. Biochim Biophys Acta.
1799:131–140. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Apetoh L, Ghiringhelli F, Tesniere A,
Obeid M, et al: Toll-like receptor 4-dependent contribution of the
immune system to anticancer chemotherapy and radiotherapy. Nat Med.
13:1050–1059. 2007. View
Article : Google Scholar
|
|
16
|
Geng H, Zhang GM, Xiao H, et al: HSP70
vaccine in combination with gene therapy with plasmid DNA encoding
sPD-1 overcomes immune resistance and suppresses the progression of
pulmonary metastatic melanoma. Int J Cancer. 118:2657–2664. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Gong W, Zhang GM, Liu Y, et al: IFN-gamma
withdrawal after immunotherapy potentiates B16 melanoma invasion
and metastasis by intensifying tumor integrin alphavbeta3
signaling. Int J Cancer. 123:702–708. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Tsan MF and Gao B: Endogenous ligands of
Toll-like receptors. J Leukoc Biol. 76:514–519. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Asea A, Rehli M, Kabingu E, Boch JA, et
al: Novel signal transduction pathway utilized by extracellular
HSP70: role of toll-like receptor (TLR) 2 and TLR4. J Biol Chem.
277:15028–15034. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Asea A: Initiation of the immune response
by extracellular Hsp72: chaperokine activity of Hsp72. Curr Immunol
Rev. 2:209–215. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Youn HS, Lee JY, Fitzgerald KA, et al:
Specific inhibition of MyD88-independent signaling pathways of TLR3
and TLR4 by resveratrol: molecular targets are TBK1 and RIP1 in
TRIF complex. J Immunol. 175:3339–3346. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Mosser DD and Morimoto RI: Molecular
chaperones and the stress of oncogenesis. Oncogene. 23:2907–2918.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Tang D, Loze MT, Zeh HJ and Kang R: the
redox protein HMGB1 regulates cell death and survival in cancer
treatment. Autophagy. 6:1181–1183. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Rohde M, Daugaard M, Jensen MH, et al:
Members of the heat-shock protein 70 family promote cancer cell
growth by distinct mechanisms. Genes Dev. 19:570–582. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Wu CX, Sun H, Liu Q, Guo H and Gong JP:
LPS induces HMGB1 relocation and release by activating the
NF-κB-CBP signal transduction pathway in the murine macrophage like
cell line RAW264.7. J Surg Res. 175:88–100. 2012.PubMed/NCBI
|
|
26
|
van Beijnum JR, Buurman WA and Griffioen
AW: Convergence and amplification of toll-like receptor (TLR) and
receptor for advanced glycation end products (RAGE) signaling
pathways via high mobility group B1 (HMGB1). Angiogenesis.
11:91–99. 2008.PubMed/NCBI
|
|
27
|
Lee JK, Kim SY, Kim YS, Lee WH, et al:
Suppression of the TRIF-dependent signaling pathway of Toll-like
receptors by luteolin. Biochem Pharmacol. 77:1391–1400. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Liliensiek B, Weigand MA, Bierhaus A,
Nicklas W, et al: Receptor for advanced glycation end products
(RAGE) regulates sepsis but not the adaptive immune response. J
Clin Invest. 113:1641–1650. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Shvartsman SY, Hagan MP, Yacoub A, Dent P,
et al: Autocrine loops with positive feedback enable
context-dependent cell signaling. Am J Physiol Cell Physiol.
282:C545–C559. 2002. View Article : Google Scholar : PubMed/NCBI
|