Introduction
Prostate cancer is the most prevalent malignancy and
the second leading cause of cancer-related mortality in men
(1–3). One protein that plays a crucial role
in development and progression of prostate cancer is the early
growth response-1 (EGR-1) which is a member of the immediate early
gene family and encodes a nuclear phosphoprotein involved in the
regulation of cell growth and differentiation in response to
signals such as mitogens, growth factors and stress stimuli
(4–9). However, in other circumstances, EGR-1
is induced very early in the apoptotic process (10,11),
where it mediates the activation of downstream regulatory genes
(8–10). It has been previously demonstrated
that EGR-1 is required for tumor formation by a variety of human
cancers (12). Another protein that
recently has been involved in cancer progression is the
cellular-Abelson tyrosine kinase (c-Abl), a 140-kDa
proto-oncoprotein non-receptor tyrosine kinase (13) that is activated by diverse genotoxic
agents inducing apoptosis (14).
The mechanisms responsible for nuclear targeting of c-Abl remain
unclear, but cytoplasmic c-Abl is targeted to the nucleus following
a DNA damage response (14,15). c-Abl has been shown to regulate the
cell cycle and to induce, under certain conditions, cell growth
arrest and apoptosis (14,16). It has been demonstrated that c-Abl
regulates EGR-1 protein in response to oxidative stress (17). In addition, c-Abl-induced apoptosis
is partially mitigated by EGR-1 activity, as cells devoid of EGR-1
expression undergo reduced rates of c-Abl-induced apoptosis
(17). Recent studies provide
evidence that implicates c-Abl as a mediator of fibrotic responses
induced by transforming growth factor-β (TGF-β), yet the precise
mechanisms underlying this novel oncogene function are unknown
(18). However, c-Abl has also been
proposed to play a role in cell cycle progression via interaction
with Rb (19), and observations
have implicated c-Abl in the process of apoptosis (20–23).
Most importantly, c-Abl is activated by DNA-damaging agents but not
by UV radiation, suggesting that it is unlikely that p53 is
involved in the c-Abl-induced apoptosis pathway. In addition, EGR-1
overexpression is correlated with loss of its co-repressor NAB2 in
primary prostate carcinoma (24,25).
Recently, we demonstrated that overexpression of EGR-1 modulates
the activity of NF-κB and AP-1 in prostate cancer cell lines
(9), while inhibition of Egr-1 by
small interfering RNA (siRNA) was able to decrease the expression
and activity of NF-κB and AP-1 (8,9).
Therefore, overexpression of EGR-1 appears to be an important
mechanism for the modulation of several types of cell responses. To
investigate the effect of the overexpression of EGR-1 on the
expression and activity of c-Abl, PC-3 and LNCaP prostate carcinoma
cell lines were transfected with an Egr-1 expression plasmid and
clones overexpressing EGR-1 were obtained. We showed that
overexpression of Egr-1 modulates the normal activity and
expression of c-Abl and inhibits the pro-apoptotic property of this
kinase, consequently promoting cell growth, independent anchorage
and survival of cells. In conclusion, the results suggest that
overexpression of EGR-1 inhibits the pro-apoptotic function of
c-Abl similarly in both wild-type p53 (LNCaP) and p53-deficient
(PC-3) prostate carcinoma cell lines.
Materials and methods
Cell lines and culture
Human prostate carcinoma cell lines PC-3 and LNCaP
were a gift from Dr Dan Mercola (The SKCC, La Jolla, CA, USA). The
cells were cultured in RPMI-1640 medium supplemented with 100 ml/l
fetal bovine serum (FBS), 8×105 U/l penicillin and 0.1
g/l streptomycin in a humidified incubator containing 50 ml/l
CO2 at 37°C (30–32).
Tris-borate-EDTA and acrylamide:bisacrylamide (29:1) were obtained
from Bio-Rad (Richmond, CA, USA). Egr-1 antibody was purchased from
Santa Cruz Biotechnology, Inc. (Santa Cruz, CA, USA). STI571 was
kindly provided by Novartis Pharma AG (Basel, Switzerland).
Lipofectamine was obtained from Life Technologies, Inc., USA, and
TNF-α was purchased from Sigma Chemical Co. (St. Louis, MO, USA).
Complete Mini-EDTA-Free Protease Inhibitor Cocktail Tablets and
Annexin-V-Fluos were purchased from Roche Diagnostics GmbH
(Mannheim, Germany). All other reagents were from Sigma Chemical
Co. (Taufkirchen, Germany).
siRNAs
To generate siRNA/Egr-1, a 19-bp sequence was
identified as unique to Egr-1, as determined by BLAST searches of
the non-redundant National Center for Biotechnology Information
(NCBI) nucleotide databases, and was flanked with 2 adenine
ribonucleotides and 2 deoxythymidines. Double-stranded siRNA
sequences were as follows: siEgr-1,
5′-AACGCAAGAGGCAUACCAAGAdTdT-3′, and
5′-AAUCUUGGUAUGCCUCUUGCGdTdT-3′. Cells were treated in parallel
with siRNA-scrambled (5′-AACUCUUCGCCGGUCAUAUCCdTdT-3′ and
5′-AAGGAUAUGACCGGCGAAGAGdTdT-3′) as the control, and were
synthesized by Shanghai GeneChem Co. These cells were cultured in
medium without antibiotics and 24 h before transfection resulting
in a confluence of the cell monolayer by 60–80%. Egr-1-siRNA or
non-silencing siRNA (70 nmol) were mixed with Lipofectamine™ 2000
(Invitrogen Life Technologies) according to the manufacturer’s
recommendation and added to the cells. After 6 h at 37°C, the
medium was replaced, and the cells were cultivated in RPMI-1640
supplemented with 10% heat-inactivated FBS (8,26).
