Introduction
It is now generally accepted that neoplastic
disorders arise through the acquisition of genomic changes by
suitably primed target cells (1).
These somatic mutations are often cytogenetically visible in the
form of balanced or unbalanced chromosome aberrations (1). Many hematologic malignancies are
characterized by balanced chromosomal abnormalities resulting in
chimeric genes of pathogenetic, diagnostic and prognostic
importance (1). In fact, more than
200 different genes are now known to be rearranged through
translocations in leukemias and leukemia-like disorders (1), with some genes being particularly
promiscuous, having numerous partners in different fusions and
disorders (1).
To date, the RUNX1 gene (previously
AML1, CBFA2 in 21q22) has been shown to fuse in-frame
with 23 different partner genes, encoding a structurally
heterogeneous group of proteins, in acute myeloid and lymphoblastic
leukemia (AML and ALL), chronic myeloid leukemia (CML; the fusion
here occurs secondarily), and myelodysplastic syndromes (MDS)
(2,3). Some of the fusions are common, such as
the ETV6/RUNX1 [t(12;21)(p13;q22)] in ALL,
RUNX1/RUNX1T1 (also known as AML1/ETO)
[t(8;21)(q22;q22)] in AML and RUNX1/MECOM [t(3;21)(q26;q22)]
in MDS, AML and CML in the blastic phase, whereas many of them have
only been reported in single cases, i.e., they have not yet been
shown to be recurrent (2,3). Whereas the prognostic impact of
frequent RUNX1 fusions is well known, corresponding
knowledge regarding infrequent chimeras is lacking (4,5).
Considering that most treatment protocols are in part based on the
presence of certain genetic changes in acute leukemias, it is of
potential clinical value to obtain further information also about
rare RUNX1 fusions, even in disease subgroups that cannot be
treated with medications specifically directed against the
leukemogenic defect. It is important to underscore that this may be
the case also for rare pathogenetic mechanisms where information is
gathered by the addition of single case reports, as recently
exemplified by the story of the rare MLL/ARHGAP26 (GRAF)
fusion in pediatric AML (6–8). For this reason, we here present the
molecular genetic and clinical features of a case of AML with
t(7;21)(p22;q22), a rare but recurrent chromosomal translocation
that was first described in 2006 by Paulsson et al(9).
Materials and methods
Case history
The study was approved by the Regional Committee for
Medical Research Ethics (REK Sør, http://helseforskning.etikkom.no), and written
informed consent was obtained from the patient.
A 52-year-old woman was admitted to the hospital
following a month of tiredness, sleepiness and symptoms of lower
airway infection. She had been treated with antibiotics without any
clinical improvement. Upon admission to the hospital, she had
fever, severe anemia (hemoglobin 5.8 g/l), thrombocytopenia
(116×109/l) and leukocytosis (34×109/l). A
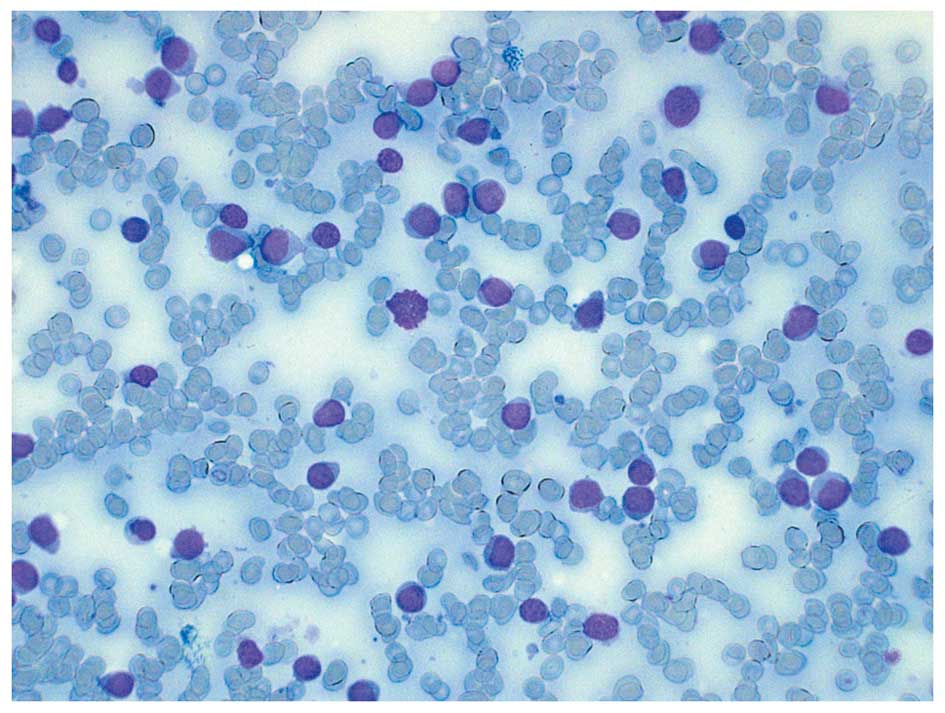
bone marrow aspirate showed >70% myeloblasts. The
immunophenotypical features of the malignant cells confirmed the
diagnosis of acute myeloblastic leukemia without differentiation.
The myeloblasts expressed CD34, CD117, HLA-DR antigens, CD13, and
