Introduction
Wilms’ tumor gene WT1 is mutated in
approximately 10–15% of sporadic Wilms’ tumors (1). Germline mutations in WT1 can
lead to developmental diseases such as WAGR (Wilms tumor, aniridia,
genitourinary abnormalities and intellectual disability),
Denys-Drash and Frasier syndromes, all of which are characterized
by glomerulonephropathy and abnormal gonad development (2). The importance of WT1 during
development was further demonstrated using mouse models. Embryos of
Wt1−/− mice die in utero with agenesis of the
kidneys, gonads, adrenal glands and spleen (3,4).
WT1 encodes a Cys2-His2 (C2H2) zinc finger protein that
functions as a transcription factor (5). WT1 has two alternative splicing
sites, three translational initiation sites, and one RNA editing
site, making possible the generation of at least 24 isoforms in the
cell. The first alternative splicing introduces 17 amino acids
encoded by exon 5 and the second alternative splicing inserts or
omits three amino acids, KTS (Lys, Thr and Ser) between the third
and fourth zinc fingers. Since KTS insertion by second alternative
splicing disrupts the spacing between two zinc fingers, the +KTS
isoform of WT1 has an altered DNA binding specificity.
Developmental processes are closely related to cell
death. During the embryonic development of the kidney, metanephric
mesenchyme serves as a precursor of glomerular and tubular
structures. Wt1-null mice develop extensive apoptosis in the
metanephric blastema (3) suggesting
that WT1 protects renal precursor cells from apoptosis. Bcl-2
expression by WT1 can explain its anti-apoptotic function during
kidney development (6). Consistent
with the anti-apoptotic effects during development, WT1 promotes
the survival of chronic leukemic cells such as K562 and MM6
(7). However, the expression of
+KTS isoforms (+Ex5/+KTS and −Ex5/+KTS) of WT1 induces
apoptosis in HepG2 cells (8).
Another isoform of WT1, −Ex5/−KTS, induces apoptosis in Saos-2
cells by upregulating BAK expression (9). These findings suggest that WT1 may
have a dichotomous effect on cancer cell death depending on the
cell types and the isoforms. One of the characteristic features of
WT1 is its interactions with several proteins that function in
apoptosis, such as p53 and HSP70 (10,11).
Maheswaran et al showed that WT1 stabilizes p53 (10) and inhibits UV-induced apoptosis
which is dependent on p53, but not p53-induced cell cycle arrest
(12). The molecular mechanisms
that are involved in the effect of WT1 on p53-induced apoptosis,
however, remain to be elucidated.
Nutlin-3 is a small molecule that binds to the p53
binding pocket of HDM2 which ubiquitylates and degrades p53
(13). Thus, nutlin-3 elevates the
level of expression of p53 and activates the p53 pathway. In cancer
cells that harbor wild-type p53, nutlin-3 can activate
p53-dependent growth arrest or an apoptosis program (14). Based on the ability of WT1 to
interact and modulate the activity of p53, we investigated whether
WT1 also modulates the anticancer effect of nutlin-3. To address
this issue, we analyzed the effect of nutlin-3 on the growth and
survival of U2OS cells which express Wt1 according to the
presence of tetracycline.
Materials and methods
Chemicals
Nutlin-3 was purchased from Sigma-Aldrich (St.
Louis, MO, USA). Z-VAD-Fmk was from Tocris Bioscience (Northpoint,
UK). Unless otherwise specified, all chemicals were of molecular
biology grade and were purchased from Sigma-Aldrich.
Cell culture
The UD29a cell line was stably established from a
U2OS cell line (human osteosarcoma cells; ATCC, Manassas, VA, USA)
by transfection of Wt1(+Ex5/+KTS) regulated by
tetracycline-repressible promoter and maintained in DMEM
(Invitrogen, Carlsbad, CA, USA) supplemented with 10%
heat-inactivated fetal bovine serum (HyClone, Logan, UT, USA), 100
U/ml of penicillin/streptomycin (Invitrogen) and 1 μg/ml of
tetracycline under a 5% CO2 and 95% humidified
atmosphere. To induce the expression of Wt1, cells were
washed three times with calcium, magnesium-free phosphate-buffered
saline (Invitrogen) and replenished with DMEM without
tetracycline.
Cell death assessment
Cell viability was measured by the MTT
[3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide]
reduction assay. UD29a cells were seeded at a density of
5×104 cells/well in 24-well plates and after 24 h,
tetracycline was removed and nutlin-3 was added. Following
incubation for the indicated periods, MTT was added and the optical
density of each well was measured at 570 nm. Data are expressed as
relative cell survival which represents the percentage of the
optical density of cells treated with nutlin-3 to that of
non-treated cells. For the measurement of apoptotic cells, UD29a
cells treated with nutlin-3 were fixed in 70% ethanol and stained
with propidium iodide (PI) followed by flow cytometric analysis
(FACSCalibur, BD Biosciences, San Jose, CA, USA) as previously
reported (15). Cell cycle
distribution was analyzed by CellQuest and ModFit software.