Western blot analysis
The prostate carcinoma cell lines PC-3 and LNCaP
transfected with the empty vector (pCMV) or with an expression
plasmid for Egr-1 (pCMV-Egr-1) (5×105) were seeded onto
6-well plates. Forty-eight hours after transfection, cells were
collected and washed twice by cold PBS, and each well was treated
with 50 ml lysis buffer [2 mmol/l Tris-HCl pH 7.4, 50 mmol/l NaCl,
25 mmol/l EDTA, 50 mmol/l NaF, 1.5 mmol/l
Na3VO4, 1% Triton X-100, 0.1% SDS,
supplemented with protease inhibitors 1 mmol/l phenylmethylsulfonyl
fluoride, 10 mg/l pepstatin, 10 mg/l aprotinin and 5 mg/l leupeptin
(all from Sigma)]. Protein concentrations were determined using the
Bradford protein assay. Equal amounts of protein (50 μg) were
separated on a 15% SDS polyacrylamide gel and transferred to
nitrocellulose membranes (Hybond C; Amersham, Freiburg, Germany).
Membranes were blocked in 5% nonfat dry milk in TBS for 1 h at room
temperature and probed with the rabbit anti-Egr-1 antibody
(dilution 1:500; Santa Cruz Biotechnology, Inc.) overnight at 4°C.
After 3 washing with TBS containing 0.1% Tween-20, membranes were
incubated with anti-rabbit IgG-horseradish-peroxidase (1:5,000;
Santa Cruz Biotechnology, Inc.) and developed by luminol-mediated
chemiluminescence (Appylgen Technologies, Inc., China). To confirm
equal protein loading, membranes were reprobed with a 1:1,000
dilution of an anti-actin antibody (Santa Cruz Biotechnology,
Inc.). Densitometric analyses were performed using Scion image
software (8,9).
Determination of caspase activity
Protein cell extracts were prepared form PC-3 and
LNCaP cells treated with either siRNA-Egr-1 or STI-571 in CE buffer
(25 mM PIPES pH 7.0, 25 mM KCl, 5 mM EGTA, 1 mM DTT, 10 μM
cytochalasin B, 0.5% NP-40 and protease inhibitor) and cleared by
centrifugation at 20,000 × g for 30 min. Protein (10–20 μg) were
diluted in a total volume of 500 ml of caspase buffer (50 mM HEPES
pH 7.4, 100 mM NaCl, 1 mM EDTA, 5 mM DTT, 0.1% CHAPS and 10%
sucrose) containing 100 μM Ac-DEVD-AFC. Reactions were incubated at
37°C, and the fluorescence at 400 nm Ex/505 nm Em was measured
every 5 min for 30–60 min. Caspase activity was calculated from the
initial slope of a standard curve with known concentrations of free
AFC. Caspase inhibitors were purchased from Enzyme Systems Products
(Livermore, CA, USA) (26).
3-(4,5-Methylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT)
assay (cell toxicity assay)
The effect of blocking c-Abl and/or Egr-1 activity
in human LNCaP and PC-3 prostate carcinoma cells was determined by
the MTT survival assay, or using a commercial MTT assay kit
(CellTiter 96® AQueous One Solution Cell Proliferation
Assay; Promega Corporation, Madison, WI, USA) according to the
manufacturer’s instructions. The MTT survival assay was performed
as described previously (Yu et al, 2000). The MTT assay is a
commonly used method for the evaluation of cell survival, based on
the ability of viable cells to convert MTT, a soluble tetrazolium
salt [3-(4,5-dimethylthiazole-2-yl)-2,5-diphenyltetrazolium bromide
(MTT)], into an insoluble formazan precipitate, which is
quantitated by spectrophotometry following solubilization in
dimethyl sulfoxide (DMSO). Briefly, LNCaP and PC-3 cells untreated
and treated with TNF-α alone, or in combination with siRNA-Egr-1 or
STI-571, were cultured in 96-well tissue culture dishes and
incubated with MTT (2 mg/ml) for 4 h. The cells were then
solubilized in 125 ml of DMSO, and absorbance readings were taken
using a 96-well Opsys MR™ microplate reader (Thermo Labsystems,
Chantilly, VA, USA). The amount of MTT dye reduction was calculated
based on the difference between absorbance at 570 and 630 nm. Cell
viability in treated cells was expressed as the amount of dye
reduction relative to that of untreated control cells. The wells
which contained only medium and 10 ml of MTT were used as blanks
for the plate reader.
Cell death assay
Cell death was assessed as previously described
(27). Annexin V-fluorescein
isothiocyanate (Annexin V-FITC) and propidium iodide (PI) labeling
was performed using the Apoptosis/Necrosis Detection kit (Abcam,
UK) according to the manufacturer’s instructions. After 24 h of
treatment, the cells were harvested and Annexin V-FITC was added to
a final concentration of 2.5 mg/ml. To detect necrotic cells, PI
was added at a concentration of 5 mg/ml. The Annexin V-FITC and
PI-labeled cells were analyzed by FACS (FACSCanto; BD Biosciences,
San Jose, CA, USA). Using flow cytometry, dot plots of Annexin
V-FITC on the x-axis against PI on the y-axis were used to
distinguish viable cells (negative for both PI and Annexin V-FITC),
early apoptotic cells (Annexin V-positive and PI-negative) and late
apoptotic or necrotic cells (positive for both PI and Annexin
V-FITC staining). Unstained cells and untreated cells were used as
negative controls. The data were analyzed using Cyflogic software
(non-commercial version, CyFlo Ltd.).
Results
In the present study we investigated the effect of
the overexpression of Egr-1 on the modulation of c-Abl in prostate
cancer cells. For this purpose, PC3 and LNCaP prostate carcinoma
cells were transfected with a plasmid expressing the Egr-1 cDNA and
subjected to clonal selection.