partly CD11b, in addition to CD7 and CD56, but were negative for
myeloperoxidase as well as B- and T-cell lineage markers. The
clinical, blood sample and bone marrow findings (Fig. 1) were conclusive for acute
myeloblastic leukemia without maturation (formerly FAB M0).
The patient was transferred to the regional hospital
and standard induction chemotherapy was administered. Following
complete remission five months later, she received an allogenic
bone marrow transplant with reduced conditioning (preferred because
of complications during initial therapy) from a sibling donor. The
patient is, at the time of the preparation of this manuscript,
still in remission with a good chimerism status nine months after
the primary diagnosis, although she is now being treated for
complications due to graft vs. host disease and cytomegalovirus
reactivation.
G-banding and fluorescence in situ
hybridization (FISH)
Bone marrow cells were cytogenetically investigated
by standard methods. Chromosome preparations were made from
metaphase cells of a 24-h culture, G-banded using Leishman’s stain
and karyotyped according to ISCN 2009 guidelines (10). As part of our standard cytogenetic
diagnosis of AML patients, interphase FISH analyses of bone marrow
cells were performed with the Cytocell Multiprobe AML/MDS panel
(Cytocell, http://www.cytocell.co.uk/) searching
for −5/del(5q), PML/RARα, del(17p) (TP53),
AML1/ETO, trisomy 8, −7/del(7q), CBFβ/MYH11 and
del(20q). The del(5q) probe contains the probe for the EGR1
gene in 5q31.1 labeled in red as well as a control probe at 5p15.31
flanking the marker D5S30 labeled in green. Fluorescent signals
were captured and analyzed using the CytoVision system (Applied
Imaging, Newcastle, UK).
PCR analyses
Total RNA (1 μg) was reverse-transcribed in a
20-μl reaction volume using iScript Advanced cDNA Synthesis
kit for RT-qPCR according to the manufacturer’s instructions
(Bio-Rad). cDNA corresponding to 50 ng total RNA was used as the
template in subsequent PCR assays. The 25-μl PCR volume
contained 12.5 μl Premix Ex Taq™ DNA Polymerase Hot Start
version (Takara), 1 μl of diluted cDNA, and 0.2 μM of
each of the forward and reverse primers. For the detection of the
RUNX1-USP42 fusion transcript, the forward RUNX1–765F
(GGATGTTCCAGATGGCACTCTGG) and the reverse USP42–562R
(ACGTCCCCAGGATTACTGAGTGCC) primers were used. For the amplification
of a possible USP42-RUNX1 fusion transcript, the primers
USP42–116F (CAGAAT CAGCCTGGCAGCTCCGA) and RUNX1–1489R (GCCGA
CATGCCGATGCCGAT) were used. The PCR was run on a C-1000 Thermal
cycler (Bio-Rad) with an initial denaturation at 94°C for 30°sec,
followed by 35 cycles of 7°sec at 98°C, 2°min at 68°C, and a final
extension for 5°min at 68°C. PCR products (4 μl) were
stained with GelRed (Biotium), analyzed by electrophoresis through
1.0% agarose gel and photographed. The remaining PCR products were
excised from the gel, purified using the Qiagen Gel extraction kit
(Qiagen), and cloned to the pCR4-TOPO vector using TOPO TA cloning
kits for sequencing (Invitrogen). Colonies were sequenced at GATC
Biotech (Germany, http://www.gatc-biotech.com/en/home.html). The BLAST
software (http://www.ncbi.nlm.nih.gov/BLAST/) was used for
computer analysis of sequence data.
Results
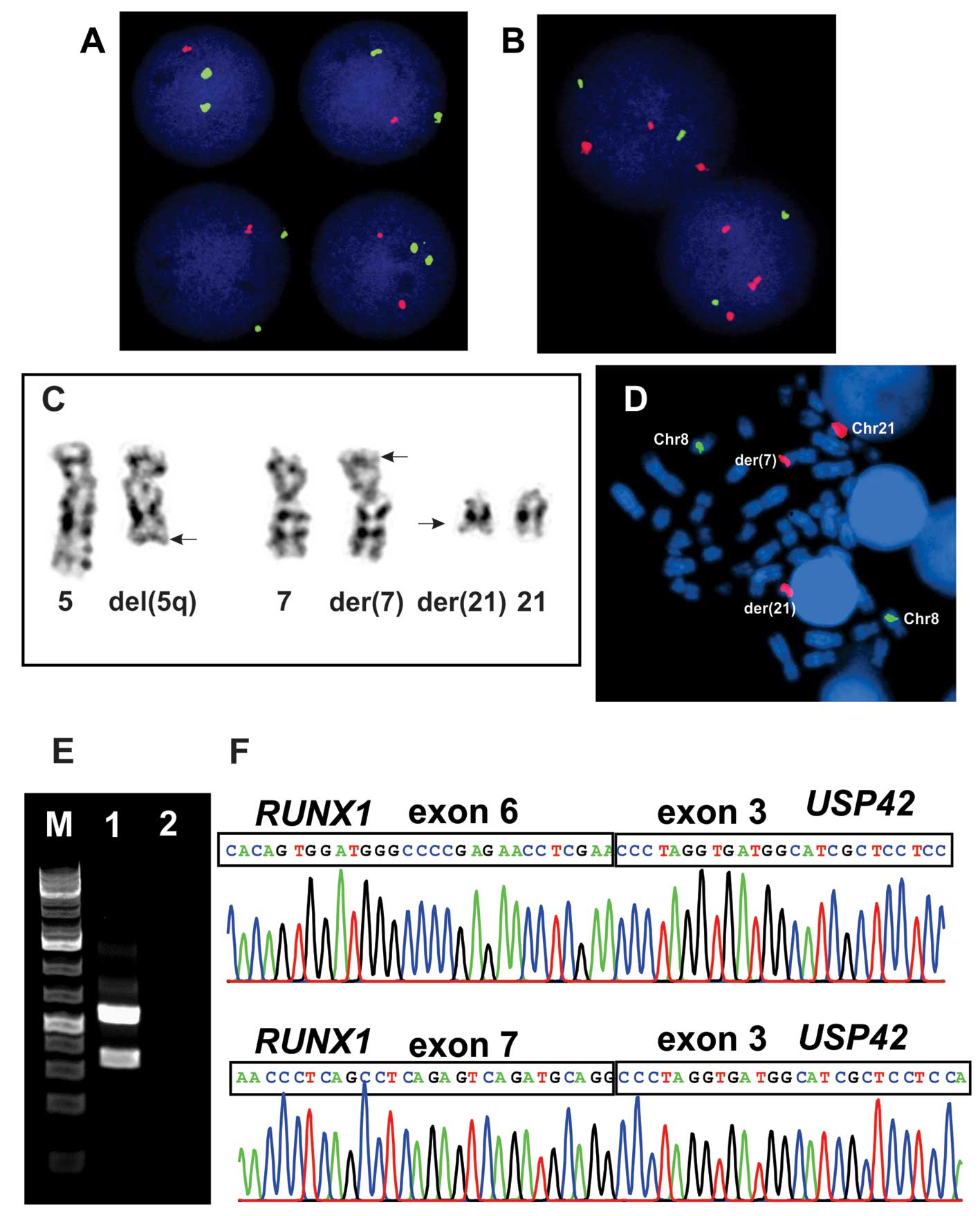
The G-banding analysis showed del(5)(q31) in all 15 cells analyzed which was
confirmed by interphase FISH (Fig. 2A
and C). The AML1/ETO probe (RUNX1/RUNX1T1) showed
abnormal signals, i.e., splitting of the RUNX1 probe was
observed in the majority of interphase nuclei examined in spite of
no cytogenetically visible rearrangement of this chromosome arm
(Fig. 2B). In the same experiment
two metaphase cells were found which demonstrated that part of the
RUNX1 probe was unexpectedly located on 7p22 (Fig. 2C and D). Other FISH analyses
detected no PML/RARα, del(17p), trisomy 8, −7/del(7q),
CBFβ/MYH11 or del(20q). Therefore, the whole karyotype was:
46,XX,del(5)(q31)[15].nuc ish
(EGR1×1)[196/206],(ETOx2,AML1×3)[186/222].ish t(7;21)(p22;q22)
(AML1+;AML1+)[2] (Fig. 1).
PCR amplification using the RUNX1–765F and
USP42–562R primers generated two RUNX1-USP42 fragments of 500- and
300-bp in size whereas PCR with primers USP42–116F and RUNX1–1489R
did not amplified any cDNA fragment (Fig. 2E). Sequencing of the two amplified
fragments showed that, in the 300-bp fragment, exon 6 of
RUNX1 (accession number NM_001754 version 3) was fused to
exon 3 of USP42 (accession number NM_032172 version 2),
whereas in the 500-bp long fragment exon 7 of RUNX1 was
fused to exon 3 of USP42 (Fig.
2F).
Discussion
The cryptic t(7;21)(p22;q22) chromosomal
translocation was first described in a 7-year-old boy with AML-M0
together with aberrant expression of T-lymphocyte-associated
markers (9). The translocation was
an unexpected finding after FISH had been performed using whole
chromosome painting probes for chromosome 7 while screening
pediatric leukemias for the t(7;12)(q36;p13) translocation
(9). In the present study, we also
detected the t(7;21) unexpectedly as a result of our standard
cytogenetic diagnosis of AML patients using interphase FISH
analyses of bone marrow cells and searching for −5/del(5q),
PML/RARα, del(17p), AML1/ETO, trisomy 8, −7/del(7q),
CBFβ/MYH11 and del(20q). The finding of a split RUNX1
probe in 186 of 222 interphase nuclei examined triggered further
investigations which led to the detection of the t(7;21). Notably,
AML with t(7;21) seems to be associated with AML-M0 [our patient
was also undifferentiated AML-M0] or myelomonocytic
differentiation. One patient was found to present with MDS RAEB-2
(11,12). Since the AML M0 subset makes up
<5% of all AMLs (14), it seems
likely that the 7;21-translocation is unusual in leading to this
particular undifferentiated myeloid leukemia in a dysproportionate
number of cases (admittedly, they may also display monocytic
differentiation according to a few reports). These leukemias are
immunophenotypically characterized by the aberrant expression of
CD7 as well as CD56 (present case and 5 previously reported cases)
(9,12,13).
Subsequent molecular genetic investigations of the
bone marrow cells showed that the result of the t(7;21)(p22;q22) is
the fusion of USP42 (on 7p22) and RUNX1 (on 21q22) to
generate a RUNX1-USP42 chimera (9). Including the present case, a
RUNX1-USP42 chimera has now been found in 8 cases while the
reciprocal USP42-RUNX1 was noted in 5 (9,11–13).
These findings suggest that RUNX1-USP42 is the leukemogenic
fusion. The incidence has hitherto been higher in males (n=6) than
in females (n=2) and all but one patient (the first described case)
were adults (age >30 years) (9,11–13).
Although recurrent, the RUNX1-USP42 fusion
seems to be rare. Paulsson et al(9) screened 35 additional AML cases, Foster
et al(11) screened 100
AML/MDS with normal karyotypes, and Giguére and Hebert (13) examined 95 leukemias without finding
additional cases of RUNX1-USP42. Jeandidier et
al(12) studied 397 AML cases
and found only 3 cases with RUNX1-USP42 fusion. An
interesting observation was that all 3 had additional 5q
abnormalities resulting in loss of material from that chromosome
arm. In total, 6 out of 8 cases (the present one included) with the
t(7;21)(p22;q22)/RUNX1-USP42 fusion had cytogenetically
visible changes of 5q resulting in loss of material (9,11–13).
In the series presented by Jeandidier et al(12), 3 out of 35 leukemias with 5q- had
the t(7;21)(p22;q22)/RUNX1-USP42 fusion gene (8.5%). In only
one case was there direct evidence as to whether 5q- or t(7;21) was
the primary cytogenetic abnormality. The patient described by
Paulsson et al(9) had only
the t(7;21) at the primary diagnosis (detected as
RUNX1-USP42 fusion), whereas a 5q- occurred secondarily as
an additional anomaly in a later sample.
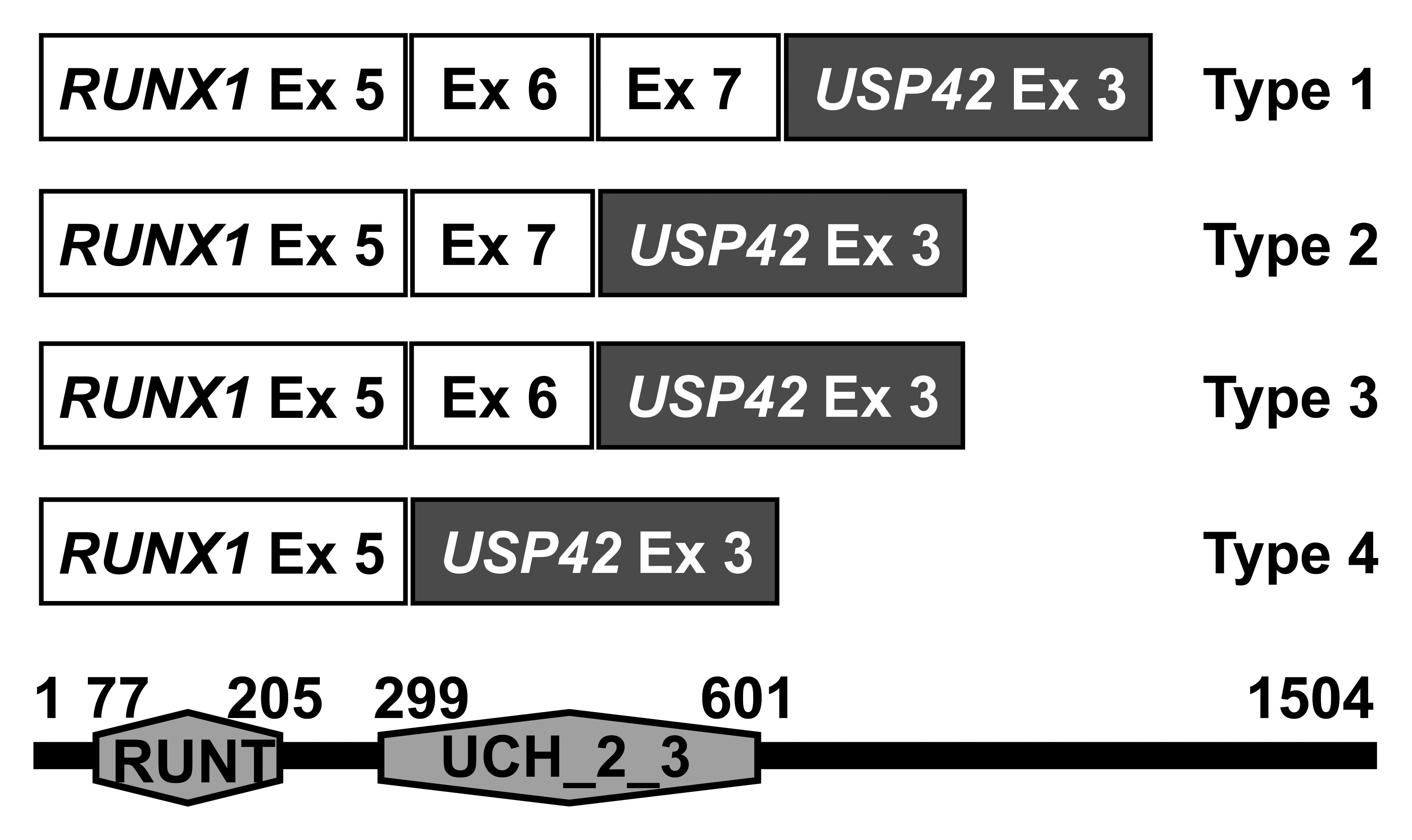
There are 4 types of RUNX1-USP42 chimeric
transcripts, defined here as types 1 to 4 (Fig. 3). The RUNX1-USP42 type 1 was
described as that in which exon 7 of RUNX1 is fused to exon
3 of USP42(9,11). The type 2 was defined as the
transcript in which exon 7 of RUNX1 is fused to exon 3 of
USP42 but exon 6 of RUNX1 is spliced out from the
fusion (9,11). In the type 3 fusion transcript, exon
6 of RUNX1 is fused to exon 3 of USP42. In type 4,
finally, exon 5 of RUNX1 is fused to exon 3 of
USP42(11). In all
RUNX1-USP42 fusion transcripts, the predicted fusion protein
would be expected to contain the Runt homology domain (RHD) which
is responsible for heterodimerization with CBFB and DNA binding,
and the catalytic UCH (ubiquitin carboxyl terminal hydroxylase)
domain of the USP42 protein (9,11). The
function of this fusion protein and its cellular consequences
leading to leukemia are unknown. It might exert its leukemogenic
effect as other RUNX1 fusions do in which the RHD is retained but
the transactivation domain of RUNX1 is removed, i.e., by acting as
a dominant-negative inhibitor of wild-type RUNX1 in transcription
activation (3). In fact, these
RUNX1 fusions mimic the RUNX1α variant which has higher affinity to
DNA binding but suppresses the transcription activation of RUNX1β
(3). RUNX1-USP42 might also affect
the regulation of TP53 since the USP42 interacts and deubiqitinates
this protein (15). More
information concerning the cellular function of the normal USP46 is
clearly needed in order to understand the role of
RUNX1-USP42 fusions in leukemias.
Acknowledgements
The authors thank Kristin Andersen and Silje
Bringsrud for their technical help. The present study was supported
by grants from the Norwegian Cancer Society and the South-Eastern
Norway Regional Healthy Authority.
References
|
1
|
Heim S and Mitelman F: Cancer
Cytogenetics. 3rd edition. John Wiley & Sons, Inc; Hoboken, NJ:
2009
|
|
2
|
Abe A, Katsumi A, Kobayashi M, et al: A
novel RUNX1-C11orf41 fusion gene in a case of acute myeloid
leukemia with a t(11;21)(p14;q22). Cancer Genet. 205:608–611. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
De Braekeleer E, Douet-Guilbert N, Morel
F, Le Bris MJ, Ferec C and De Braekeleer M: RUNX1
translocations and fusion genes in malignant hemopathies. Future
Oncol. 7:77–91. 2011.
|
|
4
|
Cho EK, Bang SM, Ahn JY, et al: Prognostic
value of AML 1/ ETO fusion transcripts in patients with
acute myelogenous leukemia. Korean J Intern Med. 18:13–20.
2003.PubMed/NCBI
|
|
5
|
Gandemer V, Chevret S, Petit A, et al:
Excellent prognosis of late relapses of ETV6/RUNX1-positive
childhood acute lymphoblastic leukemia: lessons from the FRALLE 93
protocol. Haematologica. 97:1743–1750. 2012.PubMed/NCBI
|
|
6
|
Borkhardt A, Bojesen S, Haas OA, et al:
The human GRAF gene is fused to MLL in a unique
t(5;11)(q31;q23) and both alleles are disrupted in three cases of
myelodysplastic syndrome/acute myeloid leukemia with a deletion 5q.
Proc Natl Acad Sci USA. 97:9168–9173. 2000.
|
|
7
|
Panagopoulos I, Kitagawa A, Isaksson M,
Morse H, Mitelman F and Johansson B: MLL/GRAF fusion in an
infant acute monocytic leukemia (AML M5b) with a cytogenetically
cryptic ins(5;11)(q31;q23q23). Genes Chromosomes Cancer.
41:400–404. 2004. View Article : Google Scholar
|
|
8
|
Wilda M, Perez AV, Bruch J, et al: Use of
MLL/GRAF fusion mRNA for measurement of minimal residual
disease during chemotherapy in an infant with acute monoblastic
leukemia (AML-M5). Genes Chromosomes Cancer. 43:424–426. 2005.
|
|
9
|
Paulsson K, Bekassy AN, Olofsson T,
Mitelman F, Johansson B and Panagopoulos I: A novel and
cytogenetically cryptic t(7;21)(p22;q22) in acute myeloid leukemia
results in fusion of RUNX1 with the ubiquitin-specific
protease gene USP42. Leukemia. 20:224–229. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Schaffer LG, Slovak ML and Campbell LJ:
ISCN 2009: an International System for Human Cytogenetic
Nomenclature; Karger, Basel. 2009
|
|
11
|
Foster N, Paulsson K, Sales M, et al:
Molecular characterisation of a recurrent, semi-cryptic
RUNX1 translocation t(7;21) in myelodysplastic syndrome and
acute myeloid leukaemia. Br J Haematol. 148:938–943.
2010.PubMed/NCBI
|
|
12
|
Jeandidier E, Gervais C, Radford-Weiss I,
et al: A cytogenetic study of 397 consecutive acute myeloid
leukemia cases identified three with a t(7;21) associated with 5q
abnormalities and exhibiting similar clinical and biological
features, suggesting a new, rare acute myeloid leukemia entity.
Cancer Genet. 205:365–372. 2012. View Article : Google Scholar
|
|
13
|
Giguere A and Hebert J: Microhomologies
and topoisomerase II consensus sequences identified near the
breakpoint junctions of the recurrent t(7;21)(p22;q22)
translocation in acute myeloid leukemia. Genes Chromosomes Cancer.
50:228–238. 2011.PubMed/NCBI
|
|
14
|
Bennett J, Catovsky D, Daniel M, Flandrin
G, Galton D, Gralnick H and Sultan C: Proposals for the
classification of the acute leukaemias. French-American-British
(FAB) co-operative group. Br J Haematol. 33:451–458. 1976.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Hock AK, Vigneron AM, Carter S, Ludwig RL
and Vousden KH: Regulation of p53 stability and function by the
deubiquitinating enzyme USP42. EMBO J. 30:4921–4930. 2011.
View Article : Google Scholar : PubMed/NCBI
|