Immunoblot analysis
Cell lysates, mitochondrial fractions or cytosolic
fractions were subjected to SDS-PAGE and immunoblot analysis
following a previous method (16).
The proteins were then transferred onto a nitrocellulose transfer
membrane (Whatman, Dassel, Germany). After the transfer, membranes
were blocked for 30 min in TTBS buffer (25 mM Tris, pH 7.4, 138 mM
NaCl, 0.05% Tween-20) containing 5% skim milk and then incubated
with indicated antibodies. After washing four times with TTBS, the
membranes were incubated with anti-mouse IgG-peroxidase conjugated.
The membranes were then washed four times in TTBS, followed by
chemiluminescence-based detection (GE Healthcare, Buckinghamshire,
UK).
Subcellular fractionation
Cytosolic and mitochondrial fractions were separated
as described in a previous report (17). Briefly, cells were treated as
indicated and were resuspended in ice-cold hypotonic buffer (10 mM
Hepes, 10 mM MgCl2, 42 mM KCl) containing a protease
inhibitor cocktail. Plasma membranes were ruptured by passing the
cells through a 31-gauge syringe. Following centrifugation at 1,000
rpm for 5 min at 4°C, the supernatant was collected and again
centrifuged at 13,000 rpm for 15 min at 4°C. The clear fraction was
saved as the cytosolic fraction and the pellet, the heavy membrane
fraction, was used as the mitochondrial fraction.
Assay of mitochondrial transmembrane
potential (MTP)
The change in MTP during apoptosis was analyzed
using a MitoProbe™ JC-1 Assay kit (Invitrogen), following the
manufacturer’s instructions.
Real-time quantitative RT-PCR
(qRT-PCR)
Total RNA that was extracted from cells treated as
indicated was reverse transcribed to cDNA using a random hexamer
and CycleScript Reverse Transcriptase (Bioneer Corp., Daejeon,
Korea) and subjected to qRT-PCR. qRT-PCR was performed with a
mixture containing cDNA, SYBR Premix Ex Taq (Takara Bio Inc., Otsu,
Japan) and primers using Mx3000P QPCR System (Stratagene, La Jolla,