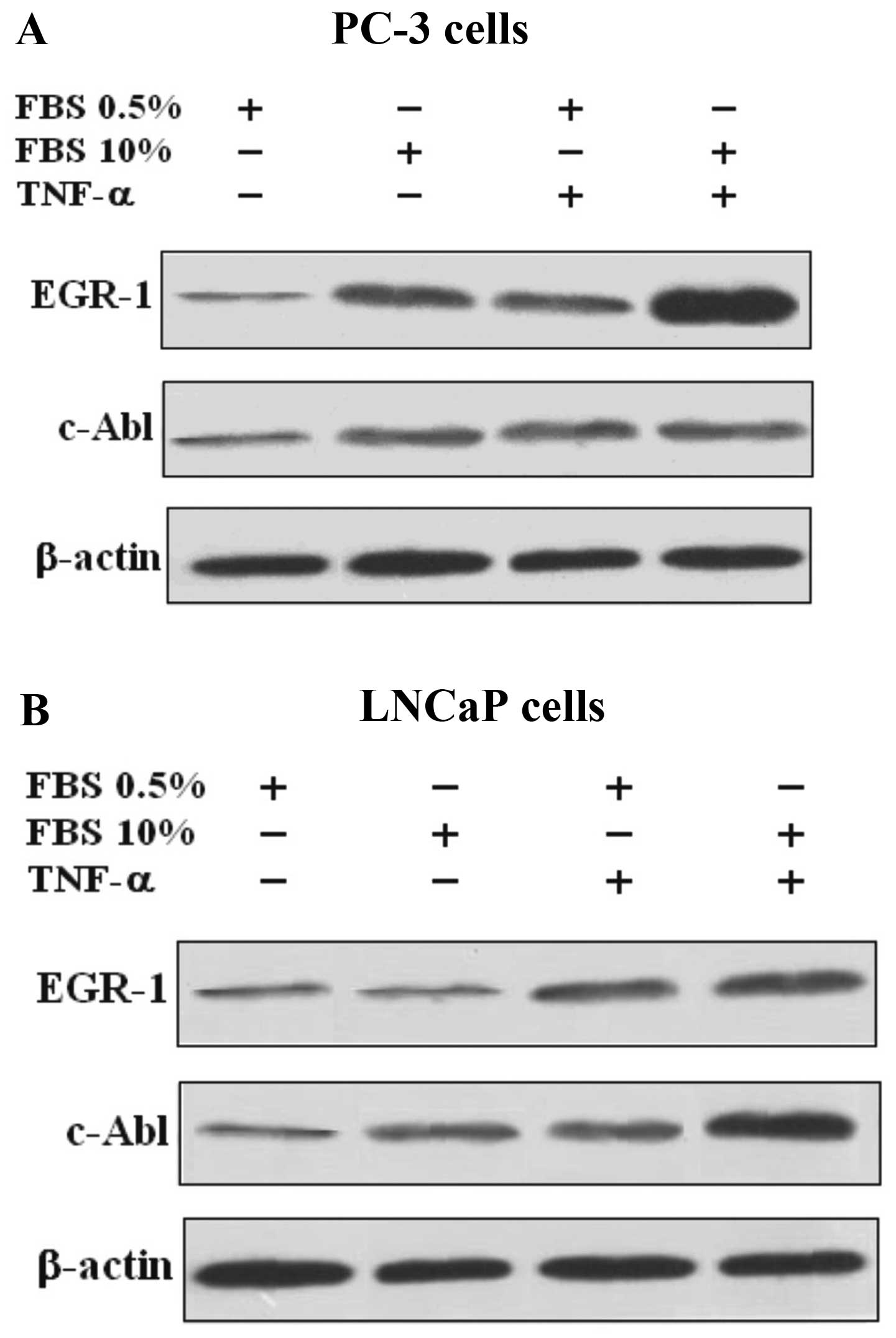
TNF-a strongly induces the expression of
Egr-1 and moderately induces the expression of c-Abl in PC-3 and
LNCaP cells
The expression of Egr-1 and c-Abl protein and mRNA
in the PC-3 and LNCaP cells were evaluated and compared. Proteins
from cells were extracted and fractionated by SDS-PAGE. The results
revealed that TNF-α caused Egr-1 activation and protein expression
in both the PC-3 and LNCaP cells, while the activation of c-Abl was
only marginal, when compared to the control cells, suggesting that
the effect of TNF-α does not involve the c-Abl pathway (Fig. 1).
Inhibition of EGR-1 suppresses the growth
of prostate carcinoma cell lines PC-3 and LNCaP, but inhibition of
c-Abl expression does not suppress growth
The involvement of Egr-1 and c-Abl was further
examined by pre-treatment of cells with siRNA-Egr-1 and Abl
inhibitor STI-571, respectively. Egr-1 and c-Abl responses were
suppressed in PC-3 and LNCaP prostate carcinoma cell lines
(Figs. 2 and 3). As shown by western blot assay,
siRNA-Egr-1 was able to strongly suppress the expression of EGR-1
and to moderately suppress the expression of c-Abl in PC-3
(Fig. 2A) and LNCaP cells (Fig. 3A), while inhibition of c-Abl
expression by STI-571 treatment did not affect the expression of
EGR-1 (Figs. 2A and 3A). To gain further insight into the role
of the overexpression of EGR-1 in prostate cells treated either
with siRNA-Egr-1 or STI-571, we overexpressed EGR-1 in PC-3 and
LNCaP cells and investigated the effect of this overexpression on
cell proliferation in cells treated either with siRNA-Egr-1 or
STI-571. Figs. 2B and 3B show the results obtained in PC-3 and
LNCaP cells after 1, 2 and 3 days of incubation in the presence of
siRNA-Egr-1 or STI-571. Cell growth was measured using an MTT
assay. A strong anti-proliferative activity was noted following
treatment with siRNA-Egr-1, while STI-571 showed only a moderate
anti-proliferative effect (Figs. 2B
and 3B). In addition, application
of TNF-α was unable to increase the cell number in the presence of
siRNA-Egr-1 (Figs. 2B and 3B). Notably, cells treated with STI-571
also showed a significant increase in the number of cells, but this
number was lower than that in the cells treated with siRNA-Egr-1
both in PC-3 (Fig. 2B) and LNCaP
cells (Fig. 3B). The results
suggest that overexpression of EGR-1 partially decreased the c-Abl
activity in a p53-independent manner since both p53-deficient
(PC-3) and wild-type p53 (LNCaP) cells were similarly affected by
overexpression of EGR-1. The results seem to contradict previous
studies, which found that Abl activation in situ, and
elevated phospho-c-Abl are correlated with increased local
expression of Egr-1. Collectively, these results position Egr-1
downstream of c-Abl in the fibrotic response.
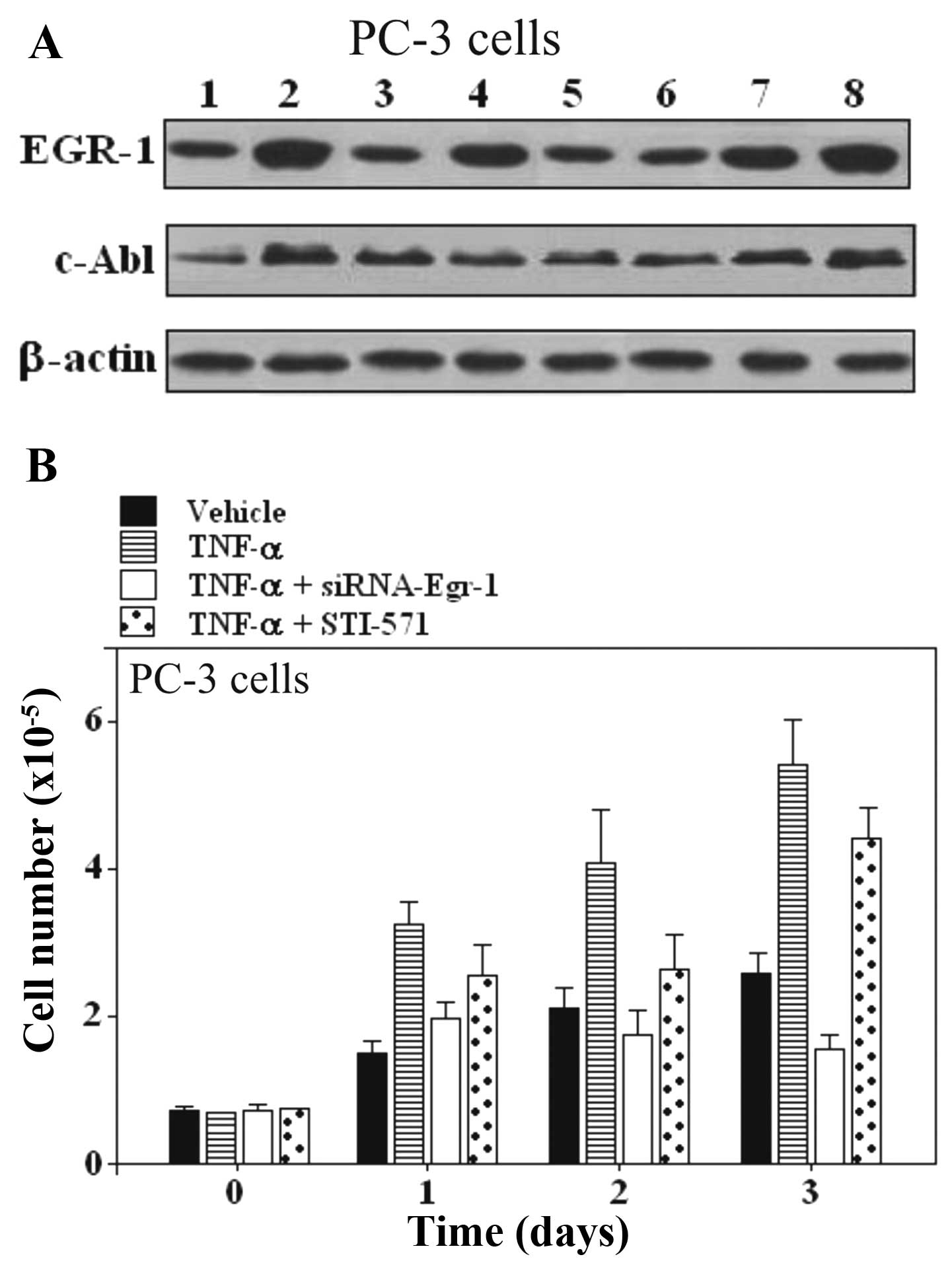 | Figure 2Effect of Egr-1-siRNA and STI-571 on
TNF-α stimulated responses in PC-3 prostate carcinoma cells. (A)
Expression of EGR-1 and c-Abl in PC-3 prostate cells. Lane 1,
untreated cells; lane 2, TNF-α; lane 3, siRNA scrambled; lane 4,
TNF-α + scrambled; lane 5, Egr-1-siRNA; lane 6, TNF-α +
Egr-1-siRNA; lane 7, STI-571; lane 8, TNF-α + STI-571. (B)
Proliferation assay in PC-3 cells treated with TNF-α and the
combination of TNF-α and Egr-1-siRNA or STI-571. One of 3 similar
experiments is shown. EGR-1, early growth response-1; siRNA, small
interfering RNA; TNF-α, tumor necrosis factor α; c-Abl,
cellular-Abelson tyrosine kinase. |
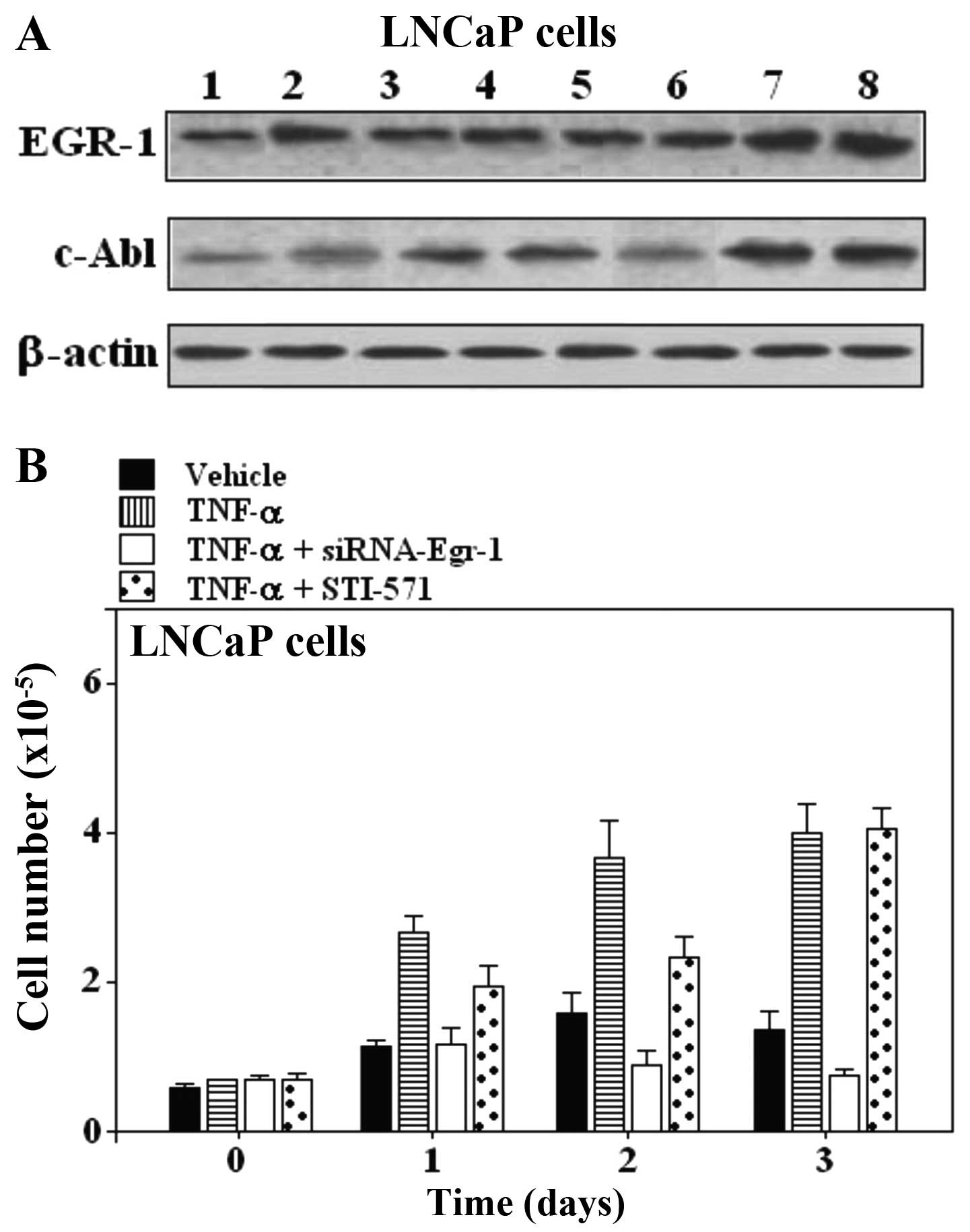 | Figure 3(A) Effect of Egr-1-siRNA and STI-571
on TNF-α stimulated responses in LNCaP prostate carcinoma cells.
Expression of EGR-1 and c-Abl in LNCaP prostate carcinoma cell
lines. Lane 1, untreated cells; lane 2, TNF-α; lane 3, siRNA
scrambled; lane 4, TNF-α + scrambled; lane 5, Egr-1-siRNA; lane 6,
TNF-α + Egr-1-siRNA; lane 7, STI-571; lane 8, TNF-α + STI-571. (B)
Proliferation assay in LNCaP cells treated with TNF-α and the
combination of TNF-α and Egr-1-siRNA or STI-571 for the indicated
periods of time. One of 3 similar experiments is shown. EGR-1,
early growth response-1; siRNA, small interfering RNA; TNF-α, tumor
necrosis factor α; c-Abl, cellular-Abelson tyrosine kinase. |
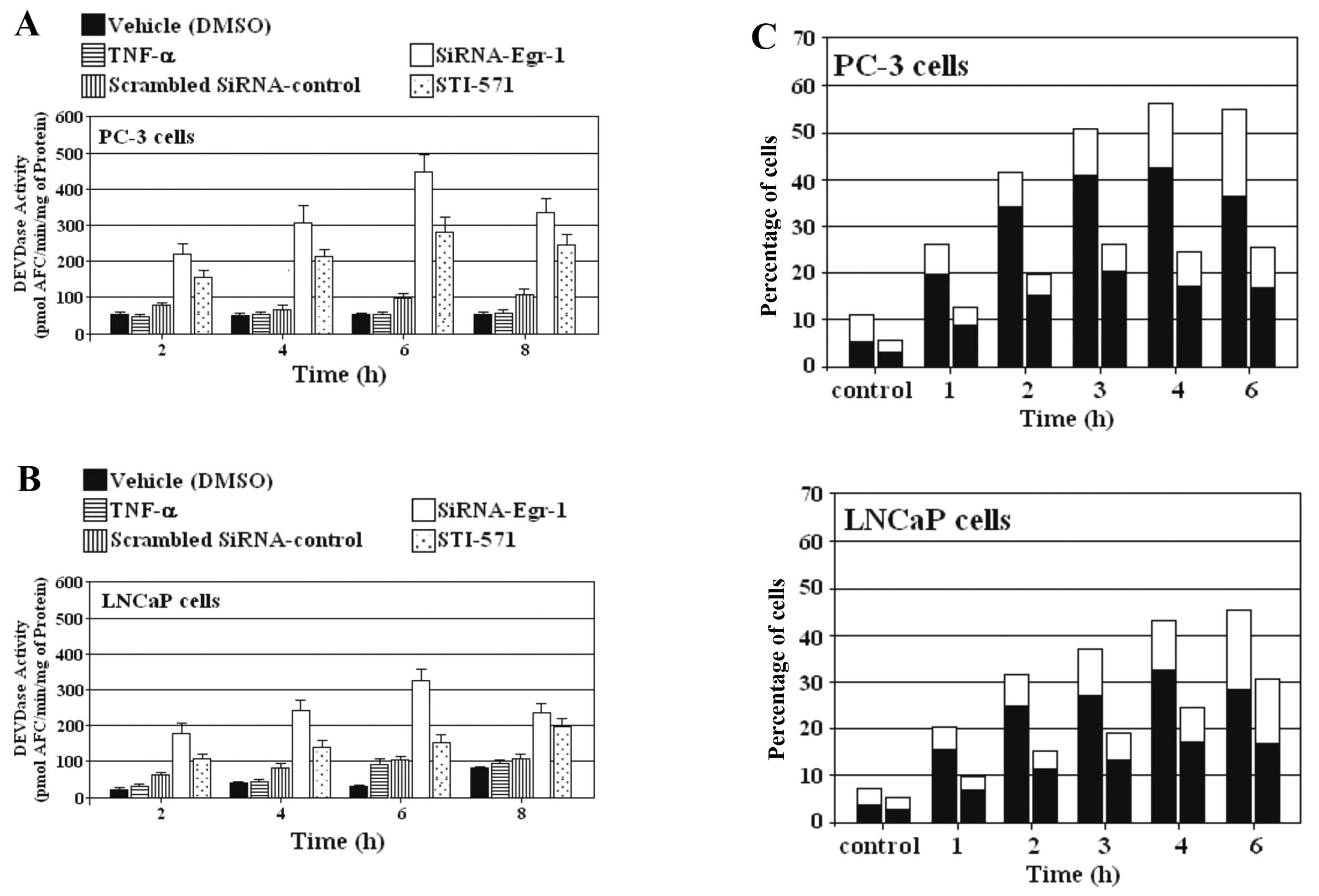
siRNA-Egr-1 and STI-571 induce apoptosis
in PC-3 and LNCaP cells
We investigated the effect of siRNA-Egr-1 and
STI-571 on the ability to induce caspase activity in PC-3 and LNCaP
cells. Cells overexpressing EGR-1 were treated or not with either
siRNA-Egr-1 or STI-571 at the indicated periods of time, and
caspase activity was subsequently assessed using DEVD-AFC as a
substrate. The induction of DEVDase activity by blocking EGR-1
expression was stronger than that induced by inhibition of c-Abl in
prostate carcinoma cell lines PC-3 and LNCaP (Fig. 4A and B). Externalization of
phosphatidylserine was also investigated as a parameter for the
induction of apoptosis by blocking EGR-1 and c-Abl expression in
prostate cancer cells. The appearance of Annexin V-positive
(apoptotic) cells was evident after only 1 h of incubation with
siRNA-Egr-1 and STI-571 and reached a maximum after 3–4 h (Fig. 4C). As expected, the percentage of
apoptotic cells was higher in the presence of cells treated with
siRNA than the percentage in cells treated with STI-571.
Discussion
EGR-1 overexpression is a transforming and
proliferative signal that contributes to tumor development
(8,9) and was firstly characterized as a
tumor-suppressor (28). However,
recent evidence suggests that EGR-1 can act both as a
tumor-suppressor and as a tumor-promoter, depending on the cell
type and the context (29). EGR-1
is expressed in limited amounts in normal cells, but is
overexpressed in tumors (30,31).
Although the mechanisms involved in its overexpression are not
clear, it is well known that it enhances resistance to apoptosis in
fibrosarcoma, prostate cancer, colon cancer and RAS-transformed
cells (32). Further studies have
demonstrated that overexpression of EGR-1 in several cancer types
directly promotes cancer progression and tumor growth by increasing
the expression and secretion of growth factors and cytokines,
extracellular matrix proteins and proteases. Similarly, the kinase
c-Abl sometimes promotes the development of certain types of cancer
and sometimes acts as a suppressor of cancer progression (23,33).
The c-Abl protein is a non-receptor tyrosine protein kinase that is
localized in the nucleus and cytoplasm. However, activation of the
transforming activity of c-Abl is associated with cytoplasmic
localization (34), that sometimes
is associated with an unknown tyrosine kinase inhibitor (35). Recent studies indicate that the
c-Abl protein is activated by the PDGF3 receptor pathway (36). In these studies, binding of PDGF to
the receptor induced c-Src to phosphorylate cytoplasmic c-Abl on
tyrosine residues, which led to the activation of c-Abl. In the
present study, we investigated whether there are any additional
mechanisms by which overexpression of EGR-1 may contribute to tumor
development, despite the presence of c-Abl protein, and whether
TNF-α, an inflammatory cytokine that is found at high levels in
cancer, could increase the c-Abl levels. We found that enhancement
of EGR-1 levels by transfection of PC-3 and LNCaP cells with a
plasmid expressing the wild-type form of the Egr-1 gene was able to
increase cell proliferation not only in the absence, but also in
the presence of c-Abl. In addition, our results demonstrated that
the treatment of PC-3 and LNCaP cells with TNF-α induced an
increase in the protein levels of endogenous EGR-1 and c-Abl-1.
However, the increase was blocked by a c-Ab1-specific inhibitor and
did not affect the expression of the overexpressed EGR-1. These
results are consistent with those of other studies suggesting that
c-Abl-induced apoptosis is partially mitigated by EGR-1 activity,
as cells devoid of EGR-1 expression undergo reduced rates of
c-Abl-induced apoptosis. It is known that EGR-1 mRNA is expressed
at higher levels in prostate tumors when compared with that in
normal tissues and is correlated with Gleason score (a measure of
prostate cancer stage). Moreover, inhibition of EGR-1 expression
reverses transformation of prostate cancer cells in vitro
and in vivo(8,9,37).
Alternatively, forced expression of EGR-1 in non-cancer cells
increases proliferation in vitro. In conclusion, we observed
that PC3 and LNCaP cells overexpressing EGR-1 did not increase the
expression of the c-Abl gene or the pro apoptotic activity induced
by this kinase. In addition, pre-treatment with the c-Abl (STI-571)
inhibitor further increased cell proliferation of PC-3 and LNCaP
cells.
Acknowledgements
We thank Dr Dan Mercola for providing the
Egr-1-expression plasmid. The present study was supported by the
Laboratory of Experimental Biomedicine, Campus Esmeralda-Iquique,
University of Tarapaca, Iquique, Chile.
References
|
1
|
American Cancer Society. Cancer Facts
& Figures 2003. American Cancer Society; Atlanta, GA: 2003
|
|
2
|
Quinn M and Babb P: Patterns and trends in
prostate cancer incidence, survival, prevalence, and mortality.
Part I: international comparisons. BJU Int. 90:162–173. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Grönberg H: Prostate cancer epidemiology.
Lancet. 361:859–864. 2003.PubMed/NCBI
|
|
4
|
Kaufmann K and Thiel G: Epidermal growth
factor and platelet-derived growth factor induce expression of
Egr-1, a zinc finger transcription factor, in human
malignant glioma cells. J Neurol Sci. 189:83–91. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Kaufmann K and Thiel G: Epidermal growth
factor and thrombin induced proliferation of immortalized human
keratinocytes is coupled to the synthesis of Egr-1, a zinc finger
transcriptional regulator. J Cell Biochem. 85:381–391. 2002.
View Article : Google Scholar
|
|
6
|
Liu C, Rangnekar VM, Adamson E and Mercola
D: Suppression of growth and transformation and induction of
apoptosis by EGR-1. Cancer Gene Ther. 5:3–28. 1998.PubMed/NCBI
|
|
7
|
Parra E, Ferreira J and Saenz L:
Inhibition of Egr-1 by siRNA in prostate carcinoma cell lines is
associated with decreased expression of AP-1 and NF-κB. Int J Mol
Med. 28:847–853. 2011.PubMed/NCBI
|
|
8
|
Parra E, Ferreira J and Ortega A:
Overexpression of EGR-1 modulates the activity of NF-κB and AP-1 in
prostate carcinoma PC-3 and LNCaP cell lines. Int J Oncol.
39:345–352. 2011.PubMed/NCBI
|
|
9
|
Parra E, Ortega A and Saenz L:
Downregulation of Egr-1 by siRNA inhibits growth of human prostate
carcinoma cell line PC-3. Oncol Rep. 22:1513–1518. 2009.PubMed/NCBI
|
|
10
|
Lee CG, Cho SJ, Kang MJ, Chapoval SP, Lee
PJ, Noble PW, Yehualaeshet T, Lu B, Flavell RA, Milbrandt J, Homer
RJ and Elias JA: Early growth response gene 1-mediated apoptosis is
essential for transforming growth factor β1-induced
pulmonary fibrosis. J Exp Med. 200:377–389. 2004.PubMed/NCBI
|
|
11
|
Parra E and Ferreira J: The effect of
siRNA- Egr-1 and camptothecin on growth and chemosensitivity of
breast cancer cell lines. Oncol Rep. 23:1159–1165. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Baron V, Adamson ED, Calogero A, Ragona G
and Mercola D: The transcription factor Egr1 is a direct regulator
of multiple tumor suppressors including TGFβ1, PTEN, p53 and
fibronectin. Cancer Gene Ther. 13:115–124. 2006.PubMed/NCBI
|
|
13
|
Shaul Y and Ben-Yehoyada M: Role of c-Abl
in the DNA damage stress response. Cell Res. 15:33–35. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Kharbanda S, Ren R, Pandey P, Shafman TD,
Feller SM, Weichselbaum RR and Kufe DW: activation of the c-Abl
tyrosine kinase in the stress response to DNA-damaging agents.
Nature. 376:785–788. 1995. View
Article : Google Scholar : PubMed/NCBI
|
|
15
|
Pandey P, Raingeaud J, Kaneli M,
Weichselbaum RR, Davis RJ, Kufe D and Kharbanda S: Activation of
p38 mitogen-activated protein kinase by c-Abl-dependent and
independent mechanisms. J Biol Chem. 271:23775–23779. 1996.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Cong F and Goff SP: c-Abl-induced
apoptosis, but not cell cycle arrest, requires mitogen-activated
protein kinase kinase 6 activation. Proc Natl Acad Sci USA.
96:13819–13824. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Stuart JR, Kawai H, Tsai KK, Chuang EY and
Yuan ZM: c-Abl regulates early growth response protein (EGR1) in
response to oxidative stress. Oncogene. 24:8085–8092.
2005.PubMed/NCBI
|
|
18
|
Daniels CE, Wilkes MC, Edens M, Kottom TJ,
Murphy SJ, Limper AH and Leof EB: Imatinib mesylate inhibits the
profibrogenic activity of TGF-β and prevents bleomycin-mediated
lung fibrosis. J Clin Invest. 114:1308–1316. 2004.PubMed/NCBI
|
|
19
|
Welch PJ and Wang JY: Abrogation of
retinoblastoma protein functions by c-Abl through tyrosine
kinase-dependent and -independent mechanisms. Mol Cell Biol.
15:5542–5551. 1995.PubMed/NCBI
|
|
20
|
Barilà D, Rufini A, Condò I, Ventura N,
Dorey K, Superti-Furga G and Testi R: Caspase-dependent cleavage of
c-Abl contributes to apoptosis. Mol Cell Biol. 23:2790–2799.
2003.PubMed/NCBI
|
|
21
|
Machuy N, Rajalingam K and Rudel T:
Requirement of caspase-mediated cleavage of c-Abl during
stress-induced apoptosis. Cell Death Differ. 11:290–300. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Huang DY, Chao Y, Tai MH, Yu YH and Lin
WW: STI571 reduces TRAIL-induced apoptosis in colon cancer cells:
c-Abl activation by the death receptor leads to stress
kinase-dependent cell death. J Biomed Sci. 19:352012. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Li B, Cong F, Tan CP, Wang SX and Goff SP:
Aph2, a protein with a zf-DHHC motif, interacts with c-Abl
and has pro-apoptotic activity. J Biol Chem. 277:28870–28876.
2002.PubMed/NCBI
|
|
24
|
Abdulkadir SA, Carbone JM, Naughton CK,
Humphrey PA, Catalona WJ and Milbrandt J: Frequent and early loss
of the EGR1 corepressor NAB2 in human prostate carcinoma. Hum
Pathol. 32:935–939. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Lucerna M, Mechtcheriakova D, Kadl A,
Schabbauer G, Schäfer R, Gruber F, Koshelnick Y, Müller HD,
Issbrücker K, Clauss M, Binder BR and Hofer E: NAB2, a corepressor
of EGR-1, inhibits vascular endothelial growth factor-mediated gene
induction and angiogenic responses of endothelial cells. J Biol
Chem. 278:11433–11440. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Parra E: Inhibition of JNK-1 by small
interference RNA induces apoptotic signalling in PC-3 prostate
cancer cells. Int J Mol Med. 30:923–930. 2012.PubMed/NCBI
|
|
27
|
Parra E and Gutiérrez L: Growth inhibition
of Tax-activated human Jurkat leukemia T cells by all-trans
retinoic acid requires JNK-1 inhibition. Oncol Rep. 29:387–393.
2013.PubMed/NCBI
|
|
28
|
Krones-Herzig A, Mittal S, Yule K, Liang
H, English C, Urcis R, Soni T, Adamson ED and Mercola D: Early
growth response 1 acts as a tumor suppressor in vivo and in vitro
via regulation of p53. Cancer Res. 65:5133–5143. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Calogero A, Arcella A, De Gregorio G,
Porcellini A, Mercola D, Liu C, Lombari V, Zani M, Giannini G,
Gagliardi FM, Caruso R, Gulino A, Frati L and Ragona G: The early
growth response gene EGR-1 behaves as a suppressor gene that
is down-regulated independent of ARF/Mdm2 but not p53 alterations
in fresh human gliomas. Clin Cancer Res. 7:2788–2796. 2001.
|
|
30
|
Egerod FL, Bartels A, Fristrup N, Borre M,
Ørntoft TF, Oleksiewicz MB, Brünner N and Dyrskj⊘t L: High
frequency of tumor cells with nuclear Egr-1 protein expression in
human bladder cancer is associated with disease progression. BMC
Cancer. 9:3852009. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Svaren J, Ehrig T, Abdulkadir SA,
Ehrengruber MU, Watson MA and Milbrandt J: EGR1 target genes in
prostate carcinoma cells identified by microarray analysis. J Biol
Chem. 275:38524–38531. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Gitenay D and Baron VT: Is EGR1 a
potential target for prostate cancer therapy? Future Oncol.
5:993–1003. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Suzuki J, Sukezane T, Akagi T, Georgescu
MM, Ohtani M, Inoue H, Jat PS, Goff SP, Hanafusa H and Shishido T:
Loss of c-abl facilitates anchorage-independent growth of
p53- and RB-deficient primary mouse embryonic fibroblasts.
Oncogene. 23:8527–8534. 2004.
|
|
34
|
Taagepera S, McDonald D, Loeb JE, Whitaker
LL, McElroy AK, Wang JY and Hope TJ: Nuclear-cytoplasmic shuttling
of C-ABL tyrosine kinase. Proc Natl Acad Sci USA. 95:7457–7462.
1998. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Ling X, Ma G, Sun T, Liu J and Arlinghaus
RB: Bcr and Abl interaction: oncogenic activation of c-Abl by
sequestering Bcr. Cancer Res. 63:298–308. 2003.PubMed/NCBI
|
|
36
|
Furstoss O, Dorey K, Simon V, Barilà D,
Superti-Furga G and Roche S: c-Abl is an effector of Src for growth
factor-induced c-myc expression and DNA synthesis. EMBO J.
21:514–524. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Baron V, De Gregorio G, Krones-Herzig A,
Virolle T, Calogero A, Urcis R and Mercola D: Inhibition of Egr-1
expression reverses transformation of prostate cancer cells in
vitro and in vivo. Oncogene. 22:4194–4204. 2003. View Article : Google Scholar : PubMed/NCBI
|