1. Introduction
The human epidermal growth factor receptor (HER or
ErbB) belongs to the tyrosine kinase receptor superfamily. It
includes 4 highly homologous members, HER-1 (ErbB1), HER-2 (ErbB2),
HER-3 (ErbB3) and HER-4 (ErbB4). The distinguishing characteristics
of the HER family are interdependent and functional complementation
between members. After the ligands bind to the receptor, it
promotes the formation of HER/ErbB receptor homodimer or
heterodimer which leads to activation of the tyrosine kinase domain
(1,2) and downstream signaling pathways
(3,4). Signal transduction networks control
cellular activities such as gene expression, mitosis, cell
differentiation, cell proliferation, cell survival and apoptosis
(1,5).
HER-3 is a distinctive member of the HER family as
its kinase domain lacks certain residues that are known to be
essential for catalytic activity in other kinases. The function of
HER-3 was previously thought to be entirely dependent on other
members of the family, the role of HER-3 was considered to be
passive and the clinical value of HER-3 was greatly underestimated.
Currently, biochemical analysis has confirmed that the kinase
domain of HER-3 is a specific allosteric activator, it acts as a
functional activator to activate the recipient kinase. Accompanied
by an in-depth understanding of the structure and function of
HER-3, recent studies have also found HER-3 is involved in the
tumorigenesis, progression, new target exploration, target therapy
resistance of several types of cancer. In the critical search of a
cure for cancer, HER-3 provides insight into the better
understanding of tumors and targeted therapy.
2. In-depth understanding of the structure
and function of HER-3
HER-3 was initially isolated by MH Kraus in 1989.
The gene of HER-3 is located on chromosome 12q13, and its 6.2 kb
transcript is expressed in normal epithelial tissues (6). Subsequently, HER-3 cDNA was isolated
from human tumor cell lines (7).
HER-3 possesses 40–50% sequence homology with HER-1 and 40–45%
homology with HER-2 (7–9). The structure of HER-3 is typical in
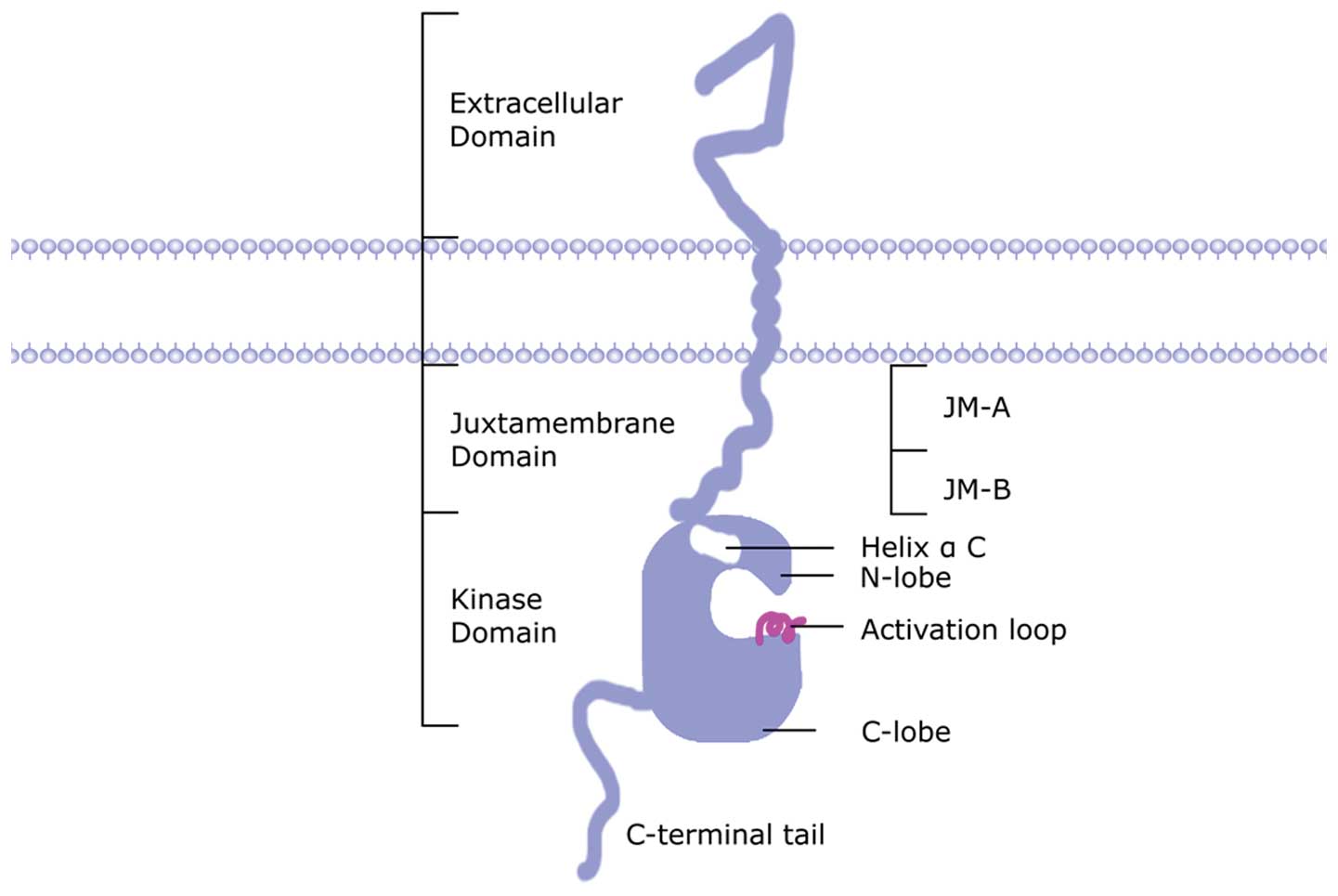
receptor tyrosine kinase family (Fig.
1). It includes an extracellular domain (ECD) with 612 amino
acid residues, a transmembrane helix domain with 32 hydrophobic
amino acids, and an intracellular tyrosine kinase domain (TKD) with
677 amino acids (7). Determination
of the 2.6-angstrom crystal structure of the entire extracellular
region of HER-3 revealed 4 type I insulin-like growth factor
receptor homologous domains, 2 specific ligand binding flanking
regions (district I and III) and 2 cysteine-rich regions (district
II and IV). The interaction between district II and IV limits the
relative direction of ligand binding domain and provides a
structural basis for understanding the varied affinity and
conformational changes of HER-3 upon ligand binding (10). The transmembrane region confers
receptor internalization and ligand-dependent calcium influx
(11). ErbB binding protein 1
(EBP-1) interacts with HER-3 and prevents the premature formation
of dimers, which prevents inappropriate activation of molecular
partners by HER-3 (12). The
intracellular domain is a continuation of the transmembrane region
and has a conserved ATP binding site
(Gly-Xaa-Gly-Xaa-Xaa-Gly-Xaa-Lys) that shares homology with other
members of the tyrosine kinase family. The intracellular region is
divided into a juxtamembrane region, a kinase domain and C-terminal
tail. The juxtamembrane region is divided into N-terminal
[juxtamembrane-A, (JM-A)] and C-terminal [juxtamembrane-B, (JM-B)]
(13). The kinase domain includes
N-lobe, helix αC, activation loop and C-lobe (13). The analysis of HER-3 dimers crystal
structure shows that recipient protein kinase (HER-1, HER-2 and
HER-4) interacts with the JM-A region of HER-3 via a conserved
amino acid sequence, and the JM-B region of recipient protein
kinase interacts with C-lobe of HER-3 by forming a stabilizing
latch (14). C-lobe of HER-3 can
combine and activate other members of the HER family, which is
consistent with the role of HER-3 as a functional activator but not
a recipient kinase. Helix αC of the kinase domain anchors activator
kinase domain to recipient kinase domain (13). In general, HER-3 is very similar to
inactivated HER-1/HER-4. Conformational changes in helix αC
sequence are highly important and may explain the difference in
function of HER-3 compared with other members of the family. For
example, Leu736 in helix αC of HER-1 is replaced by Thr738 in
HER-3, and this change stabilizes the inactivation state of HER-3.
Ile735 in hydrophobic core of HER-1 is replaced by Val737 in HER-3,
and this change weakens the ability of HER-3 to form hydrophobic
subunits. The structure of HER-3 is similar to that of an integrate
kinase, but locked in an inactive conformation similar to that of
Src/CDK. When the HER-3 sequence is activated, helix αC turns to
the active site, the activation ring center is opened and bound the
substrate peptide. There are 14 tyrosine residues in HER-3
C-terminal signal tail, including six PI3K binding sites that have
been confirmed through phosphorylation, which mediate interactions
between three regulatory subunits of PI3K and lead to activation of
downstream AKT signaling pathways (15).
At present, our understanding of the function of
HER-3 is very limited. In the 1990s, the kinase activity of HER-3
was not detected by recombinant protein technique. Therefore,
researchers believed that the kinase domain of HER-3 may be
non-functional (16,17). HER-3 kinase domain lacks several key
residues required for catalytic activity, such as Asp813 which is
present in HER-1; thus HER-3 was thought not to contain tyrosine
kinase activity and catalytic activity. The intracellular region of
HER-3 does not bind ATP and is not auto-phosphorylated (18,19).
HER-3 was considered to be functional merely as a signaling
substrate for other HER members, similar to the function of insulin
receptors, IRS1 and IRS2. However, Kornev and Taylor’s (20) study in 2009 demonstrated that HER-3
was not completely inactive; its activity was very low and was not
comparable to that of HER-1, but the kinase activity of HER-3 was
sufficient to mediate auto-phosphorylation of its intracellular
region. Shi et al(21),
using molecular mechanics simulation in 2010, revealed that the
phosphorylation catalyzed by HER-3 was mediated via the
‘inactive-like’ structure rather than the conserved catalytic
subunit, suggesting that the cytoplasmic region of HER-3 within the
receptor dimers was capable of binding ATP and promoting
auto-phosphorylation. Jura et al(13) confirmed, using biochemical analysis
in 2009, that the kinase domain of HER-3 was a specific allosteric
activator to activate the recipient protein kinase. Although these
studies challenged the traditional understanding of HER-3 function,
the exact mechanism is not fully understood and further studies
should be carried out. The function of HER-3 should be
re-inspected, the role of HER-3 in cell signaling transduction and
human cancer is becoming increasingly important. Findings with
regard to the function of HER-3 may provide insight into the
pathogenesis and therapy of human cancer.
Following ligand binding, HER-3 forms a receptor
dimer via a unique mechanism by which monomeric inactive state
changes to active state upon homo- or heterodimerization and the
tyrosine kinase and its downstream signaling pathways are activated
(22). HER-3 interacts with other
members of the HER family. First, HER-3 interacts with HER-1
directly. EGF can activate the tyrosine kinase of HER-1, and can
also cross-activate HER-3 at the same time (22). Tyrosine kinase inhibitor (TKI)
blocks downstream signaling pathways by inhibiting the interaction
of HER-1 and HER-3 (23). Second,
HER-3 interacts with HER-2 directly. Heregulin (HRG) ligand binding
causes prolonged activation of HER-3, but this process is strongly
dependent on HER-2 expression. In the HER family, HER-3 mainly
interacts with HER-2. Since HER-2 lacks ligand-binding activity and
HER-3 lacks catalytic kinase activity, both the receptors are
functionally interdependent. The relationship between HER-2 and
HER-3 has been described as ‘deaf’ and ‘dumb’ (18,24,25).
The cooperation between HER-2 and HER-3 is unique, but the
underlying molecular mechanism is poorly understood. Third, HER-3
and HER-4 can form heterodimer, but few studies have explored the
relationship between them (26).
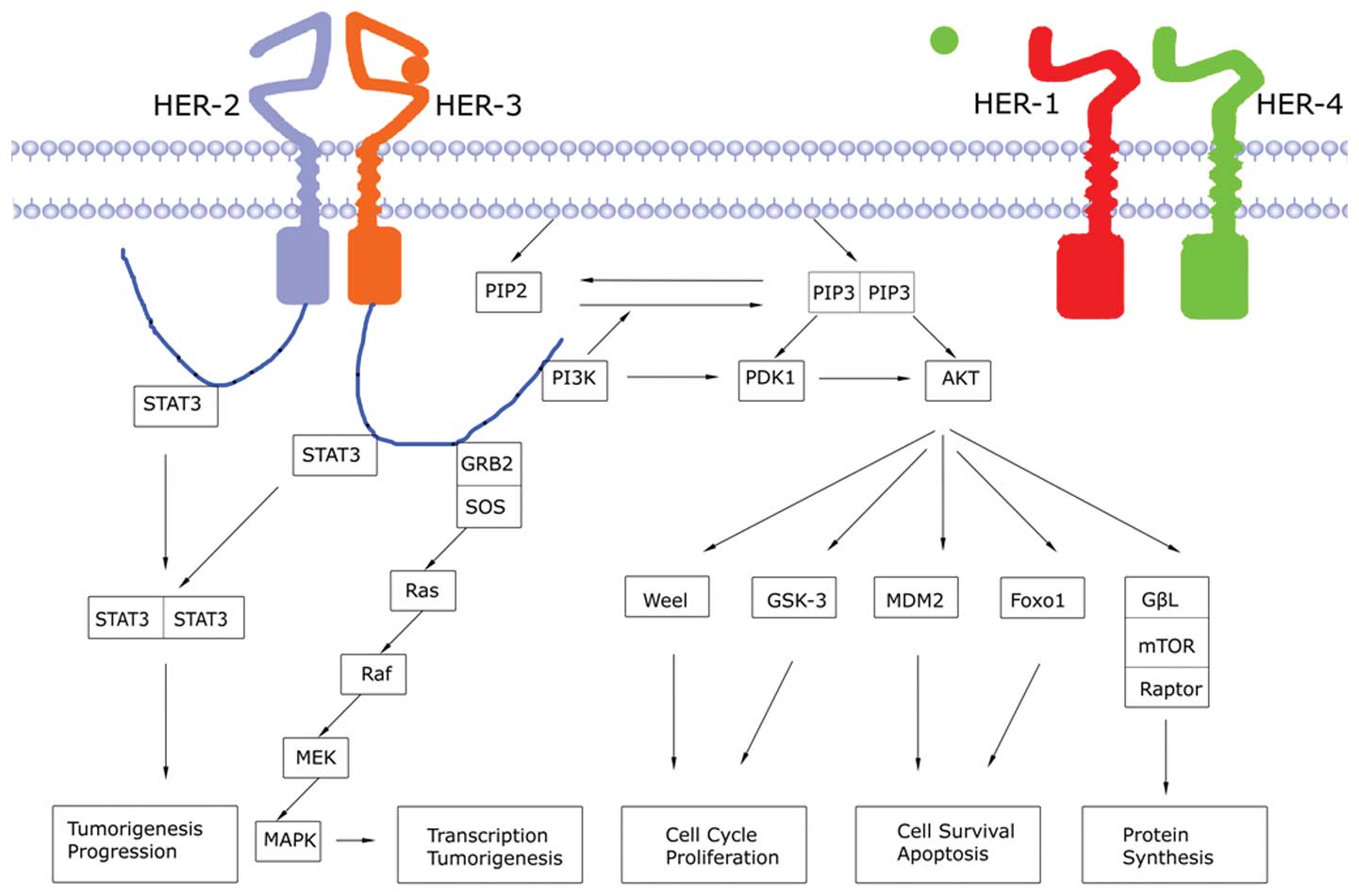
There are 2 relevant downstream signaling pathways
of HER-3 (Fig. 2). The first one is
the phosphatidylinositol 3-kinase
(PI3K)/3-phosphoinositide-dependent protein kinase (PDK) 1/protein
kinase B (AKT) pathway. Activation of PI3K is induced by the
formation of dimer between HER-3 and HER-1/2. PI3K is a dimeric
protein kinase composed of P110 catalytic subunit and P85
regulatory subunit (27). The P85
subunit binds to HER-specific anchor sites via its SH2 domain, and
the P110 subunit catalyzes the phosphorylation of
phosphatidylinositol 4,5-bisphosphate (PIP2) to 3,4,5-triphosphate
phosphatidylinositol (PIP3). The level of PIP3 is regulated by
phosphatase and tensin homologue deleted on chromosome 10 (PTEN)
(28). PI3K accumulates PDK1/AKT in
the cell membrane and activates it via phosphorylation, thus
stimulating downstream signaling. The PI3K pathway regulates cell
growth, cell apoptosis, tumor cell invasion, as well as metastasis
and chemotherapy resistance. The second pathway is the
Ras/Raf/MEK/mitogen-activated protein kinase (MAPK) pathway.
Activation of HER-3 and subsequent phosphorylation of tyrosine
kinase induce Grb2-SOS complex binding to phosphorylation anchor
sites. Then, the three-dimensional structure of SOS is altered
which enables the formation of Ras-GTP from aggregated Ras-GDP
(29,30), leading to the activation of Raf, MEK
and MAPK (31,32). Activated MAPK transduces
extracellular stimuli into the cell to regulate transcription
factors in the nucleus and induces cell migration and proliferation
(33).
3. The close relationship between HER-3 and
human tumor
The studies on HER-1 and HER-2 in tumor targeted
therapy and efficacy prediction have developed. For example, small
molecule HER-1 TKI, gefitinib, has been used in the first-line
treatment of advanced non-small cell lung cancer (NSCLC) with HER-1
mutation (34). Anti-HER-1
monoclonal antibody, cetuximab, has been used in targeted therapy
for head and neck squamous cell carcinoma, colorectal cancer and
advanced NSCLC (35–37). Anti-HER-2 monoclonal antibody,
trastuzumab, has been used in targeted therapy for HER-2 positive
advanced breast cancer and gastric cancer (38,39).
In view of the significant contribution of HER-1 and HER-2,
researchers began looking into the role of HER-3. With the new
understanding of the structure and function of HER-3, studies on
the relationship of HER-3 with tumorigenesis, progression, new
target exploration, target therapy resistance, prognosis and
efficacy prediction are increasing. Some studies have confirmed
that HER-3 plays an important role in the occurrence and
progression of lung cancer, breast cancer, colorectal cancer as
well as other types of cancer (26,40–42).
The HER-3/PI3K/AKT signal pathway plays a key role in the target
therapy resistance of NSCLC, breast cancer, head and neck squamous
cell carcinoma, prostate cancer and hepatocellular carcinoma and
other types of cancer (43–48). Inhibition of HER-3 and HER-1/HER-2
is very important for tumor treatment (49,50)
and it may be a new therapeutic target. HER-3 is also beneficial in
predicting the prognosis and treatment efficacy. In the search for
tumor treatment, HER-3 has become a focus of concern in the HER
family. The following describes the role of HER-3 in different
tumors.
Overexpression of HER-3 in NSCLC cell lines
accelerated growth and metastasis of tumor cells, and promoted
tumorigenicity of allografts in a kinase-dependent manner. By
contrast, downregulation of HER-3 inhibited proliferation and
migration of tumor cells, tumor growth and metastasis in
vivo. HER-3 silencing inhibited tumor cell growth by reducing
DNA synthesis and caspase-8-mediated apoptosis, and tumor cell
migration by increasing accumulation of focal adhesion components
(40). Lung adenocarcinoma cells
were markedly suppressed in culture by siRNAs to the receptor HER-3
or its downstream signaling partner AKT2 (51). The above studies suggest that HER-3
and its downstream signaling pathway play a crucial role in
occurrence and metastasis of lung cancer. HER-1 TKI such as
gefitinib/erlotinib is very effective treatment for NSCLC with
HER-1 mutation, but the emergence of drug resistance is difficult
to overcome. The sensitivity of HER-1 TKI is associated with
inhibition of the HER-3/PI3K/AKT signaling pathway, i.e., this
pathway will lead to TKI resistance if not effectively inhibited
(43). Gefitinib temporarily
inhibits HER-3/PI3K/AKT signaling, but in the process of
subsequently sustained inhibition of HER-1 and HER-2, recovery of
HER-3 activity and reactivation of the PI3K/AKT pathway will lead
to drug resistance. Downregulation of HER-3 led to reduction of AKT
phosphorylation level and growth inhibition in an NSCLC mutant cell
line that was sensitive to gefitinib. However, downregulation of
HER-3 did not alter the activity of AKT in resistant tumor cell
lines, suggesting that the separation of HER-3 from downstream AKT
signaling pathway was one of the important aspects of the drug
resistance (52). HER-3 and
PI3K/AKT pathways may enable tumor cells to escape TKI inhibition
through a compensatory offset of the equilibrium between HER-3
phosphorylation and dephosphorylation (43). Amplification of MET proto-oncogene
promoted HER-3-dependent PI3K activation and led to drug resistance
in gefitinib-sensitive lung cancer cell lines, inhibition of MET
proto-oncogene restored the sensitivity to gefitinib (48). In addition to HER-1 mutation, a
second mutation, T790M, is also associated with acquired drug
resistance. Introduction of exogenous HER-1 with T790M mutation
effectively blocked gefitinib activity and maintained
HER-3/PI3K/AKT signal activation in lung cancer cells (53). In addition, oncogenic mutation of
PIK3CA, p110αE545K, activated PI3K signaling and eliminated
gefitinib-induced apoptosis (54).
These studies demonstrated that HER-3 and/or PI3K/AKT signal may be
intermediate links of acquired gefitinib resistance. While
irreversible tyrosine kinase agents inhibited HER-1 in resistant
cells, they re-inhibited HER-3 and PI3K signaling at the same time,
which further highlighted the central role of HER-3 in adjusting
drug sensitivity or resistance (55). miR-22 in lung cancer cell lines
played a good antitumor effect through inhibition of HER-3
transcriptional regulation (56).
Thus, HER-3 is a potential therapeutic target and simultaneous
inhibition of HER-3 and HER-1 will bring considerable benefit to
the clinic. Due to the complexity of signaling networks, whether
the HER-3/PI3K/AKT pathway alone can induce TKI resistance in NSCLC
merits further study. The positive expression rates of HER-3 in
NSCLC are 18–67%. Study results on the response to TKI, survival
and prognosis of patients with HER-3 high expression are
inconsistent. A study including 192 surgically removed NSCLC cases
showed the expression of HER-3mRNA was higher in patients with
highly-differentiated, adenocarcinoma, mutant HER-1 than in
patients with poorly-differentiated, non-adenocarcinoma, wild-type
HER-1, and the expression was higher in females than in males
(57). Large sample studies and
more uniform and accurate research methods are required to further
clarify whether high HER-3 expression is an indicator of TKI
benefit.
The role of the HER family in tumorigenesis is most
aptly understood in breast cancer subtypes with HER-2 gene
amplification. HER-3 is required for HER-2-induced preneoplastic
changes to breast tumor formation (58). HER-2 must dimerize with HER-3 to
promote breast cancer cell proliferation; deletion of HER-3 in
HER-2 positive breast cancer cell lines produces strong
anti-proliferative effects (59).
HER-2/HER-3 heterodimer is the most powerful carcinogenic unit, the
phosphorylation state of HER-3 is critical for HER-2 positive
breast cancer cell motility and metastasis (41). A significant increase of HER-3
phosphorylation in HER-2 positive breast cancer was accompanied by
activation of downstream signaling pathways, and knockdown of HER-3
was always accompanied by tumor shrinkage in vitro(60). Dephosphorylation of HER-3 and
decoupling with PI3K lead to downregulation of AKT signaling and it
is directly related to the anti-proliferative effect of
trastuzumab. HER-3-specific affibody molecules (Z05416, Z05417)
blocked cancer cell growth by inhibiting HRG-induced HER-3
phosphorylation in the MCF-7 and SKBR-3 breast cancer cell lines
(61). The above studies clarify
that the phosphorylation state of HER-3 and downstream signaling
play a central role in the occurrence of HER-2 positive breast
cancer, and HER-3 may be a drug target. Since the process of TKI
inhibiting HER-2/HER-3 transphosphorylation and PI3K/AKT signaling
pathway activation in HER-2 positive breast cancer are transient,
the antitumor efficacy of TKI is weakened (43). The kinase function of HER-2 is
essential during tumorigenesis (62), therefore, inhibition of the
catalytic kinase activity is a key mechanism for anticancer drugs.
The buffering effect of incomplete inhibition of HER-2 kinase
activity by HER-3 may be a highly important mechanism by which
HER-2 positive breast cancer cells escape TKI treatment (43,63).
miR-205 downregulates HER-3 and recovers the sensitivity to TKI in
human breast cancer cells (64).
pHER-3 upregulation in a fulvestrant resistant cell line was mostly
accompanied by an increase of pAKT activity. However, highly
specific inhibition by HER-3 antibody (A5) significantly
downregulated pHER-3 without affecting downstream ERK/AKT
phosphorylation, suggesting that the resistant cells may produce
endogenous ligand that reactivated pHER-3. Exogenous ligands
binding to HER-3 affect AKT downstream signals, but the underlying
mechanism remains unclear (47).
Bispecific antibody (MM-111) formed trimer with HER-2/HER-3, which
effectively inhibited proliferation of HER-3 and HER-2 positive
tumor cells (50). A new selective
PI3K inhibitor (GDC-0941) combined with trastuzumab and pertuzumab
inhibited the growth of tumor cells, and led to morphological
changes of gland cells and inhibition of the HER-3/PI3K/AKT signal
pathway (65). GDC-0941 is also
effective for the treatment of trastuzumab-resistant tumor cells
(66). XL147, which is also a PI3K
inhibitor, inhibits tumor cell growth in a dose-dependent manner.
In HER-2 positive breast cancer cells, knockdown of HER-3 by siRNA
enhanced the effect of XL147 (67).
Thus, multi-target treatment provides an increase of clinical
benefit for patients with breast cancer, inhibition of HER-2, HER-3
and PI3K simultaneously may be the future treatment direction of
HER-2 positive breast cancer. The positive expression rates of
HER-3 in breast cancer are 18–75%. The expression of HER-3 was
positively correlated with high organizational classification and
lymphatic vessel invasion, suggesting it was significantly
associated with tumor progression and metastasis, and may serve as
a useful prognostic biomarker (68). Tissue microarray analysis
demonstrated that normal expression of HER-1/HER-2 and
overexpression of HER-3 in invasive breast cancer indicated poor
prognosis (69,77). Multivariate analysis showed that
HER-2 and HER-3 were independent prognostic markers, while
clustering analysis showed that coexpression of HER-1 and HER-3
suggested poor prognosis, with a 10-year survival rate of 42%
(70). However, in HER-2
negative/low expression cases or HER-2 positive breast cancer
patients treated with trastuzumab, there was no correlation between
the expression of HRG, HER-3 and survival or known clinical
prognostic factors (71,72).
There is almost no HER-3 expression in normal colon
tissue, but the positive expression rates of HER-3 in colorectal
cancer tissue are 50–89%. Knockdown of HER-3 by siRNA in colon
cancer cell lines was accompanied by absence of HER-4 expression
and elevation of tumor cell apoptosis; HER-3/HER-4 heterodimer may
be one of the precipitating factors of colon cancer (26). HRG expression was detected in colon
metastatic liver cancer cells, knockdown of integrin αv and HER-3
by siRNA significantly inhibited HRG-induced tumor cell migration
as well as liver metastasis in vivo, marked phosphorylation
of AKT was found in the process, cell migration was suppressed by
specific inhibitors of PI3K. The study indicated that
HRG/HER-3/PI3K/AKT may participate in colon cancer liver metastasis
(73). The median progression-free
survival time and the median overall survival time of the patients
with wild-type K-RAS advanced colorectal cancer receiving cetuximab
combining irinotecan treatment in the HER-3 negative group were
significantly higher than in the HER-3 positive group, suggesting
that HER-3 may be a predictor of cetuximab efficacy in patients
with wild-type K-RAS advanced colorectal cancer (74). Comprehensive analysis of HER-3 and
K-RAS may aid in identifying the most appropriate colorectal cancer
patients for cetuximab treatment and may provide an effective
treatment strategy. Studies on HER-3 and other digestive system
tumors are limited. The positive expression rate of HER-3 in
gastric cancer is 13.7%, and it correlates with late stage and poor
prognosis (75). DARPP-32 promotes
resistance of gastric cancer cells to gefitinib by stimulating
interaction between HER-1 and HER-3 and activating PI3K/AKT
signaling (76). The results of the
ToGA trial are encouraging; it is expected that routine detection
of HER-2 will be included in the diagnosis of advanced gastric
cancer (39,77). The trial investigates whether HER-3
will be of value for guiding the treatment of gastric cancer.
HER-1/2 expression is absent in normal pancreatic tissue, but
HER-3/4 are expressed (78). The
positive expression rates of HER-3 in pancreatic cancer tissues are
27–47%. Generally, HER-3 positive was prone to cause targeted
therapy drug resistance, but HER-3 increases the sensitivity of
pancreatic cancer cells to erlotinib. Knockdown of HER-3 in
erlotinib sensitive pancreatic cancer cell lines resulted in AKT
level reduction and pancreatic cancer cell proliferation,
suggesting that HER-3 may be a sensitive biomarker for erlotinib in
pancreatic cancer (79). The median
survival time in HER-3 overexpression patients with resectable
pancreatic cancer was 37.2 months, but in HER-3-negative patients
it was 58.6 months, therefore, HER-3 overexpression may be an
independent indicator of poor prognosis for patients with
curatively resected pancreatic cancer (80). A study found sHER-3 (isomers) was
more accurate than AFP in identifying early liver cancer from
chronic hepatitis; the plasma high-level was closely related to
portal venous invasion and extrahepatic metastasis (81). HER-3 restricted cell response to
sorafenib or IGF1R inhibitor in hepatocellular carcinoma cells
(46,82).
HER-3 is activated in multiple ovarian cancer cell
lines. Activation of NRG1/HER-3 autocrine loop pathway promotes the
proliferation of human ovarian cancer cells. In the mouse xenograft
model, deletion of HER-3 inhibited proliferation of OVCAR8 cells
and slowed down tumor progression, suggesting that HER-3 and/or
NRG1 play a key role in the pathogenesis of ovarian cancer and are
potential therapeutic targets for advanced ovarian cancer (83). The positive expression rates of
HER-3 in ovarian cancer are 3–53%, and some studies reported that
HER-3 expression was negatively correlated to overall survival
(84,85). Regardless of whether the
corresponding ligand NRG existed or not, HER-3 promoted prostate
cancer cells mobile in vitro and tumor formation in
vivo(86). HER-2/HER-3
heterodimer promoted aberrant activation of androgen and led to the
formation of hormone-resistant prostate cancer (45). Further studies are required to
elucidate the role of HER-3 and other HER members in reproductive
system tumors.
High expression of HER-3 and absence of HER-2
expression in melanoma indicated that HER-3 may be an allosteric
activator of HER-1 or HER-4. Disorders of NRG1/HER-3 and HRG/HER-3
signaling are correlated with development and metastasis of
melanoma (87,88). Anti-human HER-3 monoclonal antibody
promoted HER-3 receptor internalization and degradation, and
inhibited growth and migration of human melanoma cells (89). The pan-HER receptor TKI (canertinib)
inhibits HER-1, HER-2 and HER-3 receptor phosphorylation and
promotes apoptosis of malignant melanoma in vitro; it also
displays antitumor activity in vivo(90).
4. Conclusion
With protein crystallization, molecular biology has
begun to reveal the structure of HER-3. At the same time, the
kinase domain of HER-3 acting as a functional activator to activate
the recipient kinase was confirmed. Insights into the activation
mechanism of the HER family were also gradually elucidated. In the
critical search of a cure for cancer, HER-3, as a member recognized
step by step in the HER family, seems to provide some insight into
tumor therapy. Although people have known about HER-3 for several
years, the value of HER-3 was formerly ignored. Currently, research
results demonstrate that HER-3 is closely related to tumorigenesis,
progression and metastasis, which helps to clarify the mechanisms
of tumor biological behavior. HER-3 is involved in targeted therapy
resistance and may be a new therapeutic target. A further in-depth
understanding of HER-3 will play a fueling role in HER-3 associated
targeted therapies. Analyzing the relationship between the
expression of HER-3 and the effects of targeted therapy may help to
identify the most appropriate patient sub-groups for HER-3 targeted
treatment. In the search for a breakthrough in cancer treatment,
HER-3 has become an emerging protagonist in the HER family.
Acknowledgements
The authors thank Professor Xinjun Lv (Chinese
Center for Disease Control and Prevention) for direction on review
and Professor Enyun Shen (Shandong Provincial Academy of Medical
Sciences) for the helpful discussion. This study was supported by
grants from the Beijing Municipal ‘215’ High-level Health Person
Foundation Project (2011–2014 to B.C.), the Beijing Municipal ‘Ten,
Hundred, Thousand’ Person Foundation Project (2011–2013 to B.C.),
and the Capital Medical University Sciences-Clinical Research
Cooperation Foundation (2011–2012 to B.C.).
References
|
1
|
Olayioye MA, Neve RM, Lane HA and Hynes
NE: The ErbB signaling network: receptor heterodimerization in
development and cancer. EMBO J. 19:3159–3167. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Schlessinger J: Cell signaling by receptor
tyrosine kinases. Cell. 103:211–225. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Marmor MD, Skaria KB and Yarden Y: Signal
transduction and oncogenesis by ErbB/HER receptors. Int J Radiat
Oncol Biol Phys. 58:903–913. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Shepard HM, Brdlik CM and Schreiber H:
Signal integration: a framework for understanding the efficacy of
therapeutics targeting the human EGFR family. J Clin Invest.
118:3574–3581. 2008. View
Article : Google Scholar : PubMed/NCBI
|
|
5
|
Yarden Y and Sliwkowski MX: Untangling the
ErbB signalling network. Nat Rev Mol Cell Biol. 2:127–137. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Kraus MH, Issing W, Miki T, Popescu NC and
Aaronson SA: Isolation and characterization of ERBB3, a third
member of the ERBB/epidermal growth factor receptor family:
evidence for overexpression in a subset of human mammary tumors.
Proc Natl Acad Sci USA. 86:9193–9197. 1989. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Plowman GD, Whitney GS, Neubauer MG, et
al: Molecular cloning and expression of an additional epidermal
growth factor receptor-related gene. Proc Natl Acad Sci USA.
87:4905–4909. 1990. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Ullrich A, Coussens L, Hayflick JS, et al:
Human epidermal growth factor receptor cDNA sequence and aberrant
expression of the amplified gene in A431 epidermoid carcinoma
cells. Nature. 309:418–425. 1984. View
Article : Google Scholar : PubMed/NCBI
|
|
9
|
Coussens L, Yang-Feng TL, Liao YC, et al:
Tyrosine kinase receptor with extensive homology to EGF receptor
shares chromosomal location with neu oncogene. Science.
230:1132–1139. 1985. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Cho HS and Leahy DJ: Structure of the
extracellular region of HER3 reveals an interdomain tether.
Science. 297:1330–1333. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Chen WS, Lazar CS, Lund KA, et al:
Functional independence of the epidermal growth factor receptor
from a domain required for ligand-induced internalization and
calcium regulation. Cell. 59:33–43. 1989. View Article : Google Scholar
|
|
12
|
Yoo J, Wang X, Rishi A, et al: Interaction
of the PA2G4 (EBP1) protein with ErbB-3 and regulation of this
binding by heregulin. Br J Cancer. 82:683–690. 2000.PubMed/NCBI
|
|
13
|
Jura N, Shan Y, Cao X, Shaw DE and Kuriyan
J: Structural analysis of the catalytically inactive kinase domain
of the human EGF receptor 3. Proc Natl Acad Sci USA.
106:21608–21613. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Red Brewer M, Choi SH, Alvarado D, et al:
The juxtamembrane region of the EGF receptor functions as an
activation domain. Mol Cell. 34:641–651. 2009.PubMed/NCBI
|
|
15
|
Jones RB, Gordus A, Krall JA and MacBeath
G: A quantitative protein interaction network for the ErbB
receptors using protein microarrays. Nature. 439:168–174. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Sierke SL, Cheng K, Kim HH and Koland JG:
Biochemical characterization of the protein tyrosine kinase
homology domain of the ErbB3 (HER3) receptor protein. Biochem J.
322:757–763. 1997.PubMed/NCBI
|
|
17
|
Prigent SA and Gullick WJ: Identification
of c-erbB-3 binding sites for phosphatidylinositol 3′-kinase and
SHC using an EGF receptor/c-erbB-3 chimera. EMBO J. 13:2831–2841.
1994.
|
|
18
|
Citri A, Skaria KB and Yarden Y: The deaf
and the dumb: the biology of ErbB-2 and ErbB-3. Exp Cell Res.
284:54–65. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Manning G, Whyte DB, Martinez R, Hunter T
and Sudarsanam S: The protein kinase complement of the human
genome. Science. 298:1912–1934. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Kornev AP and Taylor SS: Pseudokinases:
functional insights gleaned from structure. Structure. 17:5–7.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Shi F, Telesco SE, Liu Y, Radhakrishnan R
and Lemmon MA: ErbB3/HER3 intracellular domain is competent to bind
ATP and catalyze autophosphorylation. Proc Natl Acad Sci USA.
107:7692–7697. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Dawson JP, Berger MB, Lin CC, Schlessinger
J, Lemmon MA and Ferguson KM: Epidermal growth factor receptor
dimerization and activation require ligand-induced conformational
changes in the dimer interface. Mol Cell Biol. 25:7734–7742. 2005.
View Article : Google Scholar
|
|
23
|
Carrasco-García E, Saceda M, Grasso S, et
al: Small tyrosine kinase inhibitors interrupt EGFR signaling by
interacting with erbB3 and erbB4 in glioblastoma cell lines. Exp
Cell Res. 317:1476–1489. 2011.PubMed/NCBI
|
|
24
|
Baselga J and Swain SM: Novel anticancer
targets: revisiting ERBB2 and discovering ERBB3. Nat Rev Cancer.
9:463–475. 2009. View
Article : Google Scholar : PubMed/NCBI
|
|
25
|
Zhang Y, Opresko L, Shankaran H, Chrisler
WB, Wiley HS and Resat H: HER/ErbB receptor interactions and
signaling patterns in human mammary epithelial cells. BMC Cell
Biol. 10:782009. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Lee D, Yu M, Lee E, et al: Tumor-specific
apoptosis caused by deletion of the ERBB3 pseudo-kinase in mouse
intestinal epithelium. J Clin Invest. 119:2702–2713. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Vivanco I and Sawyers CL: The
phosphatidylinositol 3-Kinase AKT pathway in human cancer. Nat Rev
Cancer. 2:489–501. 2002. View
Article : Google Scholar : PubMed/NCBI
|
|
28
|
Miller TW, Pérez-Torres M, Narasanna A, et
al: Loss of Phosphatase and Tensin homologue deleted on chromosome
10 engages ErbB3 and insulin-like growth factor-I receptor
signaling to promote antiestrogen resistance in breast cancer.
Cancer Res. 69:4192–4201. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Lowenstein EJ, Daly RJ, Batzer AG, et al:
The SH2 and SH3 domain-containing protein GRB2 links receptor
tyrosine kinases to ras signaling. Cell. 70:431–442. 1992.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Batzer AG, Rotin D, Ureña JM, Skolnik EY
and Schlessinger J: Hierarchy of binding sites for Grb2 and Shc on
the epidermal growth factor receptor. Mol Cell Biol. 14:5192–5201.
1994.PubMed/NCBI
|
|
31
|
Hallberg B, Rayter SI and Downward J:
Interaction of Ras and Raf in intact mammalian cells upon
extracellular stimulation. J Biol Chem. 269:3913–3916.
1994.PubMed/NCBI
|
|
32
|
Liebmann C: Regulation of MAP kinase
activity by peptide receptor signalling pathway: paradigms of
multiplicity. Cell Signal. 13:777–785. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Pearson G, Robinson F, Beers Gibson T, et
al: Mitogen-activated protein (MAP) kinase pathways: regulation and
physiological functions. Endocr Rev. 22:153–183. 2001.PubMed/NCBI
|
|
34
|
Gridelli C, De Marinis F, Di Maio M,
Cortinovis D, Cappuzzo F and Mok T: Gefitinib as first-line
treatment for patients with advanced non-small-cell lung cancer
with activating Epidermal Growth Factor Receptor mutation:
implications for clinical practice and open issues. Lung Cancer.
72:3–8. 2011. View Article : Google Scholar
|
|
35
|
Tejani MA, Cohen RB and Mehra R: The
contribution of cetuximab in the treatment of recurrent and/or
metastatic head and neck cancer. Biologics. 4:173–185.
2010.PubMed/NCBI
|
|
36
|
Lièvre A, Bachet JB, Boige V, et al:
KRAS mutations as an independent prognostic factor in
patients with advanced colorectal cancer treated with cetuximab. J
Clin Oncol. 26:374–379. 2008.
|
|
37
|
Pirker R, Pereira JR, Szczesna A, et al:
Cetuximab plus chemotherapy in patients with advanced
non-small-cell lung cancer (FLEX): an open-label randomised phase
III trial. Lancet. 373:1525–1531. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Slamon DJ, Leyland-Jones B, Shak S, et al:
Use of chemotherapy plus a monoclonal antibody against HER2 for
metastatic breast cancer that overexpresses HER2. N Engl J Med.
344:783–792. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Bang YJ, Van Cutsem E, Feyereislova A, et
al: Trastuzumab in combination with chemotherapy versus
chemotherapy alone for treatment of HER2-positive advanced gastric
or gastro-oesophageal junction cancer (ToGA): a phase 3,
open-label, randomised controlled trial. Lancet. 376:687–697. 2010.
View Article : Google Scholar
|
|
40
|
Ji XD, Li G, Feng YX, et al: EphB3 is
overexpressed in non-small-cell lung cancer and promotes tumor
metastasis by enhancing cell survival and migration. Cancer Res.
71:1156–1166. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Holbro T, Beerli RR, Maurer F, Koziczak M,
Barbas CF III and Hynes NE: The ErbB2/ErbB3 heterodimer functions
as an oncogenic unit: ErbB2 requires ErbB3 to drive breast tumor
cell proliferation. Proc Natl Acad Sci USA. 100:8933–8938. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Smirnova T, Zhou ZN, Flinn RJ, et al:
Phosphoinositide 3-kinase signaling is critical for ErbB3-driven
breast cancer cell motility and metastasis. Oncogene. 31:706–715.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Sergina NV, Rausch M, Wang D, et al:
Escape from HER-family tyrosine kinase inhibitor therapy by the
kinase-inactive HER3. Nature. 445:437–441. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Erjala K, Sundvall M, Junttila TT, et al:
Signaling via ErbB2 and ErbB3 associates with resistance and
epidermal growth factor receptor (EGFR) amplification with
sensitivity to EGFR inhibitor gefitinib in head and neck squamous
cell carcinoma cells. Clin Cancer Res. 12:4103–4111. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Zhang Y, Linn D, Liu Z, et al: EBP1, an
ErbB3-binding protein, is decreased in prostate cancer and
implicated in hormone resistance. Mol Cancer Ther. 7:3176–3186.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Desbois-Mouthon C, Baron A, Blivet-Van
Eggelpoël MJ, et al: Insulin-like growth factor-1 receptor
inhibition induces a resistance mechanism via the epidermal growth
factor receptor/HER3/AKT signaling pathway: rational basis for
cotargeting insulin-like growth factor-1 receptor and epidermal
growth factor receptor in hepatocellular carcinoma. Clin Cancer
Res. 15:5445–5456. 2009.
|
|
47
|
Frogne T, Benjaminsen RV, Sonne-Hansen K,
et al: Activation of ErbB3, EGFR and Erk is essential for growth of
human breast cancer cell lines with acquired resistance to
fulvestrant. Breast Cancer Res Treat. 114:263–275. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Engelman JA, Zejnullahu K, Mitsudomi T, et
al: MET amplification leads to gefitinib resistance in lung
cancer by activating ERBB3 signaling. Science. 316:1039–1043. 2007.
View Article : Google Scholar
|
|
49
|
Grøvdal LM, Kim J, Holst MR, Knudsen SLJ,
Grandal MV and van Deurs B: EGF receptor inhibitors increase ErbB3
mRNA and protein levels in breast cancer cells. Cell Signal.
24:296–301. 2012.PubMed/NCBI
|
|
50
|
McDonagh CF, Huhalov A, Harms BD, et al:
Antitumor activity of a novel bispecific antibody that targets the
ErbB2/ErbB3 oncogenic unit and inhibits heregulin-induced
activation of ErbB3. Mol Cancer Ther. 11:582–593. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Sithanandam G, Fornwald LW, Fields JR,
Morris NL and Anderson LM: Anti-tumor efficacy of naked siRNAs for
ERBB3 or AKT2 against lung adenocarcinoma cell xenografts. Int J
Cancer. 130:251–258. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Engelman JA, Jänne PA, Mermel C, et al:
ErbB-3 mediates phosphoinositide 3-kinase activity in
gefitinib-sensitive non-small cell lung cancer cell lines. Proc
Natl Acad Sci USA. 102:3788–3793. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Ogino A, Kitao H, Hirano S, et al:
Emergence of epidermal growth factor receptor T790M mutation during
chronic exposure to gefitinib in a non-small cell lung cancer cell
line. Cancer Res. 67:7807–7814. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Sequist LV, Waltman BA, Dias-Santagata D,
et al: Genotypic and histological evolution of lung cancers
acquiring resistance to EGFR inhibitors. Sci Transl Med.
3:75ra262011. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Kwak E: The role of irreversible HER
family inhibition in the treatment of patients with non-small cell
lung cancer. Oncologist. 16:1498–1507. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Ling B, Wang GX, Long G, Qiu JH and Hu ZL:
Tumor suppressor miR-22 suppresses lung cancer cell progression
through post-transcriptional regulation of ErbB3. J Cancer Res Clin
Oncol. 138:1355–1361. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Kawano O, Sasaki H, Endo K, et al: ErbB3
mRNA expression correlated with specific clinicopathologic features
of Japanese lung cancers. J Surg Res. 146:43–48. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Vaught DB, Stanford JC, Young C, et al:
HER3 is required for HER2-induced preneoplastic changes to the
breast epithelium and tumor formation. Cancer Res. 72:2672–2682.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Lee-Hoeflich ST, Crocker L, Yao E, et al:
A central role for HER3 in HER2-amplified breast cancer:
implications for targeted therapy. Cancer Res. 68:5878–5887. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Agus DB, Akita RW, Fox WD, et al:
Targeting ligand-activated ErbB2 signaling inhibits breast and
prostate tumor growth. Cancer Cell. 2:127–137. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Göstring L, Malm M, Höidén-Guthenberg I,
et al: Cellular effects of HER3-specific affibody molecules. PLoS
One. 7:e400232012.PubMed/NCBI
|
|
62
|
Weiner DB, Kokai Y, Wada T, Cohen JA,
Williams WV and Greene MI: Linkage of tyrosine kinase activity with
transforming ability of the p185neu oncoprotein. Oncogene.
4:1175–1183. 1989.PubMed/NCBI
|
|
63
|
Menendez JA and Lupu R:
Transphosphorylation of kinase-dead HER3 and breast cancer
progression: a new standpoint or an old concept revisited? Breast
Cancer Res. 9:1112007. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Iorio MV and Croce CM: MicroRNAs in
cancer: small molecules with a huge impact. J Clin Oncol.
27:5848–5856. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Yao E, Zhou W, Lee-Hoeflich ST, et al:
Suppression of HER2/HER3-mediated growth of breast cancer cells
with combinations of GDC-0941 PI3K inhibitor, trastuzumab, and
pertuzumab. Clin Cancer Res. 15:4147–4156. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Junttila TT, Akita RW, Parsons K, et al:
Ligand-independent HER2/HER3/PI3K complex is disrupted by
trastuzumab and is effectively inhibited by the PI3K inhibitor
GDC-0941. Cancer Cell. 15:429–440. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Chakrabarty A, Sánchez V, Kuba MG,
Rinehart C and Arteaga CL: Feedback upregulation of HER3 (ErbB3)
expression and activity attenuates antitumor effect of PI3K
inhibitors. Proc Natl Acad Sci USA. 109:2718–2723. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Kim JH, Im KS, Kim NH, Yhee JY, Nho WG and
Sur JH: Expression of HER-2 and nuclear localization of HER-3
protein in canine mammary tumors: histopathological and
immunohistochemical study. Vet J. 189:318–322. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Chiu CG, Masoudi H, Leung S, et al: HER-3
overexpression is prognostic of reduced breast cancer survival: a
study of 4046 patients. Ann Surg. 251:1107–1116. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Giltnane JM, Moeder CB, Camp RL and Rimm
DL: Quantitative multiplexed analysis of ErbB family coexpression
for primary breast cancer prognosis in a large retrospective
cohort. Cancer. 115:2400–2409. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Haas S, Gevensleben H, Rabstein S, et al:
Expression of heregulin, phosphorylated HER-2, HER-3 and HER-4 in
HER-2 negative breast cancers. Oncol Rep. 21:299–304.
2009.PubMed/NCBI
|
|
72
|
Gori S, Foglietta J, Mameli MG, et al:
HER-3 status by immunohistochemistry in HER-2-positive metastatic
breast cancer patients treated with trastuzumab: correlation with
clinical outcome. Tumori. 98:39–44. 2012.PubMed/NCBI
|
|
73
|
Yoshioka T, Nishikawa Y, Ito R, et al:
Significance of integrin αvβ5 and erbB3 in enhanced cell migration
and liver metastasis of colon carcinomas stimulated by
hepatocyte-derived heregulin. Cancer Sci. 101:2011–2018. 2010.
|
|
74
|
Scartozzi M, Mandolesi A, Giampieri R, et
al: The role of HER-3 expression in the prediction of clinical
outcome for advanced colorectal cancer patients receiving
irinotecan and cetuximab. Oncologist. 16:53–60. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
75
|
Zhang XL, Yang YS, Xu DP, et al:
Comparative study on overexpression of HER2/neu and HER3 in gastric
cancer. World J Surg. 33:2112–2118. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
76
|
Zhu S, Belkhiri A and El-Rifai W: DARPP-32
increases interactions between epidermal growth factor receptor and
ERBB3 to promote tumor resistance to gefitinib. Gastroenterology.
141:1738–1748. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
77
|
Moelans CB, van Diest PJ, Milne AN and
Offerhaus GJ: Her-2/neu testing and therapy in gastroesophageal
adenocarcinoma. Patholog Res Int. 2011:6741822010.PubMed/NCBI
|
|
78
|
te Velde EA, Franke AC, van Hillegersberg
R, et al: HER-family gene amplification and expression in resected
pancreatic cancer. Eur J Surg Oncol. 35:1098–1104. 2009.PubMed/NCBI
|
|
79
|
Hirakawa T, Nakata B, Amano R, et al: HER3
overexpression as an independent indicator of poor prognosis for
patients with curatively resected pancreatic cancer. Oncology.
81:192–198. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
80
|
Buck E, Eyzaguirre A, Haley JD, Gibson NW,
Cagnoni P and Iwata KK: Inactivation of Akt by the epidermal growth
factor receptor inhibitor erlotinib is mediated by HER-3 in
pancreatic and colorectal tumor cell lines and contributes to
erlotinib sensitivity. Mol Cancer Ther. 5:2051–2059. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
81
|
Hsieh SY, He JR, Yu MC, et al: Secreted
ERBB3 isoforms are serum markers for early hepatoma in patients
with chronic hepatitis and cirrhosis. J Proteome Res. 10:4715–4724.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
82
|
Blivet-Van Eggelpoël MJ, Chettouh H,
Fartoux L, et al: Epidermal growth factor receptor and HER-3
restrict cell response to sorafenib in hepatocellular carcinoma
cells. J Hepatol. 57:108–115. 2012.PubMed/NCBI
|
|
83
|
Sheng Q, Liu X, Fleming E, et al: An
activated ErbB3/NRG1 autocrine loop supports in vivo proliferation
in ovarian cancer cells. Cancer Cell. 17:298–310. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
84
|
Tanner B, Hasenclever D, Stern K, et al:
ErbB-3 predicts survival in ovarian cancer. J Clin Oncol.
24:4317–4323. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
85
|
Leng J, Lang J, Shen K and Guo L:
Overexpression of p53, EGFR, c-erbB2 and c-erbB3 in endometrioid
carcinoma of the ovary. Chin Med Sci J. 12:67–70. 1997.PubMed/NCBI
|
|
86
|
Soler M, Mancini F, Meca-Cortes O, et al:
HER3 is required for the maintenance of neuregulin-dependent and
-independent attributes of malignant progression in prostate cancer
cells. Int J Cancer. 125:2565–2575. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
87
|
Buac K, Xu M, Cronin J, Weeraratna AT,
Hewitt SM and Pavan WJ: NRG1/ERBB3 signaling in melanocyte
development and melanoma: inhibition of differentiation and
promotion of proliferation. Pigment Cell Melanoma Res. 22:773–784.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
88
|
Ueno Y, Sakurai H, Tsunoda S, et al:
Heregulin-induced activation of ErbB3 by EGFR tyrosine kinase
activity promotes tumor growth and metastasis in melanoma cells.
Int J Cancer. 123:340–347. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
89
|
Belleudi F, Marra E, Mazzetta F, et al:
Monoclonal antibody-induced ErbB3 receptor internalization and
degradation inhibits growth and migration of human melanoma cells.
Cell Cycle. 11:1455–1467. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
90
|
Djerf Severinsson EA, Trinks C, Gréen H,
et al: The pan-ErbB receptor tyrosine kinase inhibitor canertinib
promotes apoptosis of malignant melanoma in vitro and displays
antitumor activity in vivo. Biochem Biophys Res Commun.
414:563–568. 2011.PubMed/NCBI
|