CA, USA). The level of change in gene expression change was
calculated by the ΔΔCt method using GAPDH as an internal control
(18). All tests were carried out
in triplicate and repeated three times.
Expression of human BCL-XL
Coding sequence of human BCL-XL was amplified using
ExTaq DNA polymerase (Takara Bio) and inserted into pcDNA3.1-vector
(Invitrogen). After the nucleotide sequence of BCL-XL in
pcDNA3.1-/BCL-XL was confirmed to be completely matched with
published data (GenBank accession no. NM_138578), pcDNA3.1-empty
vector or pcDNA3.1-/BCL-XL was transfected into UD29a cells using
FuGENE 6 transfection reagent.
Results
Potentiation of nutlin-3-induced growth
suppression by Wt1 expression
Among all the isoforms of WT1, +Ex5/+KTS is the most
abundant, accounting for >60% of all WT1 transcripts (19). Thus, we first examined the effect of
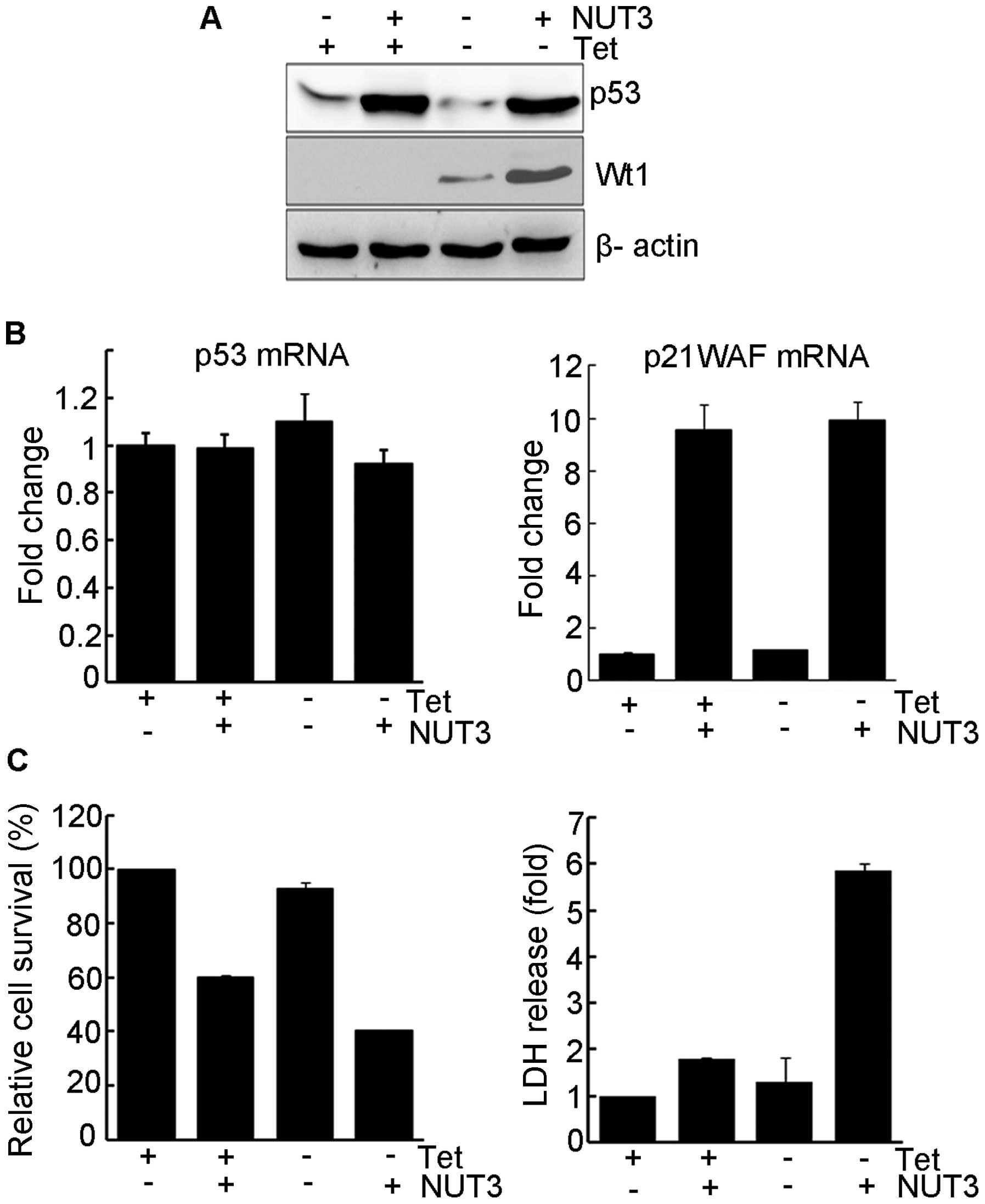
this isoform on the nutlin-3-mediated activation of p53. As shown
in Fig. 1A, the addition of
nutlin-3 led to a marked increase in the level of p53 protein and
the expression of Wt1 did not further increase p53 protein levels
induced by nutlin-3. However, the presence of nutlin-3 also caused
an increase in Wt1 levels (compare lanes 3 and 4). Consistent with
its function as an HDM2 inhibitor, nutlin-3 had no effect on p53
mRNA levels, while the level of p21WAF1 mRNA increased in parallel
with that for the p53 protein, indicating that p53 is functionally
intact in this cell line (Fig. 1B).
Nutlin-3 treatment inhibited the growth of UD29a cells, accompanied
by a marginal release of lactate dehydrogenase (LDH) (Fig. 1C). The expression of Wt1 by itself
had no negative effects on cell growth but, in the presence of
nutlin-3, growth suppression was further potentiated and the
release of LDH was markedly increased (Fig. 1C), suggesting that the combination
of WT1 expression and nutlin-3 treatment leads to enhanced cell
death.
Activation of caspase-dependent apoptosis
by nutlin-3 in the presence of Wt1
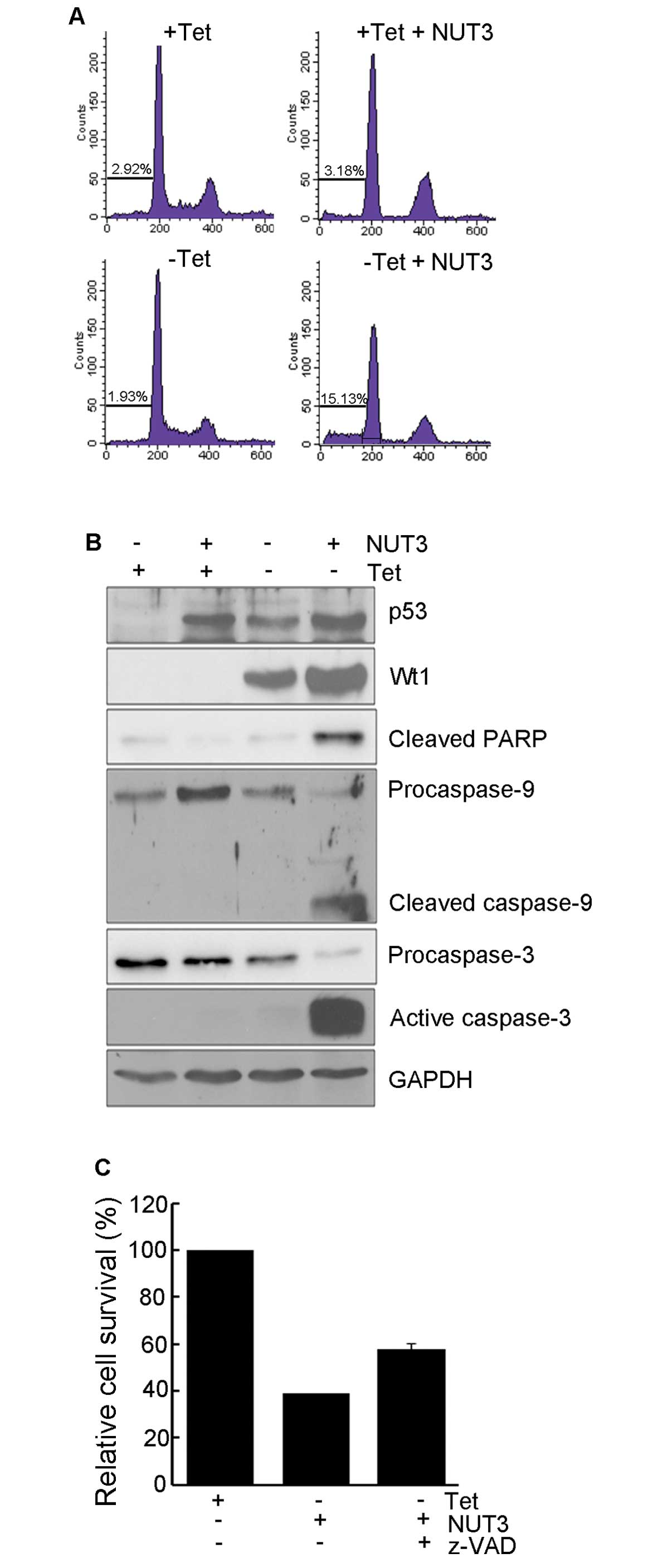
To examine the mechanism responsible for cell death
induction, we analyzed the cell cycle distribution and the
activation of caspases. As shown in Fig. 2A, nutlin-3 treatment alone induced a
decrease in S-phase cells, suggesting p53-mediated delay or arrest
of cell cycle progression. In the presence of WT1, nutlin-3 induced
a striking increase in the sub-G1 population of UD29a cells but not
non-WT1-expressing cells. Parallel to the level of the sub-G1
population, nutlin-3 induced the activation of caspase-9 and -3,
and the cleavage of poly(ADP-ribose) polymerase 1 (PARP1) (Fig. 2B). In addition, this cell death was
diminished by pretreatment with z-VAD-Fmk, a pan-caspase inhibitor
(Fig. 2C). Collectively, these
results suggest that nutlin-3 induces apoptosis via an intrinsic
apoptosis pathway in the presence of WT1.
Release of cytochrome c by nutlin-3 and
Wt1 expression
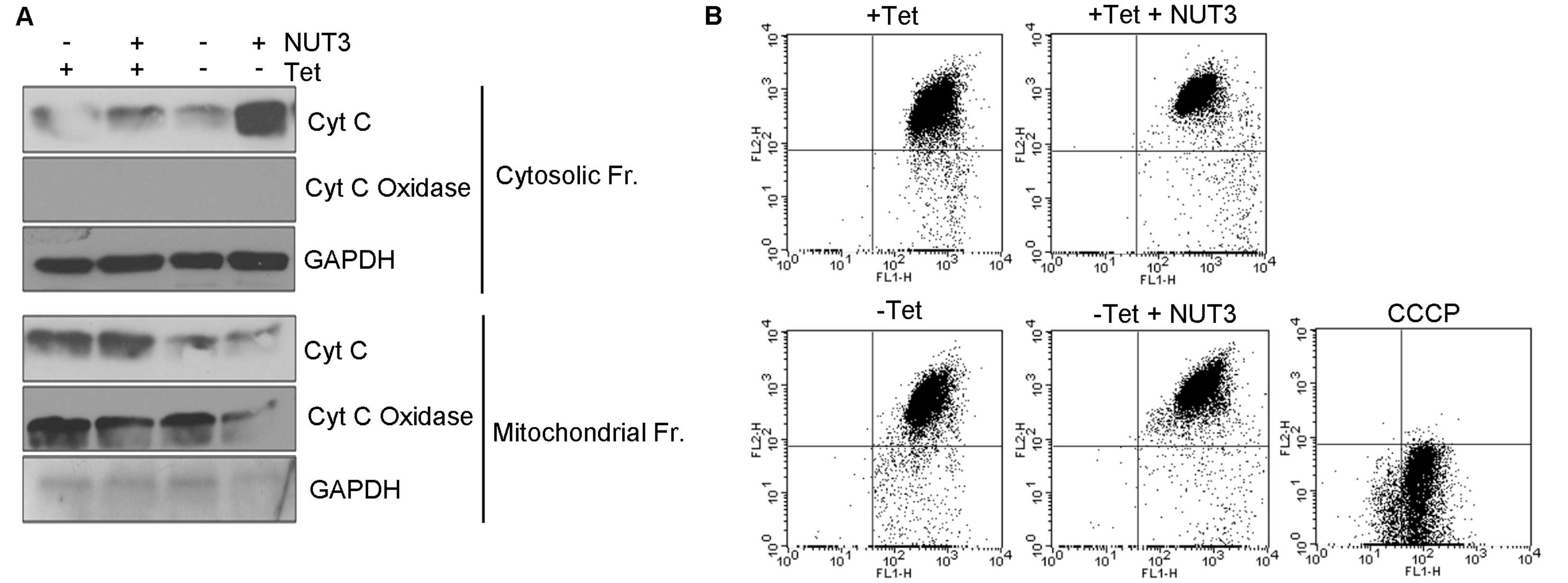
Since the intrinsic apoptotic pathway is activated
by cytochrome c released into the cytosol, we measured the
intracellular localization of cytochrome c. The addition of
nutlin-3 alone did not lead to the release of cytochrome c
into the cytosol (Fig. 3A), which
is consistent with the absence of any activation of caspases and
PARP1 cleavage (Fig. 2B). When
nutlin-3 was added in the presence of WT1 expression, however, a
marked increase in the cytosolic cytochrome c was observed.
The release of cytochrome c was not associated with the
alteration of the MTP (Fig. 3B),
indicating that the cytochrome c is released through pores
or channels formed in the mitochondrial outer membranes.
The expression of BCL-XL and BAK during
cell death
It is well known that MTP-independent cytochrome
c release occurs through the pores formed by BAX or BAK
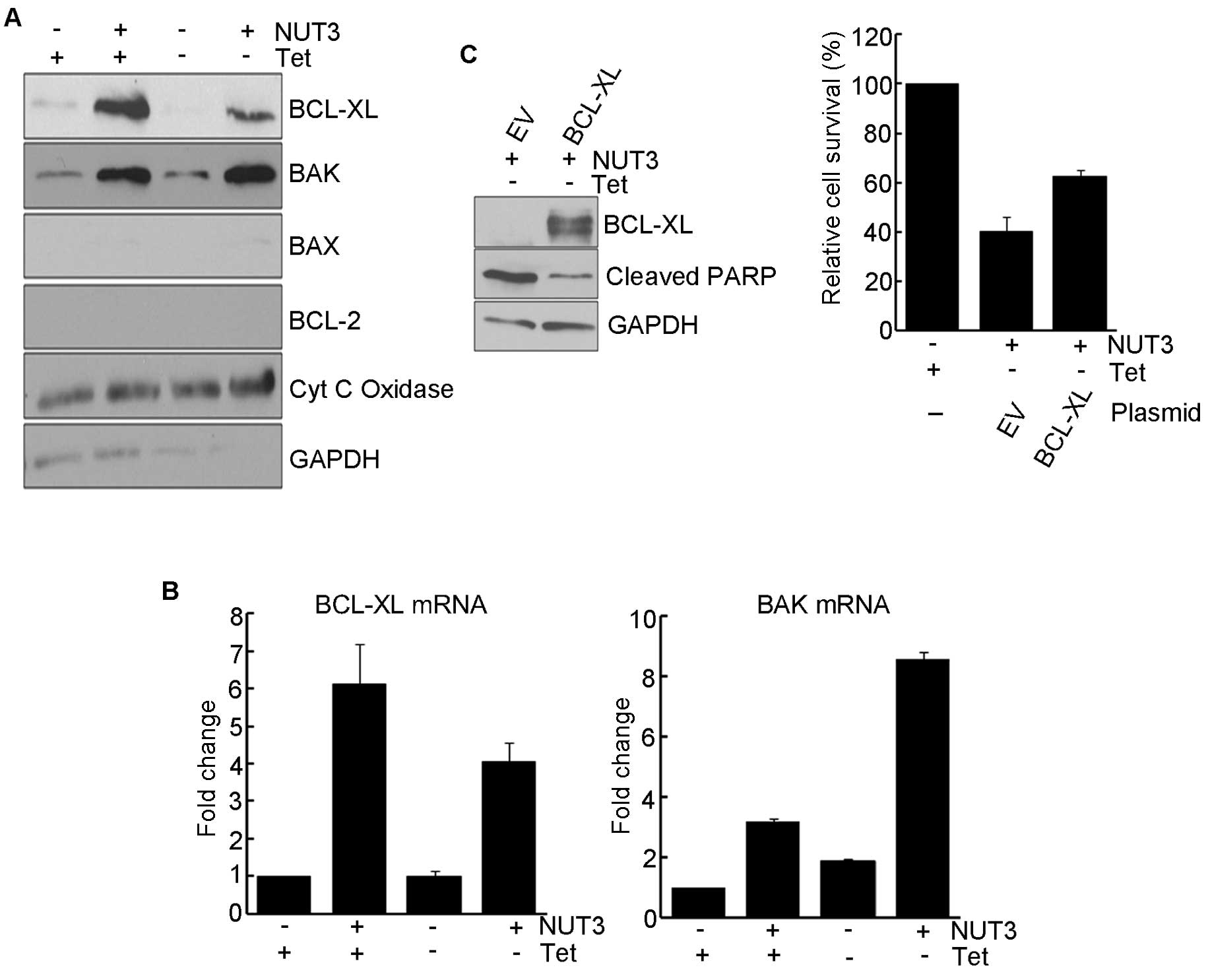
(20). Therefore, we examined the
expression of BCL-2 family proteins in mitochondria after nutlin-3
treatment. As shown in Fig. 4A,
nutlin-3 treatment resulted in an increase in the expression of
BCL-XL and BAK while the expression of BCL-2 and BAX was not
observed. In UD29a cells expressing Wt1 and treated with nutlin-3,
a sharp decrease in BCL-XL and a modest increase in BAK occurred at
the protein level. At the mRNA level, nutlin-3 treatment led to
significant increases in BCL-XL (6-fold) and BAK
(3-fold) transcripts. The expression of Wt1 alone led to a
modest induction (<2-fold) of BAK mRNA but had no effect
on the expression of BCL-XL (Fig. 4B). The expression of Wt1 in
UD29a cells treated with nutlin-3 resulted in a synergistic
increase (8-fold) in the expression of BAK transcripts while
Wt1 attenuated the nutlin-3-induced expression of BCL-XL
transcripts. The overexpression of BCL-XL in Wt1-expressing
cells attenuated nutlin-3-induced apoptosis, as evidenced by a
significant increase in cell survival and a decreased level of
cleaved PARP1. These results suggest that WT1 potentiates
nutlin-3-induced apoptosis by the synergistic induction of
BAK expression and a simultaneous decrease in BCL-XL
expression.
Discussion
p53 is a critical activator of apoptosis which is
induced by DNA damaging agents such as γ-irradiation and UV. In
contrast to the report that WT1 inhibits UV-induced apoptosis, the
+Ex5/+KTS isoform of Wt1 potentiated nutlin-3-induced apoptosis in
the present study. In the absence of Wt1 expression, cell
cycle arrest with marginal cytotoxicity was induced by nutlin-3,
but in the presence of Wt1 (+Ex5/+KTS) expression, nutlin-3
treatment provoked overt cell death (Fig. 1C). Based on an analysis of cell
cycle distribution (Fig. 2A) and
biochemical markers (Fig. 2B), cell
death induced by nutlin-3 in Wt1-expressing cells appears to be
mediated by apoptosis. During the induction of apoptosis in
Wt1-expressing cells, cytochrome c was released into the
cytosol in an MTP-independent manner (Fig. 3) and both caspase-9 and -3 were
activated (Fig. 2B), demonstrating
that the intrinsic apoptotic pathway played a major role in this
model.
BCL-2 family proteins are essential players in
MTP-independent release of cytochrome c. Both BCL-XL and BAK
were found to have accumulated in mitochondria of nutlin-3-treated
cells (Fig. 4A). Notably, when
Wt1 was expressed, nutlin-3-induced BCL-XL and
BAK expression was reduced and augmented, respectively
(Fig. 4A), which would likely
result in the activation of BAK. BAK can make pores or channels in
mitochondrial outer membranes through which cytochrome c can
exit into the cytosol, initiating the formation of apoptosome
(19). The release of cytochrome
c by a combination of nutlin-3 treatment and
Wt1(+Ex5/+KTS) expression was reversed by the overexpression
of BCL-XL (Fig. 4C), which
further supports the involvement of BAK in this process. The
increase in BCL-XL and BAK by nutlin-3 was accompanied with that of
transcripts (Fig. 4B), indicating
p53-mediated transcriptional activation. Although they are not
generally considered to be transcriptional target genes of p53, and
there has been no report showing that p53 stimulates the
transcription of BCL-XL, it was reported that the
transcription of BAK can be induced by p53, although to a
lesser extent than that for p21WAF1 and BAX(21). Therefore, it is possible that
BCL-XL and BAK genes contain p53-responsive elements,
albeit very weak, in their promoters, and a gradual accumulation of
p53 by nutlin-3 would eventually lead to a recruitment of p53 to
these promoters, finally resulting in a transcriptional increase in
this cell line.
It is noteworthy that Wt1 displayed opposing effects
on nutlin-3-induced BCL-XL and BAK expression
(Fig. 4A and B). Consistent with
the report that the −Ex5/−KTS isoform of Wt1 stimulated the
transcription of BAK in Saos-2 cells through direct binding
to a BAK promoter (9), we
observed a modest (<2-fold) induction in BAK
transcription by the Wt1 +Ex5/+KTS isoform (Fig. 4B). Moreover, the finding that the
level of Wt1 protein was upregulated by nutlin-3 (Fig. 1A) may suggest that the induction of
BAK transcription by Wt1 may be surged via upregulated Wt1
protein and thus increase of its transcriptional activity. However,
since WT1 +KTS has a lower DNA binding affinity compared to the
−KTS isoform, it is also reasonable to expect that WT1 +KTS may
modulate the p53-induced expression of BCL-XL and BAK
expression though its interaction with p53 or by regulating signal
transduction pathways that are activated by nutlin-3. Considering
that the activity of p53 has been shown to be regulated by several
coregulators and that WT1 was also reported to act as a coregulator
of p53 (10), it can be assumed
that WT1 binds to p53 and affects p53-induced BAK and
BCL-XL transcription by altering the interaction of
cofactors with p53. p53 can also activate apoptosis by a
non-transcriptional mechanism. It has been reported that p53 can be
translocated to mitochondrial membranes and interact with BAK
(22), BAX (23) or BCL-XL (24), thus enhancing the channel-forming
activities of BAK and BAX. WT1, which was originally characterized
as a nuclear protein, has been shown to shuttle between the cytosol
and the nucleus (25,26). These reports might also indicate
that, in the transcription-independent activation of apoptosis, WT1
binds to p53 or protein complexes containing p53 in the nucleus as
well as the cytosol, and moves to mitochondrial membranes to
potentiate the p53-mediated apoptotic activity. Therefore, to
identify the mechanism responsible for how WT1 potentiates
nutlin-3-induced apoptosis, the mechanisms involved in BAK
and BCL-XL induction as well as BAK activation in
mitochondria by a combination of nutlin-3 treatment and WT1
expression need to be studied further.
Although nutlin-3 can induce cell cycle arrest and
apoptosis, it has been reported to predominantly induce mitotic
arrest in solid tumor cell lines (27). Mitotic arrest can contribute to the
inhibition of cancer growth, but from the view point of
chemotherapeutic efficiency, it diminishes apoptosis and promotes
an acquired resistance to anticancer therapeutics (28). Therefore, it can be expected that
molecules such as the WT1 protein that enhance the apoptosis by
nutlin-3 can be applied to cancer treatment as combined with
nutlin-3. For the development of WT1 as an enhancer of the
anticancer effect of nutlin-3, the mechanism underlying the
potentiation of p53-induced apoptosis by WT1 and whether the effect
of WT1 is universal or cell type- or isoform-specific should be
determined first. Eventually, the molecular characterization of
relationships between WT1 and p53 could contribute to the
establishment of a carcinogenesis model and the development of a
new treatment modality against cancer in the future.
Acknowledgements
The authors thank Dr Sean B. Lee for his critical
comments and suggestions. This research was supported by a grant
(2012R1A5A2047939) and a Basic Science Research Program through the
National Research Foundation of Korea (NRF-2010-0025420).
Abbreviations:
|
HDM2
|
human double minute 2
|
|
HSP70
|
heat shock protein 70
|
|
MTP
|
mitochondrial transmembrane
potential
|
|
NUT3
|
nutlin-3
|
|
PARP1
|
poly(ADP-ribose) polymerase 1
|
|
Tet
|
tetracycline
|
|
WT1
|
Wilms’ tumor gene 1
|
References
|
1
|
Hastie ND: Wilms’ tumour gene and
function. Curr Opin Genet Dev. 3:408–413. 1993.
|
|
2
|
Call KM, Glaser T, Ito CY, et al:
Isolation and characterization of a zinc finger polypeptide gene at
the human chromosome 11 Wilms’ tumor locus. Cell. 60:509–520.
1990.
|
|
3
|
Kreidberg JA, Sariola H, Loring JM, et al:
WT-1 is required for early kidney development. Cell. 74:679–691.
1993. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Herzer U, Crocoll A, Barton D, Howells N
and Englert C: The Wilms tumor suppressor gene wt1 is required for
development of the spleen. Curr Biol. 9:837–840. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Lee SB and Haber DA: Wilms tumor and the
WT1 gene. Exp Cell Res. 264:74–99. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Mayo MW, Wang CY, Drouin SS, et al: WT1
modulates apoptosis by transcriptionally upregulating the bcl-2
proto-oncogene. EMBO J. 18:3990–4003. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Algar EM, Khromykh T, Smith SI, Blackburn
DM, Bryson GJ and Smith PJ: A WT1 antisense oligonucleotide
inhibits proliferation and induces apoptosis in myeloid leukaemia
cell lines. Oncogene. 12:1005–1014. 1996.PubMed/NCBI
|
|
8
|
Menke AL, Shvarts A, Riteco N, van Ham RC,
van der Eb AJ and Jochemsen AG: Wilms’ tumor 1-KTS isoforms induce
p53-independent apoptosis that can be partially rescued by
expression of the epidermal growth factor receptor or the insulin
receptor. Cancer Res. 57:1353–1363. 1997.
|
|
9
|
Morrison DJ, English MA and Licht JD: WT1
induces apoptosis through transcriptional regulation of the
proapoptotic Bcl-2 family member Bak. Cancer Res. 65:8174–8182.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Maheswaran S, Park S, Bernard A, et al:
Physical and functional interaction between WT1 and p53 proteins.
Proc Natl Acad Sci USA. 90:5100–5104. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Maheswaran S, Englert C, Zheng G, et al:
Inhibition of cellular proliferation by the Wilms tumor suppressor
WT1 requires association with the inducible chaperone Hsp70. Genes
Dev. 12:1108–1120. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Maheswaran S, Englert C, Bennett P,
Heinrich G and Haber DA: The WT1 gene product stabilizes p53 and
inhibits p53-mediated apoptosis. Genes Dev. 9:2143–2156. 1995.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Vassilev LT, Vu BT, Graves B, et al: In
vivo activation of the p53 pathway by small-molecule antagonists of
MDM2. Science. 303:844–848. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Shangary S and Wang S: Small-molecule
inhibitors of the MDM2-p53 protein-protein interaction to
reactivate p53 function: a novel approach for cancer therapy. Annu
Rev Pharmacol Toxicol. 49:223–241. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Lee K, Lee MH, Kang YW, Rhee KJ, Kim TU
and Kim YS: Parkin induces apoptotic cell death in TNF-α-treated
cervical cancer cells. BMB Rep. 45:526–531. 2012.PubMed/NCBI
|
|
16
|
Wang Z, Zhang Z, Wu Y, et al: Effects of
echinomycin on endothelin-2 expression and ovulation in immature
rats primed with gonadotropins. Exp Mol Med. 44:615–621. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Lee SY, Shin SJ and Kim HS: ERK1/2
activation mediated by the nutlin-3-induced mitochondrial
translocation of p53. Int J Oncol. 42:1027–1035. 2013.PubMed/NCBI
|
|
18
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2−ΔΔCt method. Methods. 25:402–408. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Haber DA, Sohn RL, Buckler AJ, Pelletier
J, Call KM and Housman DE: Alternative splicing and genomic
structure of the Wilms tumor gene WT1. Proc Natl Acad Sci USA.
88:9618–9622. 1991. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Green DR and Kroemer G: The
pathophysiology of mitochondrial cell death. Science. 305:626–629.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Zhao R, Gish K, Murphy M, et al: Analysis
of p53-regulated gene expression patterns using oligonucleotide
arrays. Genes Dev. 14:981–993. 2000.PubMed/NCBI
|
|
22
|
Leu JI, Dumont P, Hafey M, Murphy ME and
George DL: Mitochondrial p53 activates Bak and causes disruption of
a Bak-Mcl1 complex. Nat Cell Biol. 6:443–450. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Chipuk JE, Kuwana T, Bouchier-Hayes L, et
al: Direct activation of Bax by p53 mediates mitochondrial membrane
permeabilization and apoptosis. Science. 303:1010–1014. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Mihara M, Erster S, Zaika A, et al: p53
has a direct apoptogenic role at the mitochondria. Mol Cell.
11:577–590. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Vajjhala PR, Macmillan E, Gonda T and
Little M: The Wilms’ tumour suppressor protein, WT1, undergoes
CRM1-independent nucleocytoplasmic shuttling. FEBS Lett.
554:143–148. 2003.
|
|
26
|
Niksic M, Slight J, Sanford JR, Caceres JF
and Hastie ND: The Wilms’ tumour protein (WT1) shuttles between
nucleus and cytoplasm and is present in functional polysomes. Hum
Mol Genet. 13:463–471. 2004.
|
|
27
|
Tovar C, Rosinski J, Filipovic Z, et al:
Small-molecule MDM2 antagonists reveal aberrant p53 signaling in
cancer: implications for therapy. Proc Natl Acad Sci USA.
103:1888–1893. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Moreno CS, Matyunina L, Dickerson EB, et
al: Evidence that p53-mediated cell-cycle-arrest inhibits
chemotherapeutic treatment of ovarian carcinomas. PLoS One.
2:e4412007. View Article : Google Scholar : PubMed/NCBI